

Abstract
Nonalcoholic fatty liver disease (NAFLD) is the most common cause of chronic liver disease in the industrialized world. The prevalence of NAFLD is increasing, becoming a substantial public health burden. NAFLD includes a broad spectrum of disorders, from simple conditions such as steatosis to severe manifestations such as fibrosis and cirrhosis. The relationship of NAFLD with metabolic alterations such as type 2 diabetes is well described and related to insulin resistance, with NAFLD being recognized as the hepatic manifestation of metabolic syndrome. However, NAFLD may also coincide with endocrine diseases such as polycystic ovary syndrome, hypothyroidism, growth hormone deficiency or hypercortisolism. It is therefore essential to remember, when discovering altered liver enzymes or hepatic steatosis on radiological exams, that endocrine diseases can cause NAFLD. Indeed, the overall prognosis of NAFLD may be modified by treatment of the underlying endocrine pathology. In this review, we will discuss endocrine diseases that can cause NALFD. Underlying pathophysiological mechanisms will be presented and specific treatments will be reviewed.
Keywords: Endocrine diseases, Nonalcoholic fatty liver disease, Insulin resistance, Obesity, Type 2 diabetes
Core tip: The review discusses the links between nonalcoholic fatty liver disease and endocrine diseases, from common ones such as type 2 diabetes and polycystic ovary syndrome to rare disorders such as growth hormone deficiency. The pathophysiological mechanisms underlying these associations are described.
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is the most common liver disease in the Western world. The term “nonalcoholic steatohepatitis” (NASH) was introduced by Ludwig in 1980 following observations of patients, mainly obese women, with histological evidence of alcoholic hepatitis on liver biopsy without a history of alcohol abuse[1]. The term “NAFLD” was introduced in 1986 to define a spectrum ranging from hepatic steatosis to fibrosis and cirrhosis.
Given the strong association of NAFLD with metabolic syndrome and the worldwide epidemic of obesity, the prevalence of NAFLD is constantly increasing. In the United States, one-third of the overall population has NAFLD and 2%-5% have NASH[2]. Within the NAFLD spectrum, only patients with histologically proven NASH develop progressive liver disease. Progression is more likely in the setting of diabetes, insulin resistance (IR) and other pre-existing conditions[3].
As we will discuss in this review, the physiopathological mechanism common in both NAFLD and many different endocrine diseases is IR. For this reason, it is important for endocrinologists and gastroenterologists to remember that NAFLD and endocrine diseases may coexist (Figure 1).
Figure 1.
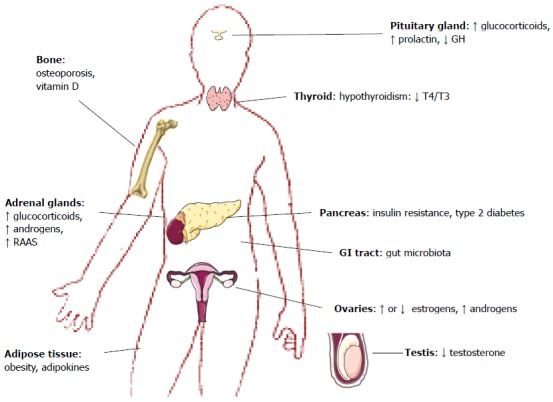
Endocrine diseases associated with nonalcoholic fatty liver disease. GH: Growth hormone; RAAS: Renin-angiotensin-aldosterone system; GI: Gastrointestinal.
EPIDEMIOLOGY
In the United States, the prevalence of NAFLD varies between 10% and 35%[2], depending on the population studied and the modality used for diagnosis. Ultimately, liver biopsy is required to make a definitive diagnosis of NASH, and estimates from biopsy series indicate that the prevalence of NASH in the United States is between 2% and 5%. NAFLD linked to metabolic syndrome is the most common cause of NASH, but NAFLD may be found in association with other diseases (e.g., Wilson disease, disorders of lipid metabolism, etc).
NAFLD is not unique to Western countries. NAFLD is also prevalent in developing countries[4], and data from the rest of the world suggest that the prevalence of NAFLD varies between 6% and 35%, with a median of 20%[3,5]. Most studies indicate that NAFLD is usually associated with metabolic syndrome, but studies in Asian countries also report NAFLD in non-obese individuals[6-9]. However, these findings may be explained by the fact that, for a given body mass index (BMI), body fat content is usually higher in Asians than in westerners[10].
Several cohorts have shown that NAFLD prevalence depends on ethnicity. Notably, Hispanics have the highest prevalence of NAFLD, hepatic steatosis, and elevated aminotransferases levels, followed by non-Hispanic whites, whereas African Americans have the lowest prevalence[5]. However, in the absence of liver biopsies, the true prevalence of NAFLD cannot be accurately estimated, and it is therefore difficult to draw clear conclusions from these analyses. Moreover, the recent MESA (Multi-Ethnic Study of Atherosclerosis) found no association between ethnicity and NAFLD[11].
NAFLD may be affected by genetic or environmental factors. Notably, 38% of Asian Indian men with the apolipoprotein C3 gene variant alleles C-482T and T-455C have NAFLD (compared to 0% amongst wild-type homozygotes). An association between these variant alleles, NAFLD and IR was therefore reported[12]. Recently, a nonsynonymous genetic variant (rs58542926) within the transmembrane 6 superfamily member 2 (TM6SF2) gene of unknown function was associated with the severity of NAFLD[13].
In summary, estimates of the prevalence of NAFLD should be considered with caution, as they may vary depending on the criteria used for diagnosis.
DIAGNOSIS
NAFLD encompasses a spectrum of diseases of different etiologies ranging from fat accumulation (steatosis) to inflammation and fibrosis (NASH) and finally cirrhosis. Formally, a diagnosis of NAFLD requires a liver biopsy with a lipid content of at least 5% of hepatocytes. In 20%-25% of cases, steatosis will evolve to NASH and, in turn, 20% of these patients will develop cirrhosis[14]. We will briefly discuss the different diagnostic methods.
Liver biopsy is the current gold standard for NASH diagnosis and staging[5], but the method is invasive and cannot be used in population-based studies. Only biopsy can assess inflammation and fibrosis. However, sampling variability may alter the accuracy of the diagnosis[15]. Several noninvasive diagnostic methods for NAFLD and NASH have been introduced recently. Notably, imaging techniques including proton magnetic resonance spectroscopy (1H-MRS), ultrasonography, computed tomography (CT), and magnetic resonance imaging (MRI) can be used[16]. 1H-MRS is considered the most accurate noninvasive method for measuring liver fat content. Ultrasonography is the most widely used method but is relatively insensitive, as it can detect steatosis only when liver fat content exceeds 33%[17].
Other studies have used elevations in the liver enzymes alanine aminotransferase (ALT) and aspartate aminotransferase (AST) as indicators of NAFLD[18-22]. However, these measurements are neither sensitive nor specific[23]. Indeed, up to 70% of subjects with NAFLD have normal levels of ALT and AST[17].
Different scoring methods have been developed for NAFLD screening, such as the Fatty Liver Index[24] and the Lipid Accumulation Product[25]. These indices are easy to use, applicable in community healthcare settings, and could contribute to better assess NAFLD prevalence. A study published by the LIDO study group tried to validate five NAFLD scoring methods (fatty liver index, NAFLD liver fat score, hepatic steatosis index, visceral adiposity index and triglyceride × glucose index) in patients with biopsy-confirmed NAFLD. All of these methods diagnosed hepatic steatosis but failed to quantify the severity[26].
More specific scoring methods using other biomarkers, such as α-2-macroglobulin, haptoglobin, apolipoprotein a1, and γ-glutamyl-transferase, have to be developed in order to better select patients for liver biopsy[27].
Clinicians should consider NAFLD in a patient with abnormal liver tests and at least one metabolic risk factor. However, clinical features are nonspecific and patients are usually asymptomatic until they progress to liver cirrhosis.
PATHOGENESIS
Historically, liver injury is thought to be the result of the ”two-hit hypothesis” involving IR and altered adipokine production, resulting in oxidative stress and apoptosis[28] (Figure 2).
Figure 2.
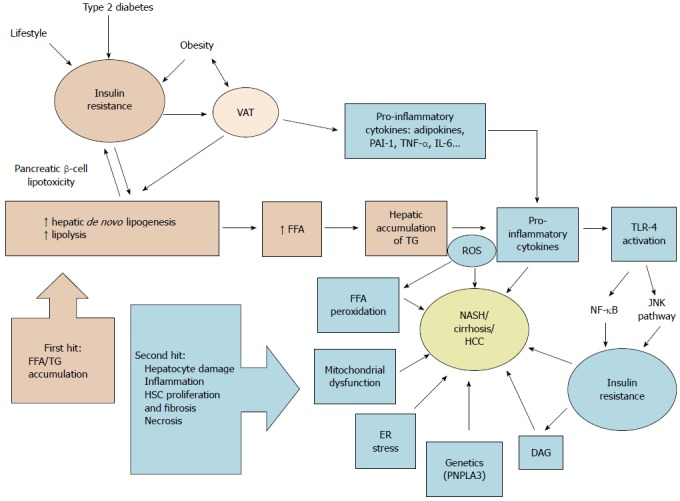
Schematic summary of nonalcoholic fatty liver disease pathophysiology according to the “two-hit hypothesis”. VAT: Visceral adipose tissue; FFA: Free fatty acid; TG: Triglycerides; PAI-1: Plasminogen activator inhibitor-1; TNF-α: Tumor necrosis factor α; IL-6: Interleukin 6; ROS: Reactive oxygen species; TLR-4: Toll-like receptor 4; DAG: Diacylglycerols; ER: Endoplasmic reticulum; NASH: Non-alcoholic steatohepatitis; HCC: Hepatocellular carcinoma; PNPLA3: Patatin-like phospholipase domain-containing protein 3; NF-κB: Nuclear factor-kappa B; JNK: c-Jun N-terminal kinases; HSC: Hepatic stellate cells.
The “two-hit hypothesis” was first described by Day et al[29] in 1998. The first hit represents accumulation of triglycerides (TG) and free fatty acids (FFA) from visceral adipose tissue in hepatocytes secondary to IR. FFA are transported to organs including the liver and undergo either β-oxidation in the mitochondria or are stored as TG. TG stored in the liver come principally from lipolysis of white adipose tissue, but also from dietary lipids and de novo lipogenesis[30]. If an imbalance is present, excessive FFA flux and accumulation induce hepatic IR.
Once hepatic steatosis is established, progression to steatohepatitis involves a “second hit”, consisting of inflammation, mitochondrial dysfunction, enhanced oxidative stress caused by reactive oxygen species, lipid oxidation and production of adipokines resulting in hepatocyte damage and fibrosis[29]. Fatty liver is susceptible to oxidative injury and lipid peroxidation[31].
In 2010 Tilg and Moschen[32] introduced the “multi-parallel hit” hypothesis to explain NAFLD pathogenesis. This hypothesis stresses the importance of gut-derived and adipose tissue-derived factors that promote liver inflammation and fibrosis. This hypothesis, based on reports that endoplasmic reticulum stress[33] and cytokine-mediated stress can induce steatosis as well as necroinflammation, suggests that multiple “hits” act together in parallel in the development of NASH[32]. The role of the gut microbiota in this process will be discussed below.
A more detailed discussion of NAFLD pathogenesis and its link with IR can be found elsewhere[34].
Gut microbiota
The gastroenterological tract contains more than 1014 microorganisms, including more than a thousand bacterial species. The role of gut microbiota in the pathogenesis of obesity is now being recognized. By regulating liver fat deposition and energy homeostasis, gut microbiota may also play a role in NAFLD pathogenesis.
The liver is supplied primarily by the portal system and is therefore exposed to metabolites originating from intestinal bacteria (such as ethanol and other volatile organic compounds) or the bacteria themselves[35]. The liver acts as a barrier between the gut and the systemic circulation by removing toxins. When Kupffer cells, the specialized macrophages in the hepatic sinusoids, are impaired, or when the gut-mucosal barrier is damaged by inflammation or portal hypertension, a metabolic endotoxinemia results. The high endotoxin level activates Kupffer cells and hepatic stellate cells (HSC). Bacteria can also produce lipopolysaccharides (LPS), which bind to Toll-like receptor 4 (TLR-4) and induce the production of pro-inflammatory cytokines[36], subsequently leading to inflammation. These events then contribute to the pathogenesis of obesity and NAFLD[37,38].
Patients with biopsy-proven NAFLD have increased gut permeability and small intestinal bacterial overgrowth, which play an important role in the alteration of hepatic fat metabolism[39]. In obese children with biopsy-proven NAFLD, expression of zonulin, a modulator of intracellular tight junctions, is increased in parallel with the severity of hepatic steatosis. However, there was no significant correlation of plasma zonulin concentrations with lobular inflammation, fibrosis or NASH[40]. These data have not been verified in adults.
Obese people have a different microbiota composition than lean people, with an increase in Firmicutes and a 50% decrease in Bacteroidetes[41,42]. This results in a change in short-chain fatty acids and an increase in intestinal energy absorption[43]. Patients with NAFLD also have different microbiota, with less Bacteroidetes and Lactobacilli and more Prevotella and Porphyromonas compared to healthy controls[44]. However, these findings are controversial with inconsistent data.
Together, bacterial overgrowth and increased intestinal permeability contribute to NAFLD pathogenesis[39]. Mouzaki et al[45], in a prospective cross-sectional study, assessed whether differences in gut microbiota could be associated with the development of NAFLD. The authors found that, independently of diet and BMI, NASH patients contained a lower ratio of Bacteriodetes to Prevotella than did healthy controls. In contrast, Raman et al[46], in an observational case-control study of obese patients with NAFLD vs healthy controls, found that Bacteriodetes representation was similar between the two groups. Interestingly, gut microbiota might contribute to the development of NAFLD through ethanol production[47]. Further studies are needed to clarify whether gut microbiota contributes to NAFLD pathogenesis or if representational differences are a result of the disease. Nevertheless, gut microbiota affects the susceptibility to NASH via metabolic endotoxinemia mediated by bacterial ethanol production, alterations in choline and bile acid metabolism, hepatocyte lipogenesis and increased intestinal permeability[43].
Probiotics modulate intestinal flora and have been proposed as a beneficial complement to NAFLD treatment[48]. Probiotics modulate gut microbiota, reduce inflammation, increase epithelial barrier function, and increase antibacterial substance production[35]. A meta-analysis of four randomized clinical trials showed that probiotic therapy decreases plasma levels of aminotransferases, total cholesterol and HDL cholesterol, and improves the Homeostasis Model Assessment of insulin resistance (HOMA-IR) index[49]. However, these studies were conducted with small group sizes without dietary control. The results should therefore be considered with caution, and the use of probiotics for NAFLD is not recommended at this time[50].
PROGNOSIS
Studies based on histological data suggest that only patients with NASH are at risk of disease progression[27]. Patients with NAFLD are, however, prone to develop type 2 diabetes. In a Swedish cohort study, most patients with NAFLD (78%) were diagnosed with diabetes or impaired glucose tolerance at follow-up. Progression to liver fibrosis occurred in 41% of the patients and was associated with marked IR and pronounced weight gain[51]. A major prognostic issue in NAFLD is hepatocellular carcinoma. Finally, NAFLD is associated with cardiovascular diseases and has emerged as a new cardiovascular risk factor (see below).
Liver transplantation is the treatment for end-stage liver disease. However, de novo NAFLD after transplantation has been reported to be common: in a retrospective study, 75% of the patients developed fatty infiltration of the graft and 38% developed NASH[52].
METABOLIC CONSEQUENCES: CARDIOVASCULAR DISEASE
NAFLD increases the incidence of cardiovascular disease (CVD) and is a predictor of CVD of other risks factors[53]. Accordingly to the review of Edens et al[54], NAFLD is linked to the CVD risk profile. After adjusting for cardiovascular risk factors, NAFLD is independently associated with markers of subclinical atherosclerosis such as impaired flow-mediated vasodilation, increased carotid artery intima-media thickness and arterial stiffness[55]. NAFLD patients are more likely than healthy individuals to have advanced high-risk coronary atherosclerosis, correlated with the severity of hepatic fibrosis[56]. Moreover, the presence of hepatic fibrosis is predictive of cardiovascular events[57]. The coronary artery calcium score is often used as a surrogate marker of coronary atherosclerosis and is considered an independent predictor of CVD[58]. Fatty liver and HOMA-IR are each associated with a high coronary artery calcium score (37.9% and 26.0%, respectively)[59]. In the MESA study, NAFLD was associated with high coronary artery calcium scores and inflammation independently of obesity and metabolic syndrome[11]. Recently, in the “Hepatic steatosis and cardiovascular disease outcomes” sub-analysis of the Framingham Heart study including 3014 participants, there was a significant association of hepatic steatosis with coronary artery calcium score. However, there was a non-significant association between hepatic steatosis and clinical CVD (non-fatal myocardial infarction, stroke, transient ischemic attack, heart failure or peripheral arterial disease)[60]. Interestingly, the increase in cardiovascular events in patients with NAFLD is almost always associated with diabetes[61-63]. NAFLD is frequently associated with dyslipidemia (high triglycerides, low HDL, high VLDL) and increased levels of pro-inflammatory cytokines which are atherogenic[64] and promote the development of CVD[65]. Finally, hepatokines such as fibroblast growth factor 21 (FGF21), fetuin-A and selenoprotein P may also play a role in the development of CVD[66].
ENDOCRINE DISEASES ASSOCIATED WITH NAFLD
Type 2 diabetes
NAFLD is more prevalent in patients with pre-existing metabolic conditions than in the general population. Specifically, type 2 diabetes and NAFLD have a particularly close relationship. A cross-sectional study of patients under 65 with type 2 diabetes found a 69% prevalence of ultrasonographic NAFLD[67] , and the prevalence varies from 30% to 70% in other studies[68,69]. In an Indian cohort, 127 of 204 diabetic patients displayed fatty liver on ultrasound. Among these, 87% were diagnosed with NAFLD after a liver biopsy[70]. Therefore, the prevalence of NAFLD is higher in patients with type 2 diabetes than in the general population, IR being the central mechanism of both diseases.
In addition to having a higher prevalence, liver disease may be more progressive in patients with type 2 diabetes. Diabetic patients with elevated BMI are at higher risk for fibrosis progression[71]. Even without diabetes, IR is a hallmark for cirrhosis[72]. A significant and independent association of degree of IR and stage of fibrosis suggests that severe IR may contribute to fibrosis development in NAFLD[14,73]. Consistent with IR, patients with NAFLD have reduced insulin sensitivity in muscle, liver and adipose tissue[74]. Finally, glucose intolerance or type 2 diabetes is found in 20%-70% of patients with NASH[75,76].
Obesity
Here the prevalence of NAFLD ranges from 57% in overweight individuals attending outpatient clinics to 98% in nondiabetic obese patients[77-79]. The median prevalence of NASH in the obese population is 33%, ranging from 10% to 56%[79-81]. Bariatric surgery is becoming a frequent treatment option and intra-operative liver biopsies are now frequently performed. For example, in a study by Boza et al[80], the prevalence of NAFLD and cirrhosis in a cohort of obese patients undergoing gastric bypass surgery was 63% and 2%, respectively.
