

Abstract
The whole mitochondrial genome of the pest fruit fly Bactrocera arecae was obtained from next-generation sequencing of genomic DNA. It had a total length of 15,900 bp, consisting of 13 protein-coding genes, 2 rRNA genes, 22 tRNA genes and a non-coding region (A + T-rich control region). The control region (952 bp) was flanked by rrnS and trnI genes. The start codons included 6 ATG, 3 ATT and 1 each of ATA, ATC, GTG and TCG. Eight TAA, two TAG, one incomplete TA and two incomplete T stop codons were represented in the protein-coding genes. The cloverleaf structure for trnS1 lacked the D-loop, and that of trnN and trnF lacked the TΨC-loop. Molecular phylogeny based on 13 protein-coding genes was concordant with 37 mitochondrial genes, with B. arecae having closest genetic affinity to B. tryoni. The subgenus Bactrocera of Dacini tribe and the Dacinae subfamily (Dacini and Ceratitidini tribes) were monophyletic. The whole mitogenome of B. arecae will serve as a useful dataset for studying the genetics, systematics and phylogenetic relationships of the many species of Bactrocera genus in particular, and tephritid fruit flies in general.
Some 200 species of tephritid fruit flies in the world are considered pests of economic importance, causing direct losses to a wide variety of fruit, vegetable and flower crops1. The larvae of about 35% of the species attack soft fruits2. Members of the Dacini tribe, particularly the Bactrocera genus, are of special economic importance in tropical Asia, Australia and the South Pacific2.
The betelnut fruit fly Bactrocera arecae (Hardy & Adachi) is a member of the Bactrocera dorsalis species complex of the Dacinae subfamily1,3. It has a predominantly orange to brown body, with a broad yellow median band on the mesonotum. Its larvae feed in the nuts of the betelnut palm (Areca catechu)3. The adult male flies are not attracted to Cue lure or methyl eugenol2. This species has a restricted distribution, from southern Thailand through Peninsular Malaysia to Singapore (the type locality)1,3.
To date, B. arecae has not received extensive attention in molecular phylogenetic studies. For example, it was not included in Smith et.al.’s study on the phylogenetic relationships among 24 Bactrocera species based on rrnL, cox2, trnK and trnD genes4, and Krosh et.al.’s study of 125 Dacini species based on rrnL, cox1, cox2 and “white-eye” genes5. More recently, it was among 47 Bactrocera species studied based on cox1 gene sequences6, and Virgilio et al.’s study of 56 Bactrocera taxa using cox1 and rrnL gene fragments7. To date there are only 7 nucleotide sequences for B. arecae in GenBank – 2 for cox1 and one each for ITS-1, cox2, nad1, rrnL and rrnS. At the mitochondrial genome (mitogenome) level, only 10 whole genomes of Bactrocera taxa are available in GenBank.
We report here the whole mitogenome of B. arecae determined using next-generation sequencing (NGS) and discuss the molecular phylogeny of Dacini tribe.
Results
Mitogenome analysis and features
Next-generation sequencing on NextSeq 500 Dekstop Sequencer generated an approximately 100 giga bases data from B. arecae library. Removal of low quality sequence (<Q20), PhiX reads and sequences shorter than 50 nucleotides resulted in 459 million paired-end reads with 57 billion bases. A total of 2,451,320 sequence reads with a total of 330,821,510 bases were mapped to full mitochondrial genome reference sequences of Bactrocera genus. De novo assembly of these mapped reads resulted in 164 contigs with maximum length of 15,925 bp and N50 of 485. The total GC content was 28.4%, with base composition of 33.8% A, 37.9% T, 17% G, and 11.4% C.
The mitogenome of B. arecae was 15,900 bp long, comprising 37 genes (13 protein-coding genes – PCGs, 2 rRNA genes, and 22 tRNA genes) and a non-coding region (A + T-rich control region) (Table 1, Fig. 1). Spacing sequences ranged from 2 to 55 bp in 15 regions, the largest was between trnQ and trnM genes. Sequences with 25, 33 and 55 bases had clear stem-loop structures (Supplementary Fig. S1). The overlaps in 9 regions ranged from 1 to 8 bp, the largest being between trnW and trnC genes (Table 1). Nine PCGs (nad2, cox1-3, atp6, atp8, nad3, nad6, cob), 14 tRNAs and the control region were located on the major J-strand (Table 1). Fourteen genes (4 PCGs – nad5, nad4, nad4l and nad1; both rRNA genes; and 8 tRNA genes) were located on the minor N-strand. The control region (952 bp) was flanked by rrnS and trnI genes (Fig. 1). It conformed to the general structure in insects, comprising two poly-T stretches with ≥7 bp (one with 25 bp and the other 7 bp). There were 7 T-stretch with 4 bp, 2 with 5 bp and 1 with 6 bp.
