

Abstract
A rhodium complex, in conjunction with commercially available Ph-BPE ligand, catalyzes the branch-selective asymmetric hydroformylation of 1-alkenes and rapidly generates α-chiral aldehydes. A wide range of terminal olefins including 1-dodecene were examined and all delivered high enantioselectivity (up to 98:2 er) as well as good branch:linear ratios (up to 15:1).
Graphical abstract
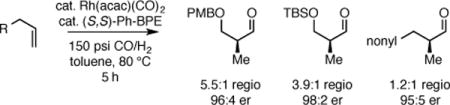
Asymmetric alkene hydroformylation (AHF) is an ideal method for the construction of carbon-carbon bonds: when accomplished in a branch-selective fashion, it converts inexpensive and abundant terminal alkenes into optically active aldehydes with perfect atom economy.1 While a number of advances in asymmetric hydroformylation have been recorded with styrenes,2 dienes,3 and vinyl acetates and amides,4 only recently has success been achieved with non-activated or lowly activated alkenes.5 Pioneered by Nozaki,5a Landis,5b Clarke,5c and Zhang,5d efficient chiral ligands, such as binaphos (1), bisdiazaphos (2), bobphos (3), and yanphos (4) have been shown to give excellent enantioselectivity and moderate regioselectivity for the AHF of a number of substrate classes (Scheme 1). However, the use of AHF in the context of total synthesis is not common, perhaps due in part to limited availability of requisite ligands.6
Scheme 1.
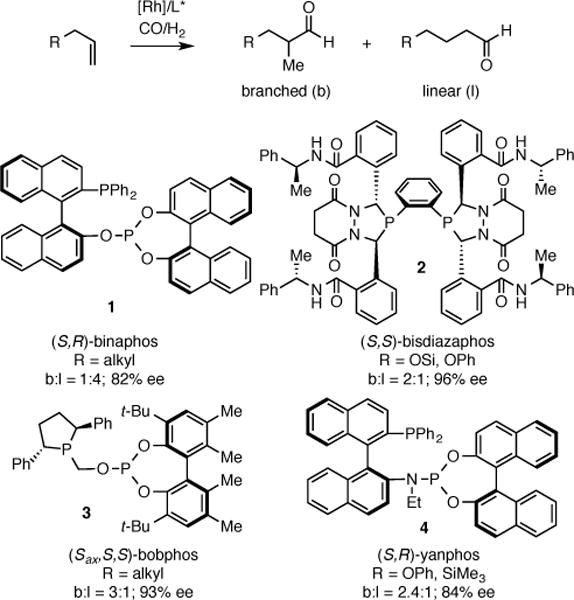
Chiral Ligands Used for AHF of 1-Alkenes.
In the planning stage for our recent total synthesis of (+)-discodermolide,7 it was considered that AHF of allyl ethers and related 1-alkenes with a readily available chiral ligand would effectively streamline the synthesis of key chiral aldehyde starting materials. While similar chiral aldehydes are often obtained from the Roche ester in several functional group interconversions, they can be obtained in just two steps from allyl alcohol when the AHF reaction is employed.5b Considering the spectacular success achieved by Landis with bisdiazaphos ligands in the hydroformylation of allyl alcohol derivatives5b, we considered that structurally-related, commercially available DuPhos and bisphospholanoethane (BPE) family of ligands might furnish competent catalysts for this substrate class.
While DuPhos and BPE ligands have proved to be excellent ligands for asymmetric hydrogenation of a broad range of substrates,8 their use for asymmetric hydroformylation reactions have been restricted to a singular report where Ph-BPE was identified as an excellent ligand for AHF of the activated substrates styrene, allyl cyanide and vinyl acetate.9 Herein we document enantioselective hydroformylation of non-activated and lowly-activated terminal alkenes with commercially available Ph-BPE ligand. The AHF reaction with Ph-BPE can occur with outstanding levels of regio- and enantioselectivity and it can be operated on preparative scale (7.5 grams) employing very low catalyst loadings (0.02 mol %).
