

Abstract
Background
Mammalian heart regenerative activity is lost before adulthood but increases after cardiac injury. Cardiac repair mechanisms, which involve both endogenous cardiac stem cells (CSCs) and cardiomyocyte cell-cycle reentry, are inadequate to achieve full recovery after myocardial infarction (MI). Mice deficient in S-nitrosoglutathione reductase (GSNOR−⁄−), an enzyme regulating S-nitrosothiol turnover, have preserved cardiac function after MI. Here, we tested the hypothesis that GSNOR activity modulates cardiac cell proliferation in the post-MI adult heart.
Methods and Results
GSNOR−⁄− and C57Bl6/J (wild-type [WT]) mice were subjected to sham operation (n=3 GSNOR−⁄−; n=3 WT) or MI (n=41 GSNOR−⁄−; n=65 WT). Compared with WT,GSNOR−⁄− mice exhibited improved survival, cardiac performance, and architecture after MI, as demonstrated by higher ejection fraction (P<0.05), lower endocardial volumes (P<0.001), and diminished scar size (P<0.05). In addition, cardiomyocytes from post-MI GSNOR−⁄− hearts exhibited faster calcium decay and sarcomeric relaxation times (P<0.001). Immunophenotypic analysis illustrated that post-MI GSNOR−⁄− hearts demonstrated enhanced neovascularization (P<0.001), c-kit+ CSC abundance (P=0.013), and a ≈3-fold increase in proliferation of adult cardiomyocytes and c-kit+/CD45− CSCs (P<0.0001 and P=0.023, respectively) as measured by using 5-bromodeoxyuridine.
Conclusions
Loss of GSNOR confers enhanced post-MI cardiac regenerative activity, characterized by enhanced turnover of cardiomyocytes and CSCs. Endogenous denitrosylases exert an inhibitory effect over cardiac repair mechanisms and therefore represents a potential novel therapeutic target.
Keywords: cardiovascular progenitor/stem cells, heart disease, heart regeneration, nitric oxide signaling, S-nitrosoglutathione reductase
The rate of myocardial renewal and regeneration in the adult mammalian heart is insufficient for recovery from damage.1,2 The mechanisms underlying this insufficiency remain highly controversial.1–3 Developmental studies in mice and human embryonic stem cells show that creation of new myocardium in mammals occurs through a multifaceted pathway that involves differentiation of cardiovascular progenitors and replication of mature cardiomyocytes.4,5 Both of these mechanisms are likely present in the postnatal mammalian heart to a limited degree,2,3,5 but their regenerative activity appears to be negatively regulated throughout adult life and are lost in the early postnatal period, possibly by mechanisms involving oxidative stress.6–12
Several pharmacologic and cell-based therapeutic agents improve the regenerative activity of postnatal cardiac progenitors and replicating cardiomyocytes.6,7,13–16 For example, infusion of thymosin-β4 into healthy mouse hearts enhances the cardiomyogenic capacity of adult epicardial progenitors after myocardial infarction (MI),15 whereas infusion of micro-RNAs 199a and 590 may selectively enhance cardiomyocyte cell-cycle reentry after MI.13 Furthermore, bone marrow–derived mesenchymal stem cells (MSCs)14 or growth hormone–releasing hormone receptor agonists17 enhance adult heart regeneration considerably by activating both cardiac progenitors and replicating cardiomyocytes.
GSNOR (alcohol dehydrogenase 3) regulates protein S-nitrosylation (SNO) turnover.18 In the adult heart, SNO modulates signaling pathways important for vasodilation, cardiomyocyte contraction, mitochondrial function,19,20 and Ca2+ handling.20–22 Interestingly, studies in mouse and rat embryos show that while GSNOR is active during induction of the mammalian cardiogenic program, the developing heart exhibits the lowest GSNOR activity compared with other embryonic tissues.23,24 Moreover, mice deficient in GSNOR develop normally25 but are characterized by a high propensity for postnatal hepatocarcinogenesis (HCC).26 Remarkably, they also exhibit a profound capacity for repairing liver27 and heart injury.21 We therefore hypothesized that GSNOR may negatively regulate the regenerative activity of cardiac progenitors and cardiomyocytes in the post-MI adult heart.
To test this hypothesis, we analyzed the cardiovascular phenotype of GSNOR−⁄− and wild-type (WT) mice in response to MI. Here, we show that, in addition to improved revascularization and cardiomyocyte Ca+2 handling, the loss of GSNOR in mice enhances the abundance and proliferative activity of adult cardiac progenitors, MSCs, and cardiomyocytes post MI. These findings have potentially important therapeutic implications because they suggest that GSNOR activity negatively regulates heart regeneration by suppressing proliferation of regenerative cardiovascular progenitors and cardiomyocytes.
Methods
This study was reviewed and approved by the University of Miami Institutional Animal Care and Use Committee and complies with all federal and state guidelines concerning the use of animals in research and teaching as defined by “The Guide for the Care and Use of Laboratory Animals” (National Institutes of Health, revised 2011).
Animals
The generation of GSNOR−⁄− mice has been reported.25 To eliminate/minimize genetic heterogeneity, GSNOR+⁄− mice were backcrossed for >10 generations into C57Bl6/J (WT) mice25 before GSNOR−⁄− mouse colonies were established at the University of Miami. Thus, our GSNOR−⁄− mice are considered to be on a C57Bl6/J background. Age-matched WT mice obtained from Jackson Laboratories were used as controls. Only male mice were used in this study. Mice received food and water ad libitum and were on a 12-hour light/dark cycle.
Experimental Model of MI
Three-month-old mice were anesthetized with isoflurane (2%) inhalation through endotracheal intubation. Body temperature was controlled during the entire procedure, and buprenorphine was provided. MI was attained through the permanent ligation of the left coronary artery (LCA) with a 7-0 Prolene suture, as previously described.21 MI was confirmed by visual blanching distal to the ligation and echocardiography at day 7 postsurgery.
Echocardiography
Noninvasive cardiac function was monitored by using a Vevo-770 imaging system (Visual Sonics Inc) 3 days before surgery (baseline) and 1, 4, and 8 weeks after surgery. Echocardiographic assessment was performed under anesthesia via isoflurane inhalation (1% to 2%) and controlled heart rates (≥500 bpm) and body temperatures (37±1°C). Endocardial volumes during diastole and systole were recorded from bidimensional long-axis parasternal views. The average of 3 consecutive cardiac cycles was calculated by using Vevo 770 3.0.0 software (Visual Sonics).
Hemodynamics
Intact heart hemodynamic analysis was performed at 2 months post MI by using miniaturized pressure-volume catheterization as previously described.28 A tipped catheter (SPR-839; Millar Instruments) was inserted into the right carotid artery and advanced retrograde into the left ventricle (LV) in the anesthetized animal (1% to 2% isoflurane inhalation). LV pressure-volume loops were recorded at steady state and at varying preloads during temporary compression of the inferior vena cava. After inferior vena cava compression, isoproterenol (ISO; 40 ng/kg per minute) was injected into left jugular vein and the analysis was repeated. All analyses were performed using LabChart 7 software (Millar Instruments).
Cardiomyocyte Activity
Calcium handling and sarcomere length (SL) shortening in isolated cardiomyocytes were analyzed at week 8 post MI. Briefly, hearts were harvested and retrograde perfused in a modified Langendorf system (at 2 mL/min) through the aorta with an isolation solution containing collagenase type 2 (Worthington Biochemical Corporation) and protease type XIV (Sigma-Aldrich Co). Cells were loaded with Fura-2, and SL and intracellular Ca2+ concentration ([Ca2+]i) were measured simultaneously in cardiomyocytes field-stimulated at 0.5, 1, 2, 3, and 4 Hz. All experiments were conducted at 37°C, and 5 cardiomyocytes were examined for each mouse (n=6).
Contractility and Calcium Measurement
Percent SL was recorded with an IonOptix iCCD camera and calculated as follows: ([resting SL−peak SL]×100/resting SL). [Ca2+]i was measured using a dual-excitation spectrofluorometer (IonOptix LLC). The “in vivo” calibration was performed by using solutions containing 10 μmol/L ionomycin (Sigma), and [Ca2+]i was calculated as described previously.29 [Ca+2]i transient (Δ[Ca+2]i) amplitude was considered as: peak [Ca+2]i−resting [Ca+2]i. ΔCa2+ decay parameters and sarcomere relaxation (τ and time to 90% decline) were analyzed by using IonWizard 6.0 software (IonOptix LLC). All resulting data were plotted and further analyzed with Prism 6 software (GraphPad Software, Inc). After Ca2+ reuptake and SL shortening were assessed under steady-state conditions, cardiomyocytes field-stimulated at 4 Hz were treated with increasing doses of ISO (Sigma-Aldrich Co). Thus, [Ca2+]i and SL were studied by superfusing 10−9, 10−8, 10−7, or 10−6 mol/L ISO. The raw data were calibrated and analyzed by using IonWizard 6.0 (IonOptix LLC), and a dose-response curve was then plotted by using Prism 6 software (GraphPad Software, Inc).
