

Abstract
Glioblastoma (GBM) is a type of tumor that is highly lethal despite maximal therapy. Standard therapeutic approaches provide modest improvement in progression-free and overall survival, necessitating the investigation of novel therapies. Oncologic therapy has recently experienced a rapid evolution toward “targeted therapy”, with drugs directed against specific targets which play essential roles in the proliferation, survival, and invasiveness of GBM cells, including numerous molecules involved in signal transduction pathways. Inhibitors of these molecules have already entered or are undergoing clinical trials. However, significant challenges in their development remain because several preclinical and clinical studies present conflicting results. In this article, we will provide an up-to-date review of the current targeted therapies in GBM.
KEYWORDS : Glioblastoma (GBM), targeted therapy, blood-brain barrier (BBB), clinical trial
Introduction
Classification and epidemiology of glioblastoma (GBM)
Gliomas are tumors that arise from glial or precursor cells and include astrocytoma, GBM, oligodendroglioma, ependymoma, mixed glioma, malignant glioma, not otherwise specified (NOS), and a few rare histologies. According to the 2007 WHO classification of central nervous system (CNS) tumors, GBM belongs to tumors of neuroepithelial tissue and could be further subdivided into giant-cell GBM and gliosarcoma. GBM, also called grade IV astrocytoma (where I refers to the least severe and IV to the most severe), is the most common type of primary malignant brain tumor in adults, accounting for 54% of all gliomas. In the United States, the incidence rate is 3.19 per 100,0001,2. Approximately 0.59 to 3.69 GBM cases per 100,000 are diagnosed annually worldwide1,3-7. GBM is also one of the most lethal brain tumors, with only one-third of patients surviving for 1 year and less than 5% living beyond 5 years8,9. GBM patients survive for 12 to 15 months on average despite aggressive surgical resection and conventional therapy10,11.
Morphological features of GBM
Compared with other tumors, GBM possesses unique characteristics associated with its poor prognosis: (I) larger dormant glioma cells with a stronger resistance to conventional radiotherapy and chemotherapy, resulting in multi-drug resistance (MDR); (II) “crab claw-like” invasion, causing unclear borders with normal cerebral structures, preventing complete surgical resection; (III) recurrence within 2 cm of the primary tumor location rather than outside the site, making the elimination of residual glioma cells critical for radical cure and improved prognosis12; (IV) “Chinese chive-like” regenerative proliferation, making it possible that simple surgical excision may stimulate and further accelerate its growth rate and degree of malignancy; and (V) protection by the blood-brain barrier (BBB) (Figure 1) and blood-brain tumor barrier (BBTB), preventing nearly all large-molecule and 98% of small-molecule drugs from entering the CNS13-15. The BBTB starts to form at the later stage of glioma and resides among the brain tumor cells and microvessels16-18, both of which limit the penetration of conventional intravenous or oral drugs into the tumor tissue.
Figure 1.
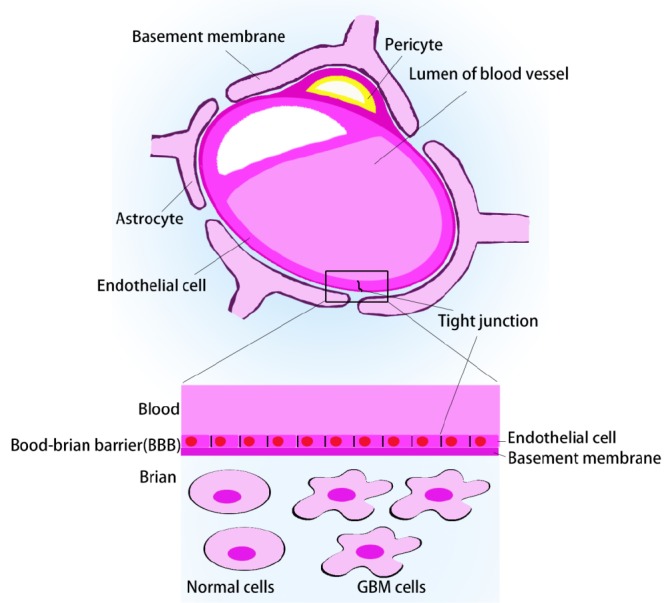
Blood-brain barrier.
According to the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines), combined chemoradiation is as a new standard of care for non-elderly patients with good performance status2. At present, the comprehensive model for the treatment of GBM consists of chemotherapy, surgery, and radiotherapy. The abovementioned morphological characteristics decisively imply the vision of advancing chemotherapeutic administration-targeted drug therapy.
Targeted strategies for GBM
Targeted therapies rationale
In targeted therapy, drugs accumulate selectively in targeted tissues, organs, cells, or intracellular structures via local drug delivery or the systemic blood circulation. These drugs could be sorted into passive and active targeting based on the delivery mechanism as follows. (I) Passive targeting: drug-loaded particles of different sizes are intercepted by different tissues because of their distinct physiological properties, such as the reticuloendothelial system (RES) in the liver and spleen or enhanced permeability and retention (EPR) effects in tumors. (II) Active targeting: drugs or carriers modified by special ligands, monoclonal antibodies, or macromolecule substances that are sensitive to certain chemicals in vivo, which act as “bullets”, are delivered directly and accumulate in target regions. Passive target-oriented microspheres could evade macrophage uptake and further reach the specific target sites after the modification of ligands, monoclonal antibodies, or other substances.
Considering the specific sites and features of GBM, a passive targeting strategy alone, such as the use or evasion of RES and the utilization of EPR, is insufficient for drug delivery to the tumor. Therefore, the following treatments emphasize active targeting or a combination of the two strategies.
Receptor-mediated targeting
Certain receptors closely correlated with tumor growth are highly expressed in BBB, GBM cells, or associated blood vessels, but not in surrounding normal tissues. Given this phenomenon, surface-functionalized or conjugated traditional drugs or drug-loaded carriers with corresponding ligands are expected to guide these receptors to cross the BBB to improve therapeutic efficacy and reduce side effects.
Transferrin receptor (TfR)
The cellular uptake of iron-loaded transferrin (Tf) occurs through receptor-mediated transcytosis (RMT). TfR, referred to as TfR1 or CD71 in literature, is expressed at low levels in most human tissues but is highly expressed on the brain capillary endothelium, which forms the BBB, and in tumor tissue. Thus, TfR functions both in mediating transport across the BBB and internalization into cancer cells. Meanwhile, TfR2, another member of the TfR family, presents considerably lower affinity for Tf than TfR1 (25-fold lower), making it a less efficient target for TfR-mediated drug or gene delivery to brain or cancer cells19,20.
Zhang et al.21 prepared Tf-modified paclitaxel-loaded micelles (TRPM), wherein Tf modification significantly enhanced cellular uptake by primary brain microvascular endothelial cells to 2.4 fold that of unmodified samples, resulting in high drug accumulation in the brain after intravenous injection. Mice bearing intracranial U-87 MG glioma treated with TRPM exhibited the longest mean survival time (42.8 days). Ying et al.22 developed liposomes conjugated with Tf. Their group found that its transport ratio across the BBB model was significantly increased up to 24.9% and that the C6 glioma spheroid volume ratio was significantly lowered to 54.7%. The inhibitory rate to C6 glioma cells after crossing the BBB was significantly enhanced up to 64.0%, and the median survival time of tumor-bearing rats after administration (22 days) was significantly longer than those of other controls. Several other studies23,24 also showed that liposomes or nanoparticles (NPs) conjugated with Tfs can target endothelial and tumor cells, penetrating tumor cells to reach the core of tumor spheroids and providing the highest brain distribution. As a result, drug-loaded formulations present the best anti-proliferative activity against GBM cells and tumor spheroids. Chiu et al.25 showed that NPs conjugated to oxalate Tf, a variant of Tf, exhibited a higher degree of cellular association compared with native Tf-conjugated NPs, because oxalate can stabilize the iron atoms in Tf, thereby decreasing Tf iron release rate in the endosome26. Accordingly, conjugates of these Tf mutants with the diphtheria toxin possessed even greater potency in cytotoxicity experiments in vitro with GBM cell lines. Moreover, intratumoral injections into xenografted glioma tumors in a mouse model resulted in near-complete tumor regression within 8 days27. Therefore, using Tf variant-based therapeutics has a potential in systemic drug delivery applications for GBM treatment.
Low-density lipoprotein (LDL) receptor
Low-density lipoprotein receptor-related proteins (LRPs), which are structurally similar to the LDL receptors, belong to the LDL receptor family. LRPs are multifunctional RMT systems with multiple ligands, such as lactoferrin, melanotransferrin, and receptor-associated protein. Moreover, LRPs are overexpressed in BBB and glioma cells. Therefore, several BBB or glioma-targeting vectors which take advantage of the LRP RMT system have been reported19,28-31.
Angiopep-2, which is derived from the Kunitz domain of aprotinin, exhibits high LRP1 binding efficiency and brain penetration capability in both the in vitro model of BBB and in situ brain perfusion in mice; several research groups used Angiopep-2 for glioma-targeting delivery31-37. Jiang et al.38,39 developed NPs and carbon nanotubes functionalized with Angiopep-2, both of which displayed higher glioma localization and penetration. The most favorable antiglioma effects both in vitro and in vivo were observed after loading with drugs. Xin et al.31 prepared paclitaxel-loaded Angiopep-NPs that exhibit a significantly higher amount of endocytosis and enhanced inhibitory effects to U87 MG cells with significantly increased transport ratios across the BBB model. The group also observed enhanced accumulation of Angiopep-NPs in the glioma bed and infiltrating margin of an intracranial U87 MG glioma tumor-bearing in vivo model. Several other studies utilized Angiopep-2 to modify the delivery system, including NPs40, gold NPs41, electro-responsive hydrogel NPs42, p-coumaric acid43, or pluronic F127-conjugated superparamagnetic iron oxide NPs44, and all exerted similar findings without exception. A study applied Angiopep-2 to GBM stem cell (GSC) in which the vector was also overexpressed and, as expected, Angiopep-2 improved anti-GSC properties, such as enhanced stability, anti-proliferation, and antitumor sphere formation abilities45. Demeule et al.46 synthesized ANG4043 by chemically conjugating the anti-HER2 mAb with Angiopep-2. Expectedly, increased BBB permeability was observed compared with unconjugated anti-HER2 mAb. Given the susceptibility to proteolysis of Angiopep-2 (here termed LAngiopep), Wei et al.47 designed a retro-inverso isomer of LAngiopep, termed DAngiopep. The latter demonstrated lower uptake efficiency in both bEnd.3 and U87 cells, suggesting lower binding affinity to LRP-1 of the Dpeptide. Moreover, DAngiopep was resistant to proteolysis in fresh rat blood serum, whereas more than 85% of LAngiopep disappeared within 2 h. This result indicates that the susceptibility to proteolysis of LAngiopep in BBB may further attenuate transcytosis efficiency. In consequence, in vivo DAngiopep-modified micelles displayed high distribution in intracranial GBM. Therefore, the proteolytically stable DAngiopep holds considerable potential in designing two-order brain tumor targeted delivery systems.