In obesity, visceral fat contributes to IR by liberating FFA that accumulates in the liver[82]. Hepatic fat content is correlated with IR as well. In some studies, hepatic fat content cancelled the correlation of visceral fat with IR[83], but in other studies there was an independent contribution of both visceral fat and IR to hepatic fat content[84]. Interestingly, perivascular and epicardial lipid deposits are correlated with atherosclerosis and metabolic syndrome[85]. Moreover, epicardial lipids are correlated with visceral fat, coronary artery disease, presence of NAFLD, and even the severity of liver fibrosis[86,87].
Due to age-related changes in body fat distribution, especially an increase in visceral fat, the prevalence of NAFLD increases with age[88]. Visceral adipose tissue produces FFA and diverse adipokines involved in NAFLD pathogenesis such as increased levels of tumor necrosis factor-α (TNF-α), resistin, and interleukin-6 (IL-6) and decreased levels of adiponectin[89]. Hypertrophied adipocytes promote, via adipokine secretion, accumulation of macrophages in the visceral fat. These macrophages produce pro-inflammatory cytokines, resulting in chronic inflammation that further exacerbates IR[90].
Altogether, these data reveal a central role of obesity in the development of IR and NAFLD.
Adipokines: Adipokines are cytokines secreted by adipose tissue that are involved in adipose homeostasis and lipid metabolism. Many adipokines are being studied as potential targets for new drugs. Adiponectin, ghrelin and leptin are adipokines that decrease IR, while TNF-α and IL-6 are cytokines that enhance IR and, subsequently, NAFLD[91]. However, IL-6 can play either a pro- or an anti-inflammatory role[92,93].
Leptin: Leptin is a protein encoded by the ob gene and produced mainly by adipocytes, but also by the skeletal muscle, stomach, ovaries and liver[94]. This peptide plays an anorexigenic role in the regulation of body weight, acting on the hypothalamus to decrease appetite and increasing energy expenditure via sympathetic stimulation of several tissues. The anti-lipogenic effect of leptin is mediated by lowering the expression of sterol regulatory element binding protein 1, which regulates genes involved in de novo lipogenesis[95]. Leptin down-regulates pre-proinsulin transcription and insulin secretion, explaining why leptin levels are high in insulin-resistant patients[91]. Leptin production is stimulated by pro-inflammatory cytokines (e.g., IL-1, TNF-α)[96]. Expression of leptin in visceral adipose tissue is associated with NAFLD features[97]. Leptin participates in NASH not only via IR but also perhaps in the regulation of HSC, contributing to the development of hepatic fibrosis[98]. In vitro, leptin has a fibrogenic effect on HSC[99] by an unknown mechanism. Mice deficient in leptin signaling are obese and have increased lipid accumulation in liver[100], and leptin infusion in wild-type mice attenuates hepatic steatosis and hyperinsulinemia[101]. In clinical studies, leptin levels are elevated in patients with NASH and correlate with fibrosis severity[102]. However, in some studies this association disappears when leptin levels are adjusted for variables such as age, gender, BMI and hyperinsulinemia, all of which influence leptin levels[14,103].
Adiponectin: Adiponectin, an anti-inflammatory cytokine, is produced predominantly by adipocytes at a level inversely correlated with visceral fat content. Low adiponectin levels are associated with IR and type 2 diabetes, dyslipidemia, hypertension, and NAFLD[104-107]. In animal studies and in vitro, adiponectin exhibits an anti-inflammatory effect by impairing NF-κB activity and inhibiting TNF-α-induced expression of endothelial adhesion molecules. Moreover, adiponectin decreases LPS-induced TNF-α production[108-110]. Anti-oxidative, anti-steatotic and anti-fibrotic effects have also been demonstrated[111]. Indeed, disruption of adiponectin receptors increases tissue triglyceride content, inflammation, oxidative stress and IR[112]. Adiponectin can prevent lipid accumulation in patients with NASH by increasing β-oxidation and by decreasing synthesis of FFA in hepatocytes[113].
In human studies, high plasma levels of adiponectin are correlated with a decreased risk of developing type 2 diabetes[114], and lower adiponectin levels have been shown to be an independent risk factor for NAFLD[115]. Adiponectin levels are correlated with NAFLD progression and are therefore a prognostic factor[116,117].
Endocrine disruptors
Endocrine disruptors (EDCs) are becoming an important health- and environment-related concern. Recent studies indicated that exposure to bisphenol A in utero increases the likelihood of adulthood hepatic steatosis by altering hepatic β-oxidation capacity, possibly through epigenetic mechanisms[118,119].
EDCs (dioxins, phtalates, bisphenol A, persistent organic pollutants) may induce IR, either directly by increasing oxidative stress or indirectly by altering gene transcription, e.g., down-regulating adiponectin[120]. For example, high bisphenol A levels are associated with increased IR and hepatic steatosis[121]. However, the time gap between fetal exposure and adult disease manifestation makes the causal relationship difficult to prove. A systematic review of observational studies demonstrated an association between EDCs and NAFLD but failed to demonstrate causality. Interventional mechanistic studies (reducing or eliminating EDC exposure) are difficult to conduct but are essential for determining the role of EDCs in NAFLD pathogenesis[122].
Sexual hormones
Polycystic ovary syndrome: Polycystic ovary syndrome (PCOS) is an endocrine syndrome frequently encountered in young women of childbearing age (prevalence 8%-15%)[123,124], hallmarked by clinical and/or biological hyperandrogenism, oligo/amenorrhea and polycystic ovarian morphology following ultrasound[124].
Genes influencing obesity and IR, β-cell dysfunction, steroid production and metabolism, androgen receptor and X-inactivation, and ovarian folliculogenesis have been studied as candidates for PCOS pathogenesis[125]. Genowe-wide association studies conducted in women with PCOS have found a relationship between the syndrome and several genes involved in type 2 diabetes, such as THADA, INSR and HMGA2[126]. In European populations, the DENND1A variant is associated with hyperandrogenism and oligomenorrhea[126]. IR occurs in about half of women with PCOS[127]. A recent meta-analysis from Ramezani-Binabaj et al[128] showed that there is a higher risk of NAFLD among women with PCOS (overall OR = 3.93). The prevalence of NAFLD in women with PCOS is between 15% and 55%[129-131], depending on the diagnostic method used. Conversely, the prevalence of PCOS in women with NAFLD is high as well, 71% in one cohort[132].
However, it is not clear whether PCOS is an independent risk factor for NAFLD. Gambarin-Gelwan et al[130] studied lean and obese women with PCOS and found a NAFLD prevalence of 39% in the lean group. Steatosis was associated with a higher BMI and HOMA-IR and a higher prevalence of glucose intolerance and type 2 diabetes. Moreover, women with PCOS had more IR than control women with the same BMI (Figure 3).
Figure 3.
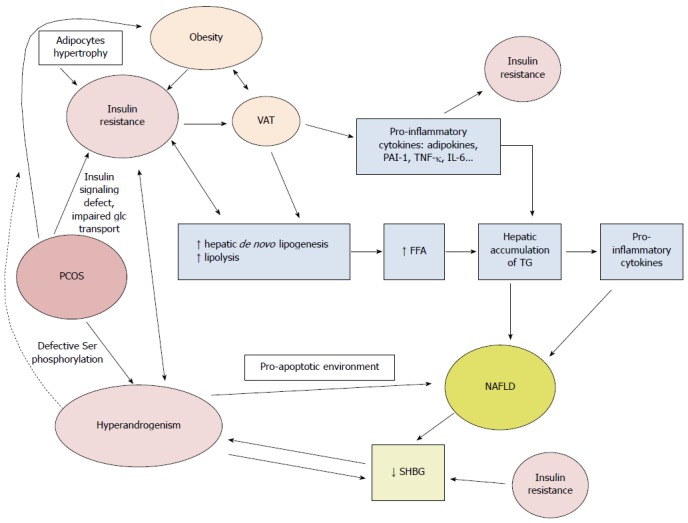
Pathophysiological mechanisms linking polycystic ovary syndrome and nonalcoholic fatty liver disease. Ser: Serine; VAT: Visceral adipose tissue; glc: Glucose; SHBG: Sex hormone binding globulin; FFA: Free fatty acids; TG: Triglycerides; PAI-1: Plasminogen activator inhibitor 1; TNF-α: Tumor necrosis factor α; IL-6: Interleukin 6.
IR is a major player in PCOS, promoting hyperandrogenism via an increased release of androstenedione and testosterone[133]. Insulin acts as a co-gonadotropin to increase luteinizing hormone-stimulated production of androgens; therefore, production of androgens is enhanced in PCOS. A concomitant decrease in sex hormone binding globulin (SHBG) by impaired liver production amplifies this phenomenon, further increasing the levels of free and active androgens. The decrease in SHBG is mediated by IR and hyperandrogenism, further increasing IR. Different hypotheses have been raised to explain why IR is present in PCOS. It is suggested that post-receptor defects in insulin receptor signal transduction are involved, because no structural abnormality in the insulin receptor has been identified in these patients[134]. Impaired glucose transport has also been suggested[135]. Defective serine phosphorylation can lead to both IR and hyperandrogenism, given that serine phosphorylation modulates the activity of key regulating enzymes of androgen biosynthesis, including the 17, 20 lyase activities of P450c17[136,137].
Whether PCOS contributes independently to NAFLD is unclear. PCOS diagnosis is significantly associated with NAFLD, after adjustments are made for age, obesity, waist circumference[138] and dyslipidemia[139]. As hyperandrogenism is a hallmark of PCOS, androgens likely play a role in the development of NAFLD. It has been hypothesized that androgens promote a pro-apoptotic environment[140] which is present in woman with PCOS[141,142]. The caspase 3-cleaved fragment of cytokeratin 18 is released from cells undergoing apoptosis and is now established as a serum marker for NASH. The levels of this fragment correlate with hepatocyte apoptosis and is elevated in women with PCOS[142]. It is not clear whether hyperandrogenism and IR act concomitantly or independently to induce NAFLD, but a synergistic action may be present[143]. Interestingly, oophorectomy in a patient with hyperthecosis reverses hirsutism but not IR[144]. However, the link between hyperandrogenism and NAFLD may be found in the down-regulation of the LDL-receptor, prolonging the half-lives of VLDL and LDL, inducing accumulation of fat in the liver and ultimately triggering NAFLD[141]. In addition, women with hyperandrogenism have higher transaminases levels (predominantly ALT) compared to control subjects, even if the women with hyperandrogenism are lean[138]. Moreover, central obesity and visceral fat are often increased in PCOS women and can be involved in the development of both IR and NAFLD. Indeed, women with PCOS have larger adipocytes, with a diameter increased by 25% (“hypertrophic obesity”), compared to obese women without PCOS (“hyperplastic obesity”)[145]. Hypertrophic obesity is associated with IR and can be mediated by androgens in vivo[146]. The role of leptin in PCOS is unclear. Compared with BMI-matched controls, lean PCOS patients have lower soluble leptin receptor levels, and PCOS per se might cause leptin resistance with higher free leptin indices[147]. Adiponectin levels are lower in women with PCOS (after controlling for BMI-related effects), in relationship with IR, but not in women with hyperandrogenism[148].
The association between PCOS and NAFLD is crucial to recognize considering the former’s young presentation age. It is important to screen young women with PCOS and an associated metabolic syndrome or IR for NAFLD, although the best screening method has not been defined[149]. Routine screening is not recommended by the Endocrine Society[150], but screening high-risk patients seems reasonable. The use of the Fatty Liver Index may be helpful and can identify PCOS patients at high risk for hepatic disturbances[149].
Estrogen deficiency/menopause: Several studies indicate that estrogens play a protective role in NAFLD. NAFLD is more prevalent in post-menopausal women than pre-menopausal women and worsens after menopause[151]. Moreover, estradiol levels in women with PCOS are lower than in women without PCOS[152].
The effects of estrogens are mediated not only through activation of estrogen receptors (ER) α and β but also by non-nuclear activities[153]. Estrogens regulate growth hormone (GH) production and energy homeostasis[154]. In murine models, estrogens have been shown to suppress hepatic fibrosis by attenuating HSC activation[155]. In ERα knockout mice (αERKO) and aromatase knockout mice (ARKO), estrogens are either not synthesized or cannot act properly. These mice contain increased amounts of visceral adipose tissue, as well as an accumulation of lipid droplets in the liver of ARKO mice, highlighting the importance of estrogens in lipid homeostasis[156]. αERKO mice manifest adipocyte hyperplasia and hypertrophy, IR and glucose intolerance in both sexes[157], and steatosis in males[158]. Interestingly, patients with an aromatase gene inactivating mutation (aromatase deficiency) exhibit estrogen deficiency, development of metabolic syndrome with IR, steatohepatitis and precocious atherogenesis. When these patients undergo estrogen treatment, their IR and liver steatohepatitis improve[159]. Moreover, estrogen replacement therapy in mice has been shown to prevent diet-induced ectopic lipid (notably diacylglycerols) deposition as well as hepatic and muscle IR[160].
Male hypogonadism: Male hypogonadism includes biochemical and clinical features such as low testosterone and/or low sperm count, erectile dysfunction, diminished libido, decrease in lean body mass and increase in visceral fat, as defined by the International Society of Andrology, the International Society for the Study of the Aging Male and the European Association of Urology.
Testosterone plays a key role in insulin sensitivity, body composition and lipid metabolism[161]. A bidirectional relationship exists between low levels of testosterone and IR[161-163]. The HERITAGE study indicated that people with lower testosterone levels have a preferential accumulation of abdominal fat and a higher visceral adipose tissue accumulation[164]. Low levels of testosterone and SHBG in men are independent predictors of the occurrence of metabolic syndrome[165]. Men with metabolic syndrome have a higher prevalence of low testosterone compared to healthy controls[166,167]. Furthermore, according to the hypogonadal-obesity-adipokine hypothesis, increased amounts of adipose tissue converts testosterone to estradiol via aromatase activity. Estradiol inhibits kisspeptin liberation and testosterone production. Moreover, adipose tissue produces leptin and pro-inflammatory cytokines that both have an effect on the gonadal axis, impairing testosterone production. Leptin has an additional effect on Leydig cells, resulting in decreased androgen production[168].
In a retrospective cohort study, hepatic steatosis, defined by sonographic criteria, was correlated with low testosterone levels (< 14.2 nmol/L) after adjusting for diverse confounders (including age, BMI, smoking, diabetes, and visceral adipose tissue)[169]. A recent cross-sectional study using data from MESA study showed that men with the highest tertile of SHBG were less likely to have a fatty liver, defined by computed tomography, than those in the lower tertile[170].
Interventional studies using testosterone replacement therapy in hypogonadal men have shown that testosterone not only improved insulin sensitivity but also decreased waist circumference[171,172] together with BMI[173]. In obese men with sleep apnea, testosterone replacement therapy led to increased insulin sensitivity and reduced liver fat content[174]. In castrated rats on a high-fat diet, testosterone replacement therapy led to a lower body fat percentage and only mild-moderate microvesicular steatosis compared to castrated rats not receiving testosterone, which displayed severe micro-and macrovesicular fat in hepatocytes[175]. However, the evolution of NAFLD during testosterone replacement therapy in men has not been studied in clinical trials.
Osteoporosis
Evidence for an important triumvirate (NAFLD, osteoporosis and metabolic syndrome) is rising[176]. A complex crosstalk of mediators coming from the liver (fetuin-A), adipose tissue (leptin, TNF-α, adiponectin) and bone (osteopontin, osteocalcin, osteoprotegerin) may contribute to the development of NAFLD and metabolic syndrome[177], and the protective effect of obesity on bone mass is progressively challenged[178]. For example, insulin can increase bone formation by binding to the insulin receptor on osteoblasts, and leptin and adiponectin can suppress bone formation or stimulate resorption. Conversely, the bone also affects glucose metabolism, by secreting cytokines, hormones and peptides like osteocalcin which increase pancreatic β-cell function[179]. Mice lacking the insulin receptor on osteoblasts develop obesity and IR that are improved after osteocalcin administration, suggesting the presence of a bone-pancreas loop[180].
In post-menopausal women with an ultrasonographic diagnosis of NAFLD, lumbar bone mass density was found to be lower (0.98 ± 0.01 g/cm² vs 1.01 ± 0.02 g/cm², P = 0.046) than in controls, after adjusting for age, BMI, ALT levels, smoking and alcohol consumption. This phenomenon was also demonstrated after adjusting for metabolic syndrome[106]. Among Asian men, NAFLD (diagnosed by ultrasound) was significantly associated with osteoporotic fractures, defined as fractures secondary to low trauma[181]; however, the association did not reach significance in women[105].
Treatment of NAFLD may also have an impact on bone. Thiazolidinediones, which are peroxisome proliferator-activated receptor γ (PPARγ) agonists, improve insulin sensitivity and reduce hepatic fibrosis progression[179], but also increase bone loss and fractures, especially vertebral fractures in males with type 2 diabetes[182].
Further studies are needed to better understand the interactions between osteoporosis and NAFLD.
Vitamin D deficiency
The pleiotropic effects of vitamin D, particularly on metabolism and the immune system, are being increasingly studied. NAFLD has been associated with low 25-OH vitamin D levels. Notably, a recent meta-analysis found that NAFLD patients are 26% more likely to be deficient in vitamin D compared with controls[183]. However, the two conditions are quite frequent and the association may be fortuitous. The use of a cross-sectional approach and the method to diagnose NAFLD are two limitations of this study. Other studies found that 25-OH vitamin D levels can predict the histological severity of NAFLD, with NASH patients having lower levels than individuals with simple steatosis, even children[184,185]. However, in the study of Dasarathy et al[184], the control group was smaller in number, had a lower BMI and was not age-matched. These differences may influence vitamin D levels, as obese patients have lower vitamin D levels.
It is unclear how vitamin D could prevent or slow the development of NAFLD. However, vitamin D has been shown to inhibit the proliferation of HSC, which express the vitamin D receptor[186], and therefore could reduce the fibrotic process. The work of Roth et al[187] demonstrates in a rat model that phototherapy can reduce fibrosis, apoptosis and inflammation, primarily by reducing hepatic expression of inflammatory genes such as TNF-α and transforming growth factor β. The authors conclude that vitamin D deficiency exacerbates inflammatory gene expression and is partially reversible[188]. In another study, treatment of adipocytes with calcitriol (1,25-OH-vitamin D) caused the GLUT4 transporter to be upregulated and translocated to the cell surface, resulting in increased glucose uptake and utilization[189]. A double-blind, placebo-controlled Iranian study[190] showed that vitamin D supplementation decreases the inflammatory marker hs-CRP, but there was no effect on liver enzymes, HOMA-IR or steatosis grade (evaluated by ultrasound). However, this study enrolled only 53 patients and was conducted for a short period of time.
Altogether, only a limited number of prospective and randomized studies have analyzed the impact of vitamin D supplementation on NAFLD. Therefore, the effect of vitamin D supplementation on NAFLD has to be further studied, as concluded in a recent review by Eliades et al[191].
Pituitary gland
Growth hormone insufficiency: GH and insulin-like growth factor-1 (IGF-1) insufficiency have recently been associated with NAFLD, progression to NASH and even liver cirrhosis. NAFLD is more common in hypopituitary patients than control subjects and patients with growth hormone deficiency (GHD) are likely to have an increased risk of developing NAFLD. In a Korean cohort of men with hypopituitarism, the frequency of NAFLD (diagnosed by abdominal ultrasonography) was significantly higher in hypopituitary men than in control subjects (32.5% vs 70.6%, P = 0.001). CRP and FFA were significantly elevated in hypopituitary patients with NAFLD compared to hypopituitary patients without NAFLD. Moreover, the severity of NAFLD correlated negatively with GH after adjusting for BMI (P = 0.020). Severe GHD in hypopituitarism was associated with more advanced NAFLD[192]. In one series NAFLD developed after 6.4 ± 7.5 years (median 3 years) in GHD patients[193].