To initiate studies, non-electronically biased 1-dodecene was chosen as a model substrate. Under conditions of 150 psi syngas (CO:H2 = 1:1) and 80 °C with 0.5 mol % of Rh(acac)(CO)2 and 0.55 mol % phosphine ligand, both Me-DuPhos and Me-BPE promoted the hydroformylation reaction converting 1-dodecene to branched aldehyde 6 with serviceable regioselectivity, but with poor enantioselectivity (Scheme 2). It was considered that the low level of asymmetric induction might be due to insufficient steric bias afforded by the small methyl groups at C2 and C5 of the ligand phospholane units. As expected, exchanging the phospholane methyl groups for larger isopropyl (i-Pr-DuPhos) and phenyl (Ph-BPE) groups provided much higher enantioselectivity (Scheme 2). Ph-BPE was identified as the most effective ligand, giving the highest enantioselectivity and slightly enhanced branch:linear ratio compared to other ligands. While lower temperatures furnish higher selectivity, the high temperature is crucial for useful reaction rates (the reaction reaches 57% conversion after 36 h at 40 °C, furnishing 1.4:1 branch to linear ratio and 98:2 er). After balancing reaction regioselectivity and reaction rate, 150 psi of syngas and 80 °C in toluene was selected as conditions for evaluation of the substrate scope with Ph-BPE as the ligand.
Scheme 2.
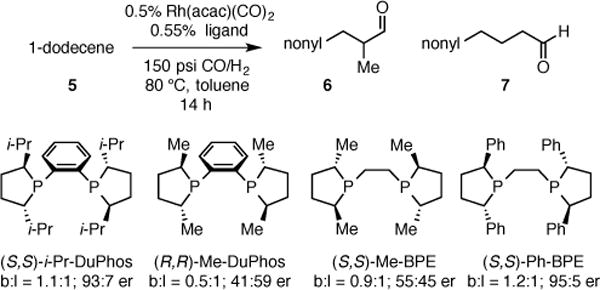
AHF of 1-Dodecene with DuPhos and BPE Ligands.a
aBranch:linear ratio (b:l) determined by 1H NMR analysis; enantiomer ratios determined by SFC analysis on a chiral stationary phase after reduction and benzoylation.
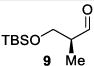
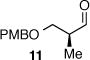
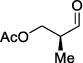
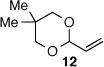
As depicted in Table 1, a collection of terminal olefins with different protecting groups and oxidation states at the allylic position were examined. TBS-protected allyl alcohol 8 gave good regioselectivity and excellent enantioselectivity (Table 1, entry 1). p-Methoxybenzyl allyl ether (10) reacted with higher regioselectivity while also maintaining a high level of enantioselectivity (Table 1, entry 2). Of note, when the allyl alcohol was protected in a manner that renders the C-O bond labile (i.e. such as acetate, mesylate and tosylate) the reaction resulted in decomposition of starting materials (Table 1, entry 3). This difference in reactivity might arise due to the formation of corresponding rhodium π-allyl intermediates derived from the activated allyl electrophile.10 Importantly, unsaturated acetal derivatives 12 and 14 provided good branch to linear ratios (>10:1) with high enantiomeric excess (Table 1, entry 4 and 5). An orthoester-containing aldehyde (17) was also obtained with excellent regio- and enantioselectivity by the AHF of 16 (Table 1, entry 6); this important aldehyde has been used in the synthesis of Prelog-Djerassi lactone.6b
Table 1.
Asymmetric Hydroformylation of Ethers, Acetals, and Orthoester.a
| |||||
|---|---|---|---|---|---|
|
| |||||
| entry | substrate | product | yield (%) | b:lb | er |
| 1b |
|
|
72 | 3.9:1 | 98:2 |
| 2 |
|
|
78 | 5.5:1 | 96:4 |
| 3 |
|
|
– | – | –c |
| 4 |
|
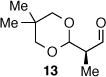
|
91 | 12:1 | 96:4 |
| 5 |
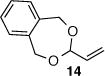
|
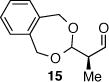
|
90 | 10:1 | 96:4 |
| 6 |
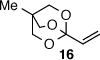
|
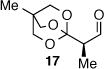
|
91 | 15:1 | 94:6 |
Reaction performed with 0.5 mol % Rh(acac)(CO)2 and 0.55 mol % (S,S)-Ph-BPE; [substrate] = 1.0 M.
Branch:linear ratio; determined by 1H NMR analysis.
Decomposition of starting material.
The asymmetric hydroformylation was conducted on larger scale both to probe the efficacy of the reaction with diminished catalyst loading and to probe the utility of the AHF for preparative organic synthesis. As shown in Scheme 3, a multigram quantity of allyl ether 8 (7.5 g) was subjected to AHF with lowered catalyst loading (0.02 mol %). As indicated by monitoring the syngas uptake, the reaction reached full conversion in 4 hours, which corresponds to a turnover frequency of 1250 h−1. Thus the AHF with Rh-Ph-BPE provides a short and attractive route to prepare large quantities of synthetically important protected β-hydroxy aldehydes (e.g. aldehyde 9, Scheme 3) in a nonracemic fashion.