GBM initiating cell (GIC) targeting
Evidently, this intrinsic resistance of GBM to current treatments is caused by a cell subpopulation with high resistance to radiation and chemotherapy. GICs or GSCs are responsible for tumor reinitiation and sustained growth, and are conceptualized as cancer’s locomotive engine48-52, making GSCs an attractive therapeutic target for GBM.
Bone morphogenetic proteins (BMP)
BMP belong to the transforming growth factor-β (TGF-β) superfamily of cytokines. This family of proteins was originally identified to induce bone and cartilage formation in ectopic skeletal sites in vivo. BMP ligands exert their activities by means of serine-threonine kinase receptors. The activation of the BMP pathway reduces glioma cell proliferation and renders GICs more susceptible to conventional therapy, so BMP treatment is considered to be a promising therapeutic tool against GBM53,54. Several studies have shown that BMPs can arrest cell cycle in GBM cells55 and suppress the tumorigenic capacity of GICs by inducing their differentiation to phenotypes with lower levels of stem cell markers51,56,57. Among all BMP ligands tested, BMP4 elicited the strongest effect, which could effectively inhibit not only GIC proliferation and self-renewal in vitro, but also tumor growth in vivo53. Mice that were intracranially injected with untreated glioma cells died after 3 to 4 months, but nearly all mice injected with BMP4-treated cells survived until the end of the experiment58. Univariate analysis showed that low BMP4 levels were correlated with high tumor grade. The Kaplan-Meier analysis indicated that patients with high BMP4 expression showed significantly better prognosis, highlighting the relevance of BMP4 as a predictor of survival59. Liu et al.60 reported that BMP4 can even reverse the MDR phenotype of tumor cells. BMP4 treatment has also been combined with bevacizumab (BEV) in GBM mouse models, and results showed that BMP4 exerted an independently favorable effect on GBM that was not synergistic with BEV treatment61. Chirasani et al.57 disclosed that endogenous neural stem cells secreted BMP-7, which acts as a paracrine suppressor of GICs. Animal experiments also showed that these cells would migrate to the borders of neoplastic foci to suppress GBM formation. Moreover, Tate et al.62 demonstrated that a BMP7 variant (BMP7v) decreased primary human GIC proliferation, angiogenesis, and stem cell marker expression while enhancing neuronal and astrocyte differentiation marker expression in vitro and in vivo. In addition, BMP7v reduced brain invasion, angiogenesis, and the associated mortality in an orthotopic glioma model. However, not all BMP levels were positively associated with a better clinical outcome of glioma patients. According to a report, BMP2 expression became significantly higher as the glioma’s grade advanced and the Karnofsky Performance Status score decreased63. Persano et al.64 reported that, besides being an effective prodifferentiation treatment for GBM-derived stem cells, BMP2 also sensitized GICs to temozolomide (TMZ) treatment by decreasing hypoxia-inducible factor 1 alpha (HIF1α) stability and consequently down-regulating O-6-methylguanine-DNA methyltransferase (MGMT), an HIF1α target, thereby promoting TMZ’s alkylating action.
CD133
The Pentaspan transmembrane glycoprotein family member CD133, also known as prominin-1 (PROM-1), is the best-validated marker of the cell subpopulation responsible for conferring stem cell properties to GBMs. CD133 as a marker of GSCs is also an attractive target for the delivery of targeting therapeutics. Shin and colleagues65 prepared CD133 antibody-conjugated immune liposomes that encapsulated gemcitabine for targeting GSCs. The in vitro cytotoxicity of gemcitabine was significantly enhanced through the endocytosis of CD133 overexpressed on GSCs. The anti-tumor effect was 15 times higher than that of free gemcitabine, thus presumably reflecting the specific targeting of the CD133 surface marker.
Telomere repeat-binding factor 2 (TRF2)
GSCs express high amounts of repressor element 1 silencing transcription (REST) factor, which may contribute to their resistance to standard therapies. Meanwhile, TRF2 stabilizes telomeres and REST to maintain the self-renewal of neural stem cells and tumor cells. Bai and coworkers66 showed that viral vector-mediated delivery of shRNAs targeting TRF2 mRNA depleted TRF2 and REST from GSCs isolated from patient specimens. As a result, GSC proliferation was reduced, and the level of proteins normally expressed by post-mitotic neurons (L1CAM and β3-tubulin) was increased. Depletion of TRF2 also sensitized GSCs to TMZ and increased the survival of mice bearing GSC xenografts. These findings reveal a role of TRF2 in the maintenance of REST-associated proliferation and the chemotherapy resistance of GSCs, suggesting that TRF2 was a potential therapeutic target for GBM.
miR-125b
Several studies67,68 have shown that miR-125b is necessary for GSC’s fission and insensitivity to chemotherapy. Chen et al.69 explored the functions and mechanisms of miR-125b action on TMZ-treated GSCs. The group found that miR-125b was up-regulated in TMZ-resistant cells, the inhibition of which caused a marked increase in TMZ-induced cytotoxicity and apoptosis, as well as a subsequent decrease in the resistance to TMZ in GSCs. Moreover, their study demonstrated that the pro-apoptotic Bcl-2 antagonist killer 1 (Bak1) was a direct target of miR-125b. In other words, miR-125b conferred TMZ resistance by targeting Bak1 expression69.
Angiogenesis targeting
Angiogenesis is known as the rate-determining process for solid tumor growth, which is also one of the main features of tumor tissues. GBM is among the most angiogenic of malignancies70. Thus, angiogenesis has emerged as a primary target of drug development for GBM over recent decades. Tumor angiogenesis is involved in many stimulating (VEGF, EGF, PDGF, etc.) and inhibiting factors. Thus, numerous promising strategies exist for targeting GBM therapy, such as down-regulating the expression of stimulating factors.
Integrins
Integrins are a family of cell-cell and cell-extracellular matrix adhesion molecules that are implicated in various cellular processes (e.g., survival, proliferation, migration, invasion, and angiogenesis) and could thus support tumor development. In particular, αvβ3 and αvβ5 integrins are speculated to be key mediators of crosstalk between tumor cells and the brain microenvironment in GBM and are overexpressed on glioma cells and the vasculature. Therefore, targeting integrins and tumor microenvironment are considered to be a promising therapeutic strategy in GBM71-74.
Arg-Gly-Asp (RGD)
Arg-Gly-Asp (RGD) is a peptide that was is widely used for neovasculature targeting delivery because of its high binding efficiency with αvβ374-77. Notably, the binding affinity of the cyclic RGD peptide [c(RGDfK)] for integrin αvβ3 is reported to be 1,000 times greater than that of the linear RGD peptide75,76. Therefore, Liu et al.76 conjugated [c(RGDfK)] to a cell-penetrating peptide R8 to develop the multifunctional peptide R8-RGD, which increased the cellular uptake of liposomes by two folds in comparison with separate R8. Liu et al.76 also displayed the effective penetration of 3D glioma spheroids and the BBB model in vitro and the glioma foci after systemic administration in C6 glioma-bearing mice. Similarly, Kibria et al.75 also selected R8/RGD to embellish PEGylated liposomes, and their results showed an enhanced cellular uptake and higher transfection efficiency in integrin αvβ3-expressing cells in comparison with versions of the single ligand. Zhang et al.21 utilized [c(RGDfK)] to modify paclitaxel-loaded micelles to target integrins overexpressed in glioma cells, which showed significantly prolonged retention in glioma tumors and peritumoral tissue.
Cilengitide
Cilengitide, an RGD peptide mimetic and a selective inhibitor of αvβ3 and αvβ5, has been tested in phase I/II trials in GBM patients74. In several other phase I/II studies in patients with recurrent or newly diagnosed GBM, cilengitide alone or in combination with TMZ chemoradiotherapy was well tolerated and showed potential antitumor activity (particularly in tumors with a methylated MGMT promoter)78-80. Eisele et al.81 analyzed the patterns of progression on MRI in 21 newly diagnosed GBM patients in a phase II trial of cilengitide added to TMZ chemoradiotherapy. Their group found that adding cilengitide did not alter patterns of progression; that is, cilengitide may not induce a more aggressive phenotype at progression, nor provide anti-invasive activity in patients with newly diagnosed GBM. Therefore, in a recent randomized phase III trial, Stupp et al.71 assessed cilengitide combined with standard treatment in a subgroup of patients with GBM with a methylated MGMT promoter. The group found that the median overall survival (OS) was 26.3 months in the cilengitide group and 26.3 months in the control group (P=0.86). Given this result, the addition of cilengitide to TMZ chemoradiotherapy did not improve outcomes, and cilengitide should not be further developed as an anticancer drug.