GHD leads to visceral adiposity, reduced lean body mass, an abnormal lipid profile and IR[194]. However, the exact pathophysiological mechanisms need to be clarified[195]. Recent data show a relationship between low IGF-1 and Sirtuin4 (Sirt4) levels. Obese patients with low levels of GH or IGF-1 have a higher waist circumference and/or metabolic syndrome. Like GH, which regulates mitochondrial oxidative capacity, Sirt4 is a mitochondrial NAD-dependent ADP-ribosyltransferase that inhibits mitochondrial glutamate dehydrogenase 1 activity, thereby down-regulating insulin secretion in response to amino acids. Sirt4 functions within the mitochondria as a negative regulator of oxidative capacity. Levels of Sirt4 are low in obese patients, in order to preserve fat oxidative capacity and mitochondrial function in liver and muscle[196]. Sirt4 reduces plasma FFA but, in turn, increases reactive oxygen species. In obese patients with NAFLD, the combination of FFA and oxidative stress products results in endothelial dysfunction and can be a coronary risk factor[197]. Oxidative stress is an important feature of the pathogenesis of NAFLD. As IGF-1 is known to have antioxidative effects and improve mitochondrial function, low IGF-1 levels may enhance oxidative stress and promote NAFLD[198,199].
Interventional studies regarding GH substitution are controversial. Some studies found an improvement after GH replacement[200], whereas others found only a reduction of abdominal and visceral fat without any impact on liver fat[201]. In one study the prevalence of NAFLD among patients with GHD was significantly higher than among controls (77% vs 12%, P < 0.001)[200]. After the introduction of GH replacement therapy, a reduction in the levels of liver enzymes and fibrosis markers (hyaluronic acid and type IV collagen) were noticed. Six months of GH-replacement therapy improved NASH and reduced oxidative stress[202]. GH replacement therapy also decreased serum levels of hsCRP and TNF-α, and drastically reversed NASH[202].
To our knowledge, there is data concerning NAFLD in acromegalic patients.
Hyperprolactinemia: Prolactin may be elevated in diverse conditions such as pituitary adenomas or by certain drugs. Hyperprolactinemia is seen in men with liver disease as well and is unrelated to the presence of gynecomastia[203]. Prolactin is not only a lactotroph hormone, but also regulates enzymes and transporters associated with glucose metabolism (stimulates insulin secretion) and lipid metabolism (suppresses lipid storage and adipokine release)[204-206]. Furthermore, adipose tissue produces prolactin in an autocrine and paracrine manner. Therefore, a potential role of prolactin in NAFLD may be evoked but has never been studied.
Bromocriptine, a dopamine agonist, has been linked to improvements in obesogenic behaviors, hepatic lipid accumulation, glucose tolerance and mitochondrial oxidative stress in rats and was therefore proposed as a therapy for NAFLD[207]. However, prolactin levels were not measured in the study.
Thyroid gland: hypothyroidism
Thyroid hormones play an important role in hepatic lipid metabolism, increasing hepatic lipogenesis and enhancing β-oxidation[208]. Increased fatty acid oxidation may produce reactive oxygen species, damaging hepatocytes[209]. Therefore, hypothyroidism is associated with reduced lipolysis and decreased liver uptake of FFA derived from triglycerides. Moreover, thyroid hormones modify hepatic fat accumulation, affecting adiponectin regulation. Hence, thyroid hormones could control the development of fibrosis through the modulation of adiponectin[209,210]. Increased leptin and FGF21 secretion may also play a role in this pathogenesis[209].
Thyroid hormones mediate their actions through thyroid hormone receptors. Thyroid receptor α (THRα) is ubiquitously expressed and THRβ is mainly expressed in the liver, brain and kidney[211]. Rodent studies show that THRβ agonists diminish hepatic lipid accumulation[212]. Mice lacking THRα do not develop high-fat diet-induced hepatic steatosis and IR[213]. Moreover, hypothyroidism has been associated with disorders of glucose and insulin metabolism involving IR[214] which can influence the development of fatty liver disease. The relationship between thyroid dysfunction and NAFLD is controversial. Both diseases share common features such as metabolic syndrome, obesity, IR and the disturbance of lipid metabolism[209]. There is, however, no proven cause-effect relationship between the two conditions. In an Indian cohort, patients with NAFLD had higher thyroid stimulating hormone (TSH) levels and lower free thyroxine levels than control subjects[215]. Overt hypothyroidism has been associated with NAFLD[216,217] with a prevalence of 30.2% vs 19.5% in control subjects, even after adjusting for age, gender, BMI, diabetes and hypertension[218]. In a Chinese study, the prevalence of NAFLD increased in parallel to the degree of hypothyroidism: 29.9% for subclinical hypothyroidism and 36.3% for overt hypothyroidism. Each 1U/L increment of TSH was associated with a 20% increase in NAFLD prevalence, independently of classical risk factors[217]. These findings were confirmed by other studies[218,219]. Several studies demonstrated that an increased TSH level is an independent risk factor for NASH in patients with NAFLD[220]. In a recent cross-sectional study, compared to the low normal range (< 2.5 mIU/L), TSH levels within the upper normal range (2.5-4.5 mIU/L) were associated with a 40% increased risk for NAFLD after adjusting for age, gender, BMI, waist circumference, triglyceride levels, HDL cholesterol levels, hypertension, and diabetes[217]. However, in an Iranian cohort, there was no statistically significant difference in serum TSH, free T4 or free T3 levels between participants with or without NAFLD[221]. Moreover, a study by Mazo et al[222] did not show any statistically significant association between NASH and hypothyroidism. Nevertheless, a recent systematic review of 11 studies on this subject suggests that hypothyroidism is an independent risk factor for NAFLD. The prevalence of hypothyroidism ranged from 15.2% to 36.3% among patients with NAFLD/NASH[209]. Although this association has not been uniformly reported, further research is needed to confirm previous findings. However, it is unclear whether a low-normal thyroid function, but still within the euthyroid range, is related to NAFLD[223]. A cross-sectional study in euthyroid elderly Chinese individuals found that the prevalence of NAFLD is negatively correlated with serum free thyroxine[224]. An Italian retrospective study showed that serum gamma-glutamyltransferase and ALT concentrations increase steadily along with TSH categories, after adjusting for gender, age, lipids and fasting glucose concentrations[225]. It is important to determine whether hypothyroidism has an impact on NAFLD pathogenesis, as hypothyroidism is easily identifiable and treatable. Conversely, hepatic steatosis may influence thyroid function[226].
The use of thyromimetics, which are thyroid hormone analogs that either have selective effects on the liver or the heart, or bind selectively to TRβ rather than to TRα without cardiac side effects, are under consideration. Such compounds could be powerful new tools to address some of the largest medical problems in developed countries, i.e., obesity and related disorders such as NAFLD[227]. Interestingly, thyroid hormones also exert non-genomic effects attributable to naturally occurring iodothyronines apart from T4 and T3[228]. Further studies are needed in this field.
To our knowledge, there is no association between hyperthyroidism and NAFLD.
Adrenal gland
Different pathologies can affect the adrenal gland, several of which appear to relate with NAFLD. One study on patients with adrenal incidentalomas found that there is no increased incidence of NAFLD in these patients[229].
Glucocorticoids - Cushing syndrome: Hypercortisolism shares metabolic features with metabolic syndrome like IR, dyslipidemia, hypertension, visceral obesity and hepatic steatosis. Cortisol is known to impair insulin sensitivity, directly by interfering with the insulin receptor pathway or indirectly by stimulating lipolysis and proteolysis, thereby increasing FFA and amino acid release. In addition, plasma glucose is increased due to stimulated gluconeogenesis[230].
However, patients with Cushing’s syndrome have a low prevalence of hepatic steatosis[231], estimated at 20%[232]. It is hypothesized that there is a local increase of available glucocorticoids through the enzymatic activity of 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1)[233]. Indeed, in obese individuals, there is increased regeneration of cortisone to cortisol mediated by increased activity of 11β-HSD1. This enzyme is expressed in the brain, adipose tissue and liver[234], and converts cortisone into active cortisol, which is able to promote metabolic changes[235]. However, not all studies have shown an increase of 11β-HSD1 activity in obese patients[236]. Another possible factor in NAFLD pathogenesis in Cushing syndrome is decreased clearance of cortisol through A-ring metabolism (5α- and 5β-reductase). Indeed, 5α-reductase type 1 deletion accelerates the development of hepatic steatosis[237]. In the early stages of NAFLD, only hepatic steatosis is observed. During this time, to protect the liver from cortisol exposure, 5α-reductase activity is increased, thus increasing cortisol clearance. Concomitantly, 11β-HSD1 activity is decreased, decreasing cortisol production, which results in hypothalamic-pituitary-adrenal axis activation[238]. In the latter stages of NAFLD, especially in NASH, there is an increase in hepatic 11β-HSD1 expression[239], which increases intra-hepatic glucocorticoid levels. In addition, increased expression of glucocorticoid receptor α and decreased activity of 5α-reductase accentuate this mechanism, resulting in hepatic lipid accumulation. Therefore, progression of NAFLD is complex, with a switch from glucocorticoid inactivation to activation[240].
11β-HSD1 inhibitors are currently being developed to impair this phenomenon. Such an approach may be beneficial during the initial phase of steatosis but deleterious afterwards, notably by increasing inflammation[240].
Low levels of dehydroepiandrosterone/dehydroepiandrosterone sulphate: The effects of low levels of dehydroepiandrosterone (DHEA), an important and abundant steroid that influences oxidative stress, insulin sensitivity and expression of PPARα, is controversial. A positive relationship between histologically advanced NAFLD and low levels of dehydroepiandrosterone sulphate (DHEA-S) has been found, but the study was performed with two different groups, one in obese patients undergoing surgery and the other in suspected NAFLD patients[241]. Indeed, low serum levels of GH and DHEA are very common in patients with NASH and more advanced fibrosis[242]. Moreover, another group found high levels of DHEA-S in patients with NAFLD, but NAFLD wasn’t histologically diagnosed[243]. Therefore, it remains unclear whether DHEA plays a role in NAFLD pathogenesis, or if this was an isolated finding.
Hyperaldosteronism: The Renin-Angiotensin-Aldosterone-System (RAAS) acts not only in the vascular system, but also in different organs such as the liver. Indeed, the angiotensin II receptor 1 (AT1) and receptor 2 (AT2) are abundant in different tissues, and in the liver the former is expressed in hepatocytes, bile duct cells, HSC, KC, myofibroblasts, and vascular endothelial cells[244]. AT1 receptor activation by angiotensin II induces HSC contraction and proliferation, causes oxidative stress, endothelial dysfunction, cell growth and inflammation[245]. The expression of AT2 in the liver has also been reported[246], with possible anti-fibrogenic effects.
An Italian cross-sectional pilot study found that in a selected population without other metabolic risk factors, patients with primary hyperaldosteronism and hypokalemia have a higher prevalence of NAFLD than normotensive controls[247]. Insulin sensitivity was lower in this group of patients, either impaired directly by aldosterone or indirectly by potassium loss[248]. Indeed, RAAS activation can increase IR. Angiotensin II stimulates phosphorylation of serine residues in the insulin receptor β-subunit and the p85 regulatory subunit of PI3-kinase, thereby inhibiting the interactions between these two components of the insulin signaling pathway[249]. Activation of NADPH oxidase subsequently generates reactive oxygen species which modulate the production of pro-inflammatory cytokines such as TNF-α and IL-6, resulting in the impairment of insulin signaling[250]. There is a growing interest in using RAAS inhibitors to treat NAFLD. Indeed, blocking RAAS reduced fibrosis in an experimental model of hepatic fibrosis[251]. Telmisartan and valsartan improved transaminases levels and insulin sensitivity, and telmisartan also significantly decreased NASH activity score and fibrosis[252]. Despite encouraging results in animal studies, RAAS inhibitors do not show consistent efficacy in NAFLD patients. A recent study by Goh et al[253] demonstrated that in a hypertensive cohort with biopsy-proven NAFLD, patients treated with RAAS inhibitors had less advanced hepatic fibrosis, indicating a beneficial effect of this class of anti-hypertensive drugs. This effect remains controversial, however, as some studies did not observe any benefit of RAAS blockers on hepatic fibrosis[251,254].
Larger randomized clinical trials are needed to directly assess the effectiveness of angiotensin converting enzyme inhibitors and angiotensin II receptor blockers in NAFLD.
TREATMENT
In endocrine disorders, the most appropriate course of action is first to treat the underlying disturbance. However, in the case of metabolic diseases such as diabetes or obesity, alternative approaches are needed.
Lifestyle is the first-line therapy[179,255]. A weight loss superior or equal to 7% improves histological disease activity[179]. Diet and exercise improve weight loss, steatosis and lobular inflammation[256]. Dietary composition is important, as reducing carbohydrate or fat intake can reduce intrahepatic lipid content[257]. It is important to emphasize that certain diets such as the low carbohydrate and high fat diet or diets rich in fatty acids or refined carbohydrates may exacerbate NAFLD[255]. Ketogenic diets impair fibroblast growth factor 21 (FGF21) signaling and enhance lipid accumulation in the liver, which may explain hepatic inflammation[258]. Dietary interventions can also modify gut microbiota, as already discussed. However, aggressive weight loss (> 1.6 kg/wk) removes lipids and fatty acids from visceral fat that can be taken up by the liver, exacerbating hepatic inflammation[259]. Some diets are more prone than others to NAFLD. Notably, diets high in saturated fatty acids, low carbohydrate diets, or diets rich in refined carbohydrates such as soft drinks, can exacerbate NAFLD[255,260], although this issue is controversial.
Insulin sensitizers
IR plays a pivotal role in NAFLD pathogenesis. Therefore, insulin sensitizers have been proposed as a treatment. Metformin, a biguanide used for type 2 diabetes, decreases hepatic gluconeogenesis and lipogenesis[179,261]. However, there is no improvement in histology, and four randomized clinical trials have failed to demonstrate a significant beneficial impact of metformin on NAFLD progression[262]. Thiazolidinediones, PPARγ agonists used for type 2 diabetes, have effects on adipose tissue and reduce liver fat deposition. They interact with metabolic regulators such as adiponectin, AMPK, Foxo1 and peroxisome proliferator-activated receptor gamma coactivator 1-α (PGC-1α)[263]. In the “Pioglitazone vs Vitamin E vs Placebo for Treatment of Non-Diabetic Patients with Nonalcoholic Steatohepatitis” (PIVENS) trial[264], pioglitazone failed to meet the primary end-point, i.e., improvement of histologic features of NASH (fibrosis score), but improved ALT/AST levels, hepatic steatosis, lobular inflammation, insulin sensitivity, and steatohepatitis. Vitamin E improved steatohepatitis but not significantly. A pilot study reported that pioglitazone improved biochemical and histological features of NAFLD (steatosis, cell injury, inflammation, Mallory bodies, fibrosis), but there was no control group[265]. Recently, Pawlak et al[266] showed that the transrepression activity of PPARα may prevent progression of NASH to liver fibrosis. A meta-analysis of seven randomized clinical trials with post-treatment histology reported that thiazolidinediones improved histological activity (steatosis, hepatic ballooning, inflammation), plasma glucose and lipid levels, and reduced the risk of fibrosis progression[179]. However, the side effects (increased weight, raised fluid retention and heart failure[267], fractures, and bladder carcinoma) may limit the use of this class of drugs. Four randomized clinical trials with either pioglitazone or rosiglitazone confirmed the improvement of steatosis, ballooning and lobular inflammation, but did not address the long-term effects[262]. Finally, incretins, neuroendocrine hormones produced by the gastrointestinal tract in response to food, stimulate insulin release and decrease glucagon levels. Glucagon-like peptide-1 agonists and dipeptidyl peptidase-4 inhibitors are currently being studied, and transaminase levels have been shown to be reduced under these treatments[268].
Lipid-lowering drugs
Two types of lipid-lowering agents have been used in clinical studies: 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors (statins) and ezetimibe. Statins have many effects, one of which is PPARγ agonism. Unfortunately, only a few pilot studies with small cohorts have used statins in monotherapy to evaluate hepatic histology in NAFLD[269,270]. Hyogo et al[270] found that atorvastatin improved transaminases levels and steatosis, but four patients had progression of fibrosis. In another study, statins improved hepatic steatosis and transaminases levels[271] but the impact on histology was not addressed[179]. Ezetimibe, an antagonist of Niemann-Pick C1-like protein, a key player in cholesterol absorption from the small intestine, may have an impact on NAFLD, but large-scale studies are needed to confirm this effect[272]. The combination of statins and ezetimibe, along with lifestyle changes, may represent a useful approach[273]. In summary, both classes of lipid-lowering drugs show promising results but need further investigation.
Anti-hypertensive drugs
Angiotensin II receptor blockers inhibit hepatic inflammation and fibrosis via inhibition of fibroblast activity and prevention of HSC proliferation. A few studies have shown an improvement in liver histology and transaminases levels with the use of angiotensin II receptor blockers[274]. Both telmisartan and valsartan were beneficial, but there was only one randomized clinical trial[252]. Details of this trial were discussed above.
Anti-oxidants and cytoprotective agents
Anti-oxidant and cytoprotective therapies have been evaluated for their effects on the inflammatory component of NAFLD. Vitamin E was studied in the PIVENS trial[264] and found to significantly improve hepatic steatosis but not fibrosis. However, the safety of large doses of vitamin E must be demonstrated, as it can increase IR and plasma triglyceride levels[179,275].
Betaine, a metabolite of choline which reduces oxidative stress, was tested but did not improve steatosis[276]. Ursodeoxycholic acid, a bile acid with antioxidant properties, failed to improve histological features[277,278]. As for pentoxyphylline, a TNF-α inhibitor, only one study showed an improvement in histology (steatosis and lobular inflammation, with only a trend for fibrosis) and transaminases levels[279].
Salsalate, a prodrug of salicylate with anti-inflammatory effects, was found to decrease steatosis and can therefore represent a new target drug if confirmed in larger studies[280].
TNF-α inhibitors like etanercept have been studied in patients with psoriasis. These inhibitors reduced transaminase and fasting insulin levels while exhibiting anti-inflammatory effects and improved insulin sensitivity.
Probiotics
Probiotics have been studied in a few trials, as previously discussed in the section on gut microbiota.
FGF21 analogs
FGF21 is an endocrine factor of the fibroblast growth factor family that improves insulin sensitivity in rodent models of IR. Administration of FGF21 decreased hepatic fat content and improved glucose homeostasis in mice[281,282]. Increased serum levels of FGF21 are found in patients with NAFLD, perhaps due to FGF21 resistance[283-286]. FGF21 analogs have been studied in humans and improve dyslipidemia, decrease body weight and fasting insulin plasma levels and increase adiponectin levels[287]. Several drugs are thought to regulate the FGF21 pathway, including resveratrol, a natural Sirtuin1 activator[288]. In diabetic rhesus monkeys, FGF21 administration improves insulin sensitivity and the lipid profile[289]. The potential beneficial effects of FGF21 in NAFLD patients warrant further investigation.
