

Abstract
Background
MYBPC3 dysfunctions have been proven to induce dilated cardiomyopathy, hypertrophic cardiomyopathy, and/or left ventricular noncompaction; however, the genotype–phenotype correlation between MYBPC3 and restrictive cardiomyopathy (RCM) has not been established. The newly developed next-generation sequencing method is capable of broad genomic DNA sequencing with high throughput and can help explore novel correlations between genetic variants and cardiomyopathies.
Methods and Results
A proband from a multigenerational family with 3 live patients and 1 unrelated patient with clinical diagnoses of RCM underwent a next-generation sequencing workflow based on a custom AmpliSeq panel, including 64 candidate pathogenic genes for cardiomyopathies, on the Ion Personal Genome Machine high-throughput sequencing benchtop instrument. The selected panel contained a total of 64 genes that were reportedly associated with inherited cardiomyopathies. All patients fulfilled strict criteria for RCM with clinical characteristics, echocardiography, and/or cardiac magnetic resonance findings. The multigenerational family with 3 adult RCM patients carried an identical nonsense MYBPC3 mutation, and the unrelated patient carried a missense mutation in the MYBPC3 gene. All of these results were confirmed by the Sanger sequencing method.
Conclusions
This study demonstrated that MYBPC3 gene mutations, revealed by next-generation sequencing, were associated with familial and sporadic RCM patients. It is suggested that the next-generation sequencing platform with a selected panel provides a highly efficient approach for molecular diagnosis of hereditary and idiopathic RCM and helps build new genotype–phenotype correlations.
Keywords: diastolic dysfunction, genotype–phenotype, restrictive cardiomyopathy
Compared with dilated cardiomyopathy (DCM) and hypertrophic cardiomyopathy (HCM), restrictive cardiomyopathy (RCM) is a much more rare nonischemic myocardial disease. RCM is characterized by restrictive ventricular-filling physiology in the presence of normal or reduced diastolic and/or systolic volumes (of 1 or both ventricles), biatrial enlargement, and normal ventricular wall thickness.1,2 Although RCM was previously believed to be idiopathic or associated with inflammatory, infiltrative, or systemic disease, recent investigations revealed that sarcomere protein gene mutations3 or desminopathy4 are responsible for primary RCM, especially in patients with familial RCM. It has been demonstrated that genetic mutations of cardiac troponin (TNNC1,TNNI3,TNNT2), β-myosin heavy chain 7 (MYH7), α-cardiac actin (ACTC), and myosin light chain 3 (MYL3) were associated with RCM.5
Previously, mutational screening of hereditary disease was performed mostly by direct Sanger capillary sequencing or with the denaturing high-performance liquid chromatography/Sanger method; however, molecular testing with the traditional individual exon-by-exon and gene-by-gene sequencing method experienced several limitations in throughput, cost-effectiveness, and time. It is particularly difficult to use this technology for the genetic diagnosis of cardiomyopathies, which are multigenic diseases, so that only the most prevalent genes associated with the phenotype are assessed routinely in clinical/and or basic medical scientific investigations. In fact, >50 genes have been reported to be related to cardiomyopathies, and most shared the same genotype groups.6
The newly developed next-generation sequencing (NGS) method has made comprehensive genetic testing possible for all known genes of cardiomyopathies. It is expected to have stronger abilities to detect genetic changes in patients with primary cardiomyopathies and to explore novel genotype–phenotype correlations, aside from predicting clinical outcomes and developing individualized treatment.7 The basic principles of these techniques include generation of colonies after incorporation of target gene–specific adapters, with further sequencing, detection, and analysis. To date, the 3 best established benchtop high-throughput sequencing instruments—the Ion Torrent Personal Genome Machine (PGM, Life Technologies; Thermo Fisher Scientific), the 454 GS Junior (Roche), and the MiSeq (Illumina)—are available, and these NGS systems have exhibited excellent quantitative performance for throughput, reads, and indel errors.8 The objective of this study was to screen for all known genes of inherited cardiomyopathies in patients with RCM, using comprehensive libraries with NGS.
Methods
Clinical Evaluation
Clinical evaluation consisted of a medical history, family history, physical examination, 12-lead ECG, and transthoracic and/or transesophageal echocardiogram. Cardiac gadolinium-enhanced magnetic resonance imaging was applied in selected cases for which it was deemed proper and necessary for the diagnosis. A clinical diagnosis of primary RCM was made by at least 2 independent experts of cardiology on the basis of the established criteria of the Contemporary Definitions and Classification of the Cardiomyopathies.1
Echocardiogram parameters were measured according to the American College of Cardiologists/American Heart Association guidelines.9 In addition to routine measurements of cardiac chamber dimensions, ventricular wall thickness, and ejection fraction, we performed a thorough evaluation of the restrictive patterns including mitral E/A ratio (when applicable), shortened deceleration time of E wave ≤160 ms, reduced early diastolic mitral annulus velocity e’ <10 cm/s (septal) or <15 cm/s (lateral), and the ratio of early transmitral flow velocity (E) to lateral e’, as E/e’ ≥13 by tissue Doppler imaging. Standard cardiac magnetic resonance (CMR) image interpretation and postprocessing were performed in accordance with the Society of Cardiovascular Magnetic Resonance guidelines.10 Late gadolinium-enhanced imaging was performed to identify myocardial fibrosis.