5-Bromo-2′-Deoxyuridine Treatment
MI was performed by using a variation of a previously described procedure.30 Two days post MI, WT or GSNOR−⁄− mice were administered daily injections of 5-bromo-2′-deoxyuridine (BrdU) (50 mg/kg IP) for 5 days, followed by 2 days with no treatment and, then, 5 additional daily treatments. Mice were killed 1 month later, and hearts were collected for immunohistochemical assessment of BrdU incorporation in c-Kit+ CSCs, coronary vessels, and cardiomyocytes.
Tissue Collection, Preparation, and Morphometric Analysis
Hearts were collected at 1, 4, and 8 weeks post MI. After each protocol, KCl (149 mg/mL) was injected into jugular vein while the animal was under sedation (2% isoflurane). The heart was harvested and fixed with formalin (10%) for histologic analysis. All hearts were cut into 4 transverse slices and stained with Masson’s trichrome. Myocardial infarct size was quantified for the circumferential extent of scar (Image J, NIH) and percentage fibrosis area (Adobe Photoshop CS3) as previously described.31
Immunohistochemistry
Paraffin sections were deparaffinized and rehydrated by immersion in xylene followed by a graded series of ethanols as previously described.14 Antigen retrieval was performed by using a heat-induced method with citrate buffer (Dako). After blocking with 10% normal donkey serum and goat anti-mouse IgG (Sigma-Aldrich) for 1 hour, sections were incubated with a primary antibody at 37°C for 1 hour, followed by the application of corresponding secondary antibody (Alexa dyes; Invitrogen) at 37°C for 1 hour. Omission of the primary antibodies on parallel sections was used as negative control. The following primary antibodies were used c-Kit (goat polyclonal; R&D systems), CD45 (rabbit polyclonal; Abcam), serine-10 phosphorylated histone-H3 (HP3; rabbit polyclonal; Abcam), anti-BrdU (biotinylated mouse monoclonal; Abcam), Aurora B kinase (Rabbit polyclonal, Abcam), activated caspase 3 (mouse monoclonal; BD), and tropomyosin (mouse monoclonal; Abcam). Nuclei were counterstained by incubation for 5 minutes with 4′,6-diamidino-2-phenylindole (DAPI, Invitrogen). The total number of positively stained cells was quantified per zone observed and corrected per area of corresponding zone (mm2). All images were obtained with a fluorescent microscope (Olympus IX81; Olympus America Inc, Center Valley, PA) or a confocal laser-scanning module (LSM710; Carl Zeiss MicroImaging).
Coronary Vascular Density
Deparaffinized sections were blocked with 10% normal donkey serum as previously described, and incubated with rhodamine wheat germ agglutinin (WGA, Invitrogen) at 37°C for 1 hour, followed by Alexa 488–conjugated isolectin-B4 (Invitrogen) for 24 hours at 4°C. Five fields of each section were randomly chosen for counting vessel profiles at the border and remote area, separately. The coronary vascular density was expressed as capillaries/high-power field (HPF).
In Vitro BrdU Cell Proliferation Assay
MSCs were plated in 96-well plates at the density of 1×104 cells/well. BrdU incorporation was measured by using a BrdU proliferation assay kit (Millipore) according to the manufacturer’s protocol. Briefly, the cells were labeled with BrdU (1:500). Then, cells were fixed, air-dried, and incubated 1 hour with anti-BrdU monoclonal antibody (1:200). The cells were washed 3 times and then incubated with peroxidase-conjugated goat anti-mouse IgG (1:2000) at room temperature. Thereafter, 100 μL of peroxidase substrate was added to each well and incubated for 30 minutes in the dark. Absorbance at a dual wavelength of 450/550 nm was measured.
GSNOR Activity in c-Kit CSCs and Whole Heart Lysates
The GSNOR activity assay was performed as previously described.18,25 Briefly, heart tissues from WT and GSNOR−⁄− mice at baseline and 8 weeks post MI were homogenized in a cell lysis buffer (Cell Signaling). CSCs were isolated from WT and GSNOR−⁄− mice ventricles, expanded, and magnetically sorted for c-Kit according to a previously described method.32 The concentration of these samples was determined using a standard Bradford assay. Heart homogenate (100 μg/mL) was incubated with Tris-HCI (2 mmol/L, pH 8.0), EDTA (0.5 mmol/L), and NADH (200 μmol/L). The reaction was started by adding S-nitrosoglutathione (GSNO) (400 μmol/L), and activity was measured as GSNO-dependent NADH consumption at absorbance of 340 nm for 5 minutes.
Statistics
Data are shown as individual points and/or mean±SE. Statistical significance was determined by unpaired Student t test, Mann–Whitney test, 1-way or 2-way ANOVA, and Kruskal–Wallis test as appropriate, to meet test assumptions. When significant differences were found, Bonferroni or Dun’s multiple comparisons post-hoc tests were carried out, as appropriate. Survival data were analyzed using Kaplan–Meier survival curves and compared using the Wilcoxon and log-rank tests.
Results
Cardiac Function is Improved While Cardiac Remodeling and Myocardial Infarct Size Are Reduced in GSNOR−⁄− Mice After MI
To study the effect of GSNOR in heart function and in response to MI, we ligated a branch of the LCA of GSNOR−⁄− (n=41) and WT (n=65) mice and followed the animals using serial echocardiography. Additionally, 3 animals from each group underwent sham operations. Survival analysis revealed that, compared with WT mice, loss of GSNOR conferred a significant decrease in mortality in response to experimental MI (Figure1A). Importantly, no differences in EF were recorded before surgery, suggesting that GSNOR−⁄− and WT mice have the same cardiac performance at baseline (Figure1B). Induction of MI was accompanied by an equal reduction in EF in both mouse strains, and these changes were not different between groups during the first 4 weeks after MI (Figure1B), indicating that the surgery was performed in a similar manner in both the GSNOR−⁄− and WT groups. However, at 8 weeks post MI, EF increased by ≈10% compared with 4 weeks earlier in GSNOR−⁄− mice and was significantly greater than in WT animals (Figure1B and Table 1, P=0.048).
Figure 1.

GSNOR−⁄− mice exhibit resilience to MI, have improved left ventricular function and less pathological remodeling at 8 weeks post MI. A, Kaplan–Meier plot illustrating significant differences in the survival of WT compared with GSNOR−⁄− mice after experimental MI. B, Both WT and GSNOR−⁄− mice have lower EF at 1 week post MI. WT EF remained low for the duration of the study, while GSNOR−⁄− mice showed significant improvement in cardiac function. C and D, GSNOR−⁄− mice exhibited preserved left ventricular remodeling during diastole (C) and systole (D) compared with WT. E, Representative Masson’s trichrome staining of histological sections of WT and GSNOR−⁄− hearts 8 weeks post MI. F, Infarcted WT hearts have more percentage of fibrosis area than sham groups and infarcted GSNOR−⁄− hearts at 8 weeks post MI. G, Infarcted WT and GSNOR−⁄− mice show increased fibrosis perimeter when compared with sham-operated animals 8 weeks post MI. WT hearts also demonstrate higher fibrosis perimeter compared with GSNOR−⁄− hearts. A, Mean±SEM (n=65 WT, n=41 GSNOR−⁄− mice). B through D, Mean±SEM (2-way ANOVA; n=15 WT, n=9 GSNOR−⁄− mice at all time points; P=difference between WT and GSNOR−⁄− mice). F and G, Individual values (1-way ANOVA; *P<0.05 compared with sham; #P<0.05 compared with MI). ANOVA indicates analysis of variance; EDV, volumes during end diastole; EF, ejection fraction; ESV, volumes during end systole; GSNOR, S-nitrosoglutathione reductase; MI, myocardial infarction; WT, wild-type.
Table 1.
Hemodynamic Measurement 8 Weeks After Left Anterior Descending Coronary Artery Ligation
| Parameters | WT (n=13) | GSNOR−⁄− (n=7) |
|---|---|---|
| HR, bpm | 448.1±17.43 | 392.3±20.83 |
| Pdev, mm Hg | 84.72±3.20 | 70.65±4.72* |
| Ped, mm Hg | 13.69±1.26 | 11.49±3.39 |
| Pes, mm Hg | 91.17±3.38 | 75.64±6.12* |
| EF, % | 22.17±3.08 | 31.29±1.21* |
Data are presented as mean±SEM. WT indiates wild-type; GSNOR, S-nitrosoglutathione reductase; HR, heart rate; Pdev, developed pressure; Ped, end-diastolic pressure; Pes, end-systolic pressure; EF, ejection fraction; MI, myocardial infarction.
P<0.05 compared with WT after MI.
Consistent with previous reports,21 these changes in EF were accompanied by significant improvements in LV remodeling. GSNOR−⁄− and WT LV volumes during end-diastole (EDV) and end-systole (ESV) did not differ at baseline and underwent similar remodeling during the initial 4-week period after MI (Figure1C and 1D). However, while LV remodeling continued to worsen in WT animals throughout the 8-week period of the study, expansion of EDV and ESV did not progress in GSNOR−⁄− mice after 4 weeks, and by 8 weeks, the EDV and ESV were significantly lower than in WT hearts (Figure1C and 1D). Consistent with echocardiographic data, hemodynamic measurements at 8 weeks post MI showed an increase in EF in the GSNOR−⁄− group (Table 1); however, the developed pressure and end-systolic pressure were lower compared with WT mice. In addition, histologic quantification of scar volumes and scar perimeters at 8 weeks post MI demonstrated that GSNOR−⁄− hearts had significantly less fibrosis compared with WT (Figure1E through 1G; P≤0.05).