Gastric bypass
Surgical procedures such as bariatric interventions (notably gastric bypass) may lead to the resolution of liver steatosis. In one study of patients biopsied at the time of bariatric surgery and at follow-up, hepatic fat content was reduced in 65 out of 91 patients, whereas increases in the steatotic score were observed in only three patients[290]. Another study of 90 biopsied bariatric surgery patients showed that 16 patients (18%) had the same degree of steatosis, 25 (28%) had improved steatosis, and 49 (54%) had normal hepatic tissue in the second biopsy[291]. A recent French prospective study of 109 patients with morbid obesity and histologically-proven NASH showed that, one year after bariatric surgery, NASH had disappeared in 85% of the patients. The results were better in patients with mild NASH before surgery (94%) than severe NASH (70%), according to the Brunt scores. Histologically, steatosis decreased from 60% of the tissue before surgery to 10%, hepatocellular ballooning was reduced in 84.2% of samples, lobular inflammation was reduced in 67.1% of samples and fibrosis was reduced in 33.8% of the patients, according to the Metavir scores[292]. However, the guidelines indicate that it is premature to consider bariatric surgery as an option to treat NASH[293].
Orlistat, a lipase inhibitor, was tested in a pilot study of 10 obese patients, resulting in weight loss and improved aminotransferase levels, steatosis, and fibrosis[294].
CONCLUSION
It is important to note that NAFLD, the most common chronic liver disease in Western countries, is intimately entangled with various endocrine diseases, sharing the keystone physiopathological mechanism of IR. In the coming years, genetics will allow us to better understand the interrelationships between these different entities in order to better target treatments. Additional studies are needed to reveal the subtle links between common diseases like NAFLD and hypothyroidism, for example, and ensure their interdependence. Regarding treatment, we have seen that many drugs are useful not only for preventing the evolution of liver disease, but also against IR found in metabolic diseases. Prevention of metabolic syndrome is still important to prevent progression of NAFLD. Reciprocally, both gastroenterologists and endocrinologists should consider the relationship between NAFLD and endocrine diseases in everyday medical practice.
ACKNOWLEDGMENTS
We want to thank Mrs. Caroline Spaight for her kind assistance in revising the English language of the present review.
Footnotes
Conflict-of-interest statement: The authors declare no conflict of interests.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: April 16, 2015
First decision: May 18, 2015
Article in press: August 28, 2015
P- Reviewer: Gnocchi D, Pacifico L, Trovato GM, Tarantino G, Vassilatou E S- Editor: Yu J L- Editor: Filipodia E- Editor: Liu XM
References
- 1.Sass DA, Chang P, Chopra KB. Nonalcoholic fatty liver disease: a clinical review. Dig Dis Sci. 2005;50:171–180. doi: 10.1007/s10620-005-1267-z. [DOI] [PubMed] [Google Scholar]
- 2.Zelber-Sagi S, Nitzan-Kaluski D, Halpern Z, Oren R. Prevalence of primary non-alcoholic fatty liver disease in a population-based study and its association with biochemical and anthropometric measures. Liver Int. 2006;26:856–863. doi: 10.1111/j.1478-3231.2006.01311.x. [DOI] [PubMed] [Google Scholar]
- 3.Vernon G, Baranova A, Younossi ZM. Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment Pharmacol Ther. 2011;34:274–285. doi: 10.1111/j.1365-2036.2011.04724.x. [DOI] [PubMed] [Google Scholar]
- 4.Karbasi-Afshar R, Saburi A, Khedmat H. Cardiovascular disorders in the context of non-alcoholic Fatty liver disease: a literature review. J Tehran Heart Cent. 2014;9:1–8. [PMC free article] [PubMed] [Google Scholar]
- 5.Williams CD, Stengel J, Asike MI, Torres DM, Shaw J, Contreras M, Landt CL, Harrison SA. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: a prospective study. Gastroenterology. 2011;140:124–131. doi: 10.1053/j.gastro.2010.09.038. [DOI] [PubMed] [Google Scholar]
- 6.Das K, Das K, Mukherjee PS, Ghosh A, Ghosh S, Mridha AR, Dhibar T, Bhattacharya B, Bhattacharya D, Manna B, et al. Nonobese population in a developing country has a high prevalence of nonalcoholic fatty liver and significant liver disease. Hepatology. 2010;51:1593–1602. doi: 10.1002/hep.23567. [DOI] [PubMed] [Google Scholar]
- 7.Jimba S, Nakagami T, Takahashi M, Wakamatsu T, Hirota Y, Iwamoto Y, Wasada T. Prevalence of non-alcoholic fatty liver disease and its association with impaired glucose metabolism in Japanese adults. Diabet Med. 2005;22:1141–1145. doi: 10.1111/j.1464-5491.2005.01582.x. [DOI] [PubMed] [Google Scholar]
- 8.Omagari K, Kadokawa Y, Masuda J, Egawa I, Sawa T, Hazama H, Ohba K, Isomoto H, Mizuta Y, Hayashida K, et al. Fatty liver in non-alcoholic non-overweight Japanese adults: incidence and clinical characteristics. J Gastroenterol Hepatol. 2002;17:1098–1105. doi: 10.1046/j.1440-1746.2002.02846.x. [DOI] [PubMed] [Google Scholar]
- 9.Fan JG, Zhu J, Li XJ, Chen L, Li L, Dai F, Li F, Chen SY. Prevalence of and risk factors for fatty liver in a general population of Shanghai, China. J Hepatol. 2005;43:508–514. doi: 10.1016/j.jhep.2005.02.042. [DOI] [PubMed] [Google Scholar]
- 10.Alberti KG, Zimmet P, Shaw J. The metabolic syndrome--a new worldwide definition. Lancet. 2005;366:1059–1062. doi: 10.1016/S0140-6736(05)67402-8. [DOI] [PubMed] [Google Scholar]
- 11.Al Rifai M, Silverman MG, Nasir K, Budoff MJ, Blankstein R, Szklo M, Katz R, Blumenthal RS, Blaha MJ. The association of nonalcoholic fatty liver disease, obesity, and metabolic syndrome, with systemic inflammation and subclinical atherosclerosis: the Multi-Ethnic Study of Atherosclerosis (MESA) Atherosclerosis. 2015;239:629–633. doi: 10.1016/j.atherosclerosis.2015.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Petersen KF, Dufour S, Hariri A, Nelson-Williams C, Foo JN, Zhang XM, Dziura J, Lifton RP, Shulman GI. Apolipoprotein C3 gene variants in nonalcoholic fatty liver disease. N Engl J Med. 2010;362:1082–1089. doi: 10.1056/NEJMoa0907295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kahali B, Liu YL, Daly AK, Day CP, Anstee QM, Speliotes EK. TM6SF2: catch-22 in the fight against nonalcoholic fatty liver disease and cardiovascular disease? Gastroenterology. 2015;148:679–684. doi: 10.1053/j.gastro.2015.01.038. [DOI] [PubMed] [Google Scholar]
- 14.Angulo P, Alba LM, Petrovic LM, Adams LA, Lindor KD, Jensen MD. Leptin, insulin resistance, and liver fibrosis in human nonalcoholic fatty liver disease. J Hepatol. 2004;41:943–949. doi: 10.1016/j.jhep.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 15.AlShaalan R, Aljiffry M, Al-Busafi S, Metrakos P, Hassanain M. Nonalcoholic fatty liver disease: Noninvasive methods of diagnosing hepatic steatosis. Saudi J Gastroenterol. 2015;21:64–70. doi: 10.4103/1319-3767.153812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Caballería L, Pera G, Auladell MA, Torán P, Muñoz L, Miranda D, Alumà A, Casas JD, Sánchez C, Gil D, et al. Prevalence and factors associated with the presence of nonalcoholic fatty liver disease in an adult population in Spain. Eur J Gastroenterol Hepatol. 2010;22:24–32. doi: 10.1097/MEG.0b013e32832fcdf0. [DOI] [PubMed] [Google Scholar]
- 17.Stefan N, Kantartzis K, Häring HU. Causes and metabolic consequences of Fatty liver. Endocr Rev. 2008;29:939–960. doi: 10.1210/er.2008-0009. [DOI] [PubMed] [Google Scholar]
- 18.Ioannou GN, Boyko EJ, Lee SP. The prevalence and predictors of elevated serum aminotransferase activity in the United States in 1999-2002. Am J Gastroenterol. 2006;101:76–82. doi: 10.1111/j.1572-0241.2005.00341.x. [DOI] [PubMed] [Google Scholar]
- 19.Ruhl CE, Everhart JE. Determinants of the association of overweight with elevated serum alanine aminotransferase activity in the United States. Gastroenterology. 2003;124:71–79. doi: 10.1053/gast.2003.50004. [DOI] [PubMed] [Google Scholar]
- 20.Clark JM, Brancati FL, Diehl AM. The prevalence and etiology of elevated aminotransferase levels in the United States. Am J Gastroenterol. 2003;98:960–967. doi: 10.1111/j.1572-0241.2003.07486.x. [DOI] [PubMed] [Google Scholar]
- 21.Patt CH, Yoo HY, Dibadj K, Flynn J, Thuluvath PJ. Prevalence of transaminase abnormalities in asymptomatic, healthy subjects participating in an executive health-screening program. Dig Dis Sci. 2003;48:797–801. doi: 10.1023/a:1022809430756. [DOI] [PubMed] [Google Scholar]
- 22.Piton A, Poynard T, Imbert-Bismut F, Khalil L, Delattre J, Pelissier E, Sansonetti N, Opolon P. Factors associated with serum alanine transaminase activity in healthy subjects: consequences for the definition of normal values, for selection of blood donors, and for patients with chronic hepatitis C. MULTIVIRC Group. Hepatology. 1998;27:1213–1219. doi: 10.1002/hep.510270505. [DOI] [PubMed] [Google Scholar]
- 23.Pacifico L, Ferraro F, Bonci E, Anania C, Romaggioli S, Chiesa C. Upper limit of normal for alanine aminotransferase: quo vadis? Clin Chim Acta. 2013;422:29–39. doi: 10.1016/j.cca.2013.03.030. [DOI] [PubMed] [Google Scholar]
- 24.Bedogni G, Bellentani S, Miglioli L, Masutti F, Passalacqua M, Castiglione A, Tiribelli C. The Fatty Liver Index: a simple and accurate predictor of hepatic steatosis in the general population. BMC Gastroenterol. 2006;6:33. doi: 10.1186/1471-230X-6-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bedogni G, Kahn HS, Bellentani S, Tiribelli C. A simple index of lipid overaccumulation is a good marker of liver steatosis. BMC Gastroenterol. 2010;10:98. doi: 10.1186/1471-230X-10-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fedchuk L, Nascimbeni F, Pais R, Charlotte F, Housset C, Ratziu V. Performance and limitations of steatosis biomarkers in patients with nonalcoholic fatty liver disease. Aliment Pharmacol Ther. 2014;40:1209–1222. doi: 10.1111/apt.12963. [DOI] [PubMed] [Google Scholar]
- 27.Sanal MG. Biomarkers in nonalcoholic fatty liver disease-the emperor has no clothes? World J Gastroenterol. 2015;21:3223–3231. doi: 10.3748/wjg.v21.i11.3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Masarone M, Federico A, Abenavoli L, Loguercio C, Persico M. Non alcoholic fatty liver: epidemiology and natural history. Rev Recent Clin Trials. 2014;9:126–133. doi: 10.2174/1574887109666141216111143. [DOI] [PubMed] [Google Scholar]
- 29.Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114:842–845. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 30.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115:1343–1351. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seki S, Kitada T, Yamada T, Sakaguchi H, Nakatani K, Wakasa K. In situ detection of lipid peroxidation and oxidative DNA damage in non-alcoholic fatty liver diseases. J Hepatol. 2002;37:56–62. doi: 10.1016/s0168-8278(02)00073-9. [DOI] [PubMed] [Google Scholar]
- 32.Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology. 2010;52:1836–1846. doi: 10.1002/hep.24001. [DOI] [PubMed] [Google Scholar]
- 33.Gariani K, Philippe J, Jornayvaz FR. Non-alcoholic fatty liver disease and insulin resistance: from bench to bedside. Diabetes Metab. 2013;39:16–26. doi: 10.1016/j.diabet.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 34.Asrih M, Jornayvaz FR. Metabolic syndrome and nonalcoholic fatty liver disease: Is insulin resistance the link? Mol Cell Endocrinol. 2015;pii:S0303–7207(15)00094-5. doi: 10.1016/j.mce.2015.02.018. [DOI] [PubMed] [Google Scholar]
- 35.Iacono A, Raso GM, Canani RB, Calignano A, Meli R. Probiotics as an emerging therapeutic strategy to treat NAFLD: focus on molecular and biochemical mechanisms. J Nutr Biochem. 2011;22:699–711. doi: 10.1016/j.jnutbio.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 36.Miura K, Seki E, Ohnishi H, Brenner DA. Role of toll-like receptors and their downstream molecules in the development of nonalcoholic Fatty liver disease. Gastroenterol Res Pract. 2010;2010:362847. doi: 10.1155/2010/362847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57:1470–1481. doi: 10.2337/db07-1403. [DOI] [PubMed] [Google Scholar]
- 38.Alisi A, Carsetti R, Nobili V. Pathogen- or damage-associated molecular patterns during nonalcoholic fatty liver disease development. Hepatology. 2011;54:1500–1502. doi: 10.1002/hep.24611. [DOI] [PubMed] [Google Scholar]
- 39.Miele L, Valenza V, La Torre G, Montalto M, Cammarota G, Ricci R, Mascianà R, Forgione A, Gabrieli ML, Perotti G, et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology. 2009;49:1877–1887. doi: 10.1002/hep.22848. [DOI] [PubMed] [Google Scholar]
- 40.Pacifico L, Bonci E, Marandola L, Romaggioli S, Bascetta S, Chiesa C. Increased circulating zonulin in children with biopsy-proven nonalcoholic fatty liver disease. World J Gastroenterol. 2014;20:17107–17114. doi: 10.3748/wjg.v20.i45.17107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444:1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- 42.Clemente JC, Ursell LK, Parfrey LW, Knight R. The impact of the gut microbiota on human health: an integrative view. Cell. 2012;148:1258–1270. doi: 10.1016/j.cell.2012.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Festi D, Schiumerini R, Eusebi LH, Marasco G, Taddia M, Colecchia A. Gut microbiota and metabolic syndrome. World J Gastroenterol. 2014;20:16079–16094. doi: 10.3748/wjg.v20.i43.16079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhu L, Baker SS, Gill C, Liu W, Alkhouri R, Baker RD, Gill SR. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology. 2013;57:601–609. doi: 10.1002/hep.26093. [DOI] [PubMed] [Google Scholar]
- 45.Mouzaki M, Comelli EM, Arendt BM, Bonengel J, Fung SK, Fischer SE, McGilvray ID, Allard JP. Intestinal microbiota in patients with nonalcoholic fatty liver disease. Hepatology. 2013;58:120–127. doi: 10.1002/hep.26319. [DOI] [PubMed] [Google Scholar]
- 46.Raman M, Ahmed I, Gillevet PM, Probert CS, Ratcliffe NM, Smith S, Greenwood R, Sikaroodi M, Lam V, Crotty P, et al. Fecal microbiome and volatile organic compound metabolome in obese humans with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2013;11:868–875.e1-e3. doi: 10.1016/j.cgh.2013.02.015. [DOI] [PubMed] [Google Scholar]
- 47.Cope K, Risby T, Diehl AM. Increased gastrointestinal ethanol production in obese mice: implications for fatty liver disease pathogenesis. Gastroenterology. 2000;119:1340–1347. doi: 10.1053/gast.2000.19267. [DOI] [PubMed] [Google Scholar]
- 48.Minemura M, Shimizu Y. Gut microbiota and liver diseases. World J Gastroenterol. 2015;21:1691–1702. doi: 10.3748/wjg.v21.i6.1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ma YY, Li L, Yu CH, Shen Z, Chen LH, Li YM. Effects of probiotics on nonalcoholic fatty liver disease: a meta-analysis. World J Gastroenterol. 2013;19:6911–6918. doi: 10.3748/wjg.v19.i40.6911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Floch MH, Walker WA, Madsen K, Sanders ME, Macfarlane GT, Flint HJ, Dieleman LA, Ringel Y, Guandalini S, Kelly CP, et al. Recommendations for probiotic use-2011 update. J Clin Gastroenterol. 2011;45 Suppl:S168–S171. doi: 10.1097/MCG.0b013e318230928b. [DOI] [PubMed] [Google Scholar]
- 51.Ekstedt M, Franzén LE, Mathiesen UL, Thorelius L, Holmqvist M, Bodemar G, Kechagias S. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology. 2006;44:865–873. doi: 10.1002/hep.21327. [DOI] [PubMed] [Google Scholar]
- 52.Siddiqui MS, Sterling RK. Posttransplant metabolic syndrome. Int J Hepatol. 2012;2012:891516. doi: 10.1155/2012/891516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Than NN, Newsome PN. A concise review of non-alcoholic fatty liver disease. Atherosclerosis. 2015;239:192–202. doi: 10.1016/j.atherosclerosis.2015.01.001. [DOI] [PubMed] [Google Scholar]
- 54.Edens MA, Kuipers F, Stolk RP. Non-alcoholic fatty liver disease is associated with cardiovascular disease risk markers. Obes Rev. 2009;10:412–419. doi: 10.1111/j.1467-789X.2009.00594.x. [DOI] [PubMed] [Google Scholar]
- 55.Pacifico L, Chiesa C, Anania C, De Merulis A, Osborn JF, Romaggioli S, Gaudio E. Nonalcoholic fatty liver disease and the heart in children and adolescents. World J Gastroenterol. 2014;20:9055–9071. doi: 10.3748/wjg.v20.i27.9055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ekstedt M, Hagström H, Nasr P, Fredrikson M, Stål P, Kechagias S, Hultcrantz R. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology. 2015;61:1547–1554. doi: 10.1002/hep.27368. [DOI] [PubMed] [Google Scholar]
