

Abstract
The transition metal-catalyzed cross-coupling of organometallic nucleophiles derived from tin, boron, and zinc with organic electrophiles enjoys a preeminent status among modern synthetic methods for the formation of carbon-carbon bonds. In recent years, organosilanes have emerged as viable alternatives to the conventional reagents, with the added benefits of low cost, low toxicity and high chemical stability. However, silicon-based cross-coupling reactions often require heating in the presence of a fluoride source, which has significantly hampered their widespread acceptance. To address the “fluoride problem”, a new paradigm for palladium-catalyzed, silicon-based cross-coupling reactions has been developed that employs a heretofore underutilized class of silicon reagents, the organosilanols. The use of organosilanols, either in the presence of Brønsted bases or as their silanolate salts, represents an operationally simple and mild alternative to the fluoride-based activation method. Organosilanols are readily available by many well-established methods for introducing carbon-silicon bonds onto alkenes, alkynes, arenes and heteroarenes. Moreover, several different protocols for the generation of alkali metal salts of, vinyl-, alkenyl-, alkynyl-, aryl-, and heteroarylsilanolates have been developed and the advantages of each of these methods have been demonstrated for a number of different coupling classes. This review will describe the development and implementation of cross-coupling reactions of organosilanols and their conjugate bases, silanolates, with a wide variety of substrate classes. In addition, application of these transformations in the total synthesis of complex natural products will also be highlighted. Finally, the unique advantages of organosilicon coupling strategies vis a vis organoboron reagents are discussed.
Keywords: cross-coupling, silicon, silanols, silanolates, Hiyama-Denmark
1. Introduction
The impact of transition metal catalyzed cross-coupling reactions cannot be overestimated. This Nobel Prize winning chemistry has transformed the practice of synthetic organic chemistry in academic and industrial laboratories alike.1,2 The ability to construct single bonds between sp3, sp2 and sp hybridized carbon atoms in myriad chemical environments has enabled the straightforward, strategic disconnection of target molecules into pairwise combinations of donors and acceptors. The recent development of high performance ligands has enabled the use of more and more acceptors such as chlorides, tosylates, phosphates and carboxylates. However, on the donor side, the boronic acid derivatives have monopolized the attention of practitioners, particularly in the pharmaceutical sector.3 This preference has stimulated the commercial production of hundreds of boronic acid derivatives, which then leads to the self-fulfilling consequence of using only these reagents and disregarding the advantages and in some cases superiority of other donors, including tin and silicon. What makes boronic acid derivatives so appealing is their versatility, stability and perceived low toxicity, all desirable features that justify their use as coupling partners in large-scale processes. However, a 2011 toxicological report unveiled the mutagenic properties of several boronic acids and esters.4 These results are further strengthened by a recent study that discloses the mutagenicity associated with a large number of boronic acids, esters, MIDA boronates, potassium trifluoroborates, as well as their precursors bis(pinacolato)diboron and bis-boronic acid.5 The newly-assessed genotoxicity of boron compounds naturally poses some serious limitations to their applications in medicinal and process chemistry, both in terms of worker exposure hazards and more stringent standards of purity to avoid the persistence of genotoxic impurities. It is foreseeable that these findings will stress the necessity of a paradigm shift and prompt the organic synthetic community to appreciate the advantages and potentials of other classes of donors in cross-coupling processes, including silicon. In this review, we will highlight the features of organosilanols and organosilanolates as easily accessible, versatile, not toxic6 cross-coupling partners.
Organosilanols (and their conjugate bases) find ubiquitous application as stabilizing ligands in coordination chemistry of main group and transition metals complexes.7 However, prior to 2000, the only application of silanols in organic synthesis was the use of KOTMS for the mild saponification of methyl esters8 and nitriles.9,10 In 2000, two independent reports demonstrated the utility of organosilanols as donors in cross-coupling reactions under activation by silver(I) oxide11 or TBAF.12 The advantages of organosilanols as coupling partners are manifold in comparison to other silanes, including: (1) ease of synthesis, (2) stability toward oxygen and moisture, (3) ease of purification, (4) diversity of synthetic methods for their preparation, and (5) functional group tolerance.13 An additional advantage of significant synthetic importance is the ability to activate the cross-coupling of organosilanols with Brønsted bases, thus avoiding the incompatibilities associated with fluoride.14,15,16 Moreover, the conjugate bases of organosilanols are stable, often free flowing powders that are “self-activating” cross-coupling partners, i.e. require no additional activators.17,18,19 These two variants of cross-coupling are presented separately because of the differences in substrate scope. Even those cross-coupling reactions that undoubtedly involve the silanolate salt through in situ generation will be presented in the silanol section; only those cross-couplings that employ the preformed, isolated silanolates are included in the subsequent section.
2. Silanols
2.1. Preparation
Many different methods are available for the preparation of silanols and silanol surrogates from a variety of precursors. The most common method for introduction of a silanol unit involves the reaction of an organometallic reagent (lithium or magnesium) with a silicon electrophile, most directly, hexamethylcyclotrisiloxane (D3) (Eq. 1).20 This method works well for aryland alkenyllithium reagents.
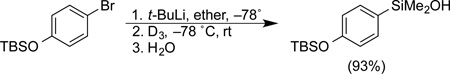 |
(Eq. 1) |
Less reactive organometallic species, or those unstable at higher temperatures, require more reactive silicon electrophiles such as dimethyldichlorosilane or dimethylchlorosilane. Whereas the former can be converted into the corresponding silanol by mild hydrolysis (acetate buffer), the latter is converted to the silanol by oxidation with water or an alcohol under catalysis by ruthenium or iridium complexes (Eq. 2).21
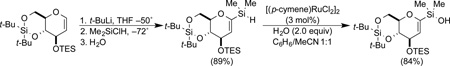 |
(Eq. 2) |
Hydrosilylation of alkynes is a powerful method for creating carbon-silicon bonds with site and stereoselectivity. For this method, silanol surrogates are needed and can be found in the many commercially available hydrosilanes bearing chloro, alkoxy, or silyloxy substituents. More robust surrogates such as benzylsilanes are also available. The steric course of the hydrosilylation is dependent upon the transition-metal catalyst: platinum catalysts (e.g. H2PtCl6, (DVDS)Pt•(t-Bu3P))22 react with terminal alkynes to give (E)-1-alkenylsilanes via a syn process, whereas [(C6H6)RuCl2]2 promotes an anti addition process to afford (Z)-alkenylsilanes.23 Remarkably, the cationic ruthenium complex [(Cp)Ru(MeCN)3]+ PF6− reacts with terminal alkynes to afford 2-alkenylsilanes.24
A palladium-catalyzed insertion method has been developed to allow the introduction of silanols on cyclic substrates wherein formation of an organometallic reagent is precluded by sensitive functionality (Eq. 3).25 For effective reaction with aryl bromides, a bulky electron-rich phosphine (JohnPhos)26 is required. The pH of the workup is critical to suppress formation of the disiloxane.
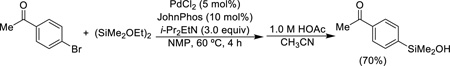 |
(Eq. 3) |
Catalytic, highly regioselective C-H silylation of arenes is achievable under mild conditions with [Rh(coe)OH]2 and HSiMe(OTMS)2 (Scheme 1).27 Unlike the multitude of cases where regiocontrol is dependent upon coordination of the catalyst to an ortho-directing group, this reaction shows exceptional meta-selectivity due to steric factors. Similarly, stereoselective C-H functionalization of alkenes has been accomplished with [Ir(coe)OH]2 and phenanthroline ligands.28
Scheme 1.