Epidermal growth factor receptor (EGFR)
EGFR, also known as HER1 or ErbB1, belongs to the ErbB family of receptor tyrosine kinases (RTKs). Ligand binding by EGF leads to in the activation of the RTK/RAS/PI3K pathway, resulting in cellular proliferation, angiogenesis, and increased local tissue invasion, as well as resistance to apoptosis82-84. EGFR is amplified in 40% to 50% of GBMs. Gain-of-function EGFRvIII mutations (EGFR variant III) in nearly half of GBMs bear amplified EGFR. EGFRvIII arises from a genomic deletion of exons 2 to 7, which encode the ligand-binding domain of the receptor, generating constitutively active oncogenic RTKs. Moreover, the signaling mechanism of EGFRvIII cells can confer resistance to EGFR inhibitors (such as erlotinib and gefitinib) and promote poor long-term survival. Recent interest has focused on an anti-EGFRVIII vaccine (known as rindopepimut), which has already entered or is undergoing clinical trials82,85-87.
Inhibitors
Preclinical results have demonstrated the ability of tyrosine kinase inhibitors (TKIs) to inhibit tumor cell growth, angiogenesis, survival, and proliferation in several different EGFR-transfected GBM cell lines. However, these results do not appear to be clinically translatable, as response rates in GBM patients for numerous inhibitors, including gefitinib and erlotinib, are poor82. Qaddoumi et al.88 conducted a new phase II trial of erlotinib and local radiotherapy in children with newly diagnosed GBM (20 patients). The 2-year progression-free survival (PFS) for patients was 19%±8%, and only five patients remained alive without tumor progression. This result indicates that erlotinib did not change the poor outcome of children with GBM. Another phase II trial reported that the combination of radiation, TMZ, erlotinib, and BEV for the initial treatment of GBM appeared to improve PFS but did not reach the primary endpoint of improved OS89. Another phase II study of erlotinib and sorafenib for patients with progressive or recurrent GBM also did not meet its objective of a 30% increase in OS time compared with historical controls90. In the case of gefitinib, in one phase II evaluation in nearly 100 patients with newly diagnosed GBM91, the OS and PFS at 1 year post-RT with gefitinib were not significantly different compared with those of the historical control population. Thus, treatment with adjuvant gefitinib post-RT was not associated with significant improvement in OS or PFS.
Monoclonal antibodies
Despite the success of antibody-based therapy in the treatment of many other cancers, these results have not been replicated in GBM82. A phase II study in 2009 stratified patients depending on their EGFR gene amplification status, and both groups were administered with cetuximab intravenously. Cetuximab exerted little effect in both groups, and the median OS was 5 months, eliciting no significant correlation between EGFR status and response or OS92. Another similar phase II clinical study in 2012 found that patients with an EGFR amplification lacking EGFRvIII expression presented a significantly superior PFS and a numerical OS following treatment with cetuximab [median PFS, 3.03 vs. 1.63 months (P=0.006); median OS, 5.57 vs. 3.97 months (P=0.12)]. Within the subgroup of patients with EGFR amplification, patients with EGFRvIII-positive GBM showed worse survival [median PFS, 1.63 vs. 3.03 months (P=0.01); median OS, 3.27 vs. 5.57 months (P=0.08)], indicating that the type of EGFR mutation may determine the outcome of GBM patients treated with cetuximab93. In Chinese patients, a study of nimotuzumab in combination with TMZ and radiotherapy for newly diagnosed GBM showed that the survival times was similar to that observed in historical data of standard therapy; that is, no correlation between efficacy and EGFR expression was found94. The newest95 phase III trial involving nimotuzumab in the treatment of newly diagnosed adult GBM also showed that EGFR amplification is not correlated with clinical efficacy of nimotuzumab. This study, albeit negative, contained hypothesis-generating signals which support the evaluation of correlative, efficacy-predicting tumor parameters for nimotuzumab in GBM treatment.
Peptide vaccines
Rindopepimut (CDX-110) is a peptide vaccine composed of a 14-mer peptide spanning the EGFRvIII-specific exon junction site conjugated to the carrier protein KLH.
Results from previous trials, namely, ACTIVATE, ACT II, and ACTIII, confirmed the safety of rindopepimut with robust EGFRvIII-specific immune responses and demonstrated a statistical increase in median PFS and OS in vaccinated patients in comparison with a cohort treated with the care standard85,96-98. Schuster et al.87 performed a phase II clinical trial (ACT III) to confirm the results mentioned above. In their study, PFS at 5.5 months (~8.5 months from diagnosis) was 66%. Relative to study entry, median OS was 21.8 months, and 36-month OS was 26%. Anti-EGFRvIII antibody titers increased ≥4 folds in 85% of patients and increased with the duration of treatment. EGFRvIII was eliminated in 4 out of 6 (67%) tumor samples obtained after >3 months of therapy. A pivotal, double-blind, randomized, phase III trial (“ACT IV”) is underway.
Platelet-derived growth factor receptor (PDGFR)
The PDGFR family constitutes the subfamily III of RTK and is formed by PDGFR-α and PDGFR-β. PDGFRs are related to cell migration, proliferation, and survival processes83. Interestingly, both PDGFRs and their ligands are co-expressed in GBM, suggesting that stimulation of autocrine PDGFRs may contribute to their growth99. Amplifications of PDGFR-α have been extensively studied in GBM. These receptors are reported to be associated with a loss of p53 function and the secondary GBMs that typically develop from low-grade astrocytoma100,101. A gene expression-based GBM molecular classification has further linked PDGFR-α aberrations in patients to the proneural subclass. This GBM subclass was identified to be nonresponsive to standard TMZ and radiotherapy. Other studies have recently found PDGFR-α mutations in a fraction of diffuse intrinsic pontine glioma, which is a pediatric brain tumor with an extremely poor prognosis101.
At present, no PDGFR-targeting agent has been approved for GBM treatment. Studies in vitro showed that imatinib could inhibit GBM cell proliferation and induce growth arrest in the G0/G1 phase of the cell cycle99, or that imatinib could reach intratumoral concentrations similar to or higher than those in plasma in GBM regions where the BBB is disrupted as indicated in contrast-enhanced MRI102. Long-term exposure to imatinib could reduce the ability of cancer stem cell through the induction of cell differentiation in GBM cells103, and all of these strategies may indicate potential in clinical applications. However, previous clinical studies using imatinib mesylate (Gleevec®) for GBM patients showed no major inhibition of tumor growth or extension of survival104. Several multicenter trials also failed to show the efficacy of imatinib alone or in combination with hydroxyurea in the treatment of recurrent GBM105,106. The molecular mechanisms of action of imatinib in GBM cells remain poorly understood. Dong et al.104 investigated the effects of imatinib on PDGFR downstream signaling pathways, as well as on other cellular functions in human GBM cells. The research group found that imatinib significantly inhibited cell migration but not cell growth. The combination of imatinib and a MEK or PI3K inhibitor resulted in significant growth inhibition but did not inhibit cell migration beyond the inhibition achieved with imatinib treatment alone. This finding indicates that the imatinib treatment of malignant glioma does not result in significant inhibitory effects and should be used with caution.
VEGF/VEGFR
Hypoxia in GBM led to HIF-1α accumulation and further activation of several hypoxia-associated genes, including VEGF. The VEGF gene family includes six secreted glycoproteins [VEGF-A, VEGF-B, VEGF-C, VEGF-D, VEGF-E, and placenta growth factor (PlGF)]. Among these glycoproteins, VEGF-A typically localizes adjacent to perinecrotic regions within glioma pseudopalisades, increases with higher glioma grade, and is associated with poor outcome among patients with GBM. The VEGF receptor (VEGFR) family includes VEGFR-1 (Flt-1), VEGFR-2 (KDR), VEGFR-3, neuropilin-1 (NRP-1), and NRP-2, which exhibit different binding affinities of VEGF homologs. Among these receptors, VEGFR-1 and VEGFR-2 regulate angiogenesis and NRP function as VEGFR tyrosine kinase co-receptors. VEGF binding to VEGFRs on tumor blood vessels markedly enhances permeability and activates endothelial cell proliferation, survival, and migration. Moreover, GBM also expresses VEGFRs, which may function in an autocrine manner to promote tumor growth107.
Inhibitors sunitinib
A phase II trial108 examined the activity of sunitinib in 12 patients with newly diagnosed, non-resectable GBM. Results showed that sunitinib had no activity as a monotherapy, and further investigation of its efficacy in this setting is unwarranted. Another trial by Hutterer et al.109 also found that continuous daily sunitinib showed minimal anti-GBM activity and substantial toxicity when given at higher doses. These are the two latest reports about trials of sunitinib in recurrent GBM to date, and both were consistent with previous trials110-112 showing that sunitinib has no significant anti-tumor efficacy alone or in combination with others in newly diagnosed GBM.
Cediranib
A phase II study113 of cediranib in patients with recurrent GBM showed that cediranib monotherapy was associated with encouraging proportions of radiographic response and 6-month PFS. Gerstner et al.114 evaluated the effects of cediranib in combination with chemoradiation on tumor blood flow and survival in newly diagnosed GBM. Improved PFS and OS compared with historical controls, particularly in those with improved perfusion were observed. This finding was confirmed by further results of improved tumor oxygenation and survival in GBM patients who showed increased blood perfusion after cediranib and chemoradiation115. Batchelor et al.116 performed a phase III trial. However, the results of the group did not meet the primary end point of PFS prolongation with cediranib either as a monotherapy or in combination with lomustine versus lomustine in patients with recurrent GBM, even though cediranib showed evidence of clinical activity on several secondary end points, including time to deterioration in neurologic status and corticosteroid-sparing effects. Similarly, a phase I study of cediranib in combination with cilengitide in patients with recurrent GBM showed that the median PFS/OS and APF6 were not very promising, notwithstanding the concluding suggestion of a low incidence of pseudoprogression in newly diagnosed GBM patients treated with cediranib in combination with chemoradiation117.