GSNOR−⁄− Cardiomyocytes Have Faster Ca2+ Transient Decay
Cardiac function depends on Ca2+ homeostasis, which is subject to intrinsic and extrinsic controls. Ca2+ transient amplitude (Δ[Ca2+]i) as well as its dynamics are important parameters for the activation of cardiac contractile proteins. Abnormalities in Ca2+ handling result in altered cardiac function. To test if better cardiac function in GSNOR−⁄− mice post MI was associated with improved Ca2+ handling, we quantified [Ca2+]i and measured SL simultaneously in cardiomyocytes collected at 8 weeks post MI. GSNOR−⁄− cardiomyocytes consistently needed less time to reuptake Ca2+ from the cytosol (Figure2A and 2B), resulting in faster sarcomeric relaxation (Figure2C and 2D). Higher cytosolic free Ca2+ levels, due to inadequate Ca2+ reuptake or higher Ca2+ leak from sarcoplasmic reticulum, are associated with the lower cardiac contraction seen in heart failure.
Figure 2.
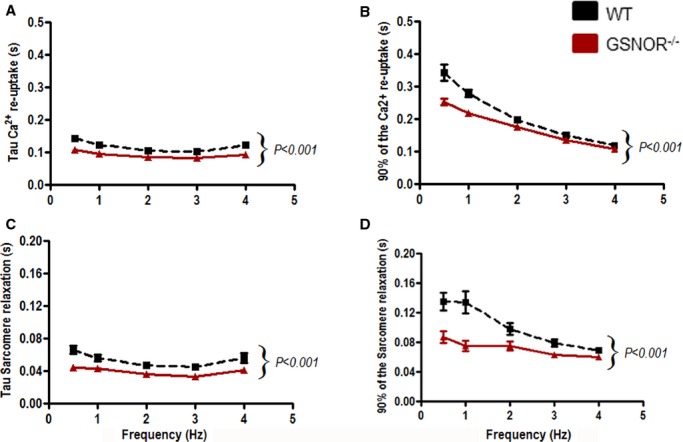
Relaxation mechanisms. GSNOR−/− and WT cardiomyocytes were stimulated at different frequencies (0.5 to 4 Hz). A, Tau, the time constant of [Ca2+]i decay (a measure of Ca2+ reuptake), and (B) the time to 90% of [Ca2+]i decay were measured and demonstrate that [Ca2+]i decay occurs faster in GSNOR−/− cardiomyocytes. C and D, Sarcomere relaxation (Tau [C] and time to 90% relaxation [D]) was improved in GSNOR−⁄− cardiomyocytes at all frequencies studied. Data are presented as mean±SEM (n=6 WT, n=6 GSNOR−⁄− mice at all time points). Two-way ANOVA;P<0.001 between WT and GSNOR−⁄− groups. ANOVA indicates analysis of variance; GSNOR, S-nitrosoglutathione reductase; WT, wild-type.
GSNOR−⁄− Hearts Exhibit Lower Adrenergic Signaling
Sympathetic hyperactivity plays an important role in cardiac dysfunction in heart failure.33 In our study, GSNOR−⁄− mice post MI exhibited depressed sensitivity to ISO in both pressure-volume loop (Figure3A) and in isolated cardiomyocyte studies (Figure3B). WT mice responded to ISO treatment with elevated heart rates and cardiac function parameters during ISO treatment, while GSNOR−⁄− hearts did not exhibit any significant changes compared with steady state. Consistently, the contractile response to increasing ISO doses was shifted to the right in GSNOR−⁄− compared with WT cardiomyocytes, evidencing lower sensitivity.
Figure 3.
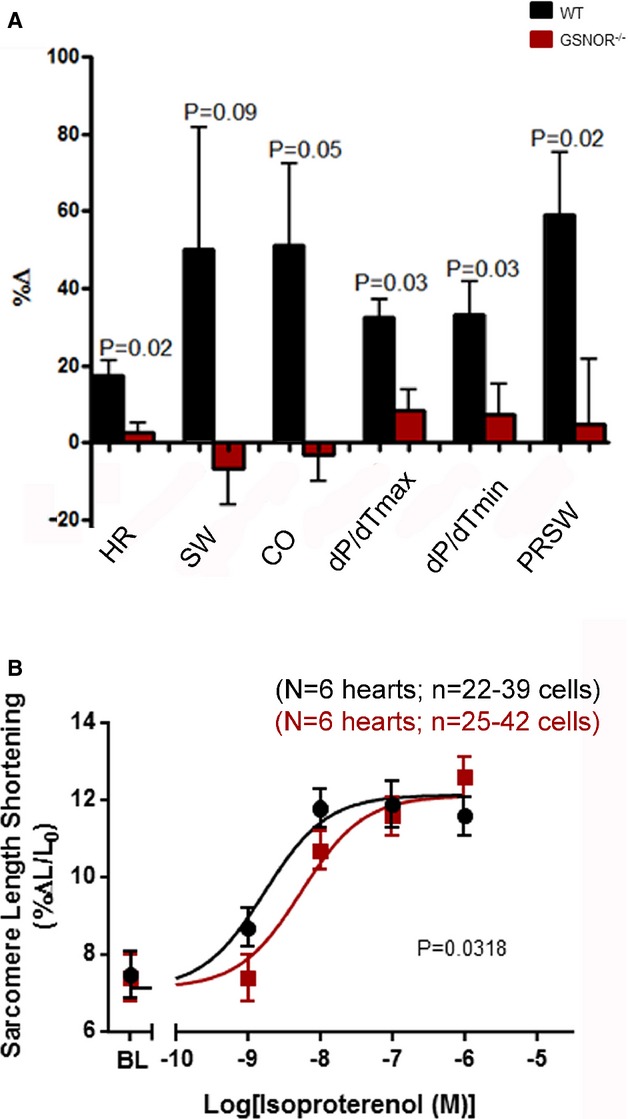
Adrenergic sensitivity. A, GSNOR−⁄− mice (n=7) post MI have lower isoproterenol sensitivity during PV loop compared with WT mice (n=9). GSNOR−⁄− mice do not exhibit changed heart parameters after 40 ng/kg per minute of isoproterenol injection into jugular vein. WT mice responded to isoproterenol infusion (1 way ANOVA). B, Dose-response to isoproterenol in GSNOR−⁄− cardiomyocytes (n=6) is right-shifted compared with WT (n=6), exhibiting a reduced sensitivity to β-adrenergic stimulation, with conserved maximal contraction (nonlinear regression dose-response fit; P=0.0318). Data are presented as mean±SEM. ANOVA indicates analysis of variance; BL, Baseline; GSNOR, S-nitrosoglutathione reductase; MI, myocardial infarction; PRSW, preload recruitable stroke work; WT, wild-type.
Enhanced Proliferative Expansion of c-Kit+ CSCs in Post-MI GSNOR−⁄− Hearts
We14,16,17,28 and others1,7,8,15,34–37 previously showed that activation of endogenous cardiac progenitors is an underlying mechanism of postnatal heart regeneration in mammals. We therefore tested whether the number of adult c-Kit+ CSCs in GSNOR−⁄− was greater than in WT mouse hearts. Accordingly, immunohistochemistry against c-Kit and CD45 was performed in the hearts of healthy GSNOR−⁄− and WT mice, as well as at 1 week, 1 month, and 2 months after experimental MI. In the absence of myocardial injury, both GSNOR−⁄− and WT hearts had equivalent numbers of endogenous c-Kit+/CD45− CSCs (Figure4A). However, in response to MI, the abundance of c-Kit+/CD45− CSCs increased significantly and this increase was greater in GSNOR−⁄− compared with WT mice (Figure4B). Importantly, compared with WT, GSNOR−⁄− infarcted hearts had significantly more c-Kit+ CSCs throughout the 2-month follow-up period (Figure4B). These findings strongly suggest that expression of GSNOR in the infarcted myocardium suppresses the activity of endogenous c-Kit+ CSCs.
Figure 4.