Panel Design and Library Preparation
Sixty-four candidate genes reported to be causative of inherited cardiomyopathy, according to Online Mendelian Inheritance in Man (http://omim.org) and PubMed literature retrieval, were selected for panel design. The gene list is shown in Table S1. The primers of overlapping amplicons covering the coding sequence region and flanking sequences (padding +25 base pairs) of each targeted gene were automatically generated by Ion AmpliSeq designer software. This design produced 3647 amplicons, which were divided into 2 primer pools (Life Technologies; Thermo Fisher Scientific). Each amplicon is about 250 base pairs. Libraries were constructed using the Ion AmpliSeq Library Kit v2.0 (Life Technologies; Thermo Fisher Scientific) and custom designed primer pools, according to the manufacturer’s instructions. DNA fragments from different samples were ligated with bar-coded sequencing adaptors using the Ion Xpress Barcode Adapter 1-16 Kit (Life Technologies; Thermo Fisher Scientific). The library was quantified with the Qubit 2.0 fluorometer (Invitrogen; Thermo Fisher Scientific).
Next-Generation Sequencing and Data Analysis
Fifteen bar-coded samples were pooled in equimolar amounts. Amplified libraries were submitted to emulsion polymerase chain reaction done on the Ion OneTouch system using the Ion OneTouch 200 Template Kit v2 DL (Life Technologies; Thermo Fisher Scientific), according to the manufacturer’s instructions. Next, ion sphere particles were recovered, and template-positive ion sphere particles were enriched using the Ion OneTouch ES system (Life Technologies; Thermo Fisher Scientific). The enriched ion sphere particles were sequenced on an Ion 318 chip using the Ion PGM 200 Sequencing Kit (Life Technologies; Thermo Fisher Scientific) on the Ion Torrent PGM (Life Technologies; Thermo Fisher Scientific). Data from the PGM runs were processed using Ion Torrent Suite 4.0 software (Life Technologies; Thermo Fisher Scientific) to generate sequence reads. After sequence alignment and extraction of SNPs and indels, all variants were filtered against dbSNP137. DNA sequences were visualized with an integrated genomics viewer. The most likely disease-causing variants were analyzed by Sanger sequencing.
This study, including both clinical evaluation and broad targeted genetic screening, was approved by the Peking Union Medical College Hospital ethics committee. Written informed consent forms were obtained from all investigated participants.
Results
Clinical Characteristics and Phenotype
In this study, 4 patients diagnosed with RCM, including 3 patients from a multigenerational family and 1 unrelated patient without obvious family history (sporadic patient) were investigated. All patients were of Chinese Han descent. Patient 1 (relative III4), proband of the RCM family (Figure1), was a woman aged 34 years who experienced persistent atrial fibrillation and congestive heart failure for 2 years before admission to our hospital. On admission, the patient presented a normal cardiac troponin I level and a significantly elevated N-terminal pro-brain natriuretic peptide level of 6672 pg/mL (normal range 0 to 125 pg/mL). Echocardiography showed significant biatrial enlargement with reduced dimensions and preserved systolic function of both ventricles (Figure2A, Table 1, and Video S1 to S4). Doppler examination demonstrated filling dysfunction of the left ventricle (LV) with an obvious restrictive pattern, with a shortened deceleration time of the E wave of 120 ms, early diastolic mitral annulus velocity e’ (lateral) of 5.1 cm/s (Figure2B), e’ (septal) of 3.2 cm/s (Figure S1), and E/e’ (lateral) of 15.7. The abnormal morphology and function were confirmed by CMR (Figure3A), which showed normal left ventricular end-diastolic volume of 51 mL/m2 (normal range 41 to 81 mL/m2) and LV mass index of 87.7 g/m2 (papillary muscles included, normal range 63 to 95 g/m2) and 83.3 g/m2 (papillary muscles excluded). When the LV wall was studied by CMR, the maximal thickness of the apical, septal, and lateral LV walls were 7.7, 11.7, and 8.6 mm, respectively (≥12 mm is considered LV hypertrophy in CMR11). The patient’s cardiac index evaluated by CMR was reduced to 1.71 L/min per m2 (normal range 1.75 to 3.80 L/min per m2). In addition, gadolinium-enhanced magnetic resonance indicated significant late enhancement of papillary muscle (Figure3B). With the diagnosis of RCM, the patient’s cardiac failure was successfully controlled by β-adrenergic receptor blocker and diuretics. In addition, long-term oral anticoagulation was prescribed for atrial fibrillation. The patient was recommended for heart transplantation.
Figure 1.