Catalytic C-H silylation of arenes and alkenes.
2.2. Cross-Coupling
2.2.1. Fluoride Activation
Arylsilanols
Only a simple phenyl group has been transferred under these conditions by the use of phenyldimethylsilanol in combination with a limited number of aryl iodides. A stoichiometric amount of silver(I) oxide is also used together with TBAT.29
Alkenylsilanols
This class represents a very large number of examples that employ acyclic and cyclic alkenylsilanes of varying substitution patterns in cross-coupling with aryl, heteroaryl and alkenyl halides. The stereospecificity of cross-coupling of simple alkenylsilanes is illustrated for a prenylating agent (Scheme 2).30 For the iodide electrophiles, no phosphine ligand is needed and the reactions take place at room temperature with electron-deficient, electron-rich and sterically hindered arenes. In a competition study, dimethyl-, diethyl-, diisopropyl- and diphenylsilanols react at approximately the same rate under activation by TBAF.31
Scheme 2.

Stereospecific cross-coupling of alkenylsilanols.
Cross-couplings with alkenyl iodides and bromides proceed with high geometrical selectivity and only minor amounts of cine substitution products (Eq. 4).12,30,32 Diisopropylsilanols give slightly higher geometrical selectivity compared to dimethylsilanols.12
 |
(Eq. 4) |
Aryl triflates can participate in cross-coupling reactions but the conditions need to be carefully adjusted. To facilitate the oxidative addition step, an electron-rich, hindered phosphine (JohnPhos) is needed and, to suppress fluoride-assisted S–O bond cleavage of the triflate, the TBAF is hydrated with 6 to 8 equiv of water (Eq. 5).33 For electron-deficient aryl triflates, TBAF•30H2O is required.
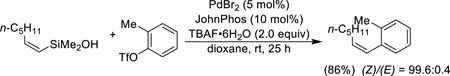 |
(Eq. 5) |
α-Alkoxyalkenylsilanols, both cyclic (pyranyl and furanyl) and acyclic undergo ready cross-coupling with aryl iodides under the standard conditions with TBAF (Eq. 6).
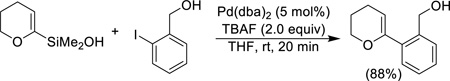 |
(Eq. 6) |
Alkynylsilanols
A limited set of simple alkynylsilanols undergo a “copper-free”, Sonogashira-type cross coupling with aryl iodides using (Ph3P)4Pd and TBAF (Eq. 7).34
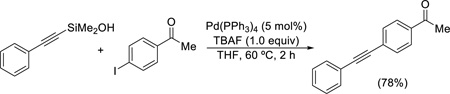 |
(Eq. 7) |
2.2.2. Brønsted Base Activation
For this disparate collection of cross-coupling reaction conditions, the only unifying characteristic is that the activators are all Brønsted bases. Thus, under this rubric is found activation by silver(I) oxide, potassium trimethylsilanolate, cesium carbonate, cesium hydroxide, potassium tert-butoxide, sodium hexamethyldisilazide, and sodium or potassium hydride. Undoubtedly, the corresponding silanolate is being formed to some extent and is likely the active component. However, because the parent silanol is the species employed in these processes, these conditions are described herein.
Arylsilanols
The first reported use of silanols in a cross-coupling reaction employed a full equivalent of silver(I) oxide as the activator in the presence of Pd(OAc)2 in warm THF.11,35,36 Phenylsilanols bearing electron-donating and withdrawing groups can be used with similarly modified aryl iodides. It is likely that silver activates the arylpalladium iodide by abstraction of an iodide to form a cationic palladium(II) species and activates the arylsilanol through formation of a hypercoordinate complex. The intermediacy of a palladium(II) silanolate was not suggested.
Phenylsilanols can also undergo cross-coupling in the presence of cesium bases (carbonate or hydroxide). However, the bases must carry 2–3 molecules of water of hydration to reverse the formation of inactive disiloxanes formed from the silanols under the elevated temperatures needed to effect cross-coupling with aryl iodides and bromides (90–110 °C).37,38 In addition, ligands play an important role in suppressing the formation of homocoupling products as well as reduction of the electrophile (Scheme 3). The failure of disiloxanes to undergo crosscoupling under Brønsted base activation sharply contrasts their behavior under fluoride activation. The limited scope of arylsilanol TG (transferable group) and harsh reaction conditions limited the utility of this method, which has been replaced by the use of the preformed potassium silanolate salts (vide infra).
Scheme 3.