Scheme 3.
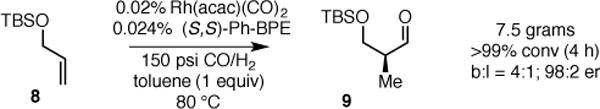
Large Scale Asymmetric Hydroformylation of Allyl Silyl Ether 8.
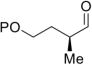
Encouraged by the outcome with allyl alcohol derivatives, we investigated less electronically biased substrates (Table 2). Chiral aldehydes were obtained with moderate regioselectivity and high levels of enantiomeric excess from homoallylic alcohol derivatives (Table 2, entries 1–5). Homoallylic alcohols with electron-withdrawing protecting groups gave higher branch to linear ratio (trifluoroacetate>benzoate>TBS). Substrates with ketone and ester oxidation states at homoallylic position (30 and 32) further enhanced regioselectivity maintaining excellent enantioselectivity (Table 2, entry 7–8). Importantly, TBS protected bishomoallylic alcohol 34 provided serviceable regioselectivity with good enantioselectivity (Table 2, entry 9).
Table 2.
Asymmetric Hydroformylation of Minimal-Electronically Biased 1-Alkenes.a
| |||||
|---|---|---|---|---|---|
|
| |||||
| entry | substrate | product | yield (%) | b:lb | erc |
|
|
|
||||
| 1 | P = TBS 18 | P = TBS 19 | 65 | 2.2:1 | 97:3 |
| 2 | P = PMB 20 | P = PMB 21 | 60 | 2.3:1 | 95:5 |
| 3 | P = Ac 22 | P = Ac 23 | 62 | 2.2:1 | 95:5 |
| 4 | P = Bz 24 | P = Bz 25 | 69 | 2.6:1 | 96:4 |
| 5 | P = THP 26 | P = THP 27 | 37 | 2:1 | 88:12 |
| 6 | P = TFA 28 | P = TFA 29 | 63 | 3:1 | NDd |
| 7 |
|
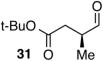
|
54 | 5.5:1 | 95:5 |
| 8 |
|
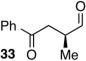
|
66 | 5.2:1 | NDd |
| 9 |
|
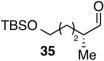
|
42 | 1.2:1 | 6:94e |
Reactions performed with 0.5 mol % Rh(acac)(CO)2 and 0.55 mol % (S,S)-Ph-BPE; [substrate] = 1.0 M.
Branch:linear ratio determined by 1H NMR analysis.
See Supporting Information for details.
ND = not determined because of instability of related products.
Ligand is (R,R)-Ph-BPE for this experiment.
Of note, the hydroformylation reactions proceed to complete conversion with <5% side-product formation such that even the 1:1 regioisomer ratio observed with compounds such as 1-dodecene represents a useful yield of chiral product from inexpensive bulk starting materials. In the particular case of 1-dodecene, the corresponding aldehyde, (S)-2-methyldodecanal (Scheme 2), was previously prepared in 6 steps from Roche ester,11 but only one catalytic step was required for the synthesis of nonracemic 6 from commercial 1-dodecene via AHF.
Internal olefin 36 was also examined in the AHF reaction with Rh-Ph-BPE and was converted to chiral aldehyde 37 with good levels of enantio- and regioselectivity (Scheme 4). Aldehyde 37 has been used in the synthesis of a novel chiral lipoxygenase inhibitor, but required four steps of synthesis including a desymmetrization of a 2-substitued 1,3-propanol; the AHF route may represent an improvement.12
Scheme 4.
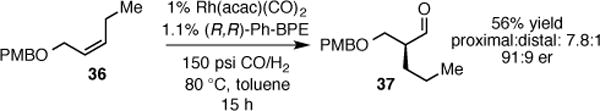
Asymmetric Hydroformylation of Internal Allyl Ether 32.