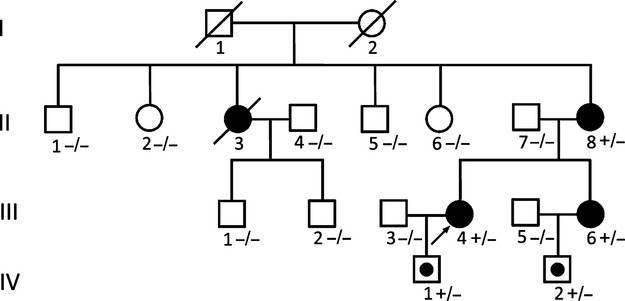
Pedigree of an RCM family. Squares represent male relatives; circles represent female relatives; filled symbols indicate RCM patients; slants indicate dead members; arrow represent proband; symbol with dots represent mutation carrier without clinical manifestation, + indicates MYBPC3 Q463X mutation positive; − indicates MYBPC3 Q463 mutation negative. MYBPC3 indicates myosin-binding protein C; RCM, restrictive cardiomyopathy.
Figure 2.
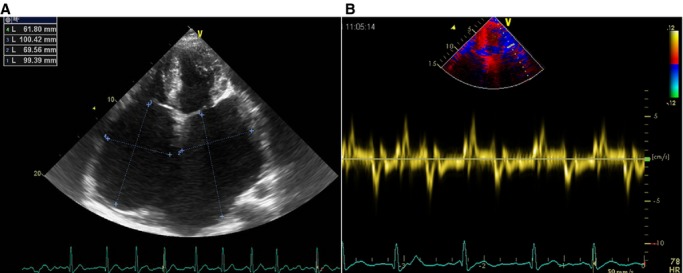
Echocardiography of relative III4 shows (A) significant biatrial enlargement and normal biventricular size and (B) reduced lateral mitral annulus velocity (e’) by tissue Doppler image.
Table 1.
Clinical Manifestation and Measurements by Echocardiogram in Studied Objects
| Subject | Age | Sex | Manifestation | LVEDD (mm) | LV Septum (mm) | LA (mm) | LVEF (%) | Mitral E Wave DT (ms) | Septal e’ (cm/s) | Lateral e’ (cm/s) | E/e’ (Lateral) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Familial | |||||||||||
| Patient 1 (III4) | 34 | F | CHF+AF | 35 | 9 | 84 | 60 | 120 | 3.2 | 5.1 | 15.7 |
| Patient 2 (II8) | 55 | F | CHF+AF | 43 | 9 | 56 | 75 | 110 | 2.7 | 3.3 | 18.2 |
| Patient 3 (III6) | 31 | F | Mild SOB after exertion | 43 | 8 | 46 | 70 | 130 | 6.5 | 8.8 | 13.2 |
| Control 1 (II1) | 79 | M | None | 52 | 9 | 40 | 70 | 184 | 11.5 | 15.6 | 8.4 |
| Control 2 (II2) | 77 | F | None | 50 | 8 | 31 | 68 | 176 | 12.2 | 16.8 | 7.7 |
| Control 3 (II5) | 68 | M | None | 50 | 9 | 38 | 73 | 160 | 12.5 | 18.5 | 6.8 |
| Control 4 (II6) | 62 | F | None | 48 | 8 | 32 | 66 | 170 | 13.2 | 16.7 | 7.3 |
| Control 5 (III1) | 45 | M | None | 44 | 8 | 29 | 71 | 172 | 11.8 | 16.5 | 6.7 |
| Control 6 (III2) | 42 | M | None | 42 | 7 | 27 | 64 | 165 | 12.0 | 15.9 | 6.3 |
| Sporadic | |||||||||||
| Patient 4 | 45 | M | CHF+AF | 45 | 9 | 81 | 52 | 115 | 3.3 | 4.6 | 16.1 |
AF indicates atrial fibrillation; CHF, congestive heart failure; DT, deceleration time; E/e’, the ratio of early transmitral flow velocity (E) to e’; e’, early diastolic mitral annulus velocity; F, female; LA, left atrium; LV, left ventricular; LVEDD, left ventricular end-diastolic dimension; LVEF, left ventricular ejection fraction; M, male; SOB, shortness of breath.
Figure 3.
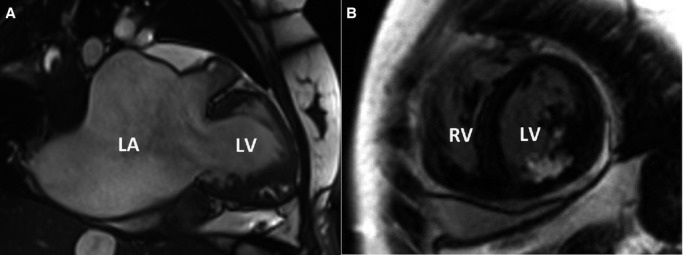
Cardiac magnetic resonance of relative III4 shows significant (A) left atrial enlargement, normal left ventricular size, and wall thickness and (B) late enhancement of papillary muscle. LA indicates left atrium; LV, left ventricle; RV, right ventricle.