Brønsted base-activated cross-coupling of arylsilanols.
Heteroarylsilanols
A significant number of π-excessive heteroaromatic silanols have been prepared and three different conditions for their cross-coupling reactions have been developed, two of which involve the use of the parent silanol.39,40,41 The cross-coupling of isolated, preformed sodium silanolate salts represent the third variant and is discussed in the next section. N-Boc- and N-methyl-2-indolyldimethylsilanol undergo smooth cross-coupling with aryl bromides and iodides in the presence of sodium tert-butoxide and 1.0 equiv of copper(I) iodide to suppress protiodesilylation (Scheme 4). The amount of copper can be reduced to 25 mol% with activated indole derivatives.
Scheme 4.

Brønsted base-activated cross-coupling of heteroarylsilanols.
A more general protocol involves the stoichiometric deprotonation of the heteroarylsilanol with either sodium hydride or sodium hexamethyldisilazide. This modification allows a number of important advances including the elimination of copper(I) salts to suppress protiodesilylation (no protons) and the ability to cross-couple with aryl and heteroaryl bromides and chlorides (Scheme 5).41,42 For the less reactive electrophiles, an appropriate phosphine ligand or precatalyst is needed to effect oxidative addition. SPhos,43 RuPhos44 and the palladium(I) dimer45 serve well in this capacity.
Scheme 5.

Copper-free, Brønsted base-activated cross-coupling of heteroarylsilanols.
In addition, 2-furyl-, 2-thienyl- and N-Boc-(2-pyrrolyl)dimethylsilanols undergo cross-coupling under these conditions with a similar scope of electrophile (Eq. 8).
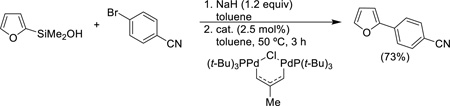 |
(Eq. 8) |
The 3-substituted-2-indolylsilanols in Scheme 5 are prepared by a variant of the Larock indole synthesis that employs alkynylsilyl ethers and places the silanol group in the 2-position for coupling (Eq. 9).42,46 2-Iodo-N-alkyl anilines are used as precursors with both isopropyl and tert-butylsilyl ethers. These silyl ethers are sufficiently robust to withstand the Larock heteroannulation conditions and can be deprotected with very mild acid hydrolysis. The sequence then allows for the controlled construction of 2,3-disubstituted indoles from anilines whereby unsymmetrical alkynes would otherwise lead to mixtures of constitutional isomers.
 |
(Eq. 9) |
Another sequential process that first constructs a silylated heterocycle, which is then used for cross-coupling, involves the [3+2] cycloaddition of nitrile oxides with alkynylsilyl ethers (Scheme 6).47 The nitrile oxides can be generated under two different conditions, both sufficiently mild to allow the use of an alkynylsilyl methyl ether as the 2π-component. The [3+2] cycloadditions proceed in modest regioselectivity (ca. 4–5:1) and the minor constitutional isomer can be removed easily. The cross-coupling reactions required extensive optimization to suppress protiodesilylation. As seen before, copper salts, in this case copper(II) acetate, are used to maximize the yield of the cross-coupling product.
Scheme 6.

Preparation of heteroarylsilanols via nitrile oxide [3+2] cycloaddition, and cross-coupling.
Alkenylsilanols
Several different methods of activation have been employed for this family of organosilanols including silver(I) oxide,11 KOTMS,48,49 sodium tert-butoxide,50 and potassium hydride51(Scheme 7). Both aromatic and olefinic iodides are active partners. Functional group compatibility is good and the reactions are stereospecific with respect to the alkenylsilanol unit. Diisopropylsilanols react significantly slower (ca. 20-fold) compared to dimethylsilanols under activation by KOTMS.
Scheme 7.