Axitinib
Axitinib is a novel orally available VEGFR-TKI. Kratzsch et al.118 conducted a study with immunodeficient mice. The group established cell line- and patient-derived GBM xenografts, which were treated with axitinib. They verified that axitinib exhibited significant effects on GBM xenografts even with primary resistance to BEV in a so far untreated tumor. Another preclinical study119 showed for the first time the antiangiogenic effect and survival prolongation provided by systemic single-agent treatment with axitinib in preclinical orthotopic GBM models, including clinically relevant GSC models. In the newest phase II study of axitinib vs. standard care performed by Duerinck and coworkers120, axitinib had single-agent activity in recurrent GBM patients. The survival of the axitinib group was comparable with that of the contemporary control arm. Tumor response on MRI is accompanied by decreased uptake of tracers on 18F-FET PET scan. Further evaluation of axitinib for recurrent GBM is warranted.
Monoclonal antibody
BEV is a recombinant humanized monoclonal antibody that could selectively bind to and neutralize the activity of VEGF-A, thereby inhibiting binding to VEGFR. BEV received accelerated FDA approval for the treatment of progressive GBM based on radiographic response rates121. Goldlust et al.121 reported that their radiographic and survival outcomes with BEV following progression after VEGFR-TKIs are similar to the data from studies of BEV as initial salvage therapy. Prior exposure to VEGFR-TKIs may not preclude response to BEV, but sensitivity to BEV may be lower following more robust VEGFR inhibition121. In a prestigious report on a randomized trial of BEV for newly diagnosed GBM, first-line use of BEV did not improve OS in patients (median, 15.7 vs. 16.1 months, BEV vs. placebo). PFS was prolonged (10.7 vs. 7.3 months) but did not reach the prespecified improvement target122. Taal et al.123 reported the results of the first phase II trial (BELOB trial) in which single-agent BEV did not support a significant role in recurrent GBM, whereas the combination of lomustine and BEV may have more activity than either drug administered alone124. This combination warrants further investigation and is currently being investigated in randomized controlled phase III EORTC trial 26101125. Nevertheless, the addition of BEV to TMZ and hypo-IMRT, or the combination of BEV/Irinotecan (IRI) did not improve OS for patients with GBM126-128. Similarly, a phase II study showed that the addition of carboplatin and IRI to BEV does not improve anti-tumor activity compared to that achieved historically with single-agent BEV among BEV-naive, recurrent GBM patients129. In another single-institution phase II trial130, the combination of BEV, erlotinib, TMZ, and radiotherapy appeared to improve PFS but did not reach the primary endpoint of improved OS. These data suggest that chemosynergy with BEV may be insufficient to enhance the benefit of BEV in recurrent GBM.
Biomarker targeting
The National Institutes of Health defines biomarkers as “characteristics that are objectively measured and evaluated as indicators of normal biologic processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention”. Biomarkers have the potential to play significant roles in the diagnosis of tumor subtypes, as well as in the identification of therapeutic targets of GBM131. A marker can consist of alterations of the genome, epigenome, or transcriptome, proteome, and aberrant microRNAs (miRNAs). Genes that are most closely associated with GBM, such as EGFR, VEGFR, and PDGFR, are previously introduced.
Herein, we emphasize the miRNAs involved in the initiation and progression of GBM. MiRNAs are a class of short non-coding RNA sequences (18 to 24 nucleotides) that repress gene expression by interacting with the 3' untranslated regions of mRNAs132. MiRNAs are predicted to target more than 50% of human protein-coding genes, enabling them to perform numerous regulated roles in physiological and developmental processes133. MiRNA-targeted therapy is still in the initial stage, and clinical trials are under recruitment or currently running. However, several miRNAs have been selected as promising tumor biomarkers, with increased potential to reduce disease progression in combination with conventional first-line therapy for GBMs.
MiR-21
Altered miRNA expression in GBMs was first reported in 2005. Chan et al.134 showed that miR-21 was highly up-regulated and exhibited anti-apoptotic capabilities in GBM cell lines. Subsequently, several reports confirmed miR-21 up-regulation in GBMs. MiR-21 functions as an oncogene in the pathogenesis of GBM, and its expression is correlated with glioma grade135-137. In addition, miR-21 and its target genes mediate radiation resistance of GBM cells138-140. Therefore, miR-21-targeted therapy is a promising alternative for GBM. Ren et al.141 showed that miR-21 inhibitors in combination with 5-FU increased glioma cell apoptosis and decreased cancer cell migration. Qian et al.142 co-delivered doxorubicin and miR-21 inhibitor (miR-21i) into glioma cells, which surprisingly exhibited an anti-proliferative efficiency. A new study demonstrated that decreased tumor cell proliferation and tumor size, as well as enhanced apoptosis activation and, to a lesser extent, improvement of animal survival, were also observed in GBM-bearing mice upon systemic delivery of targeted NP-formulated anti-miR-21 oligonucleotides and exposure to sunitinib143.
MiR-181
MiR-181 family contains a, b, c, and d isoforms. The down-regulated hsa-miR-181a and hsa-miR-181b of hsa-miR-181 family were also involved in glioma oncogenesis144. MiR-181d was also down-regulated in human glioma samples and may act as a glioma suppressor by targeting K-ras and Bcl-2Jeny145. MiR-181b and miR-181d were predictive biomarkers for TMZ response, with the former possibly enhancing TMZ sensitivity via MEK1 down-regulation and the latter partly by post-transcriptional regulation of MGMT. Therefore, a combination of miR-181b or miR-181d with TMZ may be an effective therapeutic strategy for gliomas146,147. Further studies on miR-181 are still necessary to demonstrate a therapeutic benefit in a clinical context toward GBM targeting treatment.
Conclusion and future perspectives
As chemotherapy for GBM has provided only a modest benefit in clinical outcome, the need for strategies with improved efficacy has prompted the development of current targeted therapies. However, drug delivery to the brain is hindered by the presence of the BBB. RMT is one of the transport systems used for nutrient transport to the brain for its healthy function. Thus, if appropriately targeted, RMT systems could help clinicians shuttle therapeutics into the brain in a noninvasive manner. At present, the most well-developed receptors known to undergo RMT are probably LfR and LRP. NPs or liposomes are always used as carriers to deliver corresponding ligands, antibodies, or peptide vaccines. In either case, the conjugated cargo gains access to the brain interstitium by “piggybacking” on the natural RMT system19. Notably, both LfR and LRP are overexpressed in the BBB, as well as in GBM cells, enabling the two to traverse the BBB and reach the secondary target. Therefore, achieving sequential targeting is feasible by endowing those RMTs, only expressed on either BBB or GBM cells, with an additional targeting moiety, such as RGD. The first targeting agent would allow RMT across the BBB, and the second agent would discriminate the site of action within the CNS19. Though the first agent was either LfR or LRP, we could also add another targeting moiety to improve penetration into the BBB or tumors, such as ACP, RGD, p-aminophenyl-α-D-mannopyranoside, and TLyP-1148. We could also change the structures of ligands to obtain their variant or isomer with better performance via physicochemical methods, and further accomplish a higher degree of cellular association or increased distribution in intracranial GBM, such as [c(RGDfK)], oxalate Tf, and DAngiopep. To overcome the uptake in the RES of liver or spleen, we can coat NPs or liposomes with PEG.
Another important concern associated with GBM’s poor prognosis may be recurrence, which is possibly due to the failure to eradicate GICs. Targeting GICs initiates new potential clinical therapies and interventions. TGF-β signaling could be a potential target because it has been shown to act as an oncogenic factor in GBM and could enhance the self-renewal capacity of tumor-derived spheroids in vitro149. Like BMPs, members of the TGF-β superfamily that could block proliferation and increase GIC responsiveness to chemotherapy and low BMP levels are prognostic for poor survival in human glioma, and have been proposed as promising therapeutics. The quinoline derivative LY2109761 is a TGF-β receptor I kinase inhibitor that has been found to be active against GBM alone and to enhance the antitumor efficacy of radiation both in vitro and in vivo, particularly in GICs150. Moreover, GSCs are driven by overactive signaling pathways, such as PI3K/AKT/mTOR and RAS/RAF/MAPK. Evidence has been provided that sorafenib, a member of TKIs, exhibited a selective cytotoxic effect on GSCs that is partly dependent on the inhibition of the PI3K/Akt and MAPK pathways involved in gliomagenesis151,152. The most advantageous result is the emergence of stem cell-mediated delivery, which yielded promising preclinical results. A human clinical trial utilizing this approach is currently underway, considering incomplete distribution within the entirety of GBM of small molecule inhibitors or carriers like NPs. Therapeutic agents that have been delivered to GBMs by GIC carriers include therapeutic genes, oncolytic viruses, NPs, and antibodies (for readers interested in further discussions or opinions in this area, a review has recently been published153).
GBM is also characterized by high expression levels of proangiogenic cytokines and microvascular proliferation, highlighting the potential value of treatments targeting angiogenesis. Antiangiogenic treatment likely achieves a beneficial impact through multiple mechanisms of action. However, alternative proangiogenic signal transduction pathways are activated, leading to resistance development, even in tumors that initially respond. Identifying biomarkers or imaging parameters to predict the response and herald resistance is of high priority. Despite promising phase I/II clinical trial results, adding cilengitide to TMZ chemoradiotherapy did not improve outcomes. Similarly, many other inhibitors or monoclonal antibodies tended to exert unsuccessful results, as further clinical trials are underway in newly diagnosed or recurrent GBM. Simultaneously, interesting findings were also noted, such as patients who experienced gefitinib-associated adverse effects (rash/diarrhea) exhibit improved OS. Another interesting finding is the high endothelial c-Kit expression, which may define a subgroup of patients who will benefit from sunitinib treatment by achieving prolonged PFS. Even though gefitinib reached high concentrations in tumor tissue and efficiently dephosphorylated its target, the regulation of downstream signal transducers in the EGFR pathway seemed to be dominated by regulatory circuits that are independent of EGFR phosphorylation. Therefore, future studies are still warranted. Except for biomarkers associated with angiogenesis, recent reports support the potential of miRNAs as predictive biomarkers and therapeutic targets for GBMs, despite awaiting further studies, which may allow for appropriate patient enrichment. Interdisciplinary efforts seem to be required in studying the combination among RMT approaches, GSC inhibitors, antiangiogenic treatments, biomarker targeting therapies, and cytotoxic agents, which may ultimately prove successful in improving OS and convert the “undergoing clinical trials” to “FDA-approved” therapeutics for noninvasive drug delivery to GBM.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 81472841), the basic project of the Science and Technology Commission of Shanghai Municipality (Grant No. 14JC1492500), and the medical guide project of the Science and Technology Commission of Shanghai Municipality (Grant No. 134119a1300).