Further investigation of her family history revealed that several members of this family were similarly affected. The patient’s mother (relative II8) and sister (relative III6) were also diagnosed with RCM using the same criteria. The clinical manifestation and imaging features of relative III6 were less distinct but still consistent with the diagnosis of RCM (Figure4A, Video S5). The affected sibling of relative II8 (relative II3) died in his 40s. Prior to that, he had been diagnosed with biatrial enlargement and suffered from congestive heart failure for 3 years. The autosomal domain, hereditary form, was characterized. Clinical features of studied patients are shown in Table 1.
Figure 4.
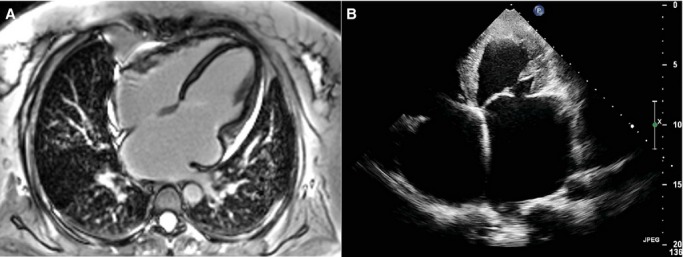
A, Cardiac magnetic resonance of relative III6 shows biatrial enlargement, normal biventricular size, and wall thickness. B, Apical 4-chamber view by echocardiography of patient 4.
Patient 4 was a man aged 45 years without obvious family history. He had symptoms of congestive heart failure for 10 years and then developed ascites before admission to our hospital. Echocardiography indicated that the patient had typical RCM phenotype with biatrial enlargement, normal LV volume and wall thickness, mildly depressed LV ejection fraction (52%), and significantly impaired LV diastolic function (Table 1, Figure4B, Video S6). For the purpose of differential diagnosis, all 4 patients received examinations of complete blood count for exclusion of eosinophilia, immune fixation electrophoresis and quantitative blood and urine light chain for exclusion of amyloidosis, autoantibody for exclusion of autoimmune diseases, and chest computed tomography scan and serum angiotensin-converting enzyme level to exclude sarcoidosis and pericardium thickness. Although we did not perform right heart catheterization for the studied participants, there was no evidence of constrictive pericarditis with the combination of all multimodality imaging methods.
Genotype–Phenotype Investigations
A nonsense mutation in myosin-binding protein C (MYBPC3) gene (c.1387C>T, p.Q463X) (Figure5A) was identified in patient III4 by NGS and confirmed by the Sanger method, as described previously. The identical mutation was also found in relatives II8 and III6 but not in any other family members without myocardial involvement (including relatives II1, II2, II5, II6, III1, and III2). The offspring (relatives IV1 and IV2) carried the same pathogenic mutation; however, they experienced no symptoms and had normal echocardiography findings. We assumed that this was due to their young ages, at 5 years and 8 months, respectively. The same mutation of MYBPC3 has already been reported in a Chinese adult population of HCM.12 Patient 4 carried a missense MYBPC3 mutation (c.1000G>A, p.E334K) (Figure5B), which was already described to be a culprit mutation of HCM.13,14 No other causative variant was detected on all genes listed in the custom panel (Table S1).
Figure 5.
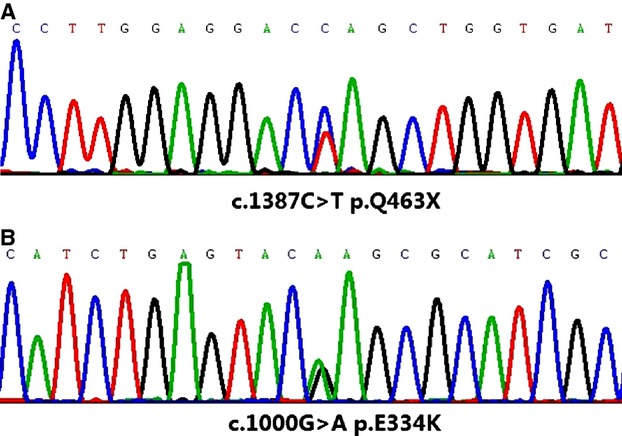
Mutation in myosin-binding protein C gene for (A) relative III4 and her family (c.1387C>T, p. Q463X) and (B) patient 4 (c.1000G>A, p.E334K).
Discussion
We report that a multigeneration family and an unrelated patient with RCM were associated with MYBPC3 gene mutations. The clinical manifestations were typical for RCM, supported by echocardiography and CMR. Aside from obvious biatrial enlargement, normal ventricular wall thickness, and normal or near-normal ejection fraction, echocardiography was extremely helpful in evaluating the LV filling dysfunction and increased left ventricular end-diastolic pressure. Patients with increased LV stiffness had more rapid rate of deceleration of early LV filling and shorter deceleration time. The e’ is critical for distinguishing between RCM and constrictive pericarditis. Such velocity decreases as LV filling pressure increases under the condition of impaired LV relaxation, whereas it will be normal or above normal in constrictive pericarditis. Furthermore, the E/e’ ratio has been identified as the best parameter for the diagnosis of diastolic function.15 Patients in this study manifested typical echocardiography findings, although CMR is more explicit in calculations of global LV volume, thickness, and mass index and detection of myocardial fibrosis with gadolinium-enhance technology. By CMR imaging, the studied patients showed normal left ventricular end-diastolic volume and LV mass index. Late enhancement in myocardium can be found in several cardiomyopathies including HCM, DCM, and RCM. It is worth noting that relatives III4 and III6 had the same enhancement of papillary muscles, indicating similar myocardial changes caused by the identical gene mutation shared in the family. An important issue to consider is whether these patients had consistent RCM or presented other types of cardiomyopathy such as HCM before evolving into the restrictive pattern. First, none of the 4 patients was diagnosed previously with HCM or ventricular hypertrophy, according to their medical records. RCM was the initial manifestation when they were first studied. Second, relative III6 was a woman aged 31 years without significant symptoms who admitted slight shortness of breath after exertion. Her RCM pattern was discovered only by clinical and genetic screening after diagnosis of relative III4. According to our findings, relative III6 is at the early stage of RCM compared with her mother and sister. Last but not least, none of the 4 patients had signs of even slight symmetric or asymmetric myocardial hypertrophy based on echocardiography and/or CMR.