- 57.Perazzo H, Munteanu M, Ngo Y, Lebray P, Seurat N, Rutka F, Couteau M, Jacqueminet S, Giral P, Monneret D, et al. Prognostic value of liver fibrosis and steatosis biomarkers in type-2 diabetes and dyslipidaemia. Aliment Pharmacol Ther. 2014;40:1081–1093. doi: 10.1111/apt.12946. [DOI] [PubMed] [Google Scholar]
- 58.Detrano R, Guerci AD, Carr JJ, Bild DE, Burke G, Folsom AR, Liu K, Shea S, Szklo M, Bluemke DA, et al. Coronary calcium as a predictor of coronary events in four racial or ethnic groups. N Engl J Med. 2008;358:1336–1345. doi: 10.1056/NEJMoa072100. [DOI] [PubMed] [Google Scholar]
- 59.VanWagner LB, Ning H, Lewis CE, Shay CM, Wilkins J, Carr JJ, Terry JG, Lloyd-Jones DM, Jacobs DR, Carnethon MR. Associations between nonalcoholic fatty liver disease and subclinical atherosclerosis in middle-aged adults: the Coronary Artery Risk Development in Young Adults Study. Atherosclerosis. 2014;235:599–605. doi: 10.1016/j.atherosclerosis.2014.05.962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mellinger JL, Pencina KM, Massaro JM, Hoffmann U, Seshadri S, Fox CS, O’Donnell CJ, Speliotes EK. Hepatic steatosis and cardiovascular disease outcomes: An analysis of the Framingham Heart Study. J Hepatol. 2015;63:470–476. doi: 10.1016/j.jhep.2015.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim D, Choi SY, Park EH, Lee W, Kang JH, Kim W, Kim YJ, Yoon JH, Jeong SH, Lee DH, et al. Nonalcoholic fatty liver disease is associated with coronary artery calcification. Hepatology. 2012;56:605–613. doi: 10.1002/hep.25593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sung KC, Wild SH, Kwag HJ, Byrne CD. Fatty liver, insulin resistance, and features of metabolic syndrome: relationships with coronary artery calcium in 10,153 people. Diabetes Care. 2012;35:2359–2364. doi: 10.2337/dc12-0515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Targher G, Bertolini L, Rodella S, Tessari R, Zenari L, Lippi G, Arcaro G. Nonalcoholic fatty liver disease is independently associated with an increased incidence of cardiovascular events in type 2 diabetic patients. Diabetes Care. 2007;30:2119–2121. doi: 10.2337/dc07-0349. [DOI] [PubMed] [Google Scholar]
- 64.Speliotes EK, Massaro JM, Hoffmann U, Vasan RS, Meigs JB, Sahani DV, Hirschhorn JN, O’Donnell CJ, Fox CS. Fatty liver is associated with dyslipidemia and dysglycemia independent of visceral fat: the Framingham Heart Study. Hepatology. 2010;51:1979–1987. doi: 10.1002/hep.23593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Verrijken A, Francque S, Mertens I, Prawitt J, Caron S, Hubens G, Van Marck E, Staels B, Michielsen P, Gaal LV. Prothrombotic factors in histologically proven NAFLD and NASH. Hepatology. 2013 doi: 10.1002/hep.26510. [DOI] [PubMed] [Google Scholar]
- 66.Yoo HJ, Choi KM. Hepatokines as a Link between Obesity and Cardiovascular Diseases. Diabetes Metab J. 2015;39:10–15. doi: 10.4093/dmj.2015.39.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Leite NC, Salles GF, Araujo AL, Villela-Nogueira CA, Cardoso CR. Prevalence and associated factors of non-alcoholic fatty liver disease in patients with type-2 diabetes mellitus. Liver Int. 2009;29:113–119. doi: 10.1111/j.1478-3231.2008.01718.x. [DOI] [PubMed] [Google Scholar]
- 68.Bellentani S, Scaglioni F, Marino M, Bedogni G. Epidemiology of non-alcoholic fatty liver disease. Dig Dis. 2010;28:155–161. doi: 10.1159/000282080. [DOI] [PubMed] [Google Scholar]
- 69.Gupte P, Amarapurkar D, Agal S, Baijal R, Kulshrestha P, Pramanik S, Patel N, Madan A, Amarapurkar A, Hafeezunnisa Non-alcoholic steatohepatitis in type 2 diabetes mellitus. J Gastroenterol Hepatol. 2004;19:854–858. doi: 10.1111/j.1440-1746.2004.03312.x. [DOI] [PubMed] [Google Scholar]
- 70.Prashanth M, Ganesh HK, Vima MV, John M, Bandgar T, Joshi SR, Shah SR, Rathi PM, Joshi AS, Thakkar H, et al. Prevalence of nonalcoholic fatty liver disease in patients with type 2 diabetes mellitus. J Assoc Physicians India. 2009;57:205–210. [PubMed] [Google Scholar]
- 71.Adams LA, Sanderson S, Lindor KD, Angulo P. The histological course of nonalcoholic fatty liver disease: a longitudinal study of 103 patients with sequential liver biopsies. J Hepatol. 2005;42:132–138. doi: 10.1016/j.jhep.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 72.Vidal J, Ferrer JP, Esmatjes E, Salmeron JM, González-Clemente JM, Gomis R, Rodés J. Diabetes mellitus in patients with liver cirrhosis. Diabetes Res Clin Pract. 1994;25:19–25. doi: 10.1016/0168-8227(94)90157-0. [DOI] [PubMed] [Google Scholar]
- 73.Bugianesi E, Manzini P, D’Antico S, Vanni E, Longo F, Leone N, Massarenti P, Piga A, Marchesini G, Rizzetto M. Relative contribution of iron burden, HFE mutations, and insulin resistance to fibrosis in nonalcoholic fatty liver. Hepatology. 2004;39:179–187. doi: 10.1002/hep.20023. [DOI] [PubMed] [Google Scholar]
- 74.Marchesini G, Brizi M, Morselli-Labate AM, Bianchi G, Bugianesi E, McCullough AJ, Forlani G, Melchionda N. Association of nonalcoholic fatty liver disease with insulin resistance. Am J Med. 1999;107:450–455. doi: 10.1016/s0002-9343(99)00271-5. [DOI] [PubMed] [Google Scholar]
- 75.Ludwig J, Viggiano TR, McGill DB, Oh BJ. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin Proc. 1980;55:434–438. [PubMed] [Google Scholar]
- 76.Bacon BR, Farahvash MJ, Janney CG, Neuschwander-Tetri BA. Nonalcoholic steatohepatitis: an expanded clinical entity. Gastroenterology. 1994;107:1103–1109. doi: 10.1016/0016-5085(94)90235-6. [DOI] [PubMed] [Google Scholar]
- 77.Colicchio P, Tarantino G, del Genio F, Sorrentino P, Saldalamacchia G, Finelli C, Conca P, Contaldo F, Pasanisi F. Non-alcoholic fatty liver disease in young adult severely obese non-diabetic patients in South Italy. Ann Nutr Metab. 2005;49:289–295. doi: 10.1159/000087295. [DOI] [PubMed] [Google Scholar]
- 78.Beymer C, Kowdley KV, Larson A, Edmonson P, Dellinger EP, Flum DR. Prevalence and predictors of asymptomatic liver disease in patients undergoing gastric bypass surgery. Arch Surg. 2003;138:1240–1244. doi: 10.1001/archsurg.138.11.1240. [DOI] [PubMed] [Google Scholar]
- 79.Machado M, Marques-Vidal P, Cortez-Pinto H. Hepatic histology in obese patients undergoing bariatric surgery. J Hepatol. 2006;45:600–606. doi: 10.1016/j.jhep.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 80.Boza C, Riquelme A, Ibañez L, Duarte I, Norero E, Viviani P, Soza A, Fernandez JI, Raddatz A, Guzman S, et al. Predictors of nonalcoholic steatohepatitis (NASH) in obese patients undergoing gastric bypass. Obes Surg. 2005;15:1148–1153. doi: 10.1381/0960892055002347. [DOI] [PubMed] [Google Scholar]
- 81.Abrams GA, Kunde SS, Lazenby AJ, Clements RH. Portal fibrosis and hepatic steatosis in morbidly obese subjects: A spectrum of nonalcoholic fatty liver disease. Hepatology. 2004;40:475–483. doi: 10.1002/hep.20323. [DOI] [PubMed] [Google Scholar]
- 82.Hocking S, Samocha-Bonet D, Milner KL, Greenfield JR, Chisholm DJ. Adiposity and insulin resistance in humans: the role of the different tissue and cellular lipid depots. Endocr Rev. 2013;34:463–500. doi: 10.1210/er.2012-1041. [DOI] [PubMed] [Google Scholar]
- 83.Kotronen A, Yki-Järvinen H, Sevastianova K, Bergholm R, Hakkarainen A, Pietiläinen KH, Juurinen L, Lundbom N, Sørensen TI. Comparison of the relative contributions of intra-abdominal and liver fat to components of the metabolic syndrome. Obesity (Silver Spring) 2011;19:23–28. doi: 10.1038/oby.2010.137. [DOI] [PubMed] [Google Scholar]
- 84.Kotronen A, Juurinen L, Tiikkainen M, Vehkavaara S, Yki-Järvinen H. Increased liver fat, impaired insulin clearance, and hepatic and adipose tissue insulin resistance in type 2 diabetes. Gastroenterology. 2008;135:122–130. doi: 10.1053/j.gastro.2008.03.021. [DOI] [PubMed] [Google Scholar]
- 85.Verhagen SN, Visseren FL. Perivascular adipose tissue as a cause of atherosclerosis. Atherosclerosis. 2011;214:3–10. doi: 10.1016/j.atherosclerosis.2010.05.034. [DOI] [PubMed] [Google Scholar]
- 86.Iacobellis G. Local and systemic effects of the multifaceted epicardial adipose tissue depot. Nat Rev Endocrinol. 2015;11:363–371. doi: 10.1038/nrendo.2015.58. [DOI] [PubMed] [Google Scholar]
- 87.Corrao S, Rinollo C, Scaglione R. Non-alcoholic fatty liver disease: severity of fibrosis and its relationships with clinical and biological variables. J Hepatol. 2015;62:1212–1213. doi: 10.1016/j.jhep.2015.01.010. [DOI] [PubMed] [Google Scholar]
- 88.Kagansky N, Levy S, Keter D, Rimon E, Taiba Z, Fridman Z, Berger D, Knobler H, Malnick S. Non-alcoholic fatty liver disease--a common and benign finding in octogenarian patients. Liver Int. 2004;24:588–594. doi: 10.1111/j.1478-3231.2004.0969.x. [DOI] [PubMed] [Google Scholar]
- 89.Ahima RS, Flier JS. Adipose tissue as an endocrine organ. Trends Endocrinol Metab. 2000;11:327–332. doi: 10.1016/s1043-2760(00)00301-5. [DOI] [PubMed] [Google Scholar]
- 90.Kamada Y, Takehara T, Hayashi N. Adipocytokines and liver disease. J Gastroenterol. 2008;43:811–822. doi: 10.1007/s00535-008-2213-6. [DOI] [PubMed] [Google Scholar]
- 91.Stojsavljević S, Gomerčić Palčić M, Virović Jukić L, Smirčić Duvnjak L, Duvnjak M. Adipokines and proinflammatory cytokines, the key mediators in the pathogenesis of nonalcoholic fatty liver disease. World J Gastroenterol. 2014;20:18070–18091. doi: 10.3748/wjg.v20.i48.18070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kristiansen OP, Mandrup-Poulsen T. Interleukin-6 and diabetes: the good, the bad, or the indifferent? Diabetes. 2005;54 Suppl 2:S114–S124. doi: 10.2337/diabetes.54.suppl_2.s114. [DOI] [PubMed] [Google Scholar]
- 93.Kamimura D, Ishihara K, Hirano T. IL-6 signal transduction and its physiological roles: the signal orchestration model. Rev Physiol Biochem Pharmacol. 2003;149:1–38. doi: 10.1007/s10254-003-0012-2. [DOI] [PubMed] [Google Scholar]
- 94.Muoio DM, Lynis Dohm G. Peripheral metabolic actions of leptin. Best Pract Res Clin Endocrinol Metab. 2002;16:653–666. doi: 10.1053/beem.2002.0223. [DOI] [PubMed] [Google Scholar]
- 95.Kakuma T, Lee Y, Higa M, Wang Zw, Pan W, Shimomura I, Unger RH. Leptin, troglitazone, and the expression of sterol regulatory element binding proteins in liver and pancreatic islets. Proc Natl Acad Sci USA. 2000;97:8536–8541. doi: 10.1073/pnas.97.15.8536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Maury E, Brichard SM. Adipokine dysregulation, adipose tissue inflammation and metabolic syndrome. Mol Cell Endocrinol. 2010;314:1–16. doi: 10.1016/j.mce.2009.07.031. [DOI] [PubMed] [Google Scholar]
- 97.Wolfs MG, Gruben N, Rensen SS, Verdam FJ, Greve JW, Driessen A, Wijmenga C, Buurman WA, Franke L, Scheja L, et al. Determining the association between adipokine expression in multiple tissues and phenotypic features of non-alcoholic fatty liver disease in obesity. Nutr Diabetes. 2015;5:e146. doi: 10.1038/nutd.2014.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tsochatzis E, Papatheodoridis GV, Archimandritis AJ. The evolving role of leptin and adiponectin in chronic liver diseases. Am J Gastroenterol. 2006;101:2629–2640. doi: 10.1111/j.1572-0241.2006.00848.x. [DOI] [PubMed] [Google Scholar]
- 99.Hui E, Xu A, Bo Yang H, Lam KS. Obesity as the common soil of non-alcoholic fatty liver disease and diabetes: Role of adipokines. J Diabetes Investig. 2013;4:413–425. doi: 10.1111/jdi.12093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Huynh FK, Levi J, Denroche HC, Gray SL, Voshol PJ, Neumann UH, Speck M, Chua SC, Covey SD, Kieffer TJ. Disruption of hepatic leptin signaling protects mice from age- and diet-related glucose intolerance. Diabetes. 2010;59:3032–3040. doi: 10.2337/db10-0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nagao K, Inoue N, Ujino Y, Higa K, Shirouchi B, Wang YM, Yanagita T. Effect of leptin infusion on insulin sensitivity and lipid metabolism in diet-induced lipodystrophy model mice. Lipids Health Dis. 2008;7:8. doi: 10.1186/1476-511X-7-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tsochatzis E, Papatheodoridis GV, Hadziyannis E, Georgiou A, Kafiri G, Tiniakos DG, Manesis EK, Archimandritis AJ. Serum adipokine levels in chronic liver diseases: association of resistin levels with fibrosis severity. Scand J Gastroenterol. 2008;43:1128–1136. doi: 10.1080/00365520802085387. [DOI] [PubMed] [Google Scholar]
- 103.Chitturi S, Farrell G, Frost L, Kriketos A, Lin R, Fung C, Liddle C, Samarasinghe D, George J. Serum leptin in NASH correlates with hepatic steatosis but not fibrosis: a manifestation of lipotoxicity? Hepatology. 2002;36:403–409. doi: 10.1053/jhep.2002.34738. [DOI] [PubMed] [Google Scholar]
- 104.Kishida K, Funahashi T, Shimomura I. Adiponectin as a routine clinical biomarker. Best Pract Res Clin Endocrinol Metab. 2014;28:119–130. doi: 10.1016/j.beem.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 105.Li M, Xu Y, Xu M, Ma L, Wang T, Liu Y, Dai M, Chen Y, Lu J, Liu J, et al. Association between nonalcoholic fatty liver disease (NAFLD) and osteoporotic fracture in middle-aged and elderly Chinese. J Clin Endocrinol Metab. 2012;97:2033–2038. doi: 10.1210/jc.2011-3010. [DOI] [PubMed] [Google Scholar]
- 106.Moon SS, Lee YS, Kim SW. Association of nonalcoholic fatty liver disease with low bone mass in postmenopausal women. Endocrine. 2012;42:423–429. doi: 10.1007/s12020-012-9639-6. [DOI] [PubMed] [Google Scholar]
- 107.Hotta K, Funahashi T, Arita Y, Takahashi M, Matsuda M, Okamoto Y, Iwahashi H, Kuriyama H, Ouchi N, Maeda K, et al. Plasma concentrations of a novel, adipose-specific protein, adiponectin, in type 2 diabetic patients. Arterioscler Thromb Vasc Biol. 2000;20:1595–1599. doi: 10.1161/01.atv.20.6.1595. [DOI] [PubMed] [Google Scholar]
- 108.Matsumoto H, Tamura S, Kamada Y, Kiso S, Fukushima J, Wada A, Maeda N, Kihara S, Funahashi T, Matsuzawa Y, et al. Adiponectin deficiency exacerbates lipopolysaccharide/D-galactosamine-induced liver injury in mice. World J Gastroenterol. 2006;12:3352–3358. doi: 10.3748/wjg.v12.i21.3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tsao TS, Murrey HE, Hug C, Lee DH, Lodish HF. Oligomerization state-dependent activation of NF-kappa B signaling pathway by adipocyte complement-related protein of 30 kDa (Acrp30) J Biol Chem. 2002;277:29359–29362. doi: 10.1074/jbc.C200312200. [DOI] [PubMed] [Google Scholar]
- 110.Ouchi N, Kihara S, Arita Y, Maeda K, Kuriyama H, Okamoto Y, Hotta K, Nishida M, Takahashi M, Nakamura T, et al. Novel modulator for endothelial adhesion molecules: adipocyte-derived plasma protein adiponectin. Circulation. 1999;100:2473–2476. doi: 10.1161/01.cir.100.25.2473. [DOI] [PubMed] [Google Scholar]
- 111.Handy JA, Saxena NK, Fu P, Lin S, Mells JE, Gupta NA, Anania FA. Adiponectin activation of AMPK disrupts leptin-mediated hepatic fibrosis via suppressors of cytokine signaling (SOCS-3) J Cell Biochem. 2010;110:1195–1207. doi: 10.1002/jcb.22634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yamauchi T, Nio Y, Maki T, Kobayashi M, Takazawa T, Iwabu M, Okada-Iwabu M, Kawamoto S, Kubota N, Kubota T, et al. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nat Med. 2007;13:332–339. doi: 10.1038/nm1557. [DOI] [PubMed] [Google Scholar]
- 113.Anania FA. Adiponectin and alcoholic fatty liver: Is it, after all, about what you eat? Hepatology. 2005;42:530–532. doi: 10.1002/hep.20861. [DOI] [PubMed] [Google Scholar]
- 114.Spranger J, Kroke A, Möhlig M, Bergmann MM, Ristow M, Boeing H, Pfeiffer AF. Adiponectin and protection against type 2 diabetes mellitus. Lancet. 2003;361:226–228. doi: 10.1016/S0140-6736(03)12255-6. [DOI] [PubMed] [Google Scholar]
- 115.Matsuzawa Y. Adiponectin: a key player in obesity related disorders. Curr Pharm Des. 2010;16:1896–1901. doi: 10.2174/138161210791208893. [DOI] [PubMed] [Google Scholar]
- 116.Lemoine M, Ratziu V, Kim M, Maachi M, Wendum D, Paye F, Bastard JP, Poupon R, Housset C, Capeau J, et al. Serum adipokine levels predictive of liver injury in non-alcoholic fatty liver disease. Liver Int. 2009;29:1431–1438. doi: 10.1111/j.1478-3231.2009.02022.x. [DOI] [PubMed] [Google Scholar]
- 117.Finelli C, Tarantino G. What is the role of adiponectin in obesity related non-alcoholic fatty liver disease? World J Gastroenterol. 2013;19:802–812. doi: 10.3748/wjg.v19.i6.802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wei J, Sun X, Chen Y, Li Y, Song L, Zhou Z, Xu B, Lin Y, Xu S. Perinatal exposure to bisphenol A exacerbates nonalcoholic steatohepatitis-like phenotype in male rat offspring fed on a high-fat diet. J Endocrinol. 2014;222:313–325. doi: 10.1530/JOE-14-0356. [DOI] [PubMed] [Google Scholar]