Table 1. Characteristics of the mitochondrial genome of Bactrocera arecae.
| Gene | Location | Strand | Size (bp) | IntergenicSequence | Start/stopcodon |
|---|---|---|---|---|---|
| trnI(gat) | 1–66 | J | 66 | −3 | |
| trnQ(ttg) | 64–132 | N | 69 | 55 | |
| trnM(cat) | 188–256 | J | 69 | ||
| nad2 | 257–1279 | J | 1023 | 12 | ATT/TAA |
| trnW(tca) | 1292–1360 | J | 69 | −8 | |
| trnC(gca) | 1353–1416 | N | 64 | 33 | |
| trnY(gta) | 1450–1516 | N | 67 | −2 | |
| cox1 | 1515–3049 | J | 1535 | TCG/TA | |
| trnL2(taa) | 3050–3115 | J | 66 | 4 | |
| cox2 | 3120–3809 | J | 690 | 3 | ATG/TAA |
| trnK(ctt) | 3813–3882 | J | 70 | ||
| trnD(gtc) | 3883–3949 | J | 67 | ||
| atp8 | 3950–4111 | J | 162 | −7 | GTG/TAA |
| atp6 | 4105 –4782 | J | 678 | −1 | ATG/TAA |
| cox3 | 4782–5570 | J | 789 | 9 | ATG/TAA |
| trnG(tcc) | 5580–5644 | J | 65 | ||
| nad3 | 5645–5996 | J | 352 | ATT/T | |
| trnA(tgc) | 5997–6061 | J | 65 | 7 | |
| trnR(tcg) | 6069–6132 | J | 64 | 25 | |
| trnN(gtt) | 6158–6222 | J | 65 | ||
| trnS1(gct) | 6223–6290 | J | 68 | ||
| trnE(ttc) | 6291–6357 | J | 67 | 18 | |
| trnF(gaa) | 6376–6440 | N | 65 | 5 | |
| nad5 | 6446–8158 | N | 1713 | 15 | ATC/TAA |
| trnH(gtg) | 8174–8239 | N | 66 | ||
| nad4 | 8240–9580 | N | 1341 | −7 | ATG/TAG |
| nad4l | 9574–9870 | N | 297 | 2 | ATG/TAA |
| trnT(tgt) | 9873–9937 | J | 65 | ||
| trnP(tgg) | 9938–10003 | N | 66 | 2 | |
| nad6 | 10006–10530 | J | 525 | −1 | ATT/TAA |
| cob | 10530–11666 | J | 1137 | −2 | ATG/TAG |
| trnS2(tga) | 11665–11731 | J | 67 | 15 | |
| nad1 | 11747–12686 | N | 940 | 10 | ATA/T |
| trnL1(tag) | 12697–12761 | N | 65 | 4 | |
| rrnL | 12766–14088 | N | 1323 | ||
| trnV(tac) | 14089–14160 | N | 72 | ||
| rrnS | 14161–14948 | N | 788 | ||
| Control region | 14949–15900 | J | 952 |
Figure 1. Complete mitogenome of Bactrocera arecae with BRIG visualization showing the protein coding genes, rRNAs, tRNAs and non-coding regions.

GC skew (−0.259) is shown on the outer surface of the ring whereas GC content (27.7%) is shown on the inner surface. The AT skew is 0.080.
The commonest start codon was ATG (in 6 PCGs – cox2, atp6, cox3, nad4l, nad4, cob), followed by three for ATT (nad2, nad3, nad6), and one each for ATA (nad1), ATC (nad5), GTG (atp8) and TCG (cox1). Eight PCGs had TAA stop codon, two had TAG while the remaining three genes had incomplete stop codons (TA in cox1; T in nad1 and nad3) (Table 1).
Table 2 summarizes the base composition of the mitochondrial whole genome, protein-coding genes, rRNA genes and control region. All were A + T rich. The A + T content for PCGs ranged from 63.3% (cox1) to 78.8% (nad4l). Eight PCGs (atp8 and all 7 nad) had A + T content of over 70%. The A + T content of the non-coding control region was 86.0%. The GC skewness values for the whole genome, PCGs, rRNA genes and control region were negative (−0.120 to −0.487) indicating bias toward the use of Cs over Gs. Although the AT skewness value was positive (0.080) for the whole genome, it was variable for individual genes.