As far as we know, the identified correlation between MYBPC3 gene mutations and RCM is novel. Cardiac myosin-binding protein C binds myosin heavy chain in thick filament and titin in elastic filaments. Mutations of MYBPC3 are strongly associated with familial HCM, DCM, and left ventricular noncompaction. As indicated previously, 3 HCM patients from different families with MYBPC3 E334K mutation, which is the same as that of patient 4 in this study, showed divergent phenotypes such as reduced ejection fraction and increased left ventricular end-diastolic diameter.14 The specific E334K mutation destabilized MYBPC3 protein through the ubiquitin–proteasome system and may contribute to cardiac dysfunction.14 It is not rare that the same myofilament gene mutation may exhibit different phenotypes in affected patients. It was reported that MYBPC3 gene mutations accounted for 15% to 25% of HCM cases.14,16 According to clinical and genetic investigations, primary RCM is increasingly recognized as overlapping with HCM in the disease spectrum and has been seen in the family members who carried genetic mutations expressed as a classic hypertrophic pattern.17,18 Importantly, all those genes associated with sarcomeric disease (eg, TNNC1,TNNI3,TNNT2,MYH7,ACTC,MYL3) and found to be associated with RCM were reported in HCM patients. Similarly, in the present study, MYBPC3—commonly related to HCM—could also be involved in RCM. MYBPC is a modular polypeptide located at the C-zone in the striated muscle. It has multiple structural and regulatory functions including sarcomere assembly and interaction with myosin heads.19 Functions of MYBPC have been investigated in depth. In an animal model of genetic knock out,20 myocytes from MYBPC3−/− mice exhibited markedly short diastolic sarcomere length and slower sarcomere shortening and relengthening in kinetics. Zebrafish with genetic knockdown of MYBPC3 showed ventricular hypertrophy and diastolic heart failure manifesting as decreased ventricular diastolic relaxation velocity, pericardial effusion, and dilatation of the atrium.21 These findings indicate that MYBPC helps complete relaxation during diastole. Our investigations also noted that phenotype of primary RCM with severe diastolic dysfunction could be ascribed to MYBPC3 mutations.
It has been presumed that primary RCM was more common in children and had an extremely ominous prognosis.22 In our study, all patients presented clinical manifestation in adulthood, whereas 2 children carrying the pathogenic mutations had not been clinically identified. Mutation-specific genetic testing is recommended as appropriate for family members following identification of an RCM-causative mutation in the index case.23 We believe it may help to differentiate affected patients early for evaluation and treatment. Long-term prognosis of MYBPC3 mutation in RCM patients is obscure. In patients with HCM, MYBPC3 gene mutations are considered to be related to late onset and to be clinically benign.24 Whether it is the same for patients with RCM is unknown.
Genetic screening of RCM patients has been extremely difficult, based on previous studies, and information on causative mutation was exiguous compared with DCM and HCM. First, RCM is perhaps the least common inherited cardiomyopathy, and many fewer studies of familial RCM patients have been conducted compared with other cardiomyopathies. It is possible that some patients from families with HCM caused by MYBPC3 mutations might present a restrictive phenotype but were unintentionally overlooked. The RCM patients in this study showed classically restrictive physiological patterns and no hypertrophic ventricular structure. In this case, unambiguous phenotype can provide more convincing evidence. Second, previous strategies based on polymerase chain reaction are insufficient for broad genomic investigations in RCM, considering the limited gene mutations reported and the difficulty in candidate gene selection. The MYBPC3 gene was not generally included in the candidate screen plan for RCM because it contained 35 exons that encoded a large multidomain protein and was not considered relevant to RCM. Nonetheless, the well-planned panel and high-throughput NGS applied in this study made it possible to screen all possible genetic variations associated with cardiomyopathy in both familial and sporadic patients. With this method, we detected the pathogenic MYBPC3 mutations and excluded defects in other cardiomyopathy-related genes.