High enantioselectivity in the AHF reaction of terminal al-kenes has been observed in most cases and the level of stereocontrol appears to be relatively independent of substrate structure. In contrast, the regioselectivity of the reaction is highly substrate dependent and ranged from 15:1 to 1:1. The data in Table 1 and Table 2, suggest that the branch:linear ratio may correlate with the electron withdrawing ability of the alkene substituent. In this regard, Sigman13 has recently observed a correlation between the regioselectivity of Pd-catalyzed Heck reactions and the difference in 13C NMR chemical shift (Δδ 13C) between the terminal and internal carbons for a given substrate. Thus the chemical shift differential can serve as an indicator of olefin polarization.14 In order to further probe the impact of inductive effects on the AHF reaction, the relative 13C chemical shift (Δδ 13C) for each terminal alkene substrate was plotted versus regioselectivity of the AHF reaction (Figure 1). Interestingly, while a strong single correlation is not observed across the entire data set, separate correlations are apparent within substrates bearing oxygenation at the allylic position, and within all other substrates. We consider it plausible that inductive effects may polarize the alkene and favor the branched product during the AHF reaction of all substrates, but that σC-Rh to σ*C-O mixing (resonance effects) may provide an additional stabilizing feature that operates for allylic ether substrates, thereby providing enhanced selectivity with these compounds. Similar orbital mixing has been proposed as an important feature in Rh-catalyzed stereoselective hydroboration of allylic ether substrates.15
Figure 1.
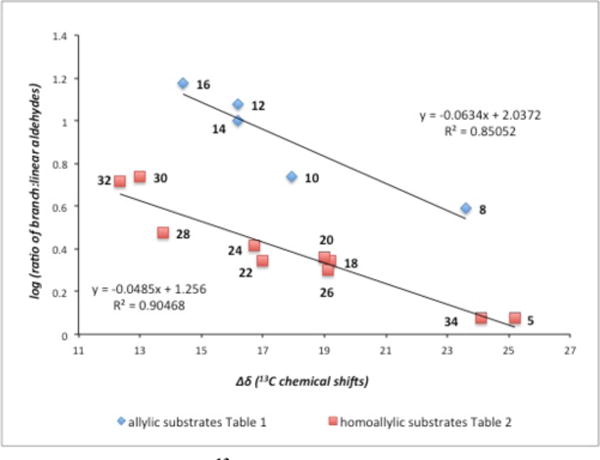
Plot of Δδ 13C Chemical Shift (Alkene Carbons) Versus Regioselectivity Derived From Data in Table 1 and Table 2.
In summary, we have developed an enantioselective hydroformylation that employs readily available Ph-BPE ligand. This methodology enables the efficient synthesis of many important and useful chiral building blocks from simple 1-alkenes. Asymmetric hydroformylation of most substrates maintained excellent enantioselectivity and moderate to good regioselectivity.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health (GM 059417 and GM 064451) Z.Y. was supported by LaMattina Fellowship. We also thank Candice Joe (Department of Chemistry, Boston College) for the assistance with the Argonaut Technologies Endeavor.
Footnotes
Supporting Information
Experimental procedure, full spectroscopic data for new compounds and chiral separations. This material is available free of charge via the Internet at http://pubs.acs.org
Notes
The authors declare no competing financial interests.
References
- 1.van Leeuwen PWNM, Claver C, editors. Rhodium Catalysed Hydroformylation. Kluwer Academic Press; Dordrecht, The Netherlands: 2000. [Google Scholar]
- 2.(a) van Leeuwen PWNM. Homogeneous Catalysis-Understanding the Art. Kluwer Academic Publishers; Dordrecht: 2004. [Google Scholar]; (b) Breit B. Aldehydes: Synthesis by Hydroformylation of Alkenes. In: Brückner R, editor. Science of Synthesis. Vol. 25 Thieme: Stuttgart; 2007. [Google Scholar]
- 3.(a) Horiuchi T, Ohta T, Nozaki K, Takaya H. Chem Commun. 1996;155 [Google Scholar]; (b) Horiuchi T, Ohta T, Shirakawa E, Nozaki K, Takaya H. Tetrahedron. 1997;53:7795. doi: 10.1021/jo9624051. [DOI] [PubMed] [Google Scholar]; (c) Watkins AL, Landis CR. Org Lett. 2011;13:164. doi: 10.1021/ol102797t. [DOI] [PubMed] [Google Scholar]