Brønsted base-activated cross-coupling of alkenylsilanols.
Alkynylsilanols
As is the case with trimethylsilylalkynes, the silanol version of the Sonogashira reaction offers little advantage under activation by fluoride, but activation by KOTMS leads to a significantly more rapid transmetalation (room temperature compared to 60–120 °C). Copper(I) iodide is essential for clean cross-coupling and in this report the substrates are limited to aryl iodides (Eq. 26).52
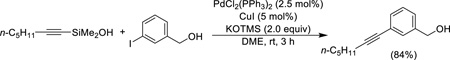 |
(Eq. 10) |
3. Silanolates
Although silanolate salts are certainly the reactive species in the cross-coupling reactions carried out under Brønsted base activation, these salts are always generated in situ from the parent silanol. Depending upon the strength of the Brønsted base employed, the formation of the silanolate may or may not be quantitative. Moreover, the conjugate acid remains in the reaction mixture (except in the case of NaH and KH). The pre-formation and isolation of the sodium or potassium silanolates (from the corresponding hydrides) offers a number of preparative advantages over silanols. First, silanols dimerize to the corresponding disiloxanes in the presence of acids or bases, and isolating the metal salt prevents this process. Second, using the isolated preformed silanolate in the reaction not only simplifies the experimental procedure (adding one reagent as opposed to the silanol and base), but also ensures that the cross-coupling partner is always present in its active form. Moreover, a metal silanolate can be generated cleanly with a stoichiometric quantity of metal hydride, without the need for an excess of an activator. The use of an excess of the activator can be problematic in the cross-coupling reaction for several reasons. Surplus activator (i.e. KOt-Bu, KOSiMe3) can compete with the silanolate for the Pd center of the arylpalladium halide. As a competitive inhibitor, the activator can form an inactive species in the cross-coupling reaction where it serves as a ligand on the Pd(II) aryl complex. This process sequesters palladium in an inactive form, and consequently slows the cross-coupling reaction.53 The silanolate must displace the activator to allow the Pd to reenter the catalytic cycle as the palladium silanolate. An excess of the activator can also limit functional group compatibility. For example, when NaOt-Bu is employed with substrates bearing ethyl esters, a competing transesterification reaction takes place.54 Furthermore, excess amounts of hydride reagents give reduction products,49 and are incompatible with substrates bearing sensitive functional groups. The direct introduction of the silanolate avoids these problems and many of these salts are stable, free-flowing powders.
Arylsilanolates
The limitations encountered in the cross-coupling of arylsilanols under Brønsted base activation (Scheme 3) are substantially overcome by the use of potassium arylsilanolate salts in combination with (t-Bu3P)2Pd.54 A wide range of arylsilanolates bearing various functional groups (alkoxy, dialkylamino, trifluoromethyl, fluoro, chloro, alkoxycarbonyl) couple effectively with aryl bromides and chlorides bearing an equally diverse set of functional groups (halo, alkoxy, silyloxy, alkoxycarbonyl) in good yield (Scheme 8). In addition, various heteroaromatic bromides function as cross-coupling partners. Other ligand/catalyst combinations show more limited generality.55
Scheme 8.

Cross-coupling of arylsilanolates.
Enantioselective biaryl cross-coupling can be achieved in the presence of chiral bishydrazone ligands containing diarylpyrrolidine groups (Eq. 11).56
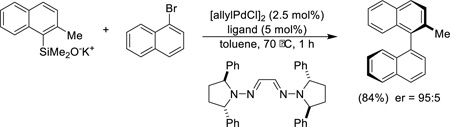 |
(Eq. 11) |
Heteroarylsilanolates
The successful cross-coupling of in situ generated sodium silanolates of π-excessive heterocyclic silanols illustrated in Schemes 4 and 5 can be extended to other heterocycles and coupling partners by the use of preformed salts. For example, the preformed N-SEM-2-indolylsilanolate can engage in cross-coupling with a variety of aryl and heteroaryl bromides and chlorides whereas the 5-bromo-N-Boc-2-indolylsilanolate couples with iodides selectively41 and finally N-Me-3-pentyl-2-indolylsilanolate (formed by a Larock annulation process)42 couples cleanly with a variety of aryl bromides (Scheme 9). Moreover, a large number of 2-benzofuranylsilanolates undergo smooth coupling under similar conditions.54
Scheme 9.

Cross-coupling of heteroarylsilanolates.
Alkenylsilanolates
Although alkenylsilanols undergo efficient cross-coupling by various methods of activation with Brønsted bases, the preformed potassium salts are highly effective in reactions with aromatic and heteroaromatic chlorides.57 These reactions display superior generality with respect to the acceptor, extremely high stereospecificity for a number of different alkenylsilanolate substitution patterns and overall higher yields than the couplings with the corresponding bromides or iodides (Scheme 10). The absence of byproducts derived from reduction or homocoupling of the electrophile accounts for the better performance of these reactions.
Scheme 10.

Cross-coupling of alkenylsilanolates.
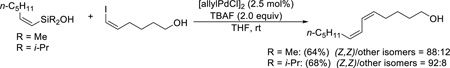
Allylsilanolates
The sodium salts of allyldimethylsilanol and 2-butenyldimethylsilanol undergo palladium-catalyzed cross-coupling with a wide variety of aryl bromides to afford allylated and crotylated arenes.58 The reaction of the allyldimethylsilanolate affords good yields of the allylation products from both electron-rich and sterically hindered bromides (Scheme 11).
Scheme 11.

Cross-coupling of sodium allyldimethylsilanolate.
Sodium 2-butenyldimethylsilanolate (E/Z = 80:20) affords good yields of the γ-substituted product with electron-rich and electron-poor bromides (Scheme 12).
Scheme 12.

Cross-coupling of sodium 2-butenyldimethylsilanolate.
In general, high γ-site selectivity is obtained with catalysts bearing π-acidic ligands such as dba. Norbornadiene assists in catalyst turnover. The scope in aromatic bromide is good and the γ-site selectivity is generally higher than 10:1. Interestingly, the use of pure sodium (E)-2-butenyldimethylsilanolate led to a noticeable improvement in the γ-site selectivity (Scheme 13). The role of π-acidic ligands and double bond geometry are interpreted in terms of a kinetically controlled, γ-selective transmetalation followed by direct reductive elimination to form the branched product. Exceptionally high γ-selectivity is obtained with sodium diethylallylsilanolates and electron rich alkyl phosphines as the catalyst.59
Scheme 13.