The NGS method developed rapidly in recent years. It allowed sequencing of information on billions of bases in a single run (high throughput) and unselective analysis of whole genomic DNA. Unlike the traditional methods, which cover only the regions of interest, NGS assessed all disease-relevant genotypes. This was extremely helpful in identifying genetic mutations of complicated hereditary diseases, including cardiomyopathies. More than 600 genetic variants have been recognized as being associated with various types of cardiomyopathy.25 Furthermore, lack of “hot” genes or genetic hot spots, variable penetrance, and incomplete expression combined with phenotype–genotype heterogeneity contributed great challenges to genetic diagnosis of cardiomyopathic disease; however, the drafted all-in-one panel, high throughput, and cost-effective techniques made NGS the right tool. The application of NGS in molecular diagnostics of cardiomyopathies was first described in 2011,26 and 6 of 10 DCM or HCM patients examined were believed to have pathogenic mutations. A total of 47 genes were included in the library design. Recently, the benchtop high-throughput sequencing instruments have proven their sensitivity and reliability. In a study of 3 patients with HCM, NGS showed significantly higher sensitivity than the denaturing high-performance liquid chromatography/Sanger sequencing method for detecting gene mutation (95.7% to 96.2% versus 53.8% to 60.9%), with similar specificity (99.9% versus 99.9%).27 In another study, a cohort of 75 patients who were previously investigated for gene mutations was used to evaluate the custom AmpliSeq panel and Ion Torrent PGM workflow.28 The high efficiency and accuracy of this method were verified repeatedly.
Conclusion
We reported that 2 MYBPC3 mutations, known to be pathogenic in HCM, DCM, and left ventricular noncompaction, were related to inherited and sporadic RCM. To date, the low-cost and highly efficient NGS strategy meets the demand required for the systematic detection of genomic variants involved in rare cardiomyopathies.
Sources of Funding
The study was supported by the youth foundation from Peking Union Medical College Hospital, Beijing.
Disclosures
None.
Supporting Information
Video S1. Parasternal long axis view of relative III4 by echocardiography: enlarged left atrium, normal left ventricle size and wall thickness, and mild pericardial effusion. Papillary muscle is easily confused with the posterior wall.
Video S2. LV short axis view with mitral valve section of relative III4 by echocardiography: normal LV size and wall thickness. LV indicates left ventricle.
Video S3. LV short axis with papillary muscle section of relative III4 by echocardiography: normal LV size and wall thickness. Papillary muscle is easily confused with the posterior wall. LV indicates left ventricle.
Video S4. Reversed apical 4-chamber view of relative III4 by echocardiography: significant biatrial enlargement, normal biventricular size. Papillary muscle can be differentiated from ventricular wall. Cloudy blood flow from the left atrium to the left ventricle indicates slow flow in the cardiac diastolic phase.
Video S5. Animated movie of cardiac magnetic resonance of relative III6: enlargement of left atrium, normal left ventricle size and wall thickness.
Video S6. Long-axis view of patient 4 by echocardiography: enlarged left atrium, normal left ventricle size and wall thickness.
Table S1. List of the genes selected to perform the custom array.
Figure S1.
References
- Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE. Young JB; American Heart Association; Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; Council on Epidemiology and Prevention. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113:1807–1816. doi: 10.1161/CIRCULATIONAHA.106.174287. [DOI] [PubMed] [Google Scholar]
- Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, Dubourg O, Kuhl U, Maisch B, McKenna WJ, Monserrat L, Pankuweit S, Rapezzi C, Seferovic P, Tavazzi L, Keren A. Classification of the cardiomyopathies: a position statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2008;29:270–276. doi: 10.1093/eurheartj/ehm342. [DOI] [PubMed] [Google Scholar]
- Kaski JP, Syrris P, Burch M, Tome-Esteban MT, Fenton M, Christiansen M, Andersen PS, Sebire N, Ashworth M, Deanfield JE, McKenna WJ, Elliott PM. Idiopathic restrictive cardiomyopathy in children is caused by mutations in cardiac sarcomere protein genes. Heart. 2008;94:1478–1484. doi: 10.1136/hrt.2007.134684. [DOI] [PubMed] [Google Scholar]