Enhanced proliferative expansion of c-Kit+ CSCs in GSNOR−⁄− hearts post MI. A, At baseline, healthy adult WT and GSNOR−⁄− mice have an equivalent number of c-Kit+ CSCs (t test; P=0.5942). B, c-Kit+ CSC at 1 and 8 weeks post MI in the infarct, border and remote zones of WT and GSNOR−⁄− mice (2-way ANOVA; *P=0.013 between WT and GSNOR−⁄−; #P=0.03 within groups). C, One month after MI,GSNOR−⁄− mice have significantly more c-Kit+ CSCs in their hearts, compared with WT. In addition, significantly more c-Kit+ CSCs have incorporated BrdU in GSNOR−⁄− mice compared with WT, suggesting that GSNOR−⁄− c-Kit+ CSCs have an enhanced proliferative capacity after MI (Mann–Whitney test; *P=0.0016 and #P=0.012). D, Representative confocal photomicrographs of BrdU incorporation by cardiac c-Kit+ CSCs in WT and GSNOR−⁄− hearts, 1 month post MI. Two GSNOR−⁄− c-Kit+ CSCs are shown, one of which has incorporated BrdU (inset) whereas the other has not (arrow). E, Principle of the GSNOR activity assay. GSNOR catalyzes the reduction of GSNO to GSSG in the presence of NADH. Thus, the activity of GSNOR can be indirectly estimated, by spectrophotometrically monitoring NADH oxidation. F, GSNOR enzymatic activity in c-Kit+ CSCs. GSNOR activity is enriched in WT, but is absent in GSNOR−⁄− progenitors. Data are presented as individual data (A) or mean±SEM (B and C) or fold-change (E); WT (n=6) GSNOR−⁄− (n=5). *P=0.01 and #P=0.02. ANOVA indicates analysis of variance; BrdU, 5-bromodeoxyuridine; BZ, border zone; CSC, cardiac stem cell; GSNOR, S-nitrosoglutathione reductase; GSSG, glutathione disulfide (oxidized GSH); IZ, infarct zone; MI, myocardial infarction; NADH, reduced form of nicotinamide adenine dinucleotide (NAD); RZ, remote zone; WT, wild-type.
To elucidate whether the difference in the abundance of post-MI c-Kit+ CSCs was due to enhanced cell-cycle activity of GSNOR−⁄− c-Kit+ CSCs, we primed infarcted GSNOR−⁄− and WT mice with BrdU, a synthetic nucleoside that substitutes thymidine in newly synthesized DNA during the S-phase of the cell cycle and therefore provides a marker for tracking cell proliferation.38 One month later, heart tissues were collected and confocal immunofluorescence analysis against BrdU and c-Kit in the myocardium illustrated that the expansion in post-MI c-Kit+ CSCs in GSNOR−/− hearts was accompanied by a significantly higher rate of BrdU incorporation (Figure4C and 4D; 10.9±4.99% of WT c-Kit+ CSCs compared with 15.9±3% of GSNOR−⁄− c-Kit+ CSCs were positive for BrdU, P=0.012). Thus, collectively these results suggest that, in response to myocardial damage, GSNOR activity negatively regulates heart regeneration by fostering important post-translational modifications to prevent proliferation and expansion of c-Kit+ CSCs.
Last, to elaborate whether the observed differences between GSNOR+/+ and GSNOR−⁄− c-Kit+ CSCs are directly or indirectly associated with GSNOR deficiency, we used an NADH-dependent GSNOR activity assay to gauge the enzymatic activity of GSNOR in culture-expanded c-Kit+ CSCs, purified from WT and GSNOR−⁄− hearts (Figure4E).18,25 No change in the absorbance of NADH was detected in the absence of GSNO between the 2 CSC strains (Figure4F). However, in the presence of GSNO, NADH oxidation produced a sharp decrease in absorbance in WT but not GSNOR−⁄− c-Kit+ lysates, over time (Figure4F). These results indicate that GSNOR activity is present in GSNOR+/+ but not in GSNOR−⁄− c-Kit+ CSCs and, therefore, the differences observed in proliferative capacity may be directly associated with GSNOR deficiency.
Enhanced Proliferative Expansion of Cardiomyocytes in Post-MI GSNOR−⁄− Hearts
Similar to c-Kit+ CSCs, the enzymatic activity of myocardial GSNOR, which is enriched in the WT heart before and after MI, is diminished in GSNOR−⁄− mice (Figure5A). Therefore, given that replication of preexisting cardiomyocytes provides an additional mechanism for cardiomyogenesis in newborn9,11,12 and adult mammals,10,14 we next asked whether, similar to c-Kit+ CSCs, GSNOR influences the cell-cycle activity in post-MI regenerative adult cardiomyocytes. Accordingly, we analyzed the expression of serine-10 phosphorylated histone H3 (Hp3) and Aurora-B kinase immunohistochemically to gauge the extent of cardiomyocyte mitosis in GSNOR−⁄− and WT mice. Compared with WT, GSNOR−⁄− mice exhibited a ≈2.4-fold increase in the abundance of mitotic cardiomyocytes at 1 week after MI (Figure5B through 5E).
Figure 5.

Enhanced mitotic activity of cardiomyocytes in GSNOR−⁄− hearts post MI. A, Absorption of NADH in whole-heart lysates from GSNOR−⁄− and WT mice, before and 8 weeks after the occurrence of MI. No NADH oxidation is recorded before the addition of GSNO in WT or GSNOR−⁄− lysates (time −2 to 0). Addition of GSNO (time 0), produces a sharp decrease in the absorbance of NADH in WT (black lines), but not in GSNOR−⁄− (red lines) heart lysates, before and after MI. These results indicate that the adult myocardium is enriched in GSNOR activity before and after MI, and that GSNOR activity is diminished in the pre- and post-MI GSNOR−⁄− heart. B and C, Confocal immunofluorescence analysis of cardiomyocyte mitosis, based on the nuclear expression of Hp3, in WT (B) and GSNOR−⁄− (C) hearts, 1 week post MI. D, Quantification of Hp3+ cardiomyocytes reveals that, compared with WT,GSNOR−⁄− hearts are characterized by a 3-fold increase in cardiomyocyte mitosis, 1 week post MI (t test; *P= 0.01). E, Representative confocal immunofluorescence image illustrating expression of Aurora-B kinase, a marker of cytokinesis (arrow), at the cleavage furrow of a mitotically dividing GSNOR−⁄− cardiomyocyte, 1 week post MI. Values are mean±SEM. cTn-T indicates cardiac troponin T; GSNOR, S-nitrosoglutathione reductase; MI, myocardial infarction; NADH, reduced form of nicotinamide adenine dinucleotide (NAD); WT, wild-type.
In order to elucidate whether the enhanced mitotic activity in GSNOR−⁄− cardiomyocytes yields newly formed cardiomyocytes in the infarcted myocardium, we studied the hearts of the GSNOR−⁄− and WT mice that we had primed with BrdU after MI (see earlier). Confocal microscopy revealed that, 1 month after MI, GSNOR−⁄− mice were characterized by ≈3-fold more BrdU+/tropomyosin+ cardiomyocytes in the infarct zone compared with the WT controls (Figure6A through 6C).
Figure 6.
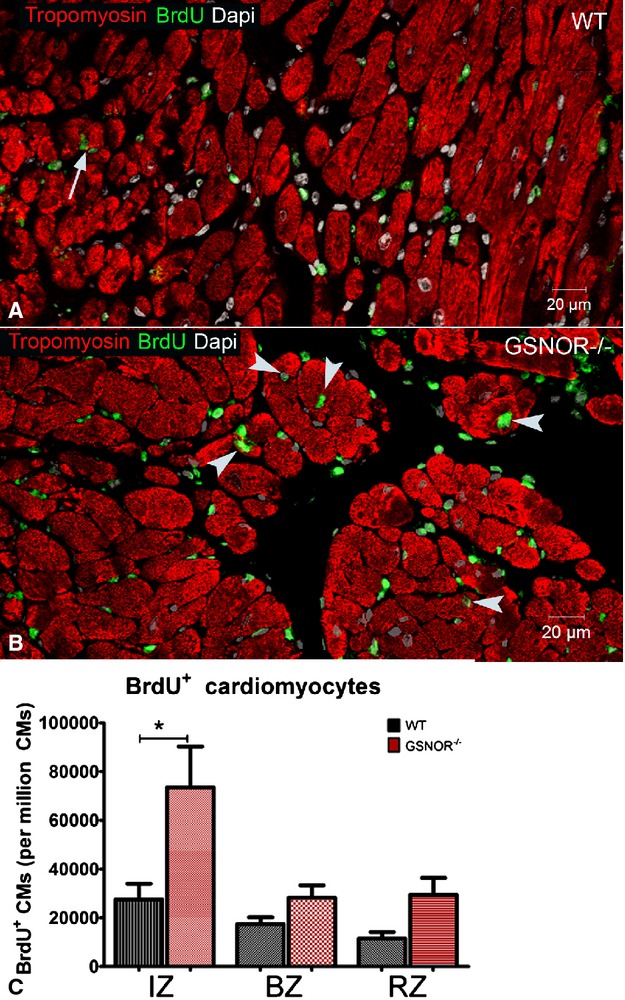
Enhanced proliferative expansion of cardiomyocytes in GSNOR−⁄− hearts post MI A and B, BrdU incorporation by cardiomyocytes in WT (A, arrows) and GSNOR−⁄− (B, arrowheads) hearts, 1 month post MI. C, Quantification of BrdU+ cardiomyocytes reveals that, compared with WT,GSNOR−⁄− hearts are characterized by a 3-fold increase in cardiomyocyte proliferation in their infarct zone (IZ) (1-way ANOVA, *P<0.0001). Values are mean±SEM. ANOVA indicates analysis of variance; BrdU, 5-bromodeoxyuridine; BZ, border zone; CMs, cardiomyocytes; GSNOR, S-nitrosoglutathione reductase; MI, myocardial infarction; RZ, remote zone; WT, wild-type.