- 119.Strakovsky RS, Wang H, Engeseth NJ, Flaws JA, Helferich WG, Pan YX, Lezmi S. Developmental bisphenol A (BPA) exposure leads to sex-specific modification of hepatic gene expression and epigenome at birth that may exacerbate high-fat diet-induced hepatic steatosis. Toxicol Appl Pharmacol. 2015;284:101–112. doi: 10.1016/j.taap.2015.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Song S, Zhang L, Zhang H, Wei W, Jia L. Perinatal BPA exposure induces hyperglycemia, oxidative stress and decreased adiponectin production in later life of male rat offspring. Int J Environ Res Public Health. 2014;11:3728–3742. doi: 10.3390/ijerph110403728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Tarantino G, Valentino R, Di Somma C, D’Esposito V, Passaretti F, Pizza G, Brancato V, Orio F, Formisano P, Colao A, et al. Bisphenol A in polycystic ovary syndrome and its association with liver-spleen axis. Clin Endocrinol (Oxf) 2013;78:447–453. doi: 10.1111/j.1365-2265.2012.04500.x. [DOI] [PubMed] [Google Scholar]
- 122.Polyzos SA, Kountouras J, Deretzi G, Zavos C, Mantzoros CS. The emerging role of endocrine disruptors in pathogenesis of insulin resistance: a concept implicating nonalcoholic fatty liver disease. Curr Mol Med. 2012;12:68–82. doi: 10.2174/156652412798376161. [DOI] [PubMed] [Google Scholar]
- 123.March WA, Moore VM, Willson KJ, Phillips DI, Norman RJ, Davies MJ. The prevalence of polycystic ovary syndrome in a community sample assessed under contrasting diagnostic criteria. Hum Reprod. 2010;25:544–551. doi: 10.1093/humrep/dep399. [DOI] [PubMed] [Google Scholar]
- 124.Rotterdam ESHRE/ASRM-Sponsored PCOS Consensus Workshop Group. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome. Fertil Steril. 2004;81:19–25. doi: 10.1016/j.fertnstert.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 125.Barber TM, Franks S. Genetics of polycystic ovary syndrome. Front Horm Res. 2013;40:28–39. doi: 10.1159/000341682. [DOI] [PubMed] [Google Scholar]
- 126.Welt CK, Duran JM. Genetics of polycystic ovary syndrome. Semin Reprod Med. 2014;32:177–182. doi: 10.1055/s-0034-1371089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kauffman RP, Baker TE, Baker V, Kauffman MM, Castracane VD. Endocrine factors associated with non-alcoholic fatty liver disease in women with polycystic ovary syndrome: do androgens play a role? Gynecol Endocrinol. 2010;26:39–46. doi: 10.3109/09513590903184084. [DOI] [PubMed] [Google Scholar]
- 128.Ramezani-Binabaj M, Motalebi M, Karimi-Sari H, Rezaee-Zavareh MS, Alavian SM. Are women with polycystic ovarian syndrome at a high risk of non-alcoholic Fatty liver disease; a meta-analysis. Hepat Mon. 2014;14:e23235. doi: 10.5812/hepatmon.23235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Schwimmer JB, Celedon MA, Lavine JE, Salem R, Campbell N, Schork NJ, Shiehmorteza M, Yokoo T, Chavez A, Middleton MS, et al. Heritability of nonalcoholic fatty liver disease. Gastroenterology. 2009;136:1585–1592. doi: 10.1053/j.gastro.2009.01.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Gambarin-Gelwan M, Kinkhabwala SV, Schiano TD, Bodian C, Yeh HC, Futterweit W. Prevalence of nonalcoholic fatty liver disease in women with polycystic ovary syndrome. Clin Gastroenterol Hepatol. 2007;5:496–501. doi: 10.1016/j.cgh.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 131.Hossain N, Stepanova M, Afendy A, Nader F, Younossi Y, Rafiq N, Goodman Z, Younossi ZM. Non-alcoholic steatohepatitis (NASH) in patients with polycystic ovarian syndrome (PCOS) Scand J Gastroenterol. 2011;46:479–484. doi: 10.3109/00365521.2010.539251. [DOI] [PubMed] [Google Scholar]
- 132.Brzozowska MM, Ostapowicz G, Weltman MD. An association between non-alcoholic fatty liver disease and polycystic ovarian syndrome. J Gastroenterol Hepatol. 2009;24:243–247. doi: 10.1111/j.1440-1746.2008.05740.x. [DOI] [PubMed] [Google Scholar]
- 133.Barbieri RL, Makris A, Randall RW, Daniels G, Kistner RW, Ryan KJ. Insulin stimulates androgen accumulation in incubations of ovarian stroma obtained from women with hyperandrogenism. J Clin Endocrinol Metab. 1986;62:904–910. doi: 10.1210/jcem-62-5-904. [DOI] [PubMed] [Google Scholar]
- 134.Conway GS, Avey C, Rumsby G. The tyrosine kinase domain of the insulin receptor gene is normal in women with hyperinsulinaemia and polycystic ovary syndrome. Hum Reprod. 1994;9:1681–1683. doi: 10.1093/oxfordjournals.humrep.a138773. [DOI] [PubMed] [Google Scholar]
- 135.Baranova A, Tran TP, Birerdinc A, Younossi ZM. Systematic review: association of polycystic ovary syndrome with metabolic syndrome and non-alcoholic fatty liver disease. Aliment Pharmacol Ther. 2011;33:801–814. doi: 10.1111/j.1365-2036.2011.04579.x. [DOI] [PubMed] [Google Scholar]
- 136.Dunaif A, Xia J, Book CB, Schenker E, Tang Z. Excessive insulin receptor serine phosphorylation in cultured fibroblasts and in skeletal muscle. A potential mechanism for insulin resistance in the polycystic ovary syndrome. J Clin Invest. 1995;96:801–810. doi: 10.1172/JCI118126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Auchus RJ, Geller DH, Lee TC, Miller WL. The regulation of human P450c17 activity: relationship to premature adrenarche, insulin resistance and the polycystic ovary syndrome. Trends Endocrinol Metab. 1998;9:47–50. doi: 10.1016/s1043-2760(98)00016-2. [DOI] [PubMed] [Google Scholar]
- 138.Vassilatou E, Lafoyianni S, Vryonidou A, Ioannidis D, Kosma L, Katsoulis K, Papavassiliou E, Tzavara I. Increased androgen bioavailability is associated with non-alcoholic fatty liver disease in women with polycystic ovary syndrome. Hum Reprod. 2010;25:212–220. doi: 10.1093/humrep/dep380. [DOI] [PubMed] [Google Scholar]
- 139.Chen MJ, Chiu HM, Chen CL, Yang WS, Yang YS, Ho HN. Hyperandrogenemia is independently associated with elevated alanine aminotransferase activity in young women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2010;95:3332–3341. doi: 10.1210/jc.2009-2698. [DOI] [PubMed] [Google Scholar]
- 140.Dai R, Yan D, Li J, Chen S, Liu Y, Chen R, Duan C, Wei M, Li H, He T. Activation of PKR/eIF2α signaling cascade is associated with dihydrotestosterone-induced cell cycle arrest and apoptosis in human liver cells. J Cell Biochem. 2012;113:1800–1808. doi: 10.1002/jcb.24051. [DOI] [PubMed] [Google Scholar]
- 141.Baranova A, Tran TP, Afendy A, Wang L, Shamsaddini A, Mehta R, Chandhoke V, Birerdinc A, Younossi ZM. Molecular signature of adipose tissue in patients with both non-alcoholic fatty liver disease (NAFLD) and polycystic ovarian syndrome (PCOS) J Transl Med. 2013;11:133. doi: 10.1186/1479-5876-11-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Tan S, Bechmann LP, Benson S, Dietz T, Eichner S, Hahn S, Janssen OE, Lahner H, Gerken G, Mann K, et al. Apoptotic markers indicate nonalcoholic steatohepatitis in polycystic ovary syndrome. J Clin Endocrinol Metab. 2010;95:343–348. doi: 10.1210/jc.2009-1834. [DOI] [PubMed] [Google Scholar]
- 143.Diamanti-Kandarakis E, Papavassiliou AG. Molecular mechanisms of insulin resistance in polycystic ovary syndrome. Trends Mol Med. 2006;12:324–332. doi: 10.1016/j.molmed.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 144.Goldman MH, Scheraldi CA, Soule WC. Ovarian hyperthecosis associated with fatty liver disease. Am J Obstet Gynecol. 1987;156:1239–1240. doi: 10.1016/0002-9378(87)90154-2. [DOI] [PubMed] [Google Scholar]
- 145.Villa J, Pratley RE. Adipose tissue dysfunction in polycystic ovary syndrome. Curr Diab Rep. 2011;11:179–184. doi: 10.1007/s11892-011-0189-8. [DOI] [PubMed] [Google Scholar]
- 146.Corbould A, Dunaif A. The adipose cell lineage is not intrinsically insulin resistant in polycystic ovary syndrome. Metabolism. 2007;56:716–722. doi: 10.1016/j.metabol.2006.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Hahn S, Haselhorst U, Quadbeck B, Tan S, Kimmig R, Mann K, Janssen OE. Decreased soluble leptin receptor levels in women with polycystic ovary syndrome. Eur J Endocrinol. 2006;154:287–294. doi: 10.1530/eje.1.02078. [DOI] [PubMed] [Google Scholar]
- 148.Yilmaz M, Bukan N, Demirci H, Oztürk C, Kan E, Ayvaz G, Arslan M. Serum resistin and adiponectin levels in women with polycystic ovary syndrome. Gynecol Endocrinol. 2009;25:246–252. doi: 10.1080/09513590802653833. [DOI] [PubMed] [Google Scholar]
- 149.Kelley CE, Brown AJ, Diehl AM, Setji TL. Review of nonalcoholic fatty liver disease in women with polycystic ovary syndrome. World J Gastroenterol. 2014;20:14172–14184. doi: 10.3748/wjg.v20.i39.14172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Legro RS, Arslanian SA, Ehrmann DA, Hoeger KM, Murad MH, Pasquali R, Welt CK. Diagnosis and treatment of polycystic ovary syndrome: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2013;98:4565–4592. doi: 10.1210/jc.2013-2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Hashimoto E, Tokushige K. Prevalence, gender, ethnic variations, and prognosis of NASH. J Gastroenterol. 2011;46 Suppl 1:63–69. doi: 10.1007/s00535-010-0311-8. [DOI] [PubMed] [Google Scholar]
- 152.Gutierrez-Grobe Y, Ponciano-Rodríguez G, Ramos MH, Uribe M, Méndez-Sánchez N. Prevalence of non alcoholic fatty liver disease in premenopausal, posmenopausal and polycystic ovary syndrome women. The role of estrogens. Ann Hepatol. 2010;9:402–409. [PubMed] [Google Scholar]
- 153.Gruber CJ, Tschugguel W, Schneeberger C, Huber JC. Production and actions of estrogens. N Engl J Med. 2002;346:340–352. doi: 10.1056/NEJMra000471. [DOI] [PubMed] [Google Scholar]
- 154.Lonardo A, Carani C, Carulli N, Loria P. ‘Endocrine NAFLD’ a hormonocentric perspective of nonalcoholic fatty liver disease pathogenesis. J Hepatol. 2006;44:1196–1207. doi: 10.1016/j.jhep.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 155.Shimizu I. Impact of oestrogens on the progression of liver disease. Liver Int. 2003;23:63–69. doi: 10.1034/j.1600-0676.2003.00811.x. [DOI] [PubMed] [Google Scholar]
- 156.Jones ME, Thorburn AW, Britt KL, Hewitt KN, Wreford NG, Proietto J, Oz OK, Leury BJ, Robertson KM, Yao S, et al. Aromatase-deficient (ArKO) mice have a phenotype of increased adiposity. Proc Natl Acad Sci USA. 2000;97:12735–12740. doi: 10.1073/pnas.97.23.12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Heine PA, Taylor JA, Iwamoto GA, Lubahn DB, Cooke PS. Increased adipose tissue in male and female estrogen receptor-alpha knockout mice. Proc Natl Acad Sci USA. 2000;97:12729–12734. doi: 10.1073/pnas.97.23.12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Hewitt KN, Pratis K, Jones ME, Simpson ER. Estrogen replacement reverses the hepatic steatosis phenotype in the male aromatase knockout mouse. Endocrinology. 2004;145:1842–1848. doi: 10.1210/en.2003-1369. [DOI] [PubMed] [Google Scholar]
- 159.Maffei L, Murata Y, Rochira V, Tubert G, Aranda C, Vazquez M, Clyne CD, Davis S, Simpson ER, Carani C. Dysmetabolic syndrome in a man with a novel mutation of the aromatase gene: effects of testosterone, alendronate, and estradiol treatment. J Clin Endocrinol Metab. 2004;89:61–70. doi: 10.1210/jc.2003-030313. [DOI] [PubMed] [Google Scholar]
- 160.Camporez JP, Jornayvaz FR, Lee HY, Kanda S, Guigni BA, Kahn M, Samuel VT, Carvalho CR, Petersen KF, Jurczak MJ, et al. Cellular mechanism by which estradiol protects female ovariectomized mice from high-fat diet-induced hepatic and muscle insulin resistance. Endocrinology. 2013;154:1021–1028. doi: 10.1210/en.2012-1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kupelian V, Page ST, Araujo AB, Travison TG, Bremner WJ, McKinlay JB. Low sex hormone-binding globulin, total testosterone, and symptomatic androgen deficiency are associated with development of the metabolic syndrome in nonobese men. J Clin Endocrinol Metab. 2006;91:843–850. doi: 10.1210/jc.2005-1326. [DOI] [PubMed] [Google Scholar]
- 162.Laaksonen DE, Niskanen L, Punnonen K, Nyyssönen K, Tuomainen TP, Valkonen VP, Salonen R, Salonen JT. Testosterone and sex hormone-binding globulin predict the metabolic syndrome and diabetes in middle-aged men. Diabetes Care. 2004;27:1036–1041. doi: 10.2337/diacare.27.5.1036. [DOI] [PubMed] [Google Scholar]
- 163.Selvin E, Feinleib M, Zhang L, Rohrmann S, Rifai N, Nelson WG, Dobs A, Basaria S, Golden SH, Platz EA. Androgens and diabetes in men: results from the Third National Health and Nutrition Examination Survey (NHANES III) Diabetes Care. 2007;30:234–238. doi: 10.2337/dc06-1579. [DOI] [PubMed] [Google Scholar]
- 164.Couillard C, Gagnon J, Bergeron J, Leon AS, Rao DC, Skinner JS, Wilmore JH, Després JP, Bouchard C. Contribution of body fatness and adipose tissue distribution to the age variation in plasma steroid hormone concentrations in men: the HERITAGE Family Study. J Clin Endocrinol Metab. 2000;85:1026–1031. doi: 10.1210/jcem.85.3.6427. [DOI] [PubMed] [Google Scholar]
- 165.Haffner SM, Shaten J, Stern MP, Smith GD, Kuller L. Low levels of sex hormone-binding globulin and testosterone predict the development of non-insulin-dependent diabetes mellitus in men. MRFIT Research Group. Multiple Risk Factor Intervention Trial. Am J Epidemiol. 1996;143:889–897. doi: 10.1093/oxfordjournals.aje.a008832. [DOI] [PubMed] [Google Scholar]
- 166.Dhindsa S, Prabhakar S, Sethi M, Bandyopadhyay A, Chaudhuri A, Dandona P. Frequent occurrence of hypogonadotropic hypogonadism in type 2 diabetes. J Clin Endocrinol Metab. 2004;89:5462–5468. doi: 10.1210/jc.2004-0804. [DOI] [PubMed] [Google Scholar]
- 167.Kapoor D, Aldred H, Clark S, Channer KS, Jones TH. Clinical and biochemical assessment of hypogonadism in men with type 2 diabetes: correlations with bioavailable testosterone and visceral adiposity. Diabetes Care. 2007;30:911–917. doi: 10.2337/dc06-1426. [DOI] [PubMed] [Google Scholar]
- 168.Rao PM, Kelly DM, Jones TH. Testosterone and insulin resistance in the metabolic syndrome and T2DM in men. Nat Rev Endocrinol. 2013;9:479–493. doi: 10.1038/nrendo.2013.122. [DOI] [PubMed] [Google Scholar]
- 169.Völzke H, Aumann N, Krebs A, Nauck M, Steveling A, Lerch MM, Rosskopf D, Wallaschofski H. Hepatic steatosis is associated with low serum testosterone and high serum DHEAS levels in men. Int J Androl. 2010;33:45–53. doi: 10.1111/j.1365-2605.2009.00953.x. [DOI] [PubMed] [Google Scholar]
- 170.Lazo M, Zeb I, Nasir K, Tracy RP, Budoff MJ, Ouyang P, Vaidya D. Association Between Endogenous Sex Hormones and Liver Fat in a Multiethnic Study of Atherosclerosis. Clin Gastroenterol Hepatol. 2015;13:1686–1693.e2. doi: 10.1016/j.cgh.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Aversa A, Bruzziches R, Francomano D, Rosano G, Isidori AM, Lenzi A, Spera G. Effects of testosterone undecanoate on cardiovascular risk factors and atherosclerosis in middle-aged men with late-onset hypogonadism and metabolic syndrome: results from a 24-month, randomized, double-blind, placebo-controlled study. J Sex Med. 2010;7:3495–3503. doi: 10.1111/j.1743-6109.2010.01931.x. [DOI] [PubMed] [Google Scholar]
- 172.Bhattacharya RK, Khera M, Blick G, Kushner H, Nguyen D, Miner MM. Effect of 12 months of testosterone replacement therapy on metabolic syndrome components in hypogonadal men: data from the Testim Registry in the US (TRiUS) BMC Endocr Disord. 2011;11:18. doi: 10.1186/1472-6823-11-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kalinchenko SY, Tishova YA, Mskhalaya GJ, Gooren LJ, Giltay EJ, Saad F. Effects of testosterone supplementation on markers of the metabolic syndrome and inflammation in hypogonadal men with the metabolic syndrome: the double-blinded placebo-controlled Moscow study. Clin Endocrinol (Oxf) 2010;73:602–612. doi: 10.1111/j.1365-2265.2010.03845.x. [DOI] [PubMed] [Google Scholar]
- 174.Hoyos CM, Yee BJ, Phillips CL, Machan EA, Grunstein RR, Liu PY. Body compositional and cardiometabolic effects of testosterone therapy in obese men with severe obstructive sleep apnoea: a randomised placebo-controlled trial. Eur J Endocrinol. 2012;167:531–541. doi: 10.1530/EJE-12-0525. [DOI] [PubMed] [Google Scholar]
- 175.Nikolaenko L, Jia Y, Wang C, Diaz-Arjonilla M, Yee JK, French SW, Liu PY, Laurel S, Chong C, Lee K, et al. Testosterone replacement ameliorates nonalcoholic fatty liver disease in castrated male rats. Endocrinology. 2014;155:417–428. doi: 10.1210/en.2013-1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Jeon YK, Lee JG, Kim SS, Kim BH, Kim SJ, Kim YK, Kim IJ. Association between bone mineral density and metabolic syndrome in pre- and postmenopausal women. Endocr J. 2011;58:87–93. doi: 10.1507/endocrj.k10e-297. [DOI] [PubMed] [Google Scholar]