Footnotes
No potential conflicts of interest are disclosed.
References
- 1.Ostrom QT, Gittleman H, Liao P, Rouse C, Chen Y, Dowling J, et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2007–2011. Neuro Oncol 2014;16:iv1-iv63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 2007;114:97-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ohgaki H, Kleihues P. Population-based studies on incidence, survival rates, and genetic alterations in astrocytic and oligodendroglial gliomas. J Neuropathol Exp Neurol 2005;64:479-489. [DOI] [PubMed] [Google Scholar]
- 4.Arora RS, Alston RD, Eden TO, Estlin EJ, Moran A, Birch JM. Age–incidence patterns of primary CNS tumors in children, adolescents, and adults in England. Neuro Oncol 2009;11:403-413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee CH, Jung KW, Yoo H, Park S, Lee SH. Epidemiology of primary brain and central nervous system tumors in Korea. J Korean Neurosurg Soc 2010;48:145-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dobes M, Khurana VG, Shadbolt B, Jain S, Smith SF, Smee R, et al. Increasing incidence of glioblastoma multiforme and meningioma, and decreasing incidence of Schwannoma (2000–2008): findings of a multicenter Australian study. Surg Neurol Int 2011;2:176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gigineishvili D, Shengelia N, Shalashvili G, Rohrmann S, Tsiskaridze A, Shakarishvili R. Primary brain tumour epidemiology in Georgia: first-year results of a population-based study. J Neurooncol 2013;112:241-246. [DOI] [PubMed] [Google Scholar]
- 8.Dunn GP, Rinne ML, Wykosky J, Genovese G, Quayle SN, Dunn IF, et al. Emerging insights into the molecular and cellular basis of glioblastoma. Genes Dev 2012;26:756-784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Network NCC. NCCN Clinical Practice Guidelines in Oncology: Central Nervous System Cancers. Version 2. 2014.
- 10.Stupp R, Mason WP, Van Den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005;352:987-996. [DOI] [PubMed] [Google Scholar]
- 11.Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med 2008;359:492-507. [DOI] [PubMed] [Google Scholar]
- 12.Wallner KE, Galicich JH, Krol G, Arbit E, Malkin MG. Patterns of failure following treatment for glioblastoma multiforme and anaplastic astrocytoma. Int J Radiat Oncol Biol Phys 1989;16:1405-1409. [DOI] [PubMed] [Google Scholar]
- 13.Serwer LP, James CD. Challenges in drug delivery to tumors of the central nervous system: an overview of pharmacological and surgical considerations. Adv Drug Deliv Rev 2012;64:590-597. [DOI] [PubMed] [Google Scholar]
- 14.Gao H, Jiang X. Progress on the diagnosis and evaluation of brain tumors. Cancer Imaging 2013;13:466-481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao H, Pang Z, Jiang X. Targeted delivery of nano-therapeutics for major disorders of the central nervous system. Pharm Res 2013;30:2485-2498. [DOI] [PubMed] [Google Scholar]
- 16.Zhan C, Wei X, Qian J, Feng L, Zhu J, Lu W. Co-delivery of TRAIL gene enhances the anti-glioblastoma effect of paclitaxel in vitro and in vivo. J Control Release 2012;160:630-636. [DOI] [PubMed] [Google Scholar]
- 17.Sarin H, Kanevsky AS, Wu H, Sousa AA, Wilson CM, Aronova MA, et al. Physiologic upper limit of pore size in the blood-tumor barrier of malignant solid tumors. J Transl Med 2009;7:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Groothuis DR. The blood-brain and blood-tumor barriers: a review of strategies for increasing drug delivery. Neuro Oncol 2000;2:45-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones AR, Shusta EV. Blood–brain barrier transport of therapeutics via receptor-mediation. Pharm Res 2007;24:1759-1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dufès C, Al Robaian M, Somani S. Transferrin and the transferrin receptor for the targeted delivery of therapeutic agents to the brain and cancer cells. Ther Deliv 2013;4:629-640. [DOI] [PubMed] [Google Scholar]
- 21.Zhang P, Hu L, Yin Q, Feng L, Li Y. Transferrin-modified c [RGDfK]-paclitaxel loaded hybrid micelle for sequential blood-brain barrier penetration and glioma targeting therapy. Mol Pharm 2012;9:1590-1598. [DOI] [PubMed] [Google Scholar]
- 22.Ying X, Wen H, Lu WL, Du J, Guo J, Tian W, et al. Dual-targeting daunorubicin liposomes improve the therapeutic efficacy of brain glioma in animals. J Control Release 2010;141:183-192. [DOI] [PubMed] [Google Scholar]
- 23.Qin L, Wang CZ, Fan HJ, Zhang CJ, Zhang HW, Lv MH, et al. A dual-targeting liposome conjugated with transferrin and arginine-glycine-aspartic acid peptide for glioma-targeting therapy. Oncol Lett 2014;8:2000-2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Porru M, Zappavigna S, Salzano G, Luce A, Stoppacciaro A, Balestrieri ML, et al. Medical treatment of orthotopic glioblastoma with transferrin-conjugated nanoparticles encapsulating zoledronic acid. Oncotarget 2014;5:10446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chiu RY, Tsuji T, Wang SJ, Wang J, Liu CT, Kamei DT. Improving the systemic drug delivery efficacy of nanoparticles using a transferrin variant for targeting. J Control Release 2014;180:33-41. [DOI] [PubMed] [Google Scholar]
- 26.Halbrooks PJ, Mason AB, Adams TE, Briggs SK, Everse SJ. The oxalate effect on release of iron from human serum transferrin explained. J Mol Biol 2004;339:217-226. [DOI] [PubMed] [Google Scholar]
- 27.Yoon DJ, Kwan BH, Chao FC, Nicolaides TP, Phillips JJ, Lam GY, et al. Intratumoral therapy of glioblastoma multiforme using genetically engineered transferrin for drug delivery. Cancer Res 2010;70:4520-4527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pan W, Kastin AJ, Zankel TC, van Kerkhof P, Terasaki T, Bu G. Efficient transfer of receptor-associated protein (RAP) across the blood-brain barrier. J Cell Sci 2004;117:5071-5078. [DOI] [PubMed] [Google Scholar]
- 29.Bell RD, Sagare AP, Friedman AE, Bedi GS, Holtzman DM, Deane R, et al. Transport pathways for clearance of human Alzheimer’s amyloid beta-peptide and apolipoproteins E and J in the mouse central nervous system. J Cereb Blood Flow Metab 2007;27:909-918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.May P, Herz J, Bock H. Molecular mechanisms of lipoprotein receptor signalling. Cell Mol Life Sci 2005;62:2325-2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xin H, Jiang X, Gu J, Sha X, Chen L, Law K, et al. Angiopep-conjugated poly(ethylene glycol)-co-poly(ε-caprolactone) nanoparticles as dual-targeting drug delivery system for brain glioma. Biomaterials 2011;32:4293-4305. [DOI] [PubMed] [Google Scholar]
- 32.Demeule M, Currie JC, Bertrand Y, Che C, Nguyen T, Regina A, et al. Involvement of the low-density lipoprotein receptor-related protein in the transcytosis of the brain delivery vector Angiopep-2. J Neurochem 2008;106:1534-1544. [DOI] [PubMed] [Google Scholar]
- 33.Ke W, Shao K, Huang R, Han L, Liu Y, Li J, et al. Gene delivery targeted to the brain using an Angiopep-conjugated polyethyleneglycol-modified polyamidoamine dendrimer. Biomaterials 2009;30:6976-6985. [DOI] [PubMed] [Google Scholar]
- 34.Shen J, Zhan C, Xie C, Meng Q, Gu B, Li C, et al. Poly (ethylene glycol)-block-poly (D, L-lactide acid) micelles anchored with angiopep-2 for brain-targeting delivery. J Drug Target 2011;19:197-203. [DOI] [PubMed] [Google Scholar]
- 35.Ché C, Yang G, Thiot C, Lacoste MC, Currie JC, Demeule M, et al. New Angiopep-modified doxorubicin (ANG1007) and etoposide (ANG1009) chemotherapeutics with increased brain penetration. J Med Chem 2010;53:2814-2824. [DOI] [PubMed] [Google Scholar]
- 36.Shao K, Huang R, Li J, Han L, Ye L, Lou J, et al. Angiopep-2 modified PE-PEG based polymeric micelles for amphotericin B delivery targeted to the brain. J Control Release 2010;147:118-126. [DOI] [PubMed] [Google Scholar]
- 37.Huang S, Li J, Han L, Liu S, Ma H, Huang R, et al. Dual targeting effect of Angiopep-2-modified, DNA-loaded nanoparticles for glioma. Biomaterials 2011;32:6832-6838. [DOI] [PubMed] [Google Scholar]
- 38.Gao H, Zhang S, Cao S, Yang Z, Pang Z, Jiang X. Angiopep-2 and activatable cell-penetrating peptide dual-functionalized nanoparticles for systemic glioma-targeting delivery. Mol Pharm 2014;11:2755-2763. [DOI] [PubMed] [Google Scholar]
- 39.Ren J, Shen S, Wang D, Xi Z, Guo L, Pang Z, et al. The targeted delivery of anticancer drugs to brain glioma by PEGylated oxidized multi-walled carbon nanotubes modified with angiopep-2. Biomaterials 2012;33:3324-3333. [DOI] [PubMed] [Google Scholar]
- 40.Mei L, Zhang Q, Yang Y, He Q, Gao H. Angiopep-2 and activatable cell penetrating peptide dual modified nanoparticles for enhanced tumor targeting and penetrating. Int J Pharm 2014;474:95-102. [DOI] [PubMed] [Google Scholar]
- 41.Ruan S, Yuan M, Zhang L, Hu G, Chen J, Cun X, et al. Tumor microenvironment sensitive doxorubicin delivery and release to glioma using angiopep-2 decorated gold nanoparticles. Biomaterials 2015;37:425-435. [DOI] [PubMed] [Google Scholar]