- 4.(a) Sakai N, Mano S, Nozaki K, Takaya H. J Am Chem Soc. 1993;115:7033. [Google Scholar]; (b) Nozaki K, Sakai N, Nanno T, Higashijima T, Mano S, Horiuchi T, Takaya H. J Am Chem Soc. 1997;119:4413. [Google Scholar]; (c) Horiuchi T, Ohta T, Shirakawa E, Nozaki K, Takaya H. J Org Chem. 1997;62:4285. doi: 10.1021/jo9624051. [DOI] [PubMed] [Google Scholar]; (d) Horiuchi T, Shirakawa E, Nozaki K, Takaya H. Organometallics. 1997;16:2981. [Google Scholar]; (e) Nozaki K, Li W, Horiuchi T, Takaya H. Tetrahedron Lett. 1997;38:4611. [Google Scholar]; (f) Nozaki K, Matsuo T, Shibahara F, Hiyama T. Adv Synth Catal. 2001;343:61. [Google Scholar]; (g) Shibahara F, Nozaki K, Hiyama T. J Am Chem Soc. 2003;125:8555. doi: 10.1021/ja034447u. [DOI] [PubMed] [Google Scholar]
- 5.(a) Nozaki K, Sakai N, Nanno T, Higashijima T, Mano S, Horiuchi T, Tskaya H. J Am Chem Soc. 1997;119:4413. [Google Scholar]; (b) McDonald RI, Wong GW, Neupane RP, Stahl SS, Landis CR. J Am Chem Soc. 2010;132:14027. doi: 10.1021/ja106674n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Noonan GM, Fuentes JA, Cobley CJ, Clarke ML. Angew Chem Int Ed. 2012;51:2477. doi: 10.1002/anie.201108203. [DOI] [PubMed] [Google Scholar]; (d) Zhang XW, Cao BN, Yu SC, Zhang XM. Angew Chem Int Ed. 2010;49:4047. doi: 10.1002/anie.201000955. [DOI] [PubMed] [Google Scholar]
- 6.For the use of AHF in total synthesis, see:; (a) Risi RM, Maza AM, Burke SD. J Org Chem. 2015;80:204. doi: 10.1021/jo502301k. [DOI] [PubMed] [Google Scholar]; (b) Ho S, Bucher C, Leighton JL. Angew Chem Int Ed. 2013;52:6757. doi: 10.1002/anie.201302565. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Risi RM, Burke SD. Org Lett. 2012;14:2572. doi: 10.1021/ol3008765. [DOI] [PubMed] [Google Scholar]; (d) Liu P, Jacobsen EN. J Am Chem Soc. 2001;123:10772. doi: 10.1021/ja016893s. [DOI] [PubMed] [Google Scholar]
- 7.Yu Z-Y, Ely RJ, Morken JP. Angew Chem Int Ed. 2014;53:9632. doi: 10.1002/anie.201405455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tang W, Zhang X. Chem Rev. 2003;103:3029. doi: 10.1021/cr020049i. [DOI] [PubMed] [Google Scholar]
- 9.Axtell AT, Cobley CJ, Klosin J, Whiteker GT, Zanotti-Gerosa A, Abboud KA. Angew Chem Int Ed. 2005;44:5834. doi: 10.1002/anie.200501478. [DOI] [PubMed] [Google Scholar]
- 10.(a) Tsuji J, Minami I, Shimidzu I. Tetrahedron Lett. 1984;25:5157. [Google Scholar]; (b) Evans PA, Nelson JD. J Am Chem Soc. 1998;120:5581. [Google Scholar]; (c) Evans PA, Robinson JE. J Am Chem Soc. 2001;123:4609. doi: 10.1021/ja015531h. [DOI] [PubMed] [Google Scholar]
- 11.(a) Wakabayashi T, Mori K, Kobayashi S. J Am Chem Soc. 2001;123:1372. doi: 10.1021/ja0057272. [DOI] [PubMed] [Google Scholar]; (b) Shirokawa S, Shinoyama M, Ooi I, Hosokawa S, Nakazaki A, Kobayashi S. Org Lett. 2007;9:849. doi: 10.1021/ol0630191. [DOI] [PubMed] [Google Scholar]
- 12.Yadav JS, Nanda S. Tetrahedron: Asymmetry. 2001;12:3223. [Google Scholar]
- 13.Mei T-S, Werner EW, Burckle AJ, Sigman MS. J Am Chem Soc. 2013;135:6830. doi: 10.1021/ja402916z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Kalinowski HO, Berger S, Braun S. Carbon-13 NMR spectroscopy. Wiley; Chichester: 1988. p. 92. Chapter 3. [Google Scholar]; (b) Sohar P. Nuclear Magnetic Resonance Spectroscopy. Vol. 2. CRC Press; Boca Raton: 1983. p. 145. Chapter 4. [Google Scholar]
- 15.Burgess K, van der Donk WA, Jarstifer MB, Ohlmeyer MJ. J Am Chem Soc. 1991;113:6139. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.