Table 2. Base composition of mitochondrial whole genome, protein-coding genes, rRNA genes and control region.
| Region | A% | C% | G% | T% | A + T% | G + C% | AT skew | GC skew |
|---|---|---|---|---|---|---|---|---|
| Whole genome | 39.0 | 17.5 | 10.3 | 33.2 | 72.3 | 27.7 | 0.080 | −0.259 |
| nad2 | 33.3 | 19.5 | 9.1 | 38.1 | 71.5 | 28.5 | −0.067 | −0.364 |
| cox1 | 29.9 | 20.5 | 16.1 | 33.4 | 63.3 | 36.7 | −0.055 | −0.120 |
| cox2 | 33.5 | 19.4 | 13.6 | 33.5 | 67.0 | 33.0 | 0 | −0.176 |
| atp8 | 35.2 | 20.4 | 9.2 | 35.2 | 70.4 | 29.6 | 0 | −0.378 |
| atp6 | 30.5 | 20.1 | 11.8 | 37.6 | 68.1 | 31.9 | −0.104 | −0.259 |
| cox3 | 29.9 | 20.6 | 15.0 | 34.5 | 64.4 | 35.6 | −0.071 | −0.157 |
| nad3 | 32.4 | 19.0 | 9.7 | 38.9 | 71.3 | 28.7 | −0.091 | −0.324 |
| nad5 | 45.3 | 18.7 | 9.2 | 26.8 | 72.2 | 27.8 | 0.257 | −0.341 |
| nad4 | 48.5 | 16.7 | 8.5 | 26.3 | 74.8 | 25.2 | 0.297 | −0.325 |
| nad4l | 51.2 | 14.8 | 6.4 | 27.6 | 78.8 | 21.2 | 0.299 | −0.396 |
| nad6 | 38.1 | 17.7 | 6.1 | 38.1 | 76.2 | 23.8 | 0 | −0.487 |
| cob | 31.3 | 21.3 | 13.3 | 34.1 | 65.4 | 34.6 | −0.043 | −0.231 |
| nad1 | 48.0 | 18.7 | 9.8 | 23.5 | 71.5 | 28.5 | 0.343 | −0.312 |
| rrnS | 43.1 | 14.3 | 6.8 | 35.8 | 78.8 | 21.2 | 0.093 | −0.355 |
| rrnL | 40.7 | 16.8 | 9.0 | 33.5 | 74.2 | 25.8 | 0.097 | −0.302 |
| Control region | 45.6 | 7.9 | 6.1 | 40.4 | 86.0 | 14.0 | 0.060 | −0.129 |
Of the tRNAs, the cloverleaf structure for trnS1 lacked the D-loop, while trnN and trnF lacked the TΨC-loop (Fig. 2). The number of base pairs in the DHU-stem ranged from 3 to 4 (Fig. 2; Supplementary Table S1). All the TΨC-stems had 5 base pairs except 4 bp in trnC, trnH, trnP and trnS1. The number of bases in the D-loop and TΨC-loop was variable.
Figure 2. Cloverleaf structure of the 22 inferred tRNAs in the mitogenome of Bactrocera arecae.

The cloverleaf structure for trnS1 lacked the D-loop, and that of trnN and trnF lacked the TΨC-loop.
Phylogenetic relationships within Dacini tribe
Figure 3 depicts the molecular phylogeny of B. arecae in relation to other taxa of the Dacini tribe of Dacinae subfamily based on 13 PCGs. The phylogram based on 37 mt-genes (13 PCGs, 2 rRNA and 22 tRNA genes) was congruent with that based on 13 PCGs (Supplementary Fig. S2). Most of the nodes were well-supported. The genus Bactrocera was monophyletic. Members of the subgenus Bactrocera formed a distinct clade from the other subgenera (Daculus, Tetradacus and Zeugodacus), with B. (B.) arecae forming a sister group to B. (B.) tryoni. The subfamily Dacinae was also monophyletic and clearly separated from Tephritinae subfamily. However, based on rrnL and rrnS genes the subfamily Dacinae was not monophyletic as Procecidochares utilis (Tephritinae subfamily) was sister to the Dacini (Fig. 4).
Figure 3. Bayesian inference and maximum likelihood tree based on 13 protein-coding genes of the whole mitogenomes of Tephritid fruit flies with Drosophilidae as outgroup.

Numeric values at the nodes are Bayesian posterior probabilities/ML bootstrap.
Figure 4. Bayesian inference and maximum likelihood tree based on rrnL and rrnS genes from whole mitogenomes of Tephritid fruit flies with Drosophilidae as outgroup.