γ-selectivity in the cross-coupling of allylsilanolates.
In the γ-site-selective cross-coupling reactions of unsymmetrically substituted allylic silanes, a new stereogenic center is created. With the aid of enantiomerically enriched and configurationally defined allylic dimethylsilanolates, the stereochemical course of the SE2’ process has been elucidated. This study has revealed a strong and consistent stereochemical correlation for the cross-coupling of enantiomerically enriched allylic dimethylsilanolates (Scheme 14).60 In the presence of [allylPdCl]2 and 4,4’-(trifluoromethyl)dibenzylideneacetone, a wide range of aryl bromides undergo highly γ-site selective cross-coupling with perfect syn SE2’ stereospecificity. These results are interpreted in terms of an intramolecular transmetalation via a chair-like transition structure. In the preferred transition structure, the Si–O–Pd linkage controls the delivery of the palladium electrophile to the γ–terminus of the allylic silane. The palladium is tricoordinate and the alkene takes up the fourth coordination site in the square-planar complex. The pseudoequatorial orientation of the iso-butyl group assures high selectivity in the formation of an E double bond in the product. In addition, the allylic methyl group is positioned orthogonal to the ligand plane of palladium to avoid unfavorable steric interactions. An alternative transition state structure that also involves an intramolecular delivery of the palladium moiety suffers from severe 1,3-diaxal steric strain between the iso-butyl and allylic methyl group.
Scheme 14.

Stereospecificity in the cross-coupling of enantiomerically enriched allylic dimethylsilanolate.
4. Total Synthesis with Silanolates61
4.1. Papulacandin D50,62,63
The papulacandins are a family of antifungal agents, isolated from the deuteromycetous fungus Papularia sphaerosperma that have demonstrated potent in vitro antifungal activity against various pathogens. All of the papulacandins are amphipathic molecules composed of an aromatic moiety linked via a spirocyclic structure to a lactose moiety with two different aliphatic acyl side-chains. The simplest member of the family, papulacandin D, lacks the O-(6'-acyl-β-galactoside) at the O-(4) position. The key strategic disconnection for the synthesis of papulacandin D requires the cross-coupling of a 2-pyranylsilanol with an aryl halide (Scheme 15). Although α-oxyalkenylsilanols are competent substrates for cross-coupling, the requirement for fluoride activation is clearly incompatible with the silyl ether protecting groups planned in the total synthesis. Therefore, fluoride-free conditions for the cross-coupling had to be developed.
Scheme 15.

Key cross-coupling step in the total synthesis of papulacandin D.
The actual synthesis required the cross-coupling of the glycal silanol 3 with the protected iodoresorcinol derivative 4. To prepare 3, silyl-protected glycal 1 was lithiated at C(1) followed by capture with chlorodimethylsilane. The resulting hydrosilane 2 was subjected to a rutheniumcatalyzed oxidative hydrolysis to afford the base-sensitive silanol 3. The key cross-coupling reaction of 4 was quite challenging because of protiodesilylation of 3, but ultimately this critical transformation could be achieved efficiently using sodium tert-butoxide as the Brønsted-base activator and Pd2(dba)3•CHCl3 as the catalyst to provide C-arylglycal 5 in good yield. Glycal 5 contains the entire carbon framework of the sugar fragment of papulacandin D.
4.2. RK-39751,64
The silicon-based cross-coupling reaction is mechanistically unique in that two different modes of transmetalation are possible.65 This mechanistic duality has significant preparative utility because the different mechanistic pathways can be accessed via complementary reaction conditions (fluoride activation or Brønsted base activation). The feasibility of using both modes of activation in a single reagent is illustrated by sequential Brønsted base/fluoride-promoted crosscoupling reactions using the linchpin reagent (E,E)-[(4-benzyldimethylsilyl)-1,3-butadienyl]dimethylsilanol 6 (Scheme 16). This bifunctional reagent can combine with two electrophiles under complementary conditions for the construction of unsymmetrical polyenes. In the first cross-coupling reaction, 6 is treated with KOTMS in the presence of an aryl iodide and Pd(dba)2 to afford the (1-aryl-1,3-butadienyl)benzylsilane 7. The benzyldimethylsilyl group is inert under these conditions. Subsequently, treatment of 7 with TBAF effects the second cross-coupling reaction which proceeds smoothly to afford the unsymmetrical 1,4-diaryl-1,3-butadiene 8.
Scheme 16.

Sequential cross-coupling strategy.
The total synthesis of the polyene-polyol antifungal agent RK-397 aptly demonstrates the power of complementary modes of activation for silicon-based cross-coupling reaction (Scheme 17). Whereas in the synthetic study shown above both electrophiles are aryl iodides, for the construction of the polyene fragment of RK-397, the cross-coupling reaction with two alkenyl iodides is required. This extension is challenging because alkenyl iodides are less reactive. Thus, for the cross-coupling of 6 with alkenyl iodide 9, NaH was employed instead of KOTMS as the Brønsted base promoter. The stoichiometric generation of the silanolate using a strong base such as NaH provides heightened reactivity. The resulting triene 10 was then combined with ethyl (E)-3-iodopropenoate under fluoride-promoted cross-coupling conditions to afford tetraene 11. This key fragment was then incorporated onto the polyol fragment, to complete the total synthesis of RK-397.
Scheme 17.

Key cross-coupling steps in the total synthesis of RK-397.
4.3. Isodomoic Acids G and H66,67
Isodomoic acids G and H belong to the family of kainoid amino acids, a series of structurally related marine natural products bearing a 3-carboxymethylproline moiety (Scheme 18). Kainoid amino acids have long been recognized as neuroexcitatory agents, thus becoming valuable tools in neuroscience and medicinal chemistry. The initial strategic disconnection of isodomoic acids G and H was based on the intention of showcasing utility of the sequential silylcarbocyclization/cross-coupling strategy developed in our research group.46,68 Surprisingly, after an extensive survey of reaction conditions, none of the key cross-coupling product 15 could be obtained from silane 13 (obtained through Rh-catalyzed carbonylative silylcarbocyclization) and iodide 14. The failure to effect this coupling led to a reversal in the roles of the donor and the acceptor. Thus, the common intermediate 12 was converted into either iodides 16 and 17 via io-dodesilylation. Cross-coupling with silanol 18 proceeded smoothly in the presence of TBAF•8H2O as an activator, affording the protected isodomoic acids 15 and 19.
Scheme 18.