S-Nitrosylation influences apoptotic activity.39,40 We also investigated the possibility that the effects of enhanced cardiomyocyte mitosis were masked by increased apoptotic cell death by immunohistochemistry staining for cleaved caspase-3, a marker for both death receptor– and mitochondrial pathway–initiated apoptotic cell death.40 There were no significant differences in the numbers of apoptotic cardiomyocytes in 1-week post-MI GSNOR−⁄− and WT mice (Figure7A through 7C). Collectively, these results suggest that in response to myocardial damage, GSNOR activity does not affect cardiomyocyte apoptosis but, rather, negatively regulates heart regeneration through post-translational modifications that prevent cardiomyocyte proliferation.
Figure 7.
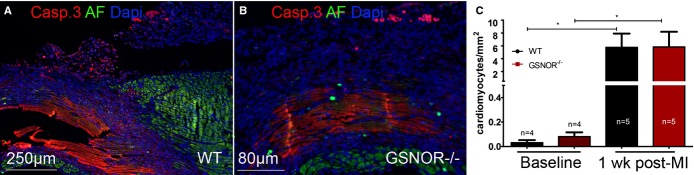
Quantification of cardiomyocyte apoptosis between WT and GSNOR−⁄− hearts post MI. A and B, Immunofluorescent images of cleaved caspase-3 (Casp. 3), a marker of apoptotic cell death, in WT (left panel) and GSNOR−⁄− (right panel) hearts, 1 week post MI. C, Quantification of activated Casp. capsase-3+ cardiomyocytes. Compared with baseline, the numbers of apoptotic cardiomyocytes expressing activated caspase-3 are dramatically increased within 1 week post MI. However, no significant differences are detected in apoptotic cardiomyocytes between WT and GSNOR−⁄− mice. (Kruskall–Wallis, *P=0.0002). Values are mean±SEM. AF indicates autofluorescence; GSNOR, S-nitrosoglutathione reductase; MI, myocardial infarction; WT, wild-type.
Enhanced Neovascularization in Post-MI GSNOR−⁄− Hearts
We previously showed that adult bone marrow–derived MSCs from GSNOR−⁄− mice form fewer capillaries and exhibited less endothelial differentiation than WT MSCs in an in vivo Matrigel-plug assay.41 We also previously showed that, compared with WT, healthy GSNOR−⁄− mice have higher coronary vascular density in their myocardium, which may partly explain their resistance to myocardial ischemia.21 To address whether GSNOR plays a role in the capacity of the myocardium to revascularize in response to MI, we analyzed the vascular density in GSNOR−⁄− and WT mice 2 months after experimental MI. Quantitative immunofluorescence analysis for isolectin-B4, a marker for identifying angiogenic endothelium,17,42 demonstrated that, compared with WT, GSNOR−⁄− mice were characterized by a significantly higher coronary vascular density in the infarct and border zones (Figure8). Furthermore, the presence of coronary vascular cells with BrdU incorporation in the myocardium of BrdU-primed GSNOR−⁄− mice 1 month after MI (Figure8B) supported that post-MI coronary vascular cell proliferation contributed to the enhanced vascularity of the damaged myocardium. The increased vascularity suggests that endothelial cells rather than MSCs are primarily responsible for blood vessel formation in healthy21 and infarcted GSNOR−⁄− mouse hearts. Thus, these findings suggest that in response to myocardial damage, GSNOR activity negatively regulates heart regeneration, limiting revascularization of the infarcted myocardium.
Figure 8.
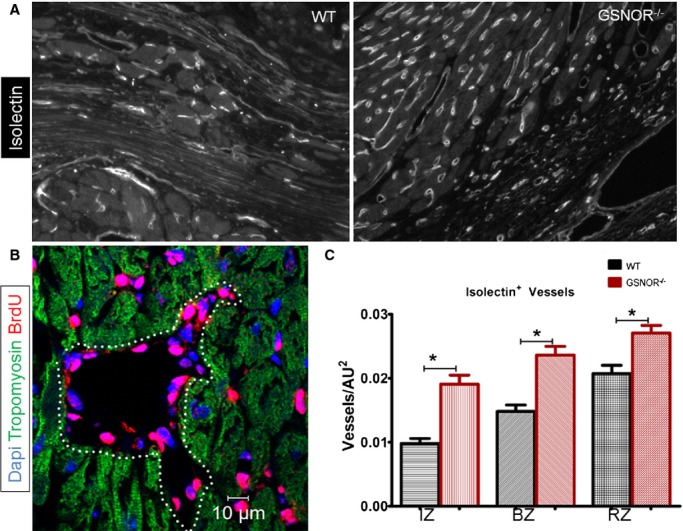
Enhanced neovascularization in post MI GSNOR−⁄− hearts. A, Representative immunofluorescent images of isolectin-IB4 in WT (left panel) and GSNOR−⁄− (right panel) hearts, 2 months post MI. B, BrdU incorporation in coronary vascular cells of a GSNOR−⁄− heart, demonstrates the presence of newly regenerated coronary vessels 1 month post MI. C, Quantification of isolectin-IB4+ vessels demonstrates that GSNOR−⁄− mice are characterized by a significantly enhanced vascular density compared with WT (1-way ANOVA, *P<0.0001). Values are mean±SEM. ANOVA indicates analysis of variance; BrdU, 5-bromodeoxyuridine; BZ, border zone; GSNOR, S-nitrosoglutathione reductase; IZ, infarct zone; MI, myocardial infarction; RZ, remote zone; WT, wild-type.
Enhanced In Vitro Proliferative Expansion of MSCs in GSNOR−⁄− Mice
In addition to studying the cell-cycle activity in the post-MI heart, we also examined whether GSNOR loss influences the proliferative activity of MSCs. MSCs are present in a wide variety of adult tissues, including the heart, and they are thought to support adult tissue homeostasis. They are also an important component of various adult stem cell niches,43 and we recently showed that GSNOR regulates their fate-choice decisions.44 Additionally, we and others have shown that MSCs comprise an important cell population for the diagnosis and treatment of cardiovascular disease.14,45 However, because MSCs are a heterogeneous population with a diverse immunophenotype, their direct in vivo identification is technically challenging. We therefore followed the approach of isolating and expanding MSCs in vitro from adult GSNOR−⁄− and WT mice, to perform our study.41,44 In vitro BrdU proliferation assay demonstrated that, similar to cardiac progenitors and cardiomyocytes, GSNOR−⁄− MSCs were characterized by a significantly higher proliferative activity compared with WT MSCs (Figure9).
Figure 9.
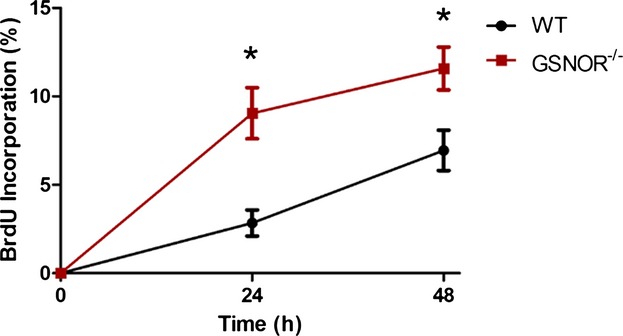
In vitro proliferation of WT and GSNOR−/− MSCs measured by BrdU cell proliferation assay. Two-way ANOVA; *P<0.0001 between groups and *P=0.0005 between WT and GSNOR−⁄− MSCs. n=3. Values are mean±SEM. ANOVA indicates analysis of variance; BrdU, 5-bromodeoxyuridine; GSNOR, S-nitrosoglutathione reductase; MSC, mesenchymal stem cell; WT, wild-type.
Discussion
The major new findings of this study are that the profound resilience to cardiac injury of mice with homozygous deletion of GSNOR involves an enhanced capacity of both c-Kit+ cardiac progenitors and mature cardiomyocytes to proliferate in response to MI. We also report that loss of GSNOR augments the capacity of the adult myocardium to turn over new coronary vessels and revascularize in response to ischemic damage. Furthermore, in agreement with the findings that GSNOR−⁄− mice are characterized by an expanded hematopoietic stem cell pool,21 we now show that the GSNOR−⁄− bone marrow is also enriched in MSCs with superior in vitro proliferative activity.