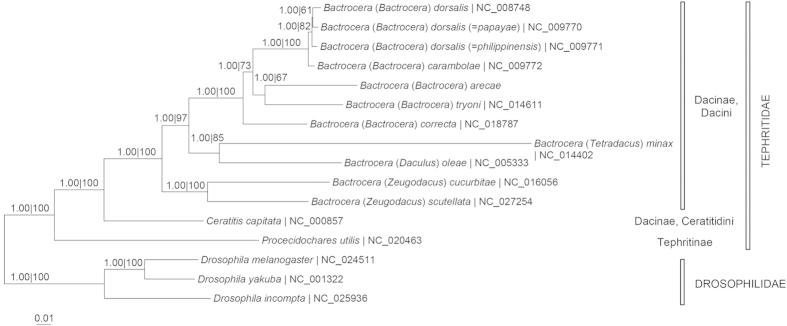
Numeric values at the nodes are Bayesian posterior probabilities/ML bootstrap.
Discussion
Mitochondrial genomes of insects have been very extensively studied8. They have been applied particularly to studies regarding phylogeny and evolution8. To date there are 12 complete mitogenomes of tephritid fruit flies in GenBank – 10 from Bactrocera (Dacinae, Dacini), and 1 each from Ceratitis (Dacinae, Ceratitidini) and Procecidochares (Tephritinae, Cecidocharini).
The mitogenome size of B. arecae (15,900 bp) is smaller than those of B. carambolae (15,915 bp), B. correcta (15,936 bp), B. dorsalis (15,915 bp), B. minax (16,043 bp), B. dorsalis (= papayae) (15,915 bp), B. dorsalis ( = philippinensis) (15,915 bp), B. scutellata (15,915 bp), B. tryoni (15,925 bp), C. capitata (15,980 bp) and P. utilis (15,922 bp) but larger than those of B. oleae (15,815 bp) and B. cucurbitae (15,825 bp).
The A + T content of the control region in B. arecae (86.0%) is higher than those of B. cucurbitae (82.4%), B. correcta (78.6%), B. minax (77.6%) and B. scutellata (72.6%) but lower than those of B. oleae (86.9%), B. tryoni (87.0%), B. carambolae (87.9%), B. dorsalis (88.1%), B. dorsalis (=papayae) (88.2%), B. dorsalis (=philippinensis) (88.2%) and C. capitata (91.1%).
In B. arecae mitogenome, in addition to 8 TAA, 2 TAG, and 3 incomplete (1 TA and 2 T) stop codons are represented in the protein-coding genes (Table 1). This differs from some tephritid mitogenomes which possess additionally truncated TA stop codon (e.g. B. dorsalis, B. oleae)9,10 or TAT stop codon (B. minax)11. Among the tephritid fruit flies, over half of the PCGs in B. dorsalis have truncated stop codons (3 TA and 4 T)9. The incomplete T–stop codons can be converted to TAA by post-translational polyadenylation12. Additionally, the nad5 gene of B. arecae has ATC instead of ATT start codon found in congeners (B. dorsalis, B. minax, B. oleae).
As in other insects, the B. arecae mitogenome has three main clusters of characteristic tRNAs (Fig. 1): (1) I-Q-M (isoleucine, glutamate and methionine); (2) W-C-Y (tryptophan, cysteine and tyrosine); and (3) A-R-N-S1-E-F (alanine, arginine, asparagine, serine S1, glutamate and phenylalanine). The atypical cloverleaf structure of trnS1 is common in all Metazoa13.
Our finding of B. arecae forming a sister group with B. tryoni and B. minax with B. oleae based on 13 PCGs and 37 mt-genes (Fig. 3, Supplementary Fig. S2) concurred with the phylogeny based on cox1 sequences6. However, based on 13 PCGs and 37 mt-genes the sister group of B. arecae plus B. tryoni was sister to the B. dorsalis lineage. B. correcta was sister to the B. dorsalis lineage based on one study of cox1 sequences6. Other studies based of cox114 and multi-gene sudies5 also indicated closer affinity of B. arecae and/or B. tryoni to the B. dorsalis complex than of B. correcta to B. dorsalis. However, the analysis based on concatenated cox1 and rrnL sequences indicated B. tryoni to be closer to B. dorsalis complex while B. arecae and B. correcta belonged to different lineages7.
