

3-Hydroxy-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors or statins are a class of medications with potent effects on reducing serum cholesterol level, and currently have been widely used in the primary and secondary prevention of atherosclerotic cardiovascular diseases (ASCVD). Previously, a substantial number of clinical studies consistently demonstrated that through decreasing serum cholesterol level, especially low-density lipoprotein (LDL) cholesterol level, statins effectively reduce the incidence of cardiovascular events such as acute myocardial infarction and ischemic stroke in subjects with dyslipidemia.1–3 Nevertheless, interestingly and importantly, results from other clinical studies in subjects with normal cholesterol level suggest that statins have additional cardioprotective effects, now universally known as pleiotropic effects, which include immunomodulation, anti-inflammation and anti-oxidation, endothelium protection, and pro-angiogenesis.4–6 Results from experimental and clinical research indicate that the pleiotropic effects of statins treatment are predominantly derived from its potent effects on inhibiting Rho-GTPase isoprenylation through reducing geranyl-geranylpyrophosphate (GGPP) generation during cholesterol biosynthesis.7,8 The Rho-GTPase family includes nearly 20 members in humans, and RhoA, Rac1, and Cdc42 are the most studied members.9 They exert complex and important effects on regulating endothelial nitric oxide synthase (eNOS) expression and nitric oxide (NO) production, vascular smooth muscle cells (VSMC) migration and proliferation, reactive oxygen species (ROS) generation, inflammatory cells infiltration, and foam cells formation.10,11 Currently, based on the attractive and beneficial effects of statins therapy on maintaining vascular homeostasis as well as improving inflammatory reaction and oxidative stress, many studies have been conducted to investigate whether the pleiotropic effects of statins regimen could contribute to better outcomes in diseases such as heart failure,12 arrhythmias,9 cardiac hypertrophy,13 and diabetes mellitus.14
With regard to the accumulating evidence that demonstrates the cardioprotective effects of statins therapy in past decades and aiming to improve our understanding about the mechanisms associated with the pleiotropic effects of statins, especially its effect on modulating Rho-GTPase and its effectors, in this review we first summarize data from previous published articles and then present our views for the direction of future studies.
Isoprenoid Intermediates and Its Associated Actions
Accordingly, HMG-CoA reductase is the rate-limiting enzyme that plays key roles in regulating HMG-CoA and l-mevalonic acid conversion during cholesterol biosynthesis (Figure1). Thereafter, farnesylpyrophosphate (FPP) and GGPP are consecutively produced by adding isopentenyl pyrophosphate into its precursor. GGPP is the major component of isoprenoid intermediates, and it serves as a crucial lipid attachment for RhoA and other Rho-GTPase. With post-transcriptional modification by GGPP, RhoA activates its main downstream effector Rho-kinase (ROCK) and thereby subsequently elicits multiple cellular effects including proliferation, apoptosis, migration, contraction, secretion, and trafficking mainly through controlling actin cytoskeletal assembly.11 Briefly, RhoA/ROCK signaling pathway has binary effects on the cardiovascular system. On the 1 hand, for example, physiologically, RhoA has been found essential for the normal formation of the cardiac structure and conduction system during embryonic cardiac development.15 In addition, through regulating endothelium and VSMC, the RhoA/ROCK signaling pathway promotes nascent vessels formation and vessel stabilization during embryonic vascular development. Intriguingly, in animal models with ischemic hindlimbs, low-dose statins enhance angiogenesis while high-dose statins inhibit angiogenesis in an eNOS-dependent manner,16,17 which indirectly suggests that excessively inhibiting RhoA isoprenylation and the RhoA/ROCK signaling pathway may be harmful for maintaining vascular homeostasis. Moreover, global ROCK1 deficiency in mice leads to reduced serine 632/635 phosphorylation of insulin receptor substrate-1 and thereby causes insulin resistance,18 which may suggest that RhoA/ROCK signaling pathway exerts fundamental roles in maintaining glycemic homeostasis. However, in morbid conditions such as diabetes mellitus18 and hypertension,19 increased ROCK activity contributes to and accelerates pathological changes of the cardiovascular system. In the following sections, we will briefly address the roles that Rho-GTPase, especially the RhoA/ROCK signaling pathway, exert on atherogenesis and ASCVD development. In addition, the pleiotropic effects of statins on atherosclerosis and ASCVD will also be concurrently described.