- 177.Musso G, Paschetta E, Gambino R, Cassader M, Molinaro F. Interactions among bone, liver, and adipose tissue predisposing to diabesity and fatty liver. Trends Mol Med. 2013;19:522–535. doi: 10.1016/j.molmed.2013.05.006. [DOI] [PubMed] [Google Scholar]
- 178.Confavreux CB. Bone: from a reservoir of minerals to a regulator of energy metabolism. Kidney Int Suppl. 2011;(121):S14–S19. doi: 10.1038/ki.2011.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Musso G, Cassader M, Rosina F, Gambino R. Impact of current treatments on liver disease, glucose metabolism and cardiovascular risk in non-alcoholic fatty liver disease (NAFLD): a systematic review and meta-analysis of randomised trials. Diabetologia. 2012;55:885–904. doi: 10.1007/s00125-011-2446-4. [DOI] [PubMed] [Google Scholar]
- 180.Fulzele K, Riddle RC, DiGirolamo DJ, Cao X, Wan C, Chen D, Faugere MC, Aja S, Hussain MA, Brüning JC, et al. Insulin receptor signaling in osteoblasts regulates postnatal bone acquisition and body composition. Cell. 2010;142:309–319. doi: 10.1016/j.cell.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Pardee PE, Dunn W, Schwimmer JB. Non-alcoholic fatty liver disease is associated with low bone mineral density in obese children. Aliment Pharmacol Ther. 2012;35:248–254. doi: 10.1111/j.1365-2036.2011.04924.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Mancini T, Mazziotti G, Doga M, Carpinteri R, Simetovic N, Vescovi PP, Giustina A. Vertebral fractures in males with type 2 diabetes treated with rosiglitazone. Bone. 2009;45:784–788. doi: 10.1016/j.bone.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 183.Eliades M, Spyrou E, Agrawal N, Lazo M, Brancati FL, Potter JJ, Koteish AA, Clark JM, Guallar E, Hernaez R. Meta-analysis: vitamin D and non-alcoholic fatty liver disease. Aliment Pharmacol Ther. 2013;38:246–254. doi: 10.1111/apt.12377. [DOI] [PubMed] [Google Scholar]
- 184.Dasarathy J, Periyalwar P, Allampati S, Bhinder V, Hawkins C, Brandt P, Khiyami A, McCullough AJ, Dasarathy S. Hypovitaminosis D is associated with increased whole body fat mass and greater severity of non-alcoholic fatty liver disease. Liver Int. 2014;34:e118–e127. doi: 10.1111/liv.12312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Manco M, Ciampalini P, Nobili V. Low levels of 25-hydroxyvitamin D(3) in children with biopsy-proven nonalcoholic fatty liver disease. Hepatology. 2010;51:2229; author reply 2230. doi: 10.1002/hep.23724. [DOI] [PubMed] [Google Scholar]
- 186.Ding N, Yu RT, Subramaniam N, Sherman MH, Wilson C, Rao R, Leblanc M, Coulter S, He M, Scott C, et al. A vitamin D receptor/SMAD genomic circuit gates hepatic fibrotic response. Cell. 2013;153:601–613. doi: 10.1016/j.cell.2013.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Roth CL, Elfers CT, Figlewicz DP, Melhorn SJ, Morton GJ, Hoofnagle A, Yeh MM, Nelson JE, Kowdley KV. Vitamin D deficiency in obese rats exacerbates nonalcoholic fatty liver disease and increases hepatic resistin and Toll-like receptor activation. Hepatology. 2012;55:1103–1111. doi: 10.1002/hep.24737. [DOI] [PubMed] [Google Scholar]
- 188.Nakano T, Cheng YF, Lai CY, Hsu LW, Chang YC, Deng JY, Huang YZ, Honda H, Chen KD, Wang CC, et al. Impact of artificial sunlight therapy on the progress of non-alcoholic fatty liver disease in rats. J Hepatol. 2011;55:415–425. doi: 10.1016/j.jhep.2010.11.028. [DOI] [PubMed] [Google Scholar]
- 189.Manna P, Jain SK. Vitamin D up-regulates glucose transporter 4 (GLUT4) translocation and glucose utilization mediated by cystathionine-γ-lyase (CSE) activation and H2S formation in 3T3L1 adipocytes. J Biol Chem. 2012;287:42324–42332. doi: 10.1074/jbc.M112.407833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Sharifi N, Amani R, Hajiani E, Cheraghian B. Does vitamin D improve liver enzymes, oxidative stress, and inflammatory biomarkers in adults with non-alcoholic fatty liver disease? A randomized clinical trial. Endocrine. 2014;47:70–80. doi: 10.1007/s12020-014-0336-5. [DOI] [PubMed] [Google Scholar]
- 191.Eliades M, Spyrou E. Vitamin D: a new player in non-alcoholic fatty liver disease? World J Gastroenterol. 2015;21:1718–1727. doi: 10.3748/wjg.v21.i6.1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Hong JW, Kim JY, Kim YE, Lee EJ. Metabolic parameters and nonalcoholic fatty liver disease in hypopituitary men. Horm Metab Res. 2011;43:48–54. doi: 10.1055/s-0030-1265217. [DOI] [PubMed] [Google Scholar]
- 193.Adams LA, Feldstein A, Lindor KD, Angulo P. Nonalcoholic fatty liver disease among patients with hypothalamic and pituitary dysfunction. Hepatology. 2004;39:909–914. doi: 10.1002/hep.20140. [DOI] [PubMed] [Google Scholar]
- 194.Thomas JD, Monson JP. Adult GH deficiency throughout lifetime. Eur J Endocrinol. 2009;161 Suppl 1:S97–S106. doi: 10.1530/EJE-09-0258. [DOI] [PubMed] [Google Scholar]
- 195.Takahashi Y. Essential roles of growth hormone (GH) and insulin-like growth factor-I (IGF-I) in the liver. Endocr J. 2012;59:955–962. doi: 10.1507/endocrj.ej12-0322. [DOI] [PubMed] [Google Scholar]
- 196.Savastano S, Di Somma C, Colao A, Barrea L, Orio F, Finelli C, Pasanisi F, Contaldo F, Tarantino G. Preliminary data on the relationship between circulating levels of Sirtuin 4, anthropometric and metabolic parameters in obese subjects according to growth hormone/insulin-like growth factor-1 status. Growth Horm IGF Res. 2015;25:28–33. doi: 10.1016/j.ghir.2014.10.006. [DOI] [PubMed] [Google Scholar]
- 197.Tarantino G, Finelli C, Scopacasa F, Pasanisi F, Contaldo F, Capone D, Savastano S. Circulating levels of sirtuin 4, a potential marker of oxidative metabolism, related to coronary artery disease in obese patients suffering from NAFLD, with normal or slightly increased liver enzymes. Oxid Med Cell Longev. 2014;2014:920676. doi: 10.1155/2014/920676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Kokoszko A, Dabrowski J, Lewiński A, Karbownik-Lewińska M. Protective effects of GH and IGF-I against iron-induced lipid peroxidation in vivo. Exp Toxicol Pathol. 2008;60:453–458. doi: 10.1016/j.etp.2008.04.012. [DOI] [PubMed] [Google Scholar]
- 199.Brown-Borg HM, Rakoczy SG, Romanick MA, Kennedy MA. Effects of growth hormone and insulin-like growth factor-1 on hepatocyte antioxidative enzymes. Exp Biol Med (Maywood) 2002;227:94–104. doi: 10.1177/153537020222700203. [DOI] [PubMed] [Google Scholar]
- 200.Nishizawa H, Iguchi G, Murawaki A, Fukuoka H, Hayashi Y, Kaji H, Yamamoto M, Suda K, Takahashi M, Seo Y, et al. Nonalcoholic fatty liver disease in adult hypopituitary patients with GH deficiency and the impact of GH replacement therapy. Eur J Endocrinol. 2012;167:67–74. doi: 10.1530/EJE-12-0252. [DOI] [PubMed] [Google Scholar]
- 201.Gardner CJ, Irwin AJ, Daousi C, McFarlane IA, Joseph F, Bell JD, Thomas EL, Adams VL, Kemp GJ, Cuthbertson DJ. Hepatic steatosis, GH deficiency and the effects of GH replacement: a Liverpool magnetic resonance spectroscopy study. Eur J Endocrinol. 2012;166:993–1002. doi: 10.1530/EJE-12-0002. [DOI] [PubMed] [Google Scholar]
- 202.Takahashi Y, Iida K, Takahashi K, Yoshioka S, Fukuoka H, Takeno R, Imanaka M, Nishizawa H, Takahashi M, Seo Y, et al. Growth hormone reverses nonalcoholic steatohepatitis in a patient with adult growth hormone deficiency. Gastroenterology. 2007;132:938–943. doi: 10.1053/j.gastro.2006.12.024. [DOI] [PubMed] [Google Scholar]
- 203.Farthing MJ, Green JR, Edwards CR, Dawson AM. Progesterone, prolactin, and gynaecomastia in men with liver disease. Gut. 1982;23:276–279. doi: 10.1136/gut.23.4.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Ben-Jonathan N, Hugo ER, Brandebourg TD, LaPensee CR. Focus on prolactin as a metabolic hormone. Trends Endocrinol Metab. 2006;17:110–116. doi: 10.1016/j.tem.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 205.Jiang XB, Li CL, He DS, Mao ZG, Liu DH, Fan X, Hu B, Zhu YH, Wang HJ. Increased carotid intima media thickness is associated with prolactin levels in subjects with untreated prolactinoma: a pilot study. Pituitary. 2014;17:232–239. doi: 10.1007/s11102-013-0495-z. [DOI] [PubMed] [Google Scholar]
- 206.Berinder K, Nyström T, Höybye C, Hall K, Hulting AL. Insulin sensitivity and lipid profile in prolactinoma patients before and after normalization of prolactin by dopamine agonist therapy. Pituitary. 2011;14:199–207. doi: 10.1007/s11102-010-0277-9. [DOI] [PubMed] [Google Scholar]
- 207.Davis LM, Pei Z, Trush MA, Cheskin LJ, Contoreggi C, McCullough K, Watkins PA, Moran TH. Bromocriptine reduces steatosis in obese rodent models. J Hepatol. 2006;45:439–444. doi: 10.1016/j.jhep.2006.03.019. [DOI] [PubMed] [Google Scholar]
- 208.Cordeiro A, Souza LL, Einicker-Lamas M, Pazos-Moura CC. Non-classic thyroid hormone signalling involved in hepatic lipid metabolism. J Endocrinol. 2013;216:R47–R57. doi: 10.1530/JOE-12-0542. [DOI] [PubMed] [Google Scholar]
- 209.Eshraghian A, Hamidian Jahromi A. Non-alcoholic fatty liver disease and thyroid dysfunction: a systematic review. World J Gastroenterol. 2014;20:8102–8109. doi: 10.3748/wjg.v20.i25.8102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Turer AT, Browning JD, Ayers CR, Das SR, Khera A, Vega GL, Grundy SM, Scherer PE. Adiponectin as an independent predictor of the presence and degree of hepatic steatosis in the Dallas Heart Study. J Clin Endocrinol Metab. 2012;97:E982–E986. doi: 10.1210/jc.2011-3305. [DOI] [PubMed] [Google Scholar]
- 211.Flamant F, Baxter JD, Forrest D, Refetoff S, Samuels H, Scanlan TS, Vennström B, Samarut J. International Union of Pharmacology. LIX. The pharmacology and classification of the nuclear receptor superfamily: thyroid hormone receptors. Pharmacol Rev. 2006;58:705–711. doi: 10.1124/pr.58.4.3. [DOI] [PubMed] [Google Scholar]
- 212.Cable EE, Finn PD, Stebbins JW, Hou J, Ito BR, van Poelje PD, Linemeyer DL, Erion MD. Reduction of hepatic steatosis in rats and mice after treatment with a liver-targeted thyroid hormone receptor agonist. Hepatology. 2009;49:407–417. doi: 10.1002/hep.22572. [DOI] [PubMed] [Google Scholar]
- 213.Jornayvaz FR, Lee HY, Jurczak MJ, Alves TC, Guebre-Egziabher F, Guigni BA, Zhang D, Samuel VT, Silva JE, Shulman GI. Thyroid hormone receptor-α gene knockout mice are protected from diet-induced hepatic insulin resistance. Endocrinology. 2012;153:583–591. doi: 10.1210/en.2011-1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.B UU, Mn S, Km S, Prashant A, Doddamani P, Sv S. Effect of insulin resistance in assessing the clinical outcome of clinical and subclinical hypothyroid patients. J Clin Diagn Res. 2015;9:OC01–OC04. doi: 10.7860/JCDR/2015/9754.5513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Misra S, Singh B. Insulin resistance and hypothyroidism: a complex relationship in non-alcoholic fatty liver disease. J Indian Med Assoc. 2013;111:324–326, 329. [PubMed] [Google Scholar]
- 216.Ittermann T, Haring R, Wallaschofski H, Baumeister SE, Nauck M, Dörr M, Lerch MM, Meyer zu Schwabedissen HE, Rosskopf D, Völzke H. Inverse association between serum free thyroxine levels and hepatic steatosis: results from the Study of Health in Pomerania. Thyroid. 2012;22:568–574. doi: 10.1089/thy.2011.0279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Chung GE, Kim D, Kim W, Yim JY, Park MJ, Kim YJ, Yoon JH, Lee HS. Non-alcoholic fatty liver disease across the spectrum of hypothyroidism. J Hepatol. 2012;57:150–156. doi: 10.1016/j.jhep.2012.02.027. [DOI] [PubMed] [Google Scholar]
- 218.Pagadala MR, Zein CO, Dasarathy S, Yerian LM, Lopez R, McCullough AJ. Prevalence of hypothyroidism in nonalcoholic fatty liver disease. Dig Dis Sci. 2012;57:528–534. doi: 10.1007/s10620-011-2006-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Tao Y, Gu H, Wu J, Sui J. Thyroid function is associated with non-alcoholic fatty liver disease in euthyroid subjects. Endocr Res. 2015;40:74–78. doi: 10.3109/07435800.2014.952014. [DOI] [PubMed] [Google Scholar]
- 220.Carulli L, Ballestri S, Lonardo A, Lami F, Violi E, Losi L, Bonilauri L, Verrone AM, Odoardi MR, Scaglioni F, et al. Is nonalcoholic steatohepatitis associated with a high-though-normal thyroid stimulating hormone level and lower cholesterol levels? Intern Emerg Med. 2013;8:297–305. doi: 10.1007/s11739-011-0609-4. [DOI] [PubMed] [Google Scholar]
- 221.Eshraghian A, Dabbaghmanesh MH, Eshraghian H, Fattahi MR, Omrani GR. Nonalcoholic fatty liver disease in a cluster of Iranian population: thyroid status and metabolic risk factors. Arch Iran Med. 2013;16:584–589. [PubMed] [Google Scholar]
- 222.Mazo DF, Lima VM, Stefano JT, Rabelo F, Faintuch J, Oliveira CP. Gluco-lipidic indices in treated hypothyroidism associated with nonalcoholic fatty liver disease. Arq Gastroenterol. 2011;48:186–189. doi: 10.1590/s0004-28032011000300006. [DOI] [PubMed] [Google Scholar]
- 223.van Tienhoven-Wind LJ, Dullaart RP. Low-normal thyroid function and the pathogenesis of common cardio-metabolic disorders. Eur J Clin Invest. 2015;45:494–503. doi: 10.1111/eci.12423. [DOI] [PubMed] [Google Scholar]
- 224.Xu C, Xu L, Yu C, Miao M, Li Y. Association between thyroid function and nonalcoholic fatty liver disease in euthyroid elderly Chinese. Clin Endocrinol (Oxf) 2011;75:240–246. doi: 10.1111/j.1365-2265.2011.04016.x. [DOI] [PubMed] [Google Scholar]
- 225.Targher G, Montagnana M, Salvagno G, Moghetti P, Zoppini G, Muggeo M, Lippi G. Association between serum TSH, free T4 and serum liver enzyme activities in a large cohort of unselected outpatients. Clin Endocrinol (Oxf) 2008;68:481–484. doi: 10.1111/j.1365-2265.2007.03068.x. [DOI] [PubMed] [Google Scholar]
- 226.Walsh JP. Setpoints and susceptibility: do small differences in thyroid function really matter? Clin Endocrinol (Oxf) 2011;75:158–159. doi: 10.1111/j.1365-2265.2011.04036.x. [DOI] [PubMed] [Google Scholar]
- 227.Baxter JD, Webb P. Thyroid hormone mimetics: potential applications in atherosclerosis, obesity and type 2 diabetes. Nat Rev Drug Discov. 2009;8:308–320. doi: 10.1038/nrd2830. [DOI] [PubMed] [Google Scholar]
- 228.Davis PJ, Lin HY, Mousa SA, Luidens MK, Hercbergs AA, Wehling M, Davis FB. Overlapping nongenomic and genomic actions of thyroid hormone and steroids. Steroids. 2011;76:829–833. doi: 10.1016/j.steroids.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 229.Papanastasiou L, Pappa T, Samara C, Apostolopoulou G, Tsiavos V, Markou A, Alexandraki K, Piaditis G, Chrousos G, Kaltsas G. Nonalcoholic fatty liver disease in subjects with adrenal incidentaloma. Eur J Clin Invest. 2012;42:1165–1172. doi: 10.1111/j.1365-2362.2012.02707.x. [DOI] [PubMed] [Google Scholar]
- 230.Pivonello R, De Leo M, Vitale P, Cozzolino A, Simeoli C, De Martino MC, Lombardi G, Colao A. Pathophysiology of diabetes mellitus in Cushing’s syndrome. Neuroendocrinology. 2010;92 Suppl 1:77–81. doi: 10.1159/000314319. [DOI] [PubMed] [Google Scholar]
- 231.Tarantino G, Finelli C. Pathogenesis of hepatic steatosis: the link between hypercortisolism and non-alcoholic fatty liver disease. World J Gastroenterol. 2013;19:6735–6743. doi: 10.3748/wjg.v19.i40.6735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Rockall AG, Sohaib SA, Evans D, Kaltsas G, Isidori AM, Monson JP, Besser GM, Grossman AB, Reznek RH. Hepatic steatosis in Cushing’s syndrome: a radiological assessment using computed tomography. Eur J Endocrinol. 2003;149:543–548. doi: 10.1530/eje.0.1490543. [DOI] [PubMed] [Google Scholar]
- 233.Sakkiah S, Meganathan C, Sohn YS, Namadevan S, Lee KW. Identification of important chemical features of 11β-hydroxysteroid dehydrogenase type1 inhibitors: application of ligand based virtual screening and density functional theory. Int J Mol Sci. 2012;13:5138–5162. doi: 10.3390/ijms13045138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Gathercole LL, Morgan SA, Bujalska IJ, Hauton D, Stewart PM, Tomlinson JW. Regulation of lipogenesis by glucocorticoids and insulin in human adipose tissue. PLoS One. 2011;6:e26223. doi: 10.1371/journal.pone.0026223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Masuzaki H, Paterson J, Shinyama H, Morton NM, Mullins JJ, Seckl JR, Flier JS. A transgenic model of visceral obesity and the metabolic syndrome. Science. 2001;294:2166–2170. doi: 10.1126/science.1066285. [DOI] [PubMed] [Google Scholar]
- 236.Tomlinson JW, Sinha B, Bujalska I, Hewison M, Stewart PM. Expression of 11beta-hydroxysteroid dehydrogenase type 1 in adipose tissue is not increased in human obesity. J Clin Endocrinol Metab. 2002;87:5630–5635. doi: 10.1210/jc.2002-020687. [DOI] [PubMed] [Google Scholar]