The phylogenetic relationships of B. dorsalis, B. dorsalis (=papayae) and B. dorsalis (=philippinensis) based on different genetic markers were not congruent. Our analysis based on 13 PCGs and 37 mt-genes indicated B. dorsalis and B. dorsalis (=papayae) as sister taxa (Fig. 3, Supplementary Fig. S2) which agreed with that based on cox1 and cox2 sequences15 as well as cox1, rrnL, trnP, nad6 and period sequences7 but differed from that based on cox1 sequences which indicated B. dorsalis and B. dorsalis (=philippinensis) as sister taxa6, and B. dorsalis (=papayae) and B. dorsalis (=philippinensis) as sister taxa based on 37 mitochondrial genes16 and rrnL, cox1, cox2 and “white-eye” genes5. In an earlier study based on 13 PCGs but included only 7 Bactrocera species17, B. dorsalis (=papayae) formed a sister group with B. dorsalis (=philippinensis) compared to B. dorsalis. Our study included B. arecae, B. correcta, B. cucurbitae, and B. scutellata as well as P. utilis (Tephritinae subfamily). The phylogeny based on rrnL and rrnS genes in our study also indicated closer affinity of B. dorsalis (=papayae) and B. dorsalis (=philippinensis) compared to B. dorsalis (Fig. 4). It is evident that gene markers form a contributory factor to the discrepancies of these results. The phylogeny based on 17 enzyme loci indicated close genetic affinity (Nei’s I = 0.99; D = 0.01) between B. dorsalis and B. dorsalis (=papayae)18. Based on our present analysis of 13 PCGs, the uncorrected genetic ‘p’-distance is 1.06 between B. dorsalis and B. dorsalis (=papayae) and 1.11 between B. dorsalis and B. dorsalis ( = philippinensis). A recent study based on six loci (cox1, nad4-3′, CAD, period, ITS1, ITS2) indicates that B. dorsalis s.s., B. papayae and B. philippinensis are the same biological species19. Another taxon B. invadens has also been synonymized with B. dorsalis20.
As in most other studies6,7,16,17,18,19, B. carambolae is closely related but distinct from B. dorsalis, B. dorsalis (=papayae) and B. dorsalis (=philippinensis) (Fig. 3, Supplementary Fig. S2). In the study based on rrnL, cox1, cox2 and “white-eye” genes, B. carambolae was closer related to B. invadens, B. dorsalis (=papayae) and B. dorsalis (=philippinensis) than B. dorsalis5. Based on 17 enzyme loci, B. carambolae has a genetic identity of I = 0.92 (genetic distance of 0.08) compared to B. dorsalis and B. dorsalis (=papayae)18. Our present study based on 13 PCGs indicates a genetic distance of ‘p’ = 1.39 between B. carambolae and B. dorsalis, ‘p’ = 1.21 between B. carambolae and B. dorsalis (=papayae), and ‘p’=1.19 between B. carambolae and B. dorsalis (=philippinensis).
In the present study, B. (Zeugodacus) cucurbitae and B. (Zeugodacus) scutellata are related but distinct from the other subgenera (Bactrocera, Daculus and Tetradacus) of Bactrocera genus (Fig. 3, Supplementary Fig. S2). It has been proposed, based on rrnL, cox1, cox2 and “white-eye” genes, that taxonomic consideration be given to raising Zeugodacus to genus level as the ‘Zeugodacus’ clade is the sister group to Dacus, not Bactrocera5. This was supported by the analysis based on cox1, rrnL, trnP, nad6 and period sequences7. However, in the study based on rrnL, cox1, cox2 and “white-eye” genes5, Anastrepha ludens and Rhagoletis pomonella (both are members of Trypetinae subfamily) were closer to Dacini (Dacinae subfamily) than to C. capitata. In the present study, the Ceratitidini tribe forms the sister group of the Dacini tribe (Fig. 3, Supplementary Fig. S2). The Tephritinae subfamily (represented by P. utilis) is distinct from Dacinae subfamily. This concurs with the finding of monophyly for tephritid subfamilies and tribes (Trypetini, Carpomyini, Tephritinae, and Dacinae) based on rrnS, rrnL, and cox2 gene sequences21. In contrast, the phylogeny based on rrnL and rrnS genes indicated closer affinity of P. utilis to Dacini tribe than C. capitata (Fig. 4). A broader taxon sampling, particularly mitogenomes of Dacus genus (Dacini tribe), Tephritinae and Trypetinae subfamilies, is needed to resolve the higher order phylogeny.
In summary, we have successfully sequenced the whole mitochondrial genome of B. arecae by next-generation sequencing. The genome features are similar to other tephritid fruit flies except the ATC start codon for nad5 gene and the absence of TΨC-loop in trnN and trnF tRNAs. The phylogenetic tree based on 13 PCGs is concordant with that based on 37 mt-genes. B. arecae shows closest genetic affinity to B. tryoni.