S-Nitrosylation/Denitrosylation of Proteins and Cell-Cycle Activity
Dynamic nitrosylation/denitrosylation of cysteine thiols (SNOs) has emerged as a ubiquitous post-translational modification system throughout biology.18,40 Similar to other post-translational modification systems (ie, phosphorylation, methylation, acetylation), SNOs regulate the expression and function of most, if not all, main classes of protein,40 including several key proteins controlling mammalian cell differentiation44 and cell-cycle activity. Tumor suppressor proteins of the retinoblastoma gene product (Rb) and p53 are directly and/or indirectly regulated post-translationally via SNOs.46,47
It is intriguing that the heart and bone marrow are not the only adult tissues that appear to exhibit an augmented proliferative phenotype in response to injury in GSNOR−⁄− mice. Recently, a higher propensity for HCC was documented in these animals,26,48 although defective DNA damage repair rather than excessive hepatocellular proliferation pathways due to SNO were primarily suggested to drive progression to malignancy. However, because in these studies cell-cycle analyses were limited to the immunohistochemical expression of a single cellular proliferation marker, Ki-67, in normal hepatocytes (which may or may not have been the cellular source of HCC), cell-cycle–based mechanisms of HCC cannot be excluded.26,48 For example, clonal analysis of BrdU incorporation by WT and GSNOR−⁄− hepatocytes and hepatic progenitors (during, before, and after the occurrence of HCC) could provide a more informative approach to determine the role of cell-cycle activity in GSNOR-mediated HCC, although this strategy is technically challenging and may also have important limitations due to the spontaneous nature of the development of HCC in GSNOR−⁄− mice. To this end, Cox et al recently studied a less complicated model of liver damage and, consistent with the findings presented here, reported how loss of GSNOR confers resistance to toxic liver injury and enhances liver regeneration through mechanisms that do involve increased proliferation of hepatic progenitors.27
Thus, it is attractive to speculate that regulation of SNO formation via denitrosylases exerts an antineoplastic role in adult mammals, as supported by Liu et al.26,48 However, in a different scenario, such as an injured tissue, our findings and those of Cox et al27 suggest that a denitrosylase-mediated cell-cycle arrest may exist and, consequently, cell proliferation that could contribute to tissue regeneration may also be regulated via SNO in the adult mammal. Our present findings and those of Cox et al27 illustrate that loss of GSNOR enhances tissue regeneration and, as a consequence, GSNOR inhibitors may stimulate regeneration of injured tissues. Future experiments will test this hypothesis and identify the mechanism(s) by which SNOs exert cell-cycle control.
S-Nitrosylation/Denitrosylation of Proteins in Response to Tissue Damage
In addition to its potential role as a post-translational regulator of cell-cycle activity in the injured adult heart, post-translational modification by SNOs modulate the expression and function of several proteins associated with damage response pathways, including NF-κB (nuclear factor κ light-chain enhancer of activated B cells), Nrf2 (nuclear factor erythroid-derived 2), and Hif-1α (hypoxia-inducible factor 1α).21,27,46 Of particular interest is the regulation of Hif-1α, 1 of the 2 subunits of the Hif-1 basic helix-loop-helix-PAS domain transcription factor. Under normal oxygen conditions, Hif-1α is expressed in the heart in an NF-κB–dependent manner but becomes degraded due to oxygen-dependent hydroxylation by prolyl hydroxylases, which mark the protein to be captured by the von Hippel–Lindau protein and undergo ubiquitination and proteasomal degradation.49 Under low oxygen conditions, the activity of hydroxylases is inhibited49 and Hif-1α is stabilized. However, GSNOR−⁄− hearts are characterized by an enhanced Hif-1α transcriptional activity and, even under normal oxygen conditions, the protein becomes stabilized due to enhanced S-nitrosylation.21 This novel mechanism of Hif-1α stabilization is thought to underlie the enhanced vasculogenic phenotype of GSNOR−⁄− mice.
Importantly, Puente et al recently reported that the rapid transition from an hypoxic to an oxygen-rich cardiac microenvironment during the first week of postnatal life in mice induces cell-cycle exit of cardiomyocytes through the production of reactive oxygen species (ROS) and DNA damage response.11 Thus, whether SNO-mediated stabilization of Hif-1α,21 resilience to ROS,22 and/or a defective DNA damage response mechanism26,48 could also underlie the enhanced proliferative phenotype we documented in adult GSNOR−⁄− mouse hearts remains to be further investigated.
S-Nitrosylation/Denitrosylation of Proteins and Cardiomyocyte Function
GSNOR metabolizes SNOs and its absence results in increased abundance of S-nitrosylated proteins.21 Important substrates of SNO are found in the heart, and they control excitation-contraction coupling.19,33 Our study expands on the knowledge that GSNOR−⁄− cardiomyocytes have faster Ca2+ decay and relaxation compared with WT cardiomyocytes post MI. Ca2+ cycling and myofilament responsiveness in cardiomyocytes underlie myocardial contractility. Thus, any alteration in intracellular Ca2+ handling is reflected as a change in heart performance. The myocardial relaxation is critical for proper diastolic function. This property is mainly governed by the affinity of myofilament for Ca2+ and by Ca2+ reuptake. It has been suggested that S-nitrosylation of certain sarcomeric proteins desensitizes myofilaments to Ca2+; however, the effect of hyper–S-nitrosylation of proteins involved in cytosolic Ca2+ removal and its cardioprotective role after myocardial injury is still unclear.50,51 Our data suggest that the cytosolic Ca2+ reuptake mechanism is preserved in GSNOR−⁄− cardiomyocytes post MI and may be associated with improved sarcoplasmic reticulum Ca2+-ATPase (SERCA2a) activity. Sun et al also found that GSNO treatment led to a reduction in cytosolic Ca2+ transients and increased SERCA2a activity.52 An increase in SERCA2a activity during MI would provide for improved Ca2+ reuptake into the sarcoplasmic reticulum and help to relax cardiac muscle during diastole. Faster Ca2+ decay and sarcomeric relaxation in GSNOR−⁄− post MI are likely involved in the cardioprotection in GSNOR−⁄− mice post MI. The lack of GSNOR decreases peripheral vascular tone and cardiac inotropic response to β-adrenergic stimulation.53 The absence of GSNOR and denitrosylation decreased cardiac function in unmanipulated GSNOR−⁄− mice, but this mechanism was masked in infarcted mice. One explanation for this controversial result is that hypernitrosylation blocks the action of ROS, which are increased in multiple cardiac disease states.11,54 Increases in superoxide results in disrupted protein S-nitrosylation seen in basal conditions, which lead to altered Ca2+ handling and impairment of cardiac function.19 In addition, Saraiva et al showed increased activity of cardiac xanthine oxidoreductase,55 a major source of superoxide in the heart, in mice that lack nitric oxide synthase 1. Thus, GSNOR deficiency may make these animals resistant to oxidant insults, preserving redox-sensitive mechanisms of Ca2+ handling, such as Ca2+ reuptake.
Our data suggest that deficiency of GSNOR protects cardiac proteins from ROS, which in turn improves Ca2+ handling homeostasis. Furthermore, the lower adrenergic sensitivity of GSNOR−⁄− mice can improve cardiac function after MI. Brum et al showed that chronic hypersympathetic conditions are associated with pathophysiologic mechanisms involved in heart failure progression.56 We showed lower ISO sensitivity in tissue (pressure-volume loop) and cells (sarcomere shortening) of GSNOR−⁄− mice post MI, which supports our previous observations.53
Cell-Cycle–Dependent Versus Cell-Cycle–Independent Effects of GSNOR in the Heart
Although the capacity of GSNOR−⁄− mice to do better after MI has been previously described,21 little is known about the mechanisms underlying this effect. Lima et al,21 suggested a potential mechanistic explanation by demonstrating that, at baseline, the uninjured, normoxic GSNOR−⁄− hearts are invested with an expanded coronary vascular bed, due to the SNO-mediated stabilization of HIF1α. In addition, we now report that GSNOR deletion produces an augmented proliferative activity in regenerative coronary vessels, cardiomyocytes, MSCs, and c-Kit+ cardiac progenitors in response to MI. Furthermore, we show that GSNOR−⁄− cardiomyocytes exhibit improved Ca2+ handling homeostasis and reduced adrenergic sensitivity due to their resilience to ROS.22 Based on the findings by Puente et al,11 it is intriguing to hypothesize that such resilience to ROS may also explain the enhanced cell turnover we documented in the GSNOR−⁄− hearts. Notably, although the degree of cardiac cell proliferation may appear insufficient to account for the post-MI functional recovery observed in the GSNOR−⁄− hearts (7.4±1.7% versus 2.7±0.7% BrdU+ cardiomyocytes in the infarct zones of GSNOR−⁄− and WT mice, respectively; Figure6), 2 recent studies reported that enhancing the degree of cardiomyocyte proliferation to a similar level may efficiently trigger heart regeneration in mice.57,58 Thus, collectively, the findings presented by us and others21,22 suggest that the mechanism by which GSNOR deletion confers cardioprotection is multifaceted, ranging from regulating damage response pathways to cardiomyocyte contractility and cardiac cell-cycle reentry. However, the relative contribution of each mechanism to tissue regeneration and functional recovery in the post-MI GSNOR−⁄− hearts remains to be determined.
In summary, our study shows augmented cardiac regenerative pathways after MI in mice with a targeted deletion of GSNOR, an enzyme that governs protein nitrosylation/denitrosylation. Both cardiac precursor cell abundance and myocyte cell cycle activity are augmented in the GSNOR−⁄− hearts after MI. Thus, collectively, our findings have therapeutic implications for the treatment of heart disease because they reveal novel pathways by which nitroso–redox balance influences cardiac repair in the adult mammal.
Sources of Funding
This study was funded by National Institutes of Health (NIH) grants (awarded to Dr Hare) R01 HL107110 and R01 HL094849. Dr Hare is also supported by the NIH grants R01 HL110737, R01 HL084275, and 5UM HL113460 and by grants from the Starr Foundation and the Soffer Family Foundation. Drs Bellio and Cao are supported by awards from the American Heart Association. Dr Kulandavelu was supported by an award from the Canadian Institutes for Health Research.
Disclosures
Drs Hare and Hatzistergos disclose a relationship with Vestion that includes equity, board membership, and consulting. Vestion did not contribute funding to this study. Dr Hare discloses equity in Kardia and a grant from Biocardia.