- 237.Dowman JK, Hopkins LJ, Reynolds GM, Armstrong MJ, Nasiri M, Nikolaou N, van Houten EL, Visser JA, Morgan SA, Lavery GG, et al. Loss of 5α-reductase type 1 accelerates the development of hepatic steatosis but protects against hepatocellular carcinoma in male mice. Endocrinology. 2013;154:4536–4547. doi: 10.1210/en.2013-1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Konopelska S, Kienitz T, Hughes B, Pirlich M, Bauditz J, Lochs H, Strasburger CJ, Stewart PM, Quinkler M. Hepatic 11beta-HSD1 mRNA expression in fatty liver and nonalcoholic steatohepatitis. Clin Endocrinol (Oxf) 2009;70:554–560. doi: 10.1111/j.1365-2265.2008.03358.x. [DOI] [PubMed] [Google Scholar]
- 239.Candia R, Riquelme A, Baudrand R, Carvajal CA, Morales M, Solís N, Pizarro M, Escalona A, Carrasco G, Boza C, et al. Overexpression of 11β-hydroxysteroid dehydrogenase type 1 in visceral adipose tissue and portal hypercortisolism in non-alcoholic fatty liver disease. Liver Int. 2012;32:392–399. doi: 10.1111/j.1478-3231.2011.02685.x. [DOI] [PubMed] [Google Scholar]
- 240.Ahmed A, Rabbitt E, Brady T, Brown C, Guest P, Bujalska IJ, Doig C, Newsome PN, Hubscher S, Elias E, et al. A switch in hepatic cortisol metabolism across the spectrum of non alcoholic fatty liver disease. PLoS One. 2012;7:e29531. doi: 10.1371/journal.pone.0029531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Charlton M, Angulo P, Chalasani N, Merriman R, Viker K, Charatcharoenwitthaya P, Sanderson S, Gawrieh S, Krishnan A, Lindor K. Low circulating levels of dehydroepiandrosterone in histologically advanced nonalcoholic fatty liver disease. Hepatology. 2008;47:484–492. doi: 10.1002/hep.22063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Koehler E, Swain J, Sanderson S, Krishnan A, Watt K, Charlton M. Growth hormone, dehydroepiandrosterone and adiponectin levels in non-alcoholic steatohepatitis: an endocrine signature for advanced fibrosis in obese patients. Liver Int. 2012;32:279–286. doi: 10.1111/j.1478-3231.2011.02637.x. [DOI] [PubMed] [Google Scholar]
- 243.Koga M, Saito H, Mukai M, Saibara T, Kasayama S. Serum dehydroepiandrosterone sulphate levels in patients with non-alcoholic fatty liver disease. Intern Med. 2011;50:1657–1661. doi: 10.2169/internalmedicine.50.4682. [DOI] [PubMed] [Google Scholar]
- 244.Warner FJ, Lubel JS, McCaughan GW, Angus PW. Liver fibrosis: a balance of ACEs? Clin Sci (Lond) 2007;113:109–118. doi: 10.1042/CS20070026. [DOI] [PubMed] [Google Scholar]
- 245.Moreira de Macêdo S, Guimarães TA, Feltenberger JD, Sousa Santos SH. The role of renin-angiotensin system modulation on treatment and prevention of liver diseases. Peptides. 2014;62:189–196. doi: 10.1016/j.peptides.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 246.Bataller R, Sancho-Bru P, Ginès P, Lora JM, Al-Garawi A, Solé M, Colmenero J, Nicolás JM, Jiménez W, Weich N, et al. Activated human hepatic stellate cells express the renin-angiotensin system and synthesize angiotensin II. Gastroenterology. 2003;125:117–125. doi: 10.1016/s0016-5085(03)00695-4. [DOI] [PubMed] [Google Scholar]
- 247.Fallo F, Dalla Pozza A, Tecchio M, Tona F, Sonino N, Ermani M, Catena C, Bertello C, Mulatero P, Sabato N, et al. Nonalcoholic fatty liver disease in primary aldosteronism: a pilot study. Am J Hypertens. 2010;23:2–5. doi: 10.1038/ajh.2009.206. [DOI] [PubMed] [Google Scholar]
- 248.Fallo F, Della Mea P, Sonino N, Bertello C, Ermani M, Vettor R, Veglio F, Mulatero P. Adiponectin and insulin sensitivity in primary aldosteronism. Am J Hypertens. 2007;20:855–861. doi: 10.1016/j.amjhyper.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 249.Folli F, Saad MJ, Velloso L, Hansen H, Carandente O, Feener EP, Kahn CR. Crosstalk between insulin and angiotensin II signalling systems. Exp Clin Endocrinol Diabetes. 1999;107:133–139. doi: 10.1055/s-0029-1212088. [DOI] [PubMed] [Google Scholar]
- 250.Liu Z. The renin-angiotensin system and insulin resistance. Curr Diab Rep. 2007;7:34–42. doi: 10.1007/s11892-007-0007-5. [DOI] [PubMed] [Google Scholar]
- 251.Paschos P, Tziomalos K. Nonalcoholic fatty liver disease and the renin-angiotensin system: Implications for treatment. World J Hepatol. 2012;4:327–331. doi: 10.4254/wjh.v4.i12.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Georgescu EF, Ionescu R, Niculescu M, Mogoanta L, Vancica L. Angiotensin-receptor blockers as therapy for mild-to-moderate hypertension-associated non-alcoholic steatohepatitis. World J Gastroenterol. 2009;15:942–954. doi: 10.3748/wjg.15.942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Goh GB, Pagadala MR, Dasarathy J, Unalp-Arida A, Sargent R, Hawkins C, Sourianarayanane A, Khiyami A, Yerian L, Pai R, et al. Renin-angiotensin system and fibrosis in non-alcoholic fatty liver disease. Liver Int. 2015;35:979–985. doi: 10.1111/liv.12611. [DOI] [PubMed] [Google Scholar]
- 254.Munshi MK, Uddin MN, Glaser SS. The role of the renin-angiotensin system in liver fibrosis. Exp Biol Med (Maywood) 2011;236:557–566. doi: 10.1258/ebm.2011.010375. [DOI] [PubMed] [Google Scholar]
- 255.Asrih M, Jornayvaz FR. Diets and nonalcoholic fatty liver disease: the good and the bad. Clin Nutr. 2014;33:186–190. doi: 10.1016/j.clnu.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 256.Promrat K, Kleiner DE, Niemeier HM, Jackvony E, Kearns M, Wands JR, Fava JL, Wing RR. Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology. 2010;51:121–129. doi: 10.1002/hep.23276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Browning JD, Davis J, Saboorian MH, Burgess SC. A low-carbohydrate diet rapidly and dramatically reduces intrahepatic triglyceride content. Hepatology. 2006;44:487–488. doi: 10.1002/hep.21264. [DOI] [PubMed] [Google Scholar]
- 258.Asrih M, Altirriba J, Rohner-Jeanrenaud F, Jornayvaz FR. Ketogenic Diet Impairs FGF21 Signaling and Promotes Differential Inflammatory Responses in the Liver and White Adipose Tissue. PLoS One. 2015;10:e0126364. doi: 10.1371/journal.pone.0126364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Andersen T, Gluud C, Franzmann MB, Christoffersen P. Hepatic effects of dietary weight loss in morbidly obese subjects. J Hepatol. 1991;12:224–229. doi: 10.1016/0168-8278(91)90942-5. [DOI] [PubMed] [Google Scholar]
- 260.Jornayvaz FR, Jurczak MJ, Lee HY, Birkenfeld AL, Frederick DW, Zhang D, Zhang XM, Samuel VT, Shulman GI. A high-fat, ketogenic diet causes hepatic insulin resistance in mice, despite increasing energy expenditure and preventing weight gain. Am J Physiol Endocrinol Metab. 2010;299:E808–E815. doi: 10.1152/ajpendo.00361.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Li Y, Liu L, Wang B, Wang J, Chen D. Metformin in non-alcoholic fatty liver disease: A systematic review and meta-analysis. Biomed Rep. 2013;1:57–64. doi: 10.3892/br.2012.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Watanabe S, Hashimoto E, Ikejima K, Uto H, Ono M, Sumida Y, Seike M, Takei Y, Takehara T, Tokushige K, et al. Evidence-based clinical practice guidelines for nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. J Gastroenterol. 2015;50:364–377. doi: 10.1007/s00535-015-1050-7. [DOI] [PubMed] [Google Scholar]
- 263.Huang YY, Gusdon AM, Qu S. Nonalcoholic fatty liver disease: molecular pathways and therapeutic strategies. Lipids Health Dis. 2013;12:171. doi: 10.1186/1476-511X-12-171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, Neuschwander-Tetri BA, Lavine JE, Tonascia J, Unalp A, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med. 2010;362:1675–1685. doi: 10.1056/NEJMoa0907929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Promrat K, Lutchman G, Uwaifo GI, Freedman RJ, Soza A, Heller T, Doo E, Ghany M, Premkumar A, Park Y, et al. A pilot study of pioglitazone treatment for nonalcoholic steatohepatitis. Hepatology. 2004;39:188–196. doi: 10.1002/hep.20012. [DOI] [PubMed] [Google Scholar]
- 266.Pawlak M, Baugé E, Bourguet W, De Bosscher K, Lalloyer F, Tailleux A, Lebherz C, Lefebvre P, Staels B. The transrepressive activity of peroxisome proliferator-activated receptor alpha is necessary and sufficient to prevent liver fibrosis in mice. Hepatology. 2014;60:1593–1606. doi: 10.1002/hep.27297. [DOI] [PubMed] [Google Scholar]
- 267.Zanchi A, Chiolero A, Maillard M, Nussberger J, Brunner HR, Burnier M. Effects of the peroxisomal proliferator-activated receptor-gamma agonist pioglitazone on renal and hormonal responses to salt in healthy men. J Clin Endocrinol Metab. 2004;89:1140–1145. doi: 10.1210/jc.2003-031526. [DOI] [PubMed] [Google Scholar]
- 268.Buse JB, Klonoff DC, Nielsen LL, Guan X, Bowlus CL, Holcombe JH, Maggs DG, Wintle ME. Metabolic effects of two years of exenatide treatment on diabetes, obesity, and hepatic biomarkers in patients with type 2 diabetes: an interim analysis of data from the open-label, uncontrolled extension of three double-blind, placebo-controlled trials. Clin Ther. 2007;29:139–153. doi: 10.1016/j.clinthera.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 269.Ekstedt M, Franzén LE, Mathiesen UL, Holmqvist M, Bodemar G, Kechagias S. Statins in non-alcoholic fatty liver disease and chronically elevated liver enzymes: a histopathological follow-up study. J Hepatol. 2007;47:135–141. doi: 10.1016/j.jhep.2007.02.013. [DOI] [PubMed] [Google Scholar]
- 270.Hyogo H, Tazuma S, Arihiro K, Iwamoto K, Nabeshima Y, Inoue M, Ishitobi T, Nonaka M, Chayama K. Efficacy of atorvastatin for the treatment of nonalcoholic steatohepatitis with dyslipidemia. Metabolism. 2008;57:1711–1718. doi: 10.1016/j.metabol.2008.07.030. [DOI] [PubMed] [Google Scholar]
- 271.Kargiotis K, Katsiki N, Athyros VG, Giouleme O, Patsiaoura K, Katsiki E, Mikhailidis DP, Karagiannis A. Effect of rosuvastatin on non-alcoholic steatohepatitis in patients with metabolic syndrome and hypercholesterolaemia: a preliminary report. Curr Vasc Pharmacol. 2014;12:505–511. doi: 10.2174/15701611113119990009. [DOI] [PubMed] [Google Scholar]
- 272.Yoneda M, Fujita K, Nozaki Y, Endo H, Takahashi H, Hosono K, Suzuki K, Mawatari H, Kirikoshi H, Inamori M, et al. Efficacy of ezetimibe for the treatment of non-alcoholic steatohepatitis: An open-label, pilot study. Hepatol Res. 2010;40:566–573. doi: 10.1111/j.1872-034X.2010.00644.x. [DOI] [PubMed] [Google Scholar]
- 273.Averna M. The effect of ezetimibe on NAFLD. Atheroscler Suppl. 2015;17:27–34. doi: 10.1016/S1567-5688(15)50007-X. [DOI] [PubMed] [Google Scholar]
- 274.Dowman JK, Armstrong MJ, Tomlinson JW, Newsome PN. Current therapeutic strategies in non-alcoholic fatty liver disease. Diabetes Obes Metab. 2011;13:692–702. doi: 10.1111/j.1463-1326.2011.01403.x. [DOI] [PubMed] [Google Scholar]
- 275.Yfanti C, Nielsen AR, Akerström T, Nielsen S, Rose AJ, Richter EA, Lykkesfeldt J, Fischer CP, Pedersen BK. Effect of antioxidant supplementation on insulin sensitivity in response to endurance exercise training. Am J Physiol Endocrinol Metab. 2011;300:E761–E770. doi: 10.1152/ajpendo.00207.2010. [DOI] [PubMed] [Google Scholar]
- 276.Abdelmalek MF, Sanderson SO, Angulo P, Soldevila-Pico C, Liu C, Peter J, Keach J, Cave M, Chen T, McClain CJ, et al. Betaine for nonalcoholic fatty liver disease: results of a randomized placebo-controlled trial. Hepatology. 2009;50:1818–1826. doi: 10.1002/hep.23239. [DOI] [PubMed] [Google Scholar]
- 277.Leuschner UF, Lindenthal B, Herrmann G, Arnold JC, Rössle M, Cordes HJ, Zeuzem S, Hein J, Berg T. High-dose ursodeoxycholic acid therapy for nonalcoholic steatohepatitis: a double-blind, randomized, placebo-controlled trial. Hepatology. 2010;52:472–479. doi: 10.1002/hep.23727. [DOI] [PubMed] [Google Scholar]
- 278.Lindor KD, Kowdley KV, Heathcote EJ, Harrison ME, Jorgensen R, Angulo P, Lymp JF, Burgart L, Colin P. Ursodeoxycholic acid for treatment of nonalcoholic steatohepatitis: results of a randomized trial. Hepatology. 2004;39:770–778. doi: 10.1002/hep.20092. [DOI] [PubMed] [Google Scholar]
- 279.Zeng T, Zhang CL, Zhao XL, Xie KQ. Pentoxifylline for the treatment of nonalcoholic fatty liver disease: a meta-analysis of randomized double-blind, placebo-controlled studies. Eur J Gastroenterol Hepatol. 2014;26:646–653. doi: 10.1097/MEG.0000000000000068. [DOI] [PubMed] [Google Scholar]
- 280.Jung TW, Youn BS, Choi HY, Lee SY, Hong HC, Yang SJ, Yoo HJ, Kim BH, Baik SH, Choi KM. Salsalate and adiponectin ameliorate hepatic steatosis by inhibition of the hepatokine fetuin-A. Biochem Pharmacol. 2013;86:960–969. doi: 10.1016/j.bcp.2013.07.034. [DOI] [PubMed] [Google Scholar]
- 281.Xu J, Lloyd DJ, Hale C, Stanislaus S, Chen M, Sivits G, Vonderfecht S, Hecht R, Li YS, Lindberg RA, et al. Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes. 2009;58:250–259. doi: 10.2337/db08-0392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Camporez JP, Jornayvaz FR, Petersen MC, Pesta D, Guigni BA, Serr J, Zhang D, Kahn M, Samuel VT, Jurczak MJ, et al. Cellular mechanisms by which FGF21 improves insulin sensitivity in male mice. Endocrinology. 2013;154:3099–3109. doi: 10.1210/en.2013-1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Li H, Fang Q, Gao F, Fan J, Zhou J, Wang X, Zhang H, Pan X, Bao Y, Xiang K, et al. Fibroblast growth factor 21 levels are increased in nonalcoholic fatty liver disease patients and are correlated with hepatic triglyceride. J Hepatol. 2010;53:934–940. doi: 10.1016/j.jhep.2010.05.018. [DOI] [PubMed] [Google Scholar]
- 284.Yilmaz Y, Eren F, Yonal O, Kurt R, Aktas B, Celikel CA, Ozdogan O, Imeryuz N, Kalayci C, Avsar E. Increased serum FGF21 levels in patients with nonalcoholic fatty liver disease. Eur J Clin Invest. 2010;40:887–892. doi: 10.1111/j.1365-2362.2010.02338.x. [DOI] [PubMed] [Google Scholar]
- 285.Yan H, Xia M, Chang X, Xu Q, Bian H, Zeng M, Rao S, Yao X, Tu Y, Jia W, et al. Circulating fibroblast growth factor 21 levels are closely associated with hepatic fat content: a cross-sectional study. PLoS One. 2011;6:e24895. doi: 10.1371/journal.pone.0024895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Samson SL, Sathyanarayana P, Jogi M, Gonzalez EV, Gutierrez A, Krishnamurthy R, Muthupillai R, Chan L, Bajaj M. Exenatide decreases hepatic fibroblast growth factor 21 resistance in non-alcoholic fatty liver disease in a mouse model of obesity and in a randomised controlled trial. Diabetologia. 2011;54:3093–3100. doi: 10.1007/s00125-011-2317-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Gaich G, Chien JY, Fu H, Glass LC, Deeg MA, Holland WL, Kharitonenkov A, Bumol T, Schilske HK, Moller DE. The effects of LY2405319, an FGF21 analog, in obese human subjects with type 2 diabetes. Cell Metab. 2013;18:333–340. doi: 10.1016/j.cmet.2013.08.005. [DOI] [PubMed] [Google Scholar]
- 288.Liu J, Xu Y, Hu Y, Wang G. The role of fibroblast growth factor 21 in the pathogenesis of non-alcoholic fatty liver disease and implications for therapy. Metabolism. 2015;64:380–390. doi: 10.1016/j.metabol.2014.11.009. [DOI] [PubMed] [Google Scholar]
- 289.Kharitonenkov A, Wroblewski VJ, Koester A, Chen YF, Clutinger CK, Tigno XT, Hansen BC, Shanafelt AB, Etgen GJ. The metabolic state of diabetic monkeys is regulated by fibroblast growth factor-21. Endocrinology. 2007;148:774–781. doi: 10.1210/en.2006-1168. [DOI] [PubMed] [Google Scholar]
- 290.Silverman EM, Sapala JA, Appelman HD. Regression of hepatic steatosis in morbidly obese persons after gastric bypass. Am J Clin Pathol. 1995;104:23–31. doi: 10.1093/ajcp/104.1.23. [DOI] [PubMed] [Google Scholar]
- 291.Mottin CC, Moretto M, Padoin AV, Kupski C, Swarowsky AM, Glock L, Duval V, da Silva JB. Histological behavior of hepatic steatosis in morbidly obese patients after weight loss induced by bariatric surgery. Obes Surg. 2005;15:788–793. doi: 10.1381/0960892054222830. [DOI] [PubMed] [Google Scholar]
- 292.Lassailly G, Caiazzo R, Buob D, Pigeyre M, Verkindt H, Labreuche J, Raverdy V, Leteurtre E, Dharancy S, Louvet A, et al. Bariatric Surgery Reduces Features of Nonalcoholic Steatohepatitis in Morbidly Obese Patients. Gastroenterology. 2015;149:379–388. doi: 10.1053/j.gastro.2015.04.014. [DOI] [PubMed] [Google Scholar]
- 293.Chavez-Tapia NC, Tellez-Avila FI, Barrientos-Gutierrez T, Mendez-Sanchez N, Lizardi-Cervera J, Uribe M. Bariatric surgery for non-alcoholic steatohepatitis in obese patients. Cochrane Database Syst Rev. 2010;(1):CD007340. doi: 10.1002/14651858.CD007340.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Harrison SA, Fincke C, Helinski D, Torgerson S, Hayashi P. A pilot study of orlistat treatment in obese, non-alcoholic steatohepatitis patients. Aliment Pharmacol Ther. 2004;20:623–628. doi: 10.1111/j.1365-2036.2004.02153.x. [DOI] [PubMed] [Google Scholar]