Methods
Ethics statement
Bactrocera arecae is not a protected or endangered species. No permission is needed to collect and study this fruit fly, a pest of A. catechu.
Specimen collection
Fallen nuts of A. catechu were collected from the University of Malaya campus. They were placed in an aquarium with suitable substrate for the larvae to hatch. Pupae were placed in plastic tubes and emerging adults were collected, preserved in absolute ethanol and stored in deep freezer until use.
Extraction of genomic DNA
Genomic DNA was extracted from thorax and legs using G-spinTM Total DNA Extraction Mini Kit (iNtRON Biotechnology, Inc, Korea) following the manufacturer’s instructions with minor modification.
Sample and library preparation
The purified genomic DNA was quantified with Qubit® 2.0 Fluorometer (Life Technologies, USA) and normalized to 2 μg. The normalized genomic DNA was fragmented to an average size of 550 bp using Covaris M220 system (Covaris, Woburn, MA, USA). Library was prepared using TruSeq DNA PCR-Free Sample Preparation Kit (Illumina, USA) following the manufacturer’s protocols. Quantification and size estimation of the library was conducted on a 2100 Bioanalyzer using High Sensitivity DNA Analysis Kit (Agilent Technologies) and quantitative real-time PCR was performed using KAPA Library Quantification Kit for Illumina sequencing platforms (KAPA Biosystems, Boston, MA, USA) on Eco Real-Time PCR System.
Genome sequencing
The library was normalized to 1.5 pM and sequenced using the NextSeq 500 Dekstop Sequencer (2 × 150 bp paired-end reads) (Illumina, USA).
Sequence and genome analysis
Raw sequences were extracted from the Illumina NextSeq 500 system in FASTQ format and the quality of sequences was evaluated using the FastQC software22. All the ambiguous nucleotides and reads with an average quality value (lower than Q20) were excluded from further analysis. The trimmed sequences were mapped against three reference mitogenomes, namely, Bactrocera dorsalis (NC_008748), Bactrocera carambolae (NC_009772) and Bactrocera dorsalis (=papayae) (NC_009770) using the CLC Genomic Workbench v.7.0.4 (Qiagen, Germany). The mapped sequences were then subjected to de novo assembly. High coverage contigs (average coverage value more than 500) greater than 15 kbp were subjected to BLAST23 alignment against the nucleotide database at National Center for Biotechnology Information (NCBI). Contigs with hits to mitochondrial genes or genomes were identified and extracted from CLC Genomic Workbench.
Mitogenome identification and annotation
A contig identified as mitogenome was manually examined for repeats at the beginning and end of the sequence to establish a circular mtDNA. It was then annotated with MITOS24 followed by manual validation of the coding regions using the NCBI ORF Finder (http://www.ncbi.nlm.nih.gov/gorf/gorf.html). The sequin file generated from MITOS was edited and submitted to NCBI according to ORF Finder result (NCBI GenBank accession number KR233259).
Mitogenome visualization
The circular mitogenome of B. arecae was visualized with Blast Ring Image Generator (BRIG)25.
Phylogenetic analysis
Mitogenome sequences (12 taxa) of Tephritidae family that were available in GenBank were used with B. arecae to reconstruct phylogenetic trees (Figs 3 and 4; Supplementary Fig. S1). Drosophila incompta NC_025936, D. melanogaster NC_024511 and D. yakuba NC_001322 of the Drosophilidae family were included as outgroups.
Nucleotide sequences of the 13 protein-coding genes (PCGs) were separately aligned using ClustalX v.1.8126 program and subsequently edited and trimmed using BioEdit v.7.0.5.327. The sequences of rrnS, rrnL and 22 mt-tRNA genes were aligned by MAFFT v.728. The best-fit nucleotide substitution models for maximum likelihood (ML) using the corrected Akaike Information Criterion29 and Bayesian (BI) analyses using the Bayesian Information Criterion30 were determined by Kakusan v.331. Phylograms of 13 concatenated PCGs, 37 mt-genes and 2 rRNA genes were constructed using TreeFinder32. Bootstrap values (BP) were generated via 1,000 ML bootstrap replicates. Bayesian analyses were conducted using the Markov chain Monte Carlo (MCMC) method via Mr. Bayes v.3.1.233, with two independent runs of 2 × 106 generations with four chains, and with trees sampled every 200th generation. Likelihood values for all post-analysis trees and parameters were evaluated for convergence and burn-in using the “sump” command in MrBayes and the computer program Tracer v.1.5 (http://tree.bio.ed.ac.uk/software/tracer/). The first 200 trees from each run were discarded as burn-in (where the likelihood values were stabilized prior to the burn-in), and the remaining trees were used for the construction of a 50% majority-rule consensus tree. Phylogenetic trees were viewed and edited by FigTree v.1.434,35,36
Additional Information
Accession Code: Bactrocera arecae mitogenome sequences are available in NCBI GenBank database (accession number: KR233259).