- 42.Ying X, Wang Y, Liang J, Yue J, Xu C, Lu L, et al. Angiopep-Conjugated Electro-Responsive Hydrogel Nanoparticles: Therapeutic Potential for Epilepsy. Angew Chem Int Ed Engl 2014;53:12436-12440. [DOI] [PubMed] [Google Scholar]
- 43.Suksrichavalit T, Prachayasittikul S, Isarankura-Na-Ayudhya C, Prachayasittikul V. Synthesis of a “clickable” Angiopep-conjugated p-coumaric acid for brain-targeted delivery. J Mater Sci 2014;49:8204-8213. [Google Scholar]
- 44.Chen GJ, Su YZ, Hsu C, Lo YL, Huang SJ, Ke JH, et al. Angiopep-pluronic F127-conjugated superparamagnetic iron oxide nanoparticles as nanotheranostic agents for BBB targeting. J Mater Chem B 2014;2:5666-5675. [DOI] [PubMed] [Google Scholar]
- 45.Xuan S, Shin DH, Kim JS. Angiopep-2-conjugated liposomes encapsulating γ-secretase inhibitor for targeting glioblastoma stem cells. Journal of Pharmaceutical Investigation 2014;44:473-483. [Google Scholar]
- 46.Demeule M, Lachowicz JE, Yang G, Das S, Ché C, Tripathy S, et al. Utilization of the Angiopep platform to enable brain penetration of therapeutic mAbs or Antibody-Drug Conjugates for treatment of brain tumors. Cancer Res 2014;74:2657. [Google Scholar]
- 47.Wei X, Zhan C, Chen X, Hou J, Xie C, Lu W. Retro-inverso isomer of angiopep-2: a stable d-peptide ligand inspires brain-targeted drug delivery. Mol Pharm 2014;11:3261-3268. [DOI] [PubMed] [Google Scholar]
- 48.Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012;488:522-526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Galderisi U, Cipollaro M, Giordano A. Stem cells and brain cancer. Cell Death Differ 2006;13:5-11. [DOI] [PubMed] [Google Scholar]
- 50.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature 2004;432:396-401. [DOI] [PubMed] [Google Scholar]
- 51.Reguera-Nuñez E, Roca C, Hardy E, De la Fuente M, Csaba N, Garcia-Fuentes M. Implantable controlled release devices for BMP-7 delivery and suppression of glioblastoma initiating cells. Biomaterials 2014;35:2859-2867. [DOI] [PubMed] [Google Scholar]
- 52.Vecchio D, Daga A, Carra E, Marubbi D, Baio G, Neumaier CE, et al. Predictability, efficacy and safety of radiosensitization of glioblastoma‐initiating cells by the ATM inhibitor KU-60019. Int J Cancer 2014;135:479-491. [DOI] [PubMed] [Google Scholar]
- 53.González-Gómez P, Anselmo NP, Mira H. BMPs as therapeutic targets and biomarkers in astrocytic glioma. Biomed Res Int 2014;2014:549742. [DOI] [PMC free article] [PubMed]
- 54.Kim M, Choe S. BMPs and their clinical potentials. BMB Rep 2011;44:619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Klose A, Waerzeggers Y, Monfared P, Vukicevic S, Kaijzel EL, Winkeler A, et al. Imaging bone morphogenetic protein 7 induced cell cycle arrest in experimental gliomas. Neoplasia 2011;13:276-285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nakano I, Saigusa K, Kornblum HI. BMPing off glioma stem cells. Cancer Cell 2008;13:3-4. [DOI] [PubMed] [Google Scholar]
- 57.Chirasani SR, Sternjak A, Wend P, Momma S, Campos B, Herrmann IM, et al. Bone morphogenetic protein-7 release from endogenous neural precursor cells suppresses the tumourigenicity of stem-like glioblastoma cells. Brain 2010;133:1961-1972. [DOI] [PubMed] [Google Scholar]
- 58.Piccirillo SG, Reynolds BA, Zanetti N, Lamorte G, Binda E, Broggi G, et al. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature 2006;444:761-765. [DOI] [PubMed] [Google Scholar]
- 59.Wu Q, Yao J. BMP4, a new prognostic factor for glioma. World J Surg Oncol 2013;11:264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu B, Chen Q, Tian D, Wu L, Dong H, Wang J, et al. BMP4 reverses multidrug resistance through modulation of BCL-2 and GDNF in glioblastoma. Brain Res 2013;1507:115-124. [DOI] [PubMed] [Google Scholar]
- 61.Rahman M, Azari H, Deleyrolle L, Millette S, Zeng H, Reynolds BA. Controlling tumor invasion: bevacizumab and BMP4 for glioblastoma. Future Oncol 2013;9:1389-1396. [DOI] [PubMed] [Google Scholar]
- 62.Tate CM, Pallini R, Ricci-Vitiani L, Dowless M, Shiyanova T, D’Alessandris GQ, et al. A BMP7 variant inhibits the tumorigenic potential of glioblastoma stem-like cells. Cell Death Differ 2012;19:1644-1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu C, Tian G, Tu Y, Fu J, Lan C, Wu N. Expression pattern and clinical prognostic relevance of bone morphogenetic protein-2 in human gliomas. Jpn J Clin Oncol 2009;39:625-631. [DOI] [PubMed] [Google Scholar]
- 64.Persano L, Pistollato F, Rampazzo E, Della Puppa A, Abbadi S, Frasson C, et al. BMP2 sensitizes glioblastoma stem-like cells to Temozolomide by affecting HIF-1α stability and MGMT expression. Cell Death Dis 2012;3:e412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shin DH, Xuan S, Kim WY, Bae GU, Kim JS. CD133 antibody-conjugated immunoliposomes encapsulating gemcitabine for targeting glioblastoma stem cells. J Mater Chem B 2014;2:3771-3781. [DOI] [PubMed] [Google Scholar]
- 66.Bai Y, Lathia JD, Zhang P, Flavahan W, Rich JN, Mattson MP. Molecular targeting of TRF2 suppresses the growth and tumorigenesis of glioblastoma stem cells. Glia 2014;62:1687-1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shi L, Zhang S, Feng K, Wu F, Wan Y, Wang Z, et al. MicroRNA-125b-2 confers human glioblastoma stem cells resistance to temozolomide through the mitochondrial pathway of apoptosis. Int J Oncol 2012;40:119-129. [DOI] [PubMed] [Google Scholar]
- 68.Shi L, Wan Y, Sun G, Zhang S, Wang Z, Zeng Y. miR-125b inhibitor may enhance the invasion-prevention activity of temozolomide in glioblastoma stem cells by targeting PIAS3. BioDrugs 2014;28:41-54. [DOI] [PubMed] [Google Scholar]
- 69.Chen J, Fu X, Wan Y, Wang Z, Jiang D, Shi L. miR-125b inhibitor enhance the chemosensitivity of glioblastoma stem cells to temozolomide by targeting Bak1. Tumour Biol 2014;35:6293-6302. [DOI] [PubMed] [Google Scholar]
- 70.Huang S, Shao K, Liu Y, Kuang Y, Li J, An S, et al. Tumor-targeting and microenvironment-responsive smart nanoparticles for combination therapy of antiangiogenesis and apoptosis. ACS Nano 2013;7:2860-2871. [DOI] [PubMed] [Google Scholar]
- 71.Stupp R, Hegi ME, Gorlia T, Erridge SC, Perry J, Hong YK, et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol 2014;15:1100-1108. [DOI] [PubMed] [Google Scholar]
- 72.Roth P, Silginer M, Goodman SL, Hasenbach K, Thies S, Maurer G, et al. Integrin control of the transforming growth factor-β pathway in glioblastoma. Brain 2013;136:564-576. [DOI] [PubMed] [Google Scholar]
- 73.Barczyk M, Carracedo S, Gullberg D. Integrins. Cell Tissue Res 2010;339:269-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer 2010;10:9-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kibria G, Hatakeyama H, Ohga N, Hida K, Harashima H. Dual-ligand modification of PEGylated liposomes shows better cell selectivity and efficient gene delivery. J Control Release 2011;153:141-148. [DOI] [PubMed] [Google Scholar]
- 76.Liu Y, Ran R, Chen J, Kuang Q, Tang J, Mei L, et al. Paclitaxel loaded liposomes decorated with a multifunctional tandem peptide for glioma targeting. Biomaterials 2014;35:4835-4847. [DOI] [PubMed] [Google Scholar]
- 77.Gao H, Yang Z, Cao S, Xiong Y, Zhang S, Pang Z, et al. Tumor cells and neovasculature dual targeting delivery for glioblastoma treatment. Biomaterials 2014;35:2374-2382. [DOI] [PubMed] [Google Scholar]
- 78.Reardon DA, Fink KL, Mikkelsen T, Cloughesy TF, O’Neill A, Plotkin S, et al. Randomized phase II study of cilengitide, an integrin-targeting arginine-glycine-aspartic acid peptide, in recurrent glioblastoma multiforme. J Clin Oncol 2008;26:5610-5617. [DOI] [PubMed] [Google Scholar]
- 79.Nabors LB, Mikkelsen T, Hegi ME, Ye X, Batchelor T, Lesser G, et al. A safety run-in and randomized phase 2 study of cilengitide combined with chemoradiation for newly diagnosed glioblastoma (NABTT 0306). Cancer 2012;118:5601-5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gilbert MR, Kuhn J, Lamborn KR, Lieberman F, Wen PY, Mehta M, et al. Cilengitide in patients with recurrent glioblastoma: the results of NABTC 03-02, a phase II trial with measures of treatment delivery. J Neurooncol 2012;106:147-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Eisele G, Wick A, Eisele AC, Clément PM, Tonn J, Tabatabai G, et al. Cilengitide treatment of newly diagnosed glioblastoma patients does not alter patterns of progression. J Neurooncol 2014;117:141-145. [DOI] [PubMed] [Google Scholar]
- 82.Padfield E, Ellis H, Kurian K. Current therapeutic advances targeting EGFR and EGFRvIII in glioblastoma. Front Oncol 2015;5:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Carrasco-García E, Saceda M, Martínez-Lacaci I. Role of receptor tyrosine kinases and their ligands in glioblastoma. Cells 2014;3:199-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Patel R, Y Leung H. Targeting the EGFR-family for therapy: biological challenges and clinical perspective. Curr Pharm Des 2012;18:2672-2679. [DOI] [PubMed] [Google Scholar]