Key cross-coupling steps in the total synthesis of isodomoic acids G and H.
5. Summary and Outlook
Boronic acid derivatives have long been considered the “gold standard” for cross-coupling reactions, finding extensive application in academic research and industrial applications. However, the recent disclosure of the genotoxicity of several classes of boron-containing compounds represents a major concern that may eventually lead to the rise of other cross-coupling techniques employing less toxic but equally reliable donors. In this review, we highlighted the versatility of organosilanols and silanolates in cross-coupling reactions. Some of the key advantages of cross-coupling reactions with organosilicon compounds include: (1) ease of introduction of the silicon moiety, (2) stability of organosilane functional groups (3) compatibility of installation and coupling conditions with various functional groups, (4) ability to engineer sequential processes to increase molecular complexity and streamline synthetic routes, (5) nontoxicity of reagents and byproducts, (6) low molecular weight overhead and (7) minimized waste stream.
One of the unique advantages (and to some extent also drawbacks) of silicon are the literally dozens of organosilane moieties that can participate in cross-coupling reactions, from the most inert trialkylsilanes to the highly reactive trichlorosilanes. This diversity allows for precise tuning of the reactivity and functional compatibility of the donor for a specific cross-coupling reaction. Of course, this diversity also presents a challenge for the experimentalist to identify which of the many options would be optimal. Fortunately, recent comprehensive reviews of the area provide a thorough treatment of the advantages of each activating group and the transformations for which it is suitable together with the reaction conditions needed to effect the cross-coupling.69,70
In addition to the structural diversity, what makes organosilanes so attractive is the ease of operation and the fact that cross-coupling reactions usually employ readily available palladium sources, without additional ligands. This is in contrast with the conditions needed for the cross-coupling of boronic acid derivatives, which often require specialized ligands and palladium precursors. Moreover, the mechanistic duality of the cross-coupling of organosilanes (fluoride activation vs Brønsted base activation) is a unique feature that allows for orthogonal functionalizations.
With the availability of more and more silicon containing building blocks and precursors, we are confident that the use of silicon-based cross-coupling processes will soon take their rightful place along with the other workhorse processes.
Supplementary Material
Acknowledgments
We are grateful to the National Institutes of Health (GM R01-63167) and the National Science Foundation (NSF CHE-1151566) for generous financial support.
References
- 1.(a) deMeijere A, Brase S, Oestreich M, editors. Metal-Catalyzed Cross-Coupling Reactions and More. Weinheim: Wiley-VCH; 2013. [Google Scholar]; (b) Molander GA, Wolfe J, Larhed M, editors. Science of Synthesis: Cross Coupling and Heck-Type Reactions. Stuttgart: Thieme; 2013. [Google Scholar]; (c) Nicolaou KC, Bulger PG, Sarlah D. Angew. Chem. Int. Ed. 2005;44:4442. doi: 10.1002/anie.200500368. [DOI] [PubMed] [Google Scholar]
- 2.Torborg C, Beller M. Adv. Synth. Catal. 2009;351:3027. [Google Scholar]
- 3.(a) Magano J, Dunetz JR. Chem. Rev. 2011;111:2177. doi: 10.1021/cr100346g. [DOI] [PubMed] [Google Scholar]; (b) Molander GA, Jean-Gérard L. Org. React. 2013;79:1. [Google Scholar]
- 4.O’Donovan MR, Mee CD, Fenner S, Teasdale A, Phillips DH. Mutat. Res. 2011;724:1. doi: 10.1016/j.mrgentox.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 5.Hansen MM, Jolly RA, Linder RJ. Org. Proc. Res. Dev. 2015 DOI: 10.1021/acs.oprd.5b00150. [Google Scholar]
- 6.The toxicology profile of silicon-containing compounds has been investigated extensively. To date, there is no known intrinsic “ element-specific” toxicity associated with silicon. See: Franz AK, Wilson SO. J. Med. Chem. 2013;56:388. doi: 10.1021/jm3010114.Tacke R, Linoh H. In: The Chemistry of Organosilicon Compounds. Patai S, Rappoport Z, editors. New York: Wiley-Interscience; 1989. Part 2; Chapt. 18. More in-depth analysis of the toxicity of organosilanols is currently underway
- 7.Lickiss P. Adv. Inorg. Chem. Vol. 42. Inc: Academic Press; 1995. p. 147. [Google Scholar]