How to cite this article: Yong, H.-S. et al. Complete mitochondrial genome of Bactrocera arecae (Insecta: Tephritidae) by next-generation sequencing and molecular phylogeny of Dacini tribe. Sci. Rep. 5, 15155; doi: 10.1038/srep15155 (2015).
Supplementary Material
Acknowledgments
We thank our institutions for providing infrastructure and other supports. This study received financial support (H-50001-00-A000025 and H-5620009) from the Ministry of Education Malaysia and University of Malaya. We thank the reviewers for their constructive suggestions to improve the manuscript.
Footnotes
Author Contributions H.-S.Y. conceived and designed the experiments in consultation with other authors. S.-L.S., W.-L.C. and H.-S.Y. performed the experiments. S.-L.S., H.-S.Y. and P.-E.L. analyzed the data. H.-S.Y., P.-E.L., K.-G.C. and P.E. contributed reagents/materials/analysis tools. H.-S.Y., S.-L.S., P.-E.L. and P.E. wrote the paper with other authors.
References
- Carroll L. E. et al. Pest fruit flies of the world. Version: 8th December 2006. Available: http://deltaintkey.com. Accessed 2015 Mar 15.
- White I. M. & Elson-Harris M. M. Fruit flies of economic significance: their identification and bionomics (CAB International, Wallingford, 1992). [Google Scholar]
- Hardy D. E. The fruits flies (Tephritidae – Diptera) of Thailand and bordering countries. Pacif. Insects Monogr. 31, 1–353 (1973). [Google Scholar]
- Smith P. T., Kambhampati S. & Armstrong K. A. Phylogenetic relationships among Bactrocera species (Diptera: Tephritidae) inferred from mitochondrial DNA sequences. Mol. Phyloget. Evol. 26, 8–17 (2003). [DOI] [PubMed] [Google Scholar]
- Krosch M. N. et al. A molecular phylogeny for the Tribe Dacini (Diptera: Tephritidae): Systematic and biogeographic implications. Mol. Phylogenet. Evol. 64, 513–523 (2012). [DOI] [PubMed] [Google Scholar]
- Liu S.-S., Zhang G.-F. & Wan F.-H. DNA barcoding and phylogenetic analysis of common species of the genus Bactrocera (Diptera: Tephritidae) based on mtDNA COI gene sequences. Acta Entomol. Sinica 57(3), 343–355 (2014). [Google Scholar]
- Virgilio M., Jordaens K., Verwimp C., White I. M. & De Meyer M. Higher phylogeny of frugivorous flies (Diptera, Tephritidae, Dacini): Localised partition conflicts and a novel generic classification. Mol. Phylogent. Evol. 85, 171–179 (2015). [DOI] [PubMed] [Google Scholar]
- Cameron S. L. Insect mitochondrial genomics: implications for evolution and phylogeny. Annu. Rev. Entomol. 59, 95–117 (2013). [DOI] [PubMed] [Google Scholar]
- Yu D. J., Xu L., Nardi F., Li J. G. & Zhang R. J. The complete nucleotide sequence of the mitochondrial genome of the oriental fruit fly, Bactrocera dorsalis (Diptera: Tephritidae). Gene 396, 66–74 (2007). [DOI] [PubMed] [Google Scholar]
- Nardi F., Carapelli A., Dallai R. & Frati F. The mitochondrial genome of the olive fly Bactrocera oleae: two haplotypes from distant geographical locations. Ins. Mol. Biol. 12(6), 605–611 (2003). [DOI] [PubMed] [Google Scholar]
- Zhang B., Nardi F., Hull-Sanders H., Wan X. & Liu Y. The complete nucleotide sequence of the mitochondrial genome of Bactrocera minax (Diptera: Tephritidae). PLOS ONE 9(6), e100558 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojala D., Montoya J. & Attardi G. tRNA punctuation model of RNA processing in human mitochondria. Nature 290, 470–474 (1981). [DOI] [PubMed] [Google Scholar]
- Jühling F. et al. Improved systematic tRNA gene annotation allows new insights into the evolution of mitochondrial tRNA structures and into the mechanisms of mitochondrial genome rearrangemnts. Nucl. Acids Res. 40(7), 2833–2845 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asokan R. et al. Molecular Identification and Phylogeny of Bactrocera Species (Diptera: Tephritidae). Fla. Entomol. 94(4), 1026–1035 (2011). [Google Scholar]
- Nakahara S. & Muraji M. Phylogenetic analyses of Bactrocera fruit flies (Diptera: Tephritidae) based on nucleotide sequences of the mitochondrial COI and COII genes. Res. Bull. Pl. Prot. Japan 44, 1–12 (2008). [Google Scholar]