- 85.Sampson JH, Aldape KD, Archer GE, Coan A, Desjardins A, Friedman AH, et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro Oncol 2011;13:324-333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cloughesy TF, Cavenee WK, Mischel PS. Glioblastoma: From molecular pathology to targeted treatment. Annu Rev Pathol 2014;9:1-25. [DOI] [PubMed] [Google Scholar]
- 87.Schuster J, Lai RK, Recht LD, Reardon DA, Paleologos NA, Groves MD, et al. A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: the ACT III study. Neuro Oncol 2015;17:854-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Qaddoumi I, Kocak M, Panandiker ASP, Armstrong GT, Wetmore C, Crawford JR, et al. Phase II trial of erlotinib during and after radiotherapy in children with newly diagnosed high-grade gliomas. Front Oncol 2014;4:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wen PY, Chang SM, Lamborn KR, Kuhn JG, Norden AD, Cloughesy TF, et al. Phase I/II study of erlotinib and temsirolimus for patients with recurrent malignant gliomas: North American Brain Tumor Consortium trial 04-02. Neuro Oncol 2014;16:567-578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Peereboom DM, Ahluwalia MS, Ye X, Supko JG, Hilderbrand SL, Phuphanich S, et al. NABTT 0502: a phase II and pharmacokinetic study of erlotinib and sorafenib for patients with progressive or recurrent glioblastoma multiforme. Neuro Oncol 2013;15:490-496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Uhm JH, Ballman KV, Wu W, Giannini C, Krauss J, Buckner JC, et al. Phase II evaluation of gefitinib in patients with newly diagnosed Grade 4 astrocytoma: Mayo/North Central Cancer Treatment Group Study N0074. Int J Radiat Oncol Biol Phys 2011;80:347-353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Neyns B, Sadones J, Joosens E, Bouttens F, Verbeke L, Baurain JF, et al. Stratified phase II trial of cetuximab in patients with recurrent high-grade glioma. Ann Oncol 2009;20:1596-1603. [DOI] [PubMed] [Google Scholar]
- 93.Lv S, Teugels E, Sadones J, De Brakeleer S, Duerinck J, Du Four S, et al. Correlation of EGFR, IDH1 and PTEN status with the outcome of patients with recurrent glioblastoma treated in a phase II clinical trial with the EGFR-blocking monoclonal antibody cetuximab. Int J Oncol 2012;41:1029-1035. [DOI] [PubMed] [Google Scholar]
- 94.Wang Y, Pan L, Sheng XF, Chen S, Dai JZ. Nimotuzumab, a humanized monoclonal antibody specific for the EGFR, in combination with temozolomide and radiation therapy for newly diagnosed glioblastoma multiforme: First results in Chinese patients. Asia Pac J Clin Oncol 2014. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 95.Westphal M, Heese O, Steinbach JP, Schnell O, Schackert G, Mehdorn M, et al. A randomised, open label phase III trial with nimotuzumab, an anti-epidermal growth factor receptor monoclonal antibody in the treatment of newly diagnosed adult glioblastoma. Eur J Cancer 2015;51:522-532. [DOI] [PubMed] [Google Scholar]
- 96.Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol 2010;28:4722-4729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lai RK, Recht LD, Reardon DA, Paleologos N, Groves M, Rosenfeld MR, et al. IM-03: Long-term follow-up of ACT III: a phase II trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma. Neuro Oncol 2011;13:ii34-ii40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Swartz AM, Batich KA, Fecci PE, Sampson JH. Peptide vaccines for the treatment of glioblastoma. J Neurooncol 2015;123:433-440. [DOI] [PubMed] [Google Scholar]
- 99.Ranza E, Mazzini G, Facoetti A, Nano R. In-vitro effects of the tyrosine kinase inhibitor imatinib on glioblastoma cell proliferation. J Neurooncol 2010;96:349-357. [DOI] [PubMed] [Google Scholar]
- 100.Nazarenko I, Hede SM, He X, Hedrén A, Thompson J, Lindström MS, et al. PDGF and PDGF receptors in glioma. Ups J Med Sci 2012;117:99-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ehnman M, Östman A. Therapeutic targeting of platelet-derived growth factor receptors in solid tumors. Expert Opin Investig Drugs 2014;23:211-226. [DOI] [PubMed] [Google Scholar]
- 102.Holdhoff M, Supko JG, Gallia GL, Hann CL, Bonekamp D, Ye X, et al. Intratumoral concentrations of imatinib after oral administration in patients with glioblastoma multiforme. J Neurooncol 2010;97:241-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dong Y, Han Q, Zou Y, Deng Z, Lu X, Wang X, et al. Long-term exposure to imatinib reduced cancer stem cell ability through induction of cell differentiation via activation of MAPK signaling in glioblastoma cells. Mol Cell Biochem 2012;370:89-102. [DOI] [PubMed] [Google Scholar]
- 104.Dong Y, Jia L, Wang X, Tan X, Xu J, Deng Z, et al. Selective inhibition of PDGFR by imatinib elicits the sustained activation of ERK and downstream receptor signaling in malignant glioma cells. Int J Oncol 2011;38:555. [DOI] [PubMed] [Google Scholar]
- 105.Dresemann G, Weller M, Rosenthal MA, Wedding U, Wagner W, Engel E, et al. Imatinib in combination with hydroxyurea versus hydroxyurea alone as oral therapy in patients with progressive pretreated glioblastoma resistant to standard dose temozolomide. J Neurooncol 2010;96:393-402. [DOI] [PubMed] [Google Scholar]
- 106.Reardon DA, Dresemann G, Taillibert S, Campone M, van den Bent M, Clement P, et al. Multicentre phase II studies evaluating imatinib plus hydroxyurea in patients with progressive glioblastoma. Br J Cancer 2009;101:1995-2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Reardon DA, Turner S, Peters KB, Desjardins A, Gururangan S, Sampson JH, et al. A review of VEGF/VEGFR-targeted therapeutics for recurrent glioblastoma. J Natl Compr Canc Netw 2011;9:414-427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Balaña C, Gil MJ, Perez P, Reynes G, Gallego O, Ribalta T, et al. Sunitinib administered prior to radiotherapy in patients with non-resectable glioblastoma: results of a phase II study. Target Oncol 2014;9:321-329. [DOI] [PubMed] [Google Scholar]
- 109.Hutterer M, Nowosielski M, Haybaeck J, Embacher S, Stockhammer F, Gotwald T, et al. A single-arm phase II Austrian/German multicenter trial on continuous daily sunitinib in primary glioblastoma at first recurrence (SURGE 01-07). Neuro Oncol 2014;16:92-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pan E, Yu D, Yue B, Potthast L, Chowdhary S, Smith P, et al. A prospective phase II single-institution trial of sunitinib for recurrent malignant glioma. J Neurooncol 2012;110:111-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kreisl TN, Smith P, Sul J, Salgado C, Iwamoto FM, Shih JH, et al. Continuous daily sunitinib for recurrent glioblastoma. J Neurooncol 2013;111:41-48. [DOI] [PubMed] [Google Scholar]
- 112.Reardon DA, Vredenburgh JJ, Coan A, Desjardins A, Peters KB, Gururangan S, et al. Phase I study of sunitinib and irinotecan for patients with recurrent malignant glioma. J Neurooncol 2011;105:621-627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Batchelor TT, Duda DG, di Tomaso E, Ancukiewicz M, Plotkin SR, Gerstner E, et al. Phase II study of cediranib, an oral pan–vascular endothelial growth factor receptor tyrosine kinase inhibitor, in patients with recurrent glioblastoma. J Clin Oncol 2010;28:2817-2823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gerstner ER, Emblem KE, Chi AS, Eichler AF, Hochberg F, Drappatz J, et al. Effects of cediranib, a VEGF signaling inhibitor, in combination with chemoradiation on tumor blood flow and survival in newly diagnosed glioblastoma. J Clin Oncol 2012;30:abstr 2009.
- 115.Batchelor TT, Gerstner ER, Emblem KE, Duda DG, Kalpathy-Cramer J, Snuderl M, et al. Improved tumor oxygenation and survival in glioblastoma patients who show increased blood perfusion after cediranib and chemoradiation. Proc Natl Acad Sci U S A 2013;110:19059-19064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Batchelor TT, Mulholland P, Neyns B, Nabors LB, Campone M, Wick A, et al. Phase III randomized trial comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, versus lomustine alone in patients with recurrent glioblastoma. J Clin Oncol 2013;31:3212-3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Pinho MC, Polaskova P, Kalpathy-Cramer J, Jennings D, Emblem KE, Jain RK, et al. Low incidence of pseudoprogression by imaging in newly diagnosed glioblastoma patients treated with cediranib in combination with chemoradiation. Oncologist 2014;19:75-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kratzsch T, Gruenwald V, Vajkoczy P, Kuhn SA. Use of axitinib, a new-generation tyrosine kinase inhibitor, to decrease glioblastoma growth despite primary resistance to the VEGF-antibody bevacizumab. J Clin Oncol 2013;31:abstr 2077.