- 8.Laganis ED, Chenard BL. Tetrahedron Lett. 1984;50:5831. [Google Scholar]
- 9.Merchant KJ. Tetrahedron Lett. 2000;41:3747. [Google Scholar]
- 10.Subsequently, KOTMS has been employed as an oxygen nucleophile in enantioselective allylic substitution. Lyothier I, Difieber C, Carreira EM. Angew. Chem. Int. Ed. 2006;45:6204. doi: 10.1002/anie.200602408. [DOI] [PubMed] [Google Scholar]
- 11.Hirabayashi E, Mori A, Kawashima J, Suguro M, Nishihara Y, Hiyama T. J. Org. Chem. 2000;65:5342. doi: 10.1021/jo000679p. [DOI] [PubMed] [Google Scholar]
- 12.Denmark SE, Wehrli D. Org. Lett. 2000;2:565. doi: 10.1021/ol005565e. [DOI] [PubMed] [Google Scholar]
- 13.(a) Denmark SE, Sweis RF. Organosilicon Compounds in Cross-Coupling Reactions. In: de Meijere A, Diederich F, editors. Metal-Catalyzed Cross-Coupling Reactions. Vol. 1. Weinheim: Wiley-VCH; 2004. Chapt. 4. [Google Scholar]; (b) Denmark SE, Sweis RF. Cross-Coupling Reactions of Organosilicon Compounds in. In: deMeijere A, Brase S, Oestreich M, editors. Metal-Catalyzed Cross-Coupling Reactions and More. Vol. 1. Weinheim: Wiley-VCH; 2013. Chapt. 4. [Google Scholar]
- 14.Denmark SE, Sweis RF. Chem. Pharm. Bull. 2002;50:1531. doi: 10.1248/cpb.50.1531. [DOI] [PubMed] [Google Scholar]
- 15.Denmark SE, Sweis RF. Acc. Chem. Res. 2002;35:835. doi: 10.1021/ar020001r. [DOI] [PubMed] [Google Scholar]
- 16.Denmark SE, Ober MH. Aldrichimica Acta. 2003;36:75. [Google Scholar]
- 17.Denmark SE, Baird JD. Chem. Eur. J. 2006;12:4954. doi: 10.1002/chem.200600034. [DOI] [PubMed] [Google Scholar]
- 18.Denmark SE, Regens CS. Acc. Chem. Res. 2008;41:1486. doi: 10.1021/ar800037p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Denmark SE. J. Org. Chem. 2009;74:2915. doi: 10.1021/jo900032x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Denmark SE, Butler CR. Hexamethylcyclotrisiloxane. e-EROS Encyclopedia of Reagents for Organic Synthesis. Published Online: 2007. [Google Scholar]
- 21.Lee M, Ko S, Chang S. J. Am. Chem. Soc. 2000;122:12011. [Google Scholar]
- 22.Chandra G, Lo PY, Hitchcock PB, Lappert MF. Organometallics. 1987;6:191. [Google Scholar]
- 23.Denmark SE, Pan WT. Org. Lett. 2002;4:4163. doi: 10.1021/ol026933c. [DOI] [PubMed] [Google Scholar]
- 24.Trost BM, Ball ZT. J. Am. Chem. Soc. 2005;127:17644. doi: 10.1021/ja0528580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Denmark SE, Kallemeyn JM. Org. Lett. 2003;5:3483. doi: 10.1021/ol035288m. [DOI] [PubMed] [Google Scholar]
- 26.Wolfe JP, Singer RA, Yang BH, Buchwald SL. J. Am. Chem. Soc. 1999;121:9550. [Google Scholar]
- 27.Cheng C, Hartwig JF. Science. 2014;343:853. doi: 10.1126/science.1248042. [DOI] [PubMed] [Google Scholar]
- 28.Cheng C, Simmons EM, Hartwig JF. Angew. Chem. Int. Ed. 2013;52:8984. doi: 10.1002/anie.201304084. [DOI] [PubMed] [Google Scholar]
- 29.Napier S, Marcuccio SM, Tye H, Whittaker M. Tetrahedron Lett. 2008;49:3939. [Google Scholar]
- 30.Denmark SE, Pan W. J. Organomet. Chem. 2002;653:98. [Google Scholar]
- 31.Denmark SE, Neuville L, Christy MEL, Tymonko SA. J. Org. Chem. 2006;71:8500. doi: 10.1021/jo061481t. [DOI] [PubMed] [Google Scholar]
- 32.Denmark SE, Neuville L. Org. Lett. 2000;2:3221. doi: 10.1021/ol0064112. [DOI] [PubMed] [Google Scholar]
- 33.Denmark SE, Sweis RF. Org. Lett. 2002;4:3771. doi: 10.1021/ol026900x. [DOI] [PubMed] [Google Scholar]
- 34.Chang S, Yang SH, Lee PH. Tetrahedron Lett. 2001;42:4833. [Google Scholar]
- 35.Hirabayashi K, Kawashima J, Nishihara Y, Mori A, Hiyama T. Org. Lett. 1999;1:299. [Google Scholar]
- 36.Hirabayashi K, Kondo T, Toriyama F, Nishihara Y, Mori A. Bull. Chem. Soc. Jpn. 2000;73:749. [Google Scholar]
- 37.Denmark SE, Ober MH. Org. Lett. 2003;5:1357. doi: 10.1021/ol034328j. [DOI] [PubMed] [Google Scholar]
- 38.Denmark SE, Ober M. Adv. Synth. Catal. 2004;346:1703. [Google Scholar]
- 39.Denmark SE, Baird JD. Org. Lett. 2004;6:3649. doi: 10.1021/ol048328a. [DOI] [PubMed] [Google Scholar]
- 40.Denmark SE, Baird JD. Org. Lett. 2006;8:793. doi: 10.1021/ol053165r. [DOI] [PubMed] [Google Scholar]
- 41.Denmark SE, Baird JD, Regens CS. J. Org. Chem. 2008;72:1440. doi: 10.1021/jo7023784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Denmark SE, Baird JD. Tetrahedron. 2009;65:3120. doi: 10.1016/j.tet.2008.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barder TE, Walker SD, Martinelli JR, Buchwald SL. J. Am. Chem. Soc. 2005;127:4685. doi: 10.1021/ja042491j. [DOI] [PubMed] [Google Scholar]