Figure 1.
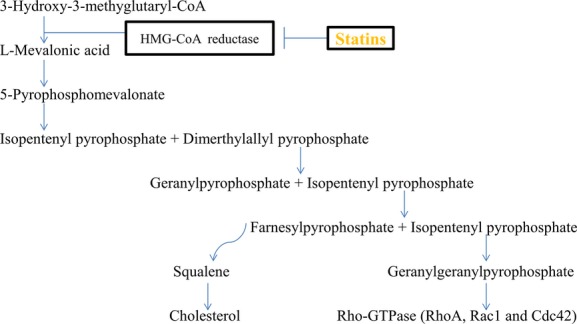
Cholesterol and isoprenoid intermediates synthesis. HMG-CoA indicates 3-hydroxy-methylglutaryl-coenzyme A.
Pharmacology of Statins
Statins were initially isolated from the metabolites of fungi20 and are a potent competitive inhibitor for the key enzyme HMG-CoA reductase during cholesterol biosynthesis. Briefly, the Ki (inhibition constant) values for the statins–enzyme complexes range between 0.1 and 2.3 nmol/L,21 whereas the Michaelis constant, Km, for HMG-CoA is 4 mmol/L.22 The underlying mechanisms ascribed to the actions of statins are broadly categorized into 2 aspects in terms of cholesterol-lowering on the 1 hand and reducing isoprenoid intermediates generation on the other hand.23 Generally, statins can be roughly divided into 2 categories (hydrophilic and lipophilic statins) owing to their differences in tissue permeability. For example, hydrophilic statins such as rosuvastatin and pravastatin are mainly targeted to hepatic tissues, while lipophilic statins such as simvastatin and atorvastatin are primarily targeted to vascular cells. Despite less potency to enter the vascular wall, hydrophilic statins still can exert cholesterol-independent pleiotropic effects, which are now considered to be due to their effects on inhibiting hepatic l-mevalonic acid generation.23 The bioavailability of statins ranges from 5% for lovastatin and simvastatin to 60% for cerivastatin.23 Both the hydrophilic and lipophilic statins excrete mainly from feces. For example, nearly 58% of simvastatin and 90% of fluvastatin undergo fecal excretion, while only 13% of simvastatin and 6% of fluvastatin excrete from urine.23 Similarly, nearly 71% of pravastatin undergoes fecal excretion and only 20% excretes from urine.23 The maximum concentrations of statins found in the blood vary broadly, with 2 ng/mL for cerivastatin to 448 ng/mL for fluvastatin.23 Overall, these differences of pharmacokinetics of statins may contribute to the somewhat different potency of each statin.
Notably, as revealed by previous research, a substantial part of the benefits statins confer to the cardiovascular system are associated with their effects on reducing the serum cholesterol level.1,3,24,25 Nevertheless, the cholesterol-independent, rapid-onset actions such as anti-inflammation and plaque stabilization of high-dose statins therapy also contribute to favorable outcomes, especially in urgent scenarios such as acute myocardial syndrome and acute ischemic stroke.26,27 Therefore, re-evaluating the pleiotropic effects and associated mechanisms of statins therapy is valuable to provide thorough evidence to help clinicians in using this armament more effectively in clinical practice and experimental research.