References
- Anversa P, Kajstura J, Rota M, Leri A. Regenerating new heart with stem cells. J Clin Invest. 2013;123:62–70. doi: 10.1172/JCI63068. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Xin M, Olson EN, Bassel-Duby R. Mending broken hearts: cardiac development as a basis for adult heart regeneration and repair. Nat Rev Mol Cell Biol. 2013;14:529–541. doi: 10.1038/nrm3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbern JC, Lee RT. Cardiac stem cell therapy and the promise of heart regeneration. Cell Stem Cell. 2013;12:689–698. doi: 10.1016/j.stem.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laflamme MA, Murry CE. Heart regeneration. Nature. 2011;473:326–335. doi: 10.1038/nature10147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans SM, Yelon D, Conlon FL, Kirby ML. Myocardial lineage development. Circ Res. 2010;107:1428–1444. doi: 10.1161/CIRCRESAHA.110.227405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, Kasahara H, Rota M, Musso E, Urbanek K, Leri A, Kajstura J, Nadal-Ginard B, Anversa P. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell. 2003;114:763–776. doi: 10.1016/s0092-8674(03)00687-1. [DOI] [PubMed] [Google Scholar]
- Ellison GM, Vicinanza C, Smith AJ, Aquila I, Leone A, Waring CD, Henning BJ, Stirparo GG, Papait R, Scarfo M, Agosti V, Viglietto G, Condorelli G, Indolfi C, Ottolenghi S, Torella D, Nadal-Ginard B. Adult c-kit(pos) cardiac stem cells are necessary and sufficient for functional cardiac regeneration and repair. Cell. 2013;154:827–842. doi: 10.1016/j.cell.2013.07.039. [DOI] [PubMed] [Google Scholar]
- Hsieh PC, Segers VF, Davis ME, MacGillivray C, Gannon J, Molkentin JD, Robbins J, Lee RT. Evidence from a genetic fate-mapping study that stem cells refresh adult mammalian cardiomyocytes after injury. Nat Med. 2007;13:970–974. doi: 10.1038/nm1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, Sadek HA. Transient regenerative potential of the neonatal mouse heart. Science. 2011;331:1078–1080. doi: 10.1126/science.1200708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senyo SE, Steinhauser ML, Pizzimenti CL, Yang VK, Cai L, Wang M, Wu TD, Guerquin-Kern JL, Lechene CP, Lee RT. Mammalian heart renewal by pre-existing cardiomyocytes. Nature. 2013;493:433–436. doi: 10.1038/nature11682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puente BN, Kimura W, Muralidhar SA, Moon J, Amatruda JF, Phelps KL, Grinsfelder D, Rothermel BA, Chen R, Garcia JA, Santos CX, Thet S, Mori E, Kinter MT, Rindler PM, Zacchigna S, Mukherjee S, Chen DJ, Mahmoud AI, Giacca M, Rabinovitch PS, Aroumougame A, Shah AM, Szweda LI, Sadek HA. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell. 2014;157:565–579. doi: 10.1016/j.cell.2014.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naqvi N, Li M, Calvert JW, Tejada T, Lambert JP, Wu J, Kesteven SH, Holman SR, Matsuda T, Lovelock JD, Howard WW, Iismaa SE, Chan AY, Crawford BH, Wagner MB, Martin DI, Lefer DJ, Graham RM, Husain A. A proliferative burst during preadolescence establishes the final cardiomyocyte number. Cell. 2014;157:795–807. doi: 10.1016/j.cell.2014.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eulalio A, Mano M, Dal Ferro M, Zentilin L, Sinagra G, Zacchigna S, Giacca M. Functional screening identifies miRNAs inducing cardiac regeneration. Nature. 2012;492:376–381. doi: 10.1038/nature11739. [DOI] [PubMed] [Google Scholar]
- Hatzistergos KE, Quevedo H, Oskouei BN, Hu Q, Feigenbaum GS, Margitich IS, Mazhari R, Boyle AJ, Zambrano JP, Rodriguez JE, Dulce R, Pattany PM, Valdes D, Revilla C, Heldman AW, McNiece I, Hare JM. Bone marrow mesenchymal stem cells stimulate cardiac stem cell proliferation and differentiation. Circ Res. 2010;107:913–922. doi: 10.1161/CIRCRESAHA.110.222703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart N, Bollini S, Dube KN, Vieira JM, Zhou B, Davidson S, Yellon D, Riegler J, Price AN, Lythgoe MF, Pu WT, Riley PR. De novo cardiomyocytes from within the activated adult heart after injury. Nature. 2011;474:640–644. doi: 10.1038/nature10188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams AR, Hatzistergos KE, Addicott B, McCall F, Carvalho D, Suncion V, Morales AR, Da Silva J, Sussman MA, Heldman AW, Hare JM. Enhanced effect of combining human cardiac stem cells and bone marrow mesenchymal stem cells to reduce infarct size and to restore cardiac function after myocardial infarction. Circulation. 2013;127:213–223. doi: 10.1161/CIRCULATIONAHA.112.131110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanashiro-Takeuchi RM, Takeuchi LM, Rick FG, Dulce R, Treuer AV, Florea V, Rodrigues CO, Paulino EC, Hatzistergos KE, Selem SM, Gonzalez DR, Block NL, Schally AV, Hare JM. Activation of growth hormone releasing hormone (GHRH) receptor stimulates cardiac reverse remodeling after myocardial infarction (MI) Proc Natl Acad Sci USA. 2012;109:559–563. doi: 10.1073/pnas.1119203109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Hausladen A, Zeng M, Que L, Heitman J, Stamler JS. A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature. 2001;410:490–494. doi: 10.1038/35068596. [DOI] [PubMed] [Google Scholar]
- Hare JM, Stamler JS. NO/redox disequilibrium in the failing heart and cardiovascular system. J Clin Invest. 2005;115:509–517. doi: 10.1172/JCI200524459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldar SM, Stamler JS. S-nitrosylation: integrator of cardiovascular performance and oxygen delivery. J Clin Invest. 2013;123:101–110. doi: 10.1172/JCI62854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima B, Lam GK, Xie L, Diesen DL, Villamizar N, Nienaber J, Messina E, Bowles D, Kontos CD, Hare JM, Stamler JS, Rockman HA. Endogenous S-nitrosothiols protect against myocardial injury. Proc Natl Acad Sci USA. 2009;106:6297–6302. doi: 10.1073/pnas.0901043106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulce R, Mayo V, Rangel EB, Balkan W, Hare JM. Interaction between neuronal NOS signaling and temperature influences SR Ca2+ leak: role of nitroso-redox balance. Circ Res. 2014;116:46–55. doi: 10.1161/CIRCRESAHA.116.305172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris C, Wang SW, Lauchu JJ, Hansen JM. Methanol metabolism and embryotoxicity in rat and mouse conceptuses: comparisons of alcohol dehydrogenase (ADH1), formaldehyde dehydrogenase (ADH3), and catalase. Reprod Toxicol. 2003;17:349–357. doi: 10.1016/s0890-6238(03)00013-3. [DOI] [PubMed] [Google Scholar]
- Thompson CM, Sonawane B, Grafstrom RC. The ontogeny, distribution, and regulation of alcohol dehydrogenase 3: implications for pulmonary physiology. Drug Metab Dispos. 2009;37:1565–1571. doi: 10.1124/dmd.109.027904. [DOI] [PubMed] [Google Scholar]
- Liu L, Yan Y, Zeng M, Zhang J, Hanes MA, Ahearn G, McMahon TJ, Dickfeld T, Marshall HE, Que LG, Stamler JS. Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell. 2004;116:617–628. doi: 10.1016/s0092-8674(04)00131-x. [DOI] [PubMed] [Google Scholar]