- Pruszczyk P, Kostera-Pruszczyk A, Shatunov A, Goudeau B, Draminska A, Takeda K, Sambuughin N, Vicart P, Strelkov SV, Goldfarb LG, Kaminska A. Restrictive cardiomyopathy with atrioventricular conduction block resulting from a desmin mutation. Int J Cardiol. 2007;117:244–253. doi: 10.1016/j.ijcard.2006.05.019. [DOI] [PubMed] [Google Scholar]
- Arbustini E, Narula N, Dec GW, Reddy KS, Greenberg B, Kushwaha S, Marwick T, Pinney S, Bellazzi R, Favalli V, Kramer C, Roberts R, Zoghbi WA, Bonow R, Tavazzi L, Fuster V, Narula J. The MOGE(S) classification for a phenotype-genotype nomenclature of cardiomyopathy: endorsed by the World Heart Federation. J Am Coll Cardiol. 2013;62:2046–2072. doi: 10.1016/j.jacc.2013.08.1644. [DOI] [PubMed] [Google Scholar]
- Teekakirikul P, Kelly MA, Rehm HL, Lakdawala NK, Funke BH. Inherited cardiomyopathies: molecular genetics and clinical genetic testing in the postgenomic era. J Mol Diagn. 2013;15:158–170. doi: 10.1016/j.jmoldx.2012.09.002. [DOI] [PubMed] [Google Scholar]
- Haas J, Katus HA, Meder B. Next-generation sequencing entering the clinical arena. Mol Cell Probes. 2011;25:206–211. doi: 10.1016/j.mcp.2011.08.005. [DOI] [PubMed] [Google Scholar]
- Loman NJ, Misra RV, Dallman TJ, Constantinidou C, Gharbia SE, Wain J, Pallen MJ. Performance comparison of benchtop high-throughput sequencing platforms. Nat Biotechnol. 2012;30:434–439. doi: 10.1038/nbt.2198. [DOI] [PubMed] [Google Scholar]
- Cheitlin MD, Armstrong WF, Aurigemma GP, Beller GA, Bierman FZ, Davis JL, Douglas PS, Faxon DP, Gillam LD, Kimball TR, Kussmaul WG, Pearlman AS, Philbrick JT, Rakowski H, Thys DM. ACC/AHA/ASE 2003 guideline update for the clinical application of echocardiography–summary article: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (ACC/AHA/ASE Committee to Update the 1997 Guidelines for the Clinical Application of Echocardiography) J Am Coll Cardiol. 2003;42:954–970. doi: 10.1016/s0735-1097(03)01065-9. [DOI] [PubMed] [Google Scholar]
- Schulz-Menger J, Bluemke DA, Bremerich J, Flamm SD, Fogel MA, Friedrich MG, Kim RJ, von Knobelsdorff-Brenkenhoff F, Kramer CM, Pennell DJ, Plein S, Nagel E. Standardized image interpretation and post processing in cardiovascular magnetic resonance: society for cardiovascular magnetic resonance (SCMR) board of trustees task force on standardized post processing. J Cardiovasc Magn Reson. 2013;15:35. doi: 10.1186/1532-429X-15-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogaert J, Olivotto I. MR imaging in hypertrophic cardiomyopathy: from magnet to bedside. Radiology. 2014;273:329–348. doi: 10.1148/radiol.14131626. [DOI] [PubMed] [Google Scholar]
- Zou Y, Wang J, Liu X, Wang Y, Chen Y, Sun K, Gao S, Zhang C, Wang Z, Zhang Y, Feng X, Song Y, Wu Y, Zhang H, Jia L, Wang H, Wang D, Yan C, Lu M, Zhou X, Song L, Hui R. Multiple gene mutations, not the type of mutation, are the modifier of left ventricle hypertrophy in patients with hypertrophic cardiomyopathy. Mol Biol Rep. 2013;40:3969–3976. doi: 10.1007/s11033-012-2474-2. [DOI] [PubMed] [Google Scholar]
- Anan R, Niimura H, Takenaka T, Hamasaki S, Tei C. Mutations in the genes for sarcomeric proteins in Japanese patients with onset sporadic hypertrophic cardiomyopathy after age 40 years. Am J Cardiol. 2007;99:1750–1754. doi: 10.1016/j.amjcard.2007.01.066. [DOI] [PubMed] [Google Scholar]
- Bahrudin U, Morisaki H, Morisaki T, Ninomiya H, Higaki K, Nanba E, Igawa O, Takashima S, Mizuta E, Miake J, Yamamoto Y, Shirayoshi Y, Kitakaze M, Carrier L, Hisatome I. Ubiquitin-proteasome system impairment caused by a missense cardiac myosin-binding protein C mutation and associated with cardiac dysfunction in hypertrophic cardiomyopathy. J Mol Biol. 2008;384:896–907. doi: 10.1016/j.jmb.2008.09.070. [DOI] [PubMed] [Google Scholar]
- Nagueh SF, Appleton CP, Gillebert TC, Marino PN, Oh JK, Smiseth OA, Waggoner AD, Flachskampf FA, Pellikka PA, Evangelista A. Recommendations for the evaluation of left ventricular diastolic function by echocardiography. J Am Soc Echocardiogr. 2009;22:107–133. doi: 10.1016/j.echo.2008.11.023. [DOI] [PubMed] [Google Scholar]
- Bos JM, Towbin JA, Ackerman MJ. Diagnostic, prognostic, and therapeutic implications of genetic testing for hypertrophic cardiomyopathy. J Am Coll Cardiol. 2009;54:201–211. doi: 10.1016/j.jacc.2009.02.075. [DOI] [PubMed] [Google Scholar]
- Kubo T, Gimeno JR, Bahl A, Steffensen U, Steffensen M, Osman E, Thaman R, Mogensen J, Elliott PM, Doi Y, McKenna WJ. Prevalence, clinical significance, and genetic basis of hypertrophic cardiomyopathy with restrictive phenotype. J Am Coll Cardiol. 2007;49:2419–2426. doi: 10.1016/j.jacc.2007.02.061. [DOI] [PubMed] [Google Scholar]