- 44.Milne JE, Buchwald SL. J. Am. Chem. Soc. 2004;126:13028. doi: 10.1021/ja0474493. [DOI] [PubMed] [Google Scholar]
- 45.Werner H, Kühn A. J. Organomet. Chem. 1979;179:439. [Google Scholar]
- 46.For a review of this and other sequential processes in silicon-based cross-coupling, see: Denmark SE, Liu JH-C. Isr. J. Chem. 2010;50:577. doi: 10.1002/ijch.201000036.
- 47.Denmark SE, Kallemeyn JM. J. Org. Chem. 2005;70:2839. doi: 10.1021/jo047755z. [DOI] [PubMed] [Google Scholar]
- 48.Denmark SE, Tymonko SA. J. Am. Chem. Soc. 2005;127:8004. doi: 10.1021/ja0518373. [DOI] [PubMed] [Google Scholar]
- 49.Denmark SE, Sweis RF. J. Am. Chem. Soc. 2001;123:6439. doi: 10.1021/ja016021q. [DOI] [PubMed] [Google Scholar]
- 50.Denmark SE, Regens CS, Kobayashi T. J. Am. Chem. Soc. 2007;129:2774. doi: 10.1021/ja070071z. [DOI] [PubMed] [Google Scholar]
- 51.Denmark SE, Fujimori S. J. Am. Chem. Soc. 2005;127:8971. doi: 10.1021/ja052226d. [DOI] [PubMed] [Google Scholar]
- 52.Denmark SE, Tymonko SA. J. Org. Chem. 2003;68:9151. doi: 10.1021/jo0351771. [DOI] [PubMed] [Google Scholar]
- 53.Denmark SE, Sweis RF. J. Am. Chem. Soc. 2004;126:4876. doi: 10.1021/ja0372356. [DOI] [PubMed] [Google Scholar]
- 54.Denmark SE, Smith RC, Chang W-TT, Muhuhi JM. J. Am. Chem. Soc. 2009;131:3104. doi: 10.1021/ja8091449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Denmark SE, Smith RC, Tymonko SA. Tetrahedron. 2007;63:5730. doi: 10.1016/j.tet.2007.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Denmark SE, Chang W-TT, Houk KN, Liu P. J. Org. Chem. 2015;80:313. doi: 10.1021/jo502388r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Denmark SE, Kallemeyn JM. J. Am. Chem. Soc. 2006;128:15958. doi: 10.1021/ja065988x. [DOI] [PubMed] [Google Scholar]
- 58.Denmark SE, Werner NS. J. Am. Chem. Soc. 2008;130:16382. doi: 10.1021/ja805951j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Denmark SE, Werner NS. Org. Lett. 2011;13:4596. doi: 10.1021/ol2017998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Denmark SE, Werner NS. J. Am. Chem. Soc. 2010;132:3612. doi: 10.1021/ja910804u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Denmark SE, Liu JH-C. Angew. Chem. Int. Ed. 2010;49:2978. doi: 10.1002/anie.200905657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Denmark SE, Kobayashi T, Regens CS. Tetrahedron. 2010;66:4745. doi: 10.1016/j.tet.2010.03.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kobayashi T, Regens CS, Denmark SE. In: Strategies and Tactics in Organic Synthesis. Harmata MA, editor. Vol. 8 Amsterdam: Elsevier; 2012. [Google Scholar]
- 64.Denmark SE, Fujimori S. In: Strategies and Tactics in Organic Synthesis. Harmata MA, editor. Vol. 7 Amsterdam: Elsevier; 2008. [Google Scholar]
- 65.(a) Tymonko SA, Smith RC, Ambrosi A, Denmark SE. J. Am. Chem. Soc. 2015;137:6192. doi: 10.1021/jacs.5b02515. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tymonko SA, Smith RC, Ambrosi A, Ober MH, Wang H, Denmark SE. J. Am. Chem. Soc. 2015;137:6200. doi: 10.1021/jacs.5b02518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Denmark SE, Liu JH-C, Muhuhi JM. J. Am. Chem. Soc. 2009;131:14188. doi: 10.1021/ja9063475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Denmark SE, Liu JH-C, Muhuhi JM. J. Org. Chem. 2010:75. doi: 10.1021/jo101790z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Denmark SE, Liu JHC. J. Am. Chem. Soc. 2007;129:3737. doi: 10.1021/ja067854p. [DOI] [PubMed] [Google Scholar]
- 69.Chang W-tT, Smith RC, Regens CS, Bailey AD, Werner NS, Denmark SE. Org. React. 2011;75:214. [Google Scholar]
- 70.(a) Chang W-tT, Denmark SE. Cross-Coupling with Silicon Reagents: Arylsilanes. In: Molander GA, editor. Science of Synthesis: Cross Coupling and Heck-Type Reactions. Vol. 1. Stuttgart: Thieme; 2013. p. 383. [Google Scholar]; (b) Chang W-tT, Denmark SE. Cross-Coupling with Silicon Reagents: Alkenylsilanes. In: Molander GA, editor. Science of Synthesis: Cross Coupling and Heck-Type Reactions. Vol. 1. Stuttgart: Thieme; 2013. p. 431. [Google Scholar]; (c) Chang W-tT, Denmark SE. Cross-Coupling with Silicon Reagents: Alkylsilanes. In: Molander GA, editor. Science of Synthesis: Cross Coupling and Heck-Type Reactions. Vol. 1. Stuttgart: Thieme; 2013. p. 443. [Google Scholar]; (d) Chang W-tT, Denmark SE. Cross-Coupling with Silicon Reagents: Heteroarylsilanes. In: Molander GA, editor. Science of Synthesis: Cross Coupling and Heck-Type Reactions. Vol. 1. Stuttgart: Thieme; 2013. p. 495. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.