ROCK Isoforms and ROCK Inhibitors
Owing to the potential cardioprotective effects of ROCKs inhibitor and the potential additive benefits regarding the combined therapy of ROCKs inhibitor and statins, we herein briefly detail the pharmacology of ROCKs inhibitor as well as the isoforms of ROCKs. More information about the pharmacology of ROCKs inhibitor is available in more comprehensive reviews.28,29 Accordingly, ROCKs are generally classified into 2 types, ROCK1 and ROCK2, and they share nearly 65% homology in amino-acid sequence and ≈92% homology in the kinase domains.30 ROCK1 is predominantly expressed in lung, liver, spleen, and kidney, while ROCK2 is preferentially expressed in cardiac tissue and vascular wall.31 Generally, the ROCK isoforms have overlapping and nonredundant functions. With the animal models of haploinsufficiency of ROCK1 or ROCK2, the effects of ROCK1 and ROCK2 on the cardiovascular system can now be partially distinguished. For example, it is reported that haploinsufficient-ROCK1 mice exhibit reduced cardiac fibrosis but not hypertrophy in response to angiotensin II (AngII) stimulation.32 In contrast, cardiac hypertrophy is associated with ROCK2 as suggested by the finding that compared to wild-type mice, cardiac-specific deletion of ROCK2 prevents cardiac hypertrophy induced by transverse aortic constriction.33 In addition, with regard to arterial remodeling, ROCK1 seems to play a prominent role as revealed by the finding that neointima formation induced by carotid ligation is markedly reduced in ROCK1-haploinsufficiency mice compared to the ROCK2-haploinsufficiency counterpart,34 suggesting that ROCK1 but not ROCK2 has a more prominent role in arterial remodeling after carotid ligation. Currently, no ROCK1- or ROCK2-specific inhibitor has been developed, and fasudil and Y27632 are the 2 most-investigated inhibitors.29 Through unselectively inhibiting ROCK1 and ROCK2 activity, both fasudil and Y27632 confer cardio- and vascular-protective effects.29 In the following section, we will further detail the benefits of ROCKs inhibition by fasudil or Y27632, and discuss the potential additive benefits regarding combined therapy of statins plus ROCKs inhibitor.
Rho-GTPase and Endothelium: Effects of Statins
Endothelial dysfunction as defined by reduced bioavailability of endothelium-derived NO is the hallmark and early indicator of atherogenesis. Previously, a large number of studies revealed that ROCK activity is significantly associated with eNOS expression and NO production. For example, in in vitro study, direct antagonist of Rho by Clostridium botulinum C3 transferase35 or overexpression of a dominant-negative mutant of RhoA36 leads to increased eNOS expression. In addition, inhibiting ROCK activity by its specific antagonist fasudil37 or Y2763238 contributes to increased eNOS expression, further confirming that endothelial dysfunction is associated with the RhoA/ROCK signaling pathway. Moreover, through enhancing ROS production and thereby diminishing NO bioavailability, Rac1 activation may also lead to endothelial dysfunction.39 The mechanism is partially associated with the generation of the nonradical derivative peroxynitrite (ONOO-), which is relatively stable and has a long half-life, when NO reacts with superoxide.39 In addition, through activation of protein tyrosine phosphorylation and extracellular-signal regulated kinase, Rac1 enhances ROS production in bovine endothelial cells stimulated with shear stress.40 Moreover, depolarization of endothelium results in increased ROS production in a Rac1-dependent fashion.41 Since both the RhoA and Rac1 activation requires post-translational modification by GGPP, it is therefore probable that statins therapy could protect endothelium, and both the experimental and clinical research support this notion. For example, Laufs et al report that simvastatin enhances eNOS expression in human saphenous vein endothelial cells treated with oxidized LDL (ox-LDL). Also, nuclear run-on assays and transient transfection studies with a −1.6-kb eNOS promoter construct indicate that simvastatin does not affect eNOS gene transcription,42 which suggests that statins-increased eNOS expression is due to a post-translational mechanism. In subjects with stable atherosclerosis, 10 mg of rosuvastatin or 40 mg of atorvastatin daily results in similar improvement in flow-mediated dilation as well as attenuation in ROCK activity in a cholesterol-independent manner.43 Moreover, as compared to low-dose simvastatin plus ezetimibe, high-dose simvastatin exerts greater effects on attenuating ROCK activity and improving endothelial function, despite a comparable LDL cholesterol level being achieved by either regimen.44 These results compellingly support the concept that endothelium protection by statins therapy is beyond cholesterol-lowering, but significantly dependent upon Rho-GTPase.
Inducible nitric oxide synthase is also involved in endothelial dysfunction,45 and through inhibiting inducible nitric oxide synthase expression may be one of the mechanisms contributing to statins’ pleiotropic effects. For example, Wagner et al report that atorvastatin reduces inducible nitric oxide synthase expression in native endothelial cells in situ and the underlying mechanism is associated with inhibition of tumor necrosis factor-α and interferon-γ expressions.46 Furthermore, through normalization of eNOS/inducible nitric oxide synthase ratio, atorvastatin improves age-related endothelial dysfunction as reported by Gong and colleagues.47
Other than increasing eNOS expression, enhancing eNOS phosphorylation is another important mechanism by which statins render their pleiotropic effects. For example, Kureishi et al report that in normocholesterolemic animals, serine-threonine protein kinase Akt activated by simvastatin therapy enhances eNOS phosphorylation and thereby promotes angiogenesis.48 In addition, results from our previous research indicate that in rats with ischemia injury, atorvastatin enhances cardiac eNOS and p-eNOS expression in a PI3K/Akt-dependent manner.49 Promoting endothelial progenitor cells (EPC) migration and thereby replenishing injured endothelium is another mechanism by which statins therapy confers benefits to the vascular system,50,51 and this effect is also dependent upon the activation of the PI3K/Akt signaling pathway. However, whether Rho-GTPase is involved in these processes is not yet fully investigated.