- Sen-Chowdhry S, Syrris P, McKenna WJ. Genetics of restrictive cardiomyopathy. Heart Fail Clin. 2010;6:179–186. doi: 10.1016/j.hfc.2009.11.005. [DOI] [PubMed] [Google Scholar]
- Oakley CE, Hambly BD, Curmi PM, Brown LJ. Myosin binding protein C: structural abnormalities in familial hypertrophic cardiomyopathy. Cell Res. 2004;14:95–110. doi: 10.1038/sj.cr.7290208. [DOI] [PubMed] [Google Scholar]
- Pohlmann L, Kroger I, Vignier N, Schlossarek S, Kramer E, Coirault C, Sultan KR, El-Armouche A, Winegrad S, Eschenhagen T, Carrier L. Cardiac myosin-binding protein C is required for complete relaxation in intact myocytes. Circ Res. 2007;101:928–938. doi: 10.1161/CIRCRESAHA.107.158774. [DOI] [PubMed] [Google Scholar]
- Chen YH, Pai CW, Huang SW, Chang SN, Lin LY, Chiang FT, Lin JL, Hwang JJ, Tsai CT. Inactivation of myosin binding protein C homolog in zebrafish as a model for human cardiac hypertrophy and diastolic dysfunction. J Am Heart Assoc. 2013;2:e000231. doi: 10.1161/JAHA.113.000231. doi: 10.1161/JAHA.113.000231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller RJ, Weintraub R, Addonizio LJ, Chrisant MR, Gersony WM, Hsu DT. Outcome of idiopathic restrictive cardiomyopathy in children. Am J Cardiol. 2002;90:501–506. doi: 10.1016/s0002-9149(02)02522-5. [DOI] [PubMed] [Google Scholar]
- Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, Camm AJ, Ellinor PT, Gollob M, Hamilton R, Hershberger RE, Judge DP, Le Marec H, McKenna WJ, Schulze-Bahr E, Semsarian C, Towbin JA, Watkins H, Wilde A, Wolpert C, Zipes DP. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA) Heart Rhythm. 2011;8:1308–1339. doi: 10.1016/j.hrthm.2011.05.020. [DOI] [PubMed] [Google Scholar]
- Page SP, Kounas S, Syrris P, Christiansen M, Frank-Hansen R, Andersen PS, Elliott PM, McKenna WJ. Cardiac myosin binding protein-C mutations in families with hypertrophic cardiomyopathy: disease expression in relation to age, gender, and long term outcome. Circ Cardiovasc Genet. 2012;5:156–166. doi: 10.1161/CIRCGENETICS.111.960831. [DOI] [PubMed] [Google Scholar]
- Jacoby D, McKenna WJ. Genetics of inherited cardiomyopathy. Eur Heart J. 2012;33:296–304. doi: 10.1093/eurheartj/ehr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meder B, Haas J, Keller A, Heid C, Just S, Borries A, Boisguerin V, Scharfenberger-Schmeer M, Stahler P, Beier M, Weichenhan D, Strom TM, Pfeufer A, Korn B, Katus HA, Rottbauer W. Targeted next-generation sequencing for the molecular genetic diagnostics of cardiomyopathies. Circ Cardiovasc Genet. 2011;4:110–122. doi: 10.1161/CIRCGENETICS.110.958322. [DOI] [PubMed] [Google Scholar]
- D’Argenio V, Frisso G, Precone V, Boccia A, Fienga A, Pacileo G, Limongelli G, Paolella G, Calabro R, Salvatore F. DNA sequence capture and next-generation sequencing for the molecular diagnosis of genetic cardiomyopathies. J Mol Diagn. 2014;16:32–44. doi: 10.1016/j.jmoldx.2013.07.008. [DOI] [PubMed] [Google Scholar]
- Millat G, Chanavat V, Rousson R. Evaluation of a new NGS method based on a custom AmpliSeq library and Ion Torrent PGM sequencing for the fast detection of genetic variations in cardiomyopathies. Clin Chim Acta. 2014;433:266–271. doi: 10.1016/j.cca.2014.03.032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. Parasternal long axis view of relative III4 by echocardiography: enlarged left atrium, normal left ventricle size and wall thickness, and mild pericardial effusion. Papillary muscle is easily confused with the posterior wall.
Video S2. LV short axis view with mitral valve section of relative III4 by echocardiography: normal LV size and wall thickness. LV indicates left ventricle.
Video S3. LV short axis with papillary muscle section of relative III4 by echocardiography: normal LV size and wall thickness. Papillary muscle is easily confused with the posterior wall. LV indicates left ventricle.
Video S4. Reversed apical 4-chamber view of relative III4 by echocardiography: significant biatrial enlargement, normal biventricular size. Papillary muscle can be differentiated from ventricular wall. Cloudy blood flow from the left atrium to the left ventricle indicates slow flow in the cardiac diastolic phase.
Video S5. Animated movie of cardiac magnetic resonance of relative III6: enlargement of left atrium, normal left ventricle size and wall thickness.
Video S6. Long-axis view of patient 4 by echocardiography: enlarged left atrium, normal left ventricle size and wall thickness.
Table S1. List of the genes selected to perform the custom array.
Figure S1.