- 119.Lu L, Saha D, Martuza RL, Rabkin SD, Wakimoto H. Single agent efficacy of the VEGFR kinase inhibitor axitinib in preclinical models of glioblastoma. J Neurooncol 2015;121:91-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Duerinck J, Du Four S, Bouttens F, Verschaeve V, Everaert H, De Raedt S, et al. O10. 07 Randomezed phase II study of axitinib vs. standard of care in patients with recurrent glioblastoma. Neuro Oncol 2014;16:ii24-ii25. [DOI] [PubMed] [Google Scholar]
- 121.Goldlust SA, Cavaliere R, Newton HB, Hsu M, DeAngelis LM, Batchelor TT, et al. Bevacizumab for glioblastoma refractory to vascular endothelial growth factor receptor inhibitors. J Neurooncol 2012;107:407-411. [DOI] [PubMed] [Google Scholar]
- 122.Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med 2014;370:699-708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Taal W, Oosterkamp HM, Walenkamp AM, Dubbink HJ, Beerepoot LV, Hanse MC, et al. Single-agent bevacizumab or lomustine versus a combination of bevacizumab plus lomustine in patients with recurrent glioblastoma (BELOB trial): a randomised controlled phase 2 trial. Lancet Oncol 2014;15:943-953. [DOI] [PubMed] [Google Scholar]
- 124.Taal W, Oosterkamp HM, Walenkamp AM, Dubbink HJ, Beerepoot LV, Hanse M, et al. O10. 05 Final analysis of the BELOB trial (a randomised phase II study on bevacizumab versus bevacizumab plus lomustine versus lomustine single agent in recurrent glioblastoma) and first radiology review results. Neuro Oncol 2014;16:ii24. [Google Scholar]
- 125.Taal W, Enting R, Taphoorn M, Smits M, Dubbink H, Beerepoot L, et al. AT-55 Final analysis of the BELOB trial (a randomised phase II study on bevacizumab versus bevacizumab plus lomustine versus lomustine single agent in recurrent glioblastoma) and first radiology review results. Neuro Oncol 2014;16:v20-v21. [Google Scholar]
- 126.Carlson JA, Reddy K, Gaspar LE, Ney D, Kavanagh BD, Damek D, et al. Hypofractionated–intensity modulated radiation therapy (Hypo-IMRT) and temozolomide (TMZ) with and without bevacizumab (BEV) for glioblastoma multiforme (GBM): a comparison of 2 prospective phase 2 trials. Int J Radiat Oncol Biol Phys 2014;90:S284. [DOI] [PubMed] [Google Scholar]
- 127.Herrlinger U, Schäfer N, Steinbach JP, Weyerbrock A, Hau P, Goldbrunner R, et al. O10. 04 The randomised, multicenter GLARIUS trial in vestigating bevacizumab/ irinotecan vs standard temozolomide in newly diagnosed, MGMT-non-methylated glioblastoma patients: final survival results and quality of life. Neuro Oncol 2014;16:ii23-ii24. [Google Scholar]
- 128.Chauffert B, Feuvret L, Bonnetain F, Taillandier L, Frappaz D, Taillia H, et al. Randomized phase II trial of irinotecan and bevacizumab as neo-adjuvant and adjuvant to temozolomide-based chemoradiation compared to temozolomide-chemoradiation for unresectable glioblastoma: final results of the TEMAVIR study from ANOCEF. Ann Oncol 2014;25:1442-1447. [DOI] [PubMed] [Google Scholar]
- 129.Reardon DA, Desjardins A, Peters KB, Gururangan S, Sampson JH, McLendon RE, et al. Phase II study of carboplatin, irinotecan, and bevacizumab for bevacizumab naive, recurrent glioblastoma. J Neurooncol 2012;107:155-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Clarke JL, Molinaro AM, Phillips JJ, Butowski NA, Chang SM, Perry A, et al. A single-institution phase II trial of radiation, temozolomide, erlotinib, and bevacizumab for initial treatment of glioblastoma. Neuro Oncol 2014;16:984-990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Farias-Eisner G, Bank AM, Hwang BY, Appelboom G, Piazza MA, Bruce SS, et al. Glioblastoma biomarkers from bench to bedside: advances and challenges. Br J Neurosurg 2012;26:189-194. [DOI] [PubMed] [Google Scholar]
- 132.Hermansen SK, Kristensen BW. MicroRNA biomarkers in glioblastoma. J Neurooncol 2013;114:13-23. [DOI] [PubMed] [Google Scholar]
- 133.Qian X, Ren Y, Shi Z, Long L, Pu P, Sheng J, et al. Sequence-dependent synergistic inhibition of human glioma cell lines by combined temozolomide and miR-21 inhibitor gene therapy. Mol Pharm 2012;9:2636-2645. [DOI] [PubMed] [Google Scholar]
- 134.Chan JA, Krichevsky AM, Kosik KS. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res 2005;65:6029-6033. [DOI] [PubMed] [Google Scholar]
- 135.Ciafrè SA, Galardi S, Mangiola A, Ferracin M, Liu CG, Sabatino G, et al. Extensive modulation of a set of microRNAs in primary glioblastoma. Biochem Biophys Res Commun 2005;334:1351-1358. [DOI] [PubMed] [Google Scholar]
- 136.Gaur AB, Holbeck SL, Colburn NH, Israel MA. Downregulation of Pdcd4 by mir-21 facilitates glioblastoma proliferation in vivo. Neuro Oncol 2011;13:580-590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lawler S, Chiocca EA. Emerging functions of microRNAs in glioblastoma. J Neurooncol 2009;92:297-306. [DOI] [PubMed] [Google Scholar]
- 138.Chao TF, Xiong HH, Liu W, Chen Y, Zhang JX. MiR-21 mediates the radiation resistance of glioblastoma cells by regulating PDCD4 and hMSH2. J Huazhong Univ Sci Technolog Med Sci 2013;33:525-529. [DOI] [PubMed] [Google Scholar]
- 139.Gwak HS, Kim TH, Jo GH, Kim YJ, Kwak HJ, Kim JH, et al. Silencing of microRNA-21 confers radio-sensitivity through inhibition of the PI3K/AKT pathway and enhancing autophagy in malignant glioma cell lines. PLoS One 2012;7:e47449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Liu J, Zhu H, Yang X, Ge Y, Zhang C, Qin Q, et al. MicroRNA-21 is a novel promising target in cancer radiation therapy. Tumour Biol 2014;35:3975-3979. [DOI] [PubMed] [Google Scholar]
- 141.Ren Y, Kang CS, Yuan XB, Zhou X, Xu P, Han L, et al. Co-delivery of as-miR-21 and 5-FU by poly (amidoamine) dendrimer attenuates human glioma cell growth in vitro. J Biomater Sci Polym Ed 2010;21:303-314. [DOI] [PubMed] [Google Scholar]
- 142.Qian X, Long L, Shi Z, Liu C, Qiu M, Sheng J, et al. Star-branched amphiphilic PLA-b-PDMAEMA copolymers for co-delivery of miR-21 inhibitor and doxorubicin to treat glioma. Biomaterials 2014;35:2322-2335. [DOI] [PubMed] [Google Scholar]
- 143.Costa PM, Cardoso AL, Custódia C, Cunha P, Pereira de Almeida L, Pedroso de Lima MC. MiRNA-21 silencing mediated by tumor-targeted nanoparticles combined with sunitinib: A new multimodal gene therapy approach for glioblastoma. J Control Release 2015;207:31-39. [DOI] [PubMed] [Google Scholar]
- 144.She X, Yu Z, Cui Y, Lei Q, Wang Z, Xu G, et al. miR-181 subunits enhance the chemosensitivity of temozolomide by Rap1B-mediated cytoskeleton remodeling in glioblastoma cells. Med Oncol 2014;31:892. [DOI] [PubMed] [Google Scholar]
- 145.Wang XF, Shi ZM, Wang XR, Cao L, Wang YY, Zhang JX, et al. MiR-181d acts as a tumor suppressor in glioma by targeting K-ras and Bcl-2. J Cancer Res Clin Oncol 2012;138:573-584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wang J, Sai K, Chen FR, Chen ZP. miR-181b modulates glioma cell sensitivity to temozolomide by targeting MEK1. Cancer Chemother Pharmacol 2013;72:147-158. [DOI] [PubMed] [Google Scholar]
- 147.Zhang W, Zhang J, Hoadley K, Kushwaha D, Ramakrishnan V, Li S, et al. miR-181d: a predictive glioblastoma biomarker that downregulates MGMT expression. Neuro Oncol 2012;14:712-719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Yang ZZ, Li JQ, Wang ZZ, Dong DW, Qi XR. Tumor-targeting dual peptides-modified cationic liposomes for delivery of siRNA and docetaxel to gliomas. Biomaterials 2014;35:5226-5239. [DOI] [PubMed] [Google Scholar]
- 149.Genovese G, Ergun A, Shukla SA, Campos B, Hanna J, Ghosh P, et al. microRNA regulatory network inference identifies miR-34a as a novel regulator of TGF-β signaling in glioblastoma. Cancer Discov 2012;2:736-749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Zhang M, Kleber S, Röhrich M, Timke C, Han N, Tuettenberg J, et al. Blockade of TGF-β signaling by the TGFßR-I kinase inhibitor LY2109761 enhances radiation response and prolongs survival in glioblastoma. Cancer Res 2011;71:7155-7167. [DOI] [PubMed] [Google Scholar]
- 151.Carra E, Barbieri F, Marubbi D, Pattarozzi A, Favoni RE, Florio T, et al. Sorafenib selectively depletes human glioblastoma tumor-initiating cells from primary cultures. Cell Cycle 2013;12:491-500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Aldea MD, Petrushev B, Soritau O, Tomuleasa CI, Berindan-Neagoe I, Filip AG, et al. Metformin plus sorafenib highly impacts temozolomideresistant glioblastoma stem-like cells. J BUON 2014;19:502-511. [PubMed] [Google Scholar]
- 153.Young JS, Morshed RA, Kim JW, Balyasnikova IV, Ahmed AU, Lesniak MS. Advances in stem cells, induced pluripotent stem cells, and engineered cells: delivery vehicles for anti-glioma therapy. Expert Opin Drug Deliv 2014;11:1733-1746. [DOI] [PubMed] [Google Scholar]