Endothelial activation as defined by increased endothelial expression of cell-surface adhesion molecules such as intracellular adhesion molecule (ICAM), vascular cell adhesion molecule (VCAM), and E-selectin in response to a variety of stimuli also contributes to vascular inflammation and atherosclerosis. Decreased adhesion molecule expression leads to decreased leukocyte infiltration into the artery wall. Therefore, through promoting NO production, statins should play important roles in ameliorating vascular inflammation. Since there is a close cross-talk between endothelial dysfunction and endothelial activation, it is possible that Rho-GTPase is involved in endothelial activation. Indeed, previous research suggests that increased NO production by statins therapy leads to reduced ICAM, VCAM, and E-selectin expressions.52,53 Furthermore, Seye et al report that dominant-negative RhoA inhibits UTP-induced VCAM expression in coronary artery endothelium,54 and dominant-negative Rac1 attenuates ICAM expression induced by tumor necrosis factor-α in aortic endothelium.55 Results from our previous research also suggest that in rats with ischemia, atorvastatin therapy diminishes cardiac VCAM expression and inflammatory cells infiltration.56 These data consistently demonstrate that through regulating Rho-GTPase, statins diminish endothelial activation and ameliorate vascular inflammation.
Taken together, through improving endothelial function and attenuating endothelial activation, statins exert great effects on endothelium protection and anti-inflammation, and the mechanisms operating in these processes are at least partially attributable to RhoA/ROCK and Rac1 modulation. Since the RhoA/ROCK signaling pathway is significantly associated with endothelial dysfunction and activation, it is clinically relevant and warranted to investigate whether statins combined with ROCK inhibitor therapy could confer additive benefits for the vascular system.
Rho-GTPase and VSMC: Effects of Statins
VSMC is the key component of the vascular wall and critical for arteriogenesis in physiological conditions. Nonetheless, in past decades, experimental and clinical research indicate that VSMC proliferation and migration leads to stent restenosis and venous graft occlusion,57–59 and a substantial part of the adverse effects are associated with Rho-GTPase. For example, Laufs et al report that inhibition of platelet-derived growth factor–induced DNA synthesis in VSMC is reversed by GGPP but not cholesterol, and the mechanism is associated with downregulation of the cyclin-dependent kinase inhibitor p27Kip1.60 In rats with balloon-injured arteries, increased ROCK activity contributes to neointimal formation and these detrimental effects are significantly suppressed by the ROCK-specific antagonist Y27632.61 Moreover, increased ROCK activity induced by AngII in murine VSMC is substantially attenuated by exogenous NO in both in vitro and in vivo studies.62 Overall, these data strongly suggest that the RhoA/ROCK signaling pathway is implicated in VSMC proliferation as well as involved in the pathogenesis of vascular proliferative disease. Indeed, attenuating ROCK activity results in inhibiting VSMC proliferation and reducing the incidence of stent restenosis. For example, simvastatin therapy significantly attenuates VSMC proliferation by preventing Rho-GTPase-induced downregulation of p27Kip1.60 In LDL receptor-deficient mice with experimental angioplasty, simvastatin therapy reduces neointimal thickening beyond cholesterol-lowering,63 and atorvastatin treatment inhibits interleukin-18-induced human coronary artery VSMC migration.58 Other than statins, inhibiting ROCK activity by fasudil or through gene transfer of a dominant-negative mutant of ROCK also exerts great effects on inhibiting VSMC migration and reducing the incidence of vascular proliferative disease in experimental models.61,34,64 Taken together, these data consistently reveal that VSMC proliferation and migration mediated by Rho-GTPase is a key mechanism contributing to vascular proliferative diseases, and statins combined with ROCK antagonist therapy should confer synergistic effects on preventing stent restenosis.