- Nardi F. et al. Domestication of olive fly through a multi-regional host shift to cultivated olives: Comparative dating using complete mitochondrial genomes. Mol. Phylogenet. Evol. 57, 678–686 (2010). [DOI] [PubMed] [Google Scholar]
- Li Q.-Q. et al. Phylogenetic analysis within Tephritidae of Diptera based on the concatenated 13 mitochondrial protein coding genes of mt genomes. Asian J. Anim. Vet. Adv. 8(3), 542–547 (2013). [Google Scholar]
- Yong H. S. Genetic differentiation and relationships in five taxa of the Bactrocera dorsalis complex (Insecta: Diptera: Tephritidae). Bull. Ent. Res. 85, 431–435 (1995). [Google Scholar]
- Boykin L. M. et al. Multi-gene phylogenetic analysis of south-east Asian pest members of the Bactrocera dorsalis species complex (Diptera: Tephritidae) does not support current taxonomy. J. Appl. Entomol. 138, 235–253 (2014). [Google Scholar]
- Schutze M. K. et al. Synonymization of key pest species within the Bactrocera dorsalis species complex (Diptera: Tephritidae): taxonomic changes based on a review of 20 years of integrative morphological, molecular, cytogenetic, behavioural and chemoecological data. Syst. Entomol. 40, 456–471 (2014). [Google Scholar]
- Han H.-Y. & Ro K.-E. Molecular phylogeny of the family Tephritidae (Insecta: Diptera): New insight from combined analysis of the mitochondrial 12S, 16S, and COII genes. Mol. Cells 27, 55–66 (2009). [DOI] [PubMed] [Google Scholar]
- Andrews S. FastQC: a quality control tool for high throughput sequence data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (2010).
- Altschul S. F., Gish W., Miller W., Myers E. W. & Lipman D. J. Basic local alignment search tool. J. Mol. Biol. 215(3), 403–410 (1990). [DOI] [PubMed] [Google Scholar]
- Bernt M. et al. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69, 313–319 (2012). [DOI] [PubMed] [Google Scholar]
- Alikhan N. F., Petty N. K., Ben Zakour N. L. & Beatson S. A. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12, 402 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J. D., Gibson T. J., Plewniak F., Jeanmougin F. & Higgins D. G. The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucl. Acids Res. 24, 4876–4882 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall T. A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl. Acids Symp. Ser. 41, 95–98 (1999). [Google Scholar]
- Katoh K. & Standley D. M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in performance and usability. Mol. Biol. Evol. 30(4), 772–780 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akaike H. Information Theory and an Extension of the Maximum Likelihood Principle. Proceedings of the 2nd International Symposium on Information Theory. Petrov B. N. & Csaki F. (eds.) 267–281 (Akademia Kiado, Budapest, 1973). [Google Scholar]
- Schwarz G. Estimating the dimension of a model. Ann. Stat. 6, 461–464 (1978). [Google Scholar]
- Tanabe A. S. Kakusan: a computer program to automate the selection of a nucleotide substitution model and the configuration of a mixed model on multilocus data. Mol. Ecol. Notes 7, 962–964 (2007). [Google Scholar]
- Jobb G., von Haeseler A. & Strimmer K. Treefinder: a powerful graphical analysis environment for molecular phylogenetics. BMC Evol. Biol. 4, 18 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Huelsenbeck J. P. & Ronquist F. MrBayes: Bayesian Inference of phylogenetic trees. Bioinformatics 17, 754–755 (2001). [DOI] [PubMed] [Google Scholar]
- Rambaut A. FigTree (version 1.4.0). Available at http://tree.bio.ed.ac.uk/software/figtree/ (2012).
- Wu P. F. et al. The complete mitochondrial genome of the melon fly Bactrocera cucurbitae (Diptera: Tephritidae). Mitochondrial DNA 24(1), 6–7 (2013). [DOI] [PubMed] [Google Scholar]
- Spanos L., Koutroumbas G., Kotsyfakis M. & Louis C. The mitochondrial genome of the Mediterranean fruit fly, Ceratitis capitata. Ins. Mol. Biol. 9(2), 139–144 (2000). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.