- Wei W, Li B, Hanes MA, Kakar S, Chen X, Liu L. S-nitrosylation from GSNOR deficiency impairs DNA repair and promotes hepatocarcinogenesis. Sci Transl Med. 2010;2:19ra13. doi: 10.1126/scitranslmed.3000328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox AG, Saunders DC, Kelsey PB, Jr, Conway AA, Tesmenitsky Y, Marchini JF, Brown KK, Stamler JS, Colagiovanni DB, Rosenthal GJ, Croce KJ, North TE, Goessling W. S-nitrosothiol signaling regulates liver development and improves outcome following toxic liver injury. Cell Rep. 2014;6:56–69. doi: 10.1016/j.celrep.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oskouei BN, Lamirault G, Joseph C, Treuer AV, Landa S, Da Silva J, Hatzistergos K, Dauer M, Balkan W, McNiece I, Hare JM. Increased potency of cardiac stem cells compared with bone marrow mesenchymal stem cells in cardiac repair. Stem Cells Transl Med. 2012;1:116–124. doi: 10.5966/sctm.2011-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulce RA, Yiginer O, Gonzalez DR, Goss G, Feng N, Zheng M, Hare JM. Hydralazine and organic nitrates restore impaired excitation-contraction coupling by reducing calcium leak associated with nitroso-redox imbalance. J Biol Chem. 2013;288:6522–6533. doi: 10.1074/jbc.M112.412130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beigi F, Schmeckpeper J, Pow-Anpongkul P, Payne JA, Zhang L, Zhang Z, Huang J, Mirotsou M, Dzau VJ. C3orf58, a novel paracrine protein, stimulates cardiomyocyte cell-cycle progression through the PI3K-AKT-CDK7 pathway. Circ Res. 2013;113:372–380. doi: 10.1161/CIRCRESAHA.113.301075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanashiro-Takeuchi RM, Heidecker B, Lamirault G, Dharamsi JW, Hare JM. Sex-specific impact of aldosterone receptor antagonism on ventricular remodeling and gene expression after myocardial infarction. Clin Transl Sci. 2009;2:134–142. doi: 10.1111/j.1752-8062.2009.00094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran JM, Makarewich CA, Sharp TE, Starosta T, Zhu F, Hoffman NE, Chiba Y, Madesh M, Berretta RM, Kubo H, Houser SR. Bone-derived stem cells repair the heart after myocardial infarction through transdifferentiation and paracrine signaling mechanisms. Circ Res. 2013;113:539–552. doi: 10.1161/CIRCRESAHA.113.301202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez DR, Treuer A, Sun QA, Stamler JS, Hare JM. S-Nitrosylation of cardiac ion channels. J Cardiovasc Pharmacol. 2009;54:188–195. doi: 10.1097/FJC.0b013e3181b72c9f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loffredo FS, Steinhauser ML, Gannon J, Lee RT. Bone marrow-derived cell therapy stimulates endogenous cardiomyocyte progenitors and promotes cardiac repair. Cell Stem Cell. 2011;8:389–398. doi: 10.1016/j.stem.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolli R, Tang XL, Sanganalmath SK, Rimoldi O, Mosna F, Abdel-Latif A, Jneid H, Rota M, Leri A, Kajstura J. Intracoronary delivery of autologous cardiac stem cells improves cardiac function in a porcine model of chronic ischemic cardiomyopathy. Circulation. 2013;128:122–131. doi: 10.1161/CIRCULATIONAHA.112.001075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandolfi F, Vanelli A, Pennarossa G, Rahaman M, Acocella F, Brevini TA. Large animal models for cardiac stem cell therapies. Theriogenology. 2011;75:1416–1425. doi: 10.1016/j.theriogenology.2011.01.026. [DOI] [PubMed] [Google Scholar]
- Welt FG, Gallegos R, Connell J, Kajstura J, D’Amario D, Kwong RY, Coelho-Filho O, Shah R, Mitchell R, Leri A, Foley L, Anversa P, Pfeffer MA. Effect of cardiac stem cells on left-ventricular remodeling in a canine model of chronic myocardial infarction. Circ Heart Fail. 2013;6:99–106. doi: 10.1161/CIRCHEARTFAILURE.112.972273. [DOI] [PubMed] [Google Scholar]
- Angert D, Berretta RM, Kubo H, Zhang H, Chen X, Wang W, Ogorek B, Barbe M, Houser SR. Repair of the injured adult heart involves new myocytes potentially derived from resident cardiac stem cells. Circ Res. 2011;108:1226–1237. doi: 10.1161/CIRCRESAHA.110.239046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Wang L, Wong CC, Scott FL, Eckelman BP, Han X, Tzitzilonis C, Meng F, Gu Z, Holland EA, Clemente AT, Okamoto S, Salvesen GS, Riek R, Yates JR, III, Lipton SA. Transnitrosylation of XIAP regulates caspase-dependent neuronal cell death. Mol Cell. 2010;39:184–195. doi: 10.1016/j.molcel.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- Gomes SA, Rangel EB, Premer C, Dulce RA, Cao Y, Florea V, Balkan W, Rodrigues CO, Schally AV, Hare JM. S-nitrosoglutathione reductase (GSNOR) enhances vasculogenesis by mesenchymal stem cells. Proc Natl Acad Sci USA. 2013;110:2834–2839. doi: 10.1073/pnas.1220185110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton RL, Maddie MA, Minnillo DR, Hagg T, Whittemore SR. Griffonia simplicifolia isolectin B4 identifies a specific subpopulation of angiogenic blood vessels following contusive spinal cord injury in the adult mouse. J Comp Neurol. 2008;507:1031–1052. doi: 10.1002/cne.21570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nombela-Arrieta C, Ritz J, Silberstein LE. The elusive nature and function of mesenchymal stem cells. Nat Rev Mol Cell Biol. 2011;12:126–131. doi: 10.1038/nrm3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Gomes SA, Rangel EB, Paulino EC, Fonseca TL, Li J, Teixeira MB, Gouveia CH, Bianco AC, Kapiloff MS, Balkan W, Hare JM. S-nitrosoglutathione reductase-dependent PPARγ denitrosylation participates in MSC-derived adipogenesis and osteogenesis. J Clin Invest. 2015;125:1679–1691. doi: 10.1172/JCI73780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karantalis V, Balkan W, Schulman IH, Hatzistergos KE, Hare JM. Cell-based therapy for prevention and reversal of myocardial remodeling. Am J Physiol Heart Circ Physiol. 2012;303:H256–H270. doi: 10.1152/ajpheart.00221.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess DT, Stamler JS. Regulation by S-nitrosylation of protein post-translational modification. J Biol Chem. 2012;287:4411–4418. doi: 10.1074/jbc.R111.285742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol. 2001;3:193–197. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]
- Tang CH, Wei W, Hanes MA, Liu L. Hepatocarcinogenesis driven by GSNOR deficiency is prevented by iNOS inhibition. Cancer Res. 2013;73:2897–2904. doi: 10.1158/0008-5472.CAN-12-3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. Hypoxia-inducible factor 1 (HIF-1) pathway. Sci STKE. 2007;2007:cm8. doi: 10.1126/stke.4072007cm8. [DOI] [PubMed] [Google Scholar]
- Nogueira L, Figueiredo-Freitas C, Casimiro-Lopes G, Magdesian MH, Assreuy J, Sorenson MM. Myosin is reversibly inhibited by S-nitrosylation. Biochem J. 2009;424:221–231. doi: 10.1042/BJ20091144. [DOI] [PubMed] [Google Scholar]
- Shoji H, Takahashi S, Okabe E. Intracellular effects of nitric oxide on force production and Ca2+ sensitivity of cardiac myofilaments. Antioxid Redox Signal. 1999;1:509–521. doi: 10.1089/ars.1999.1.4-509. [DOI] [PubMed] [Google Scholar]
- Sun J, Morgan M, Shen RF, Steenbergen C, Murphy E. Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circ Res. 2007;101:1155–1163. doi: 10.1161/CIRCRESAHA.107.155879. [DOI] [PubMed] [Google Scholar]
- Beigi F, Gonzalez DR, Minhas KM, Sun QA, Foster MW, Khan SA, Treuer AV, Dulce RA, Harrison RW, Saraiva RM, Premer C, Schulman IH, Stamler JS, Hare JM. Dynamic denitrosylation via S-nitrosoglutathione reductase regulates cardiovascular function. Proc Natl Acad Sci USA. 2012;109:4314–4319. doi: 10.1073/pnas.1113319109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmet JM, Hare JM. Nitroso-redox interactions in the cardiovascular system. Circulation. 2006;114:1531–1544. doi: 10.1161/CIRCULATIONAHA.105.605519. [DOI] [PubMed] [Google Scholar]
- Saraiva RM, Minhas KM, Raju SV, Barouch LA, Pitz E, Schuleri KH, Vandegaer K, Li D, Hare JM. Deficiency of neuronal nitric oxide synthase increases mortality and cardiac remodeling after myocardial infarction: role of nitroso-redox equilibrium. Circulation. 2005;112:3415–3422. doi: 10.1161/CIRCULATIONAHA.105.557892. [DOI] [PubMed] [Google Scholar]
- Brum PC, Kosek J, Patterson A, Bernstein D, Kobilka B. Abnormal cardiac function associated with sympathetic nervous system hyperactivity in mice. Am J Physiol Heart Circ Physiol. 2002;283:H1838–H1845. doi: 10.1152/ajpheart.01063.2001. [DOI] [PubMed] [Google Scholar]
- D’Uva G, Aharonov A, Lauriola M, Kain D, Yahalom-Ronen Y, Carvalho S, Weisinger K, Bassat E, Rajchman D, Yifa O, Lysenko M, Konfino T, Hegesh J, Brenner O, Neeman M, Yarden Y, Leor J, Sarig R, Harvey RP, Tzahor E. ERBB2 triggers mammalian heart regeneration by promoting cardiomyocyte dedifferentiation and proliferation. Nat Cell Biol. 2015;17:627–638. doi: 10.1038/ncb3149. [DOI] [PubMed] [Google Scholar]
- Polizzotti BD, Ganapathy B, Walsh S, Choudhury S, Ammanamanchi N. Bennett DG, Dos Remedios CG, Haubner BJ, Penninger JM, Kuhn B. Neuregulin stimulation of cardiomyocyte regeneration in mice and human myocardium reveals a therapeutic window. Sci Transl Med. 2015;7:281ra45. doi: 10.1126/scitranslmed.aaa5171. , . . [DOI] [PMC free article] [PubMed] [Google Scholar]