In addition to regulating VSMC proliferation and migration, Rho-GTPase also plays central roles in VSMC contractility,65 differentiation,66 and ROS generation of VSMC.67 For example, through Ca2+-sensitizing effect of vasoconstrictor, the RhoA/ROCK signaling pathway phosphorylates myosin light-chain phosphatase and thereby contributes to VSMC constriction and blood pressure elevation.65 Indeed, in an in vivo animal study, inhibition of ROCK activity by Y2763265 or fasudil68 lowers blood pressure. Moreover, in an in vitro study, through Arhgef1, a RhoA guanine exchange factor, AngII activates RhoA and thereby leads to VSMC constriction.69 Collectively, both in vitro and in vivo studies consistently support the notion that RhoA/ROCK signaling is involved in VSMC constriction and blood pressure elevation. Although both ROCK1 and ROCK2 are expressed in VSMC, however, compared to ROCK1, ROCK2 plays profound roles in VSMC contractility.70 Other than ROCK inhibitor, statins have also been found beneficial for reducing VSCM constriction. Li et al report that through inhibiting myocardin gene expression, atorvastatin inhibits VSMC constriction, and this mechanism is dependent on RhoA/ROCK signaling pathway modulation.71 In contrast to RhoA, the effects of Rac1 on VSMC constriction remain unclear. For example, Rac1 and its effector Pak1 has been found beneficial for decreasing VSMC constriction through reducing myosin light chain phosphorylation.72 In contrast, coupling with Pak3, Rac1 enhances VSMC Ca2+ sensitivity and contractility through phosphorylation of the thin filament regulatory protein caldesmon.73 Regarding VSMC differentiation, through modulating serum response factor–dependent skeletal and smooth muscle gene expression, the RhoA/ROCK signaling pathway regulates VSMC differentiation.66 Results from Mack and colleagues also indicate that RhoA/ROCK signaling may serve as a convergence point for the multiple signaling pathways that regulate VSMC differentiation.74 With regard to ROS generation of VSMC, Rho-GTPase also exerts crucial roles, and statins therapy may confer protective effects on VSMC through decreasing ROS production. For example, Wassmann et al report that inhibition of Rac1 isoprenylation by atorvastatin therapy reduces AngII-mediated free-radical production in VSMC.67 Taken together, through different mechanisms, Rho-GTPase plays complex roles in VSMC, and statins combined with ROCK inhibitor should have additive effects on VSMC and the vascular wall.
Rho-GTPase and Inflammatory Cells: Effects of Statins
It is known that atherosclerosis is a chronic inflammatory disease characterized by inflammatory cells such as macrophages that accumulate in the vascular wall and thereby elicit a cascade of inflammatory reaction.75 Endothelial activation plays fundamental roles in the initiation of vascular inflammation. Briefly, as mentioned before, after activation by stimuli, endothelium increases the expressions of VCAM, ICAM, and E-selectin and thereby promotes leukocytes–endothelium adhesion. Other than exerting roles in augmenting these cell-surface adhesion molecules expressions, Rho-GTPase such as RhoA and Rac1 additionally play pivotal roles in promoting inflammatory cells transendothelium migration, and the mechanisms are partially associated with their effects on regulating cell morphology, adhesion, and motility. For example, Rho mediates endothelial stress fiber formation in response to a variety of stimuli such as tumor necrosis factor-α76 and shear stress,77 and thereby increases endothelial permeability, which is ready for inflammatory cells infiltration and accumulation. Through regulating the clustering of monocyte-binding receptors ICAM, VCAM, and E-selectin in human endothelium, Rho promotes monocytes transendothelium migration.78 Moreover, activated by shear stress, Rac1 mediates endothelial cytoskeletal reorganization and enhances VCAM expression, which thereby recruits leukocytes into atherosclerotic plaque.79 Overall, these data suggest that Rho-GTPase exerts fundamental roles in enhancing leukocytes–endothelium adhesion and leukocytes infiltration, and statins may have a role in diminishing inflammatory cells infiltration and ameliorating vascular inflammation. Indeed, results from our previous research support this notion.56 We observe that in rats with acute myocardial infarction, atorvastatin therapy significantly reduces cardiac VCAM expression as well as leukocytes infiltration, which is beneficial for improving the cardiac microenvironment and cardiac function. Moreover, Zheng et al report that pitavastatin and atorvastatin suppress apolipoprotein CIII–induced VCAM expression and thereby reduce monocytes adhesion in an in vitro and in vivo study,80 which strongly suggests that statins have an important role in ameliorating vascular inflammation. In addition, De Caterina and colleagues report that endothelial VCAM expression induced by interleukin-1 could be suppressed by NO, which indirectly suggests that statins therapy should be beneficial for endothelium protection and anti-inflammation.53
Other than regulating transendothelium migration, the RhoA/ROCK signaling pathway also plays complex roles in biological functions of inflammatory cells. For example, Mallat and colleagues report that Y27632 limits early atherosclerotic plaque development, and the underlying mechanism is associated with the inhibition of T-lymphocyte proliferation through blocking the RhoA/ROCK signaling pathway.81 In addition, in LDL-receptor knock-out mice, ROCK1-deficient macrophages exhibit impaired chemotaxis to monocyte chemotactic protein-1 and appear to have reduced potential to uptake lipids and to develop into foam cells when exposed to modified low-density lipoprotein.82 These findings indicate that ROCK1 should play fundamental roles in pro-atherosclerotic features of macrophages. As the RhoA/ROCK signaling pathway, Rac1 activation is also essential for macrophage movement through modulating actin cytoskeleton reorganization.83 For example, Ridley reported that macrophage migration triggered by monocyte chemotactic protein-1 is dependent on the Rac1/Pak1 signaling pathway,84 and Pak1-deficient macrophages spread more rapidly and have more lamellipodia than wild-type cells,85 which suggests that the Rac1/Pak1 signaling pathway exerts a central role in lamellipodial stability and macrophage motility. Owing to its potent effects on inhibiting RhoA and Rac1 isoprenylation, statins should also have a role in regulating inflammatory cells biological functions. Indeed, some previous studies have revealed the benefits of statins therapy in modulating inflammatory cells biological functions. For example, Kwak and coworkers report that statins suppress major histocompatibility complex class II (MHC-II) expression on monocyte-macrophages and thereby inhibit MHC-II mediated T-cell activation.86 Moreover, as reported by Senokuchi et al, by inactivation of the small G protein-p38 MAPK pathway, cerivastatin and simvastatin suppress oxidized LDL -induced macrophage proliferation.87 Moreover, Argmann et al report that atorvastatin treatment inhibits cholesteryl ester accumulation in macrophages and thereby reduces foam cell formation through a RhoA-ABCA1-mediated mechanism.88
Taken together, with regard to the adverse effects inflammatory cells exert on the vascular wall and the profound benefits statins confer to inhibiting endothelial activation, it is convincing that statins therapy could prevent atherosclerosis development beyond cholesterol lowering. Moreover, it is highly possible that added ROCKs inhibitor should provide additive benefit. However, whether excessively inhibiting leukocytes transendothelium migration by statins or ROCKs inhibitor therapy will result in diminishment of biological functions of inflammatory cells such as engulfing pathogens is unknown and deserves further investigation.
Rho-GTPase and Platelet: Effects of Statins
Platelet activation and aggregation in response to a variety of stimuli such as atherosclerotic plaque fissure or rupture is the key step in the initiation and aggravation of coronary artery thrombus formation. Briefly, these abnormalities are mainly associated with increased thromboxane A2 synthesis and release, reduced platelet-derived NO production, and increase in the ratio of cholesterol-to-phospholipid within platelet.89,90 Owing to the effects of Rho-GTPase on NO production and ROS generation, it is possible that Rho-GTPase may contribute to platelet activation and aggregation.89,90 Indeed, data from previous research support this concept. For example, Kaneider et al report that through inhibiting Rho-GTPase, statins reduce platelet ADP and ATP release and the consecutive augmentation of neutrophil oxygen–free radical release.91 In addition, results from Aslan et al show that Rho-GTPase plays a central role in regulating platelet activation, lamellipodia formation, and aggregate formation under shear stress, and these effects are associated with Pak in coordination with platelet collagen receptor glycoprotein receptor VI.92 Moreover, in an in vivo study, Iida et al report that ROCK regulates thromboxane A2–induced human platelet activation independently of p38 MAPK pathway, and these effects are blocked by fasudil and Y27632.93 Importantly, other than through modulating the Rho-GTPase signaling pathway, statins are capable of regulating platelet activation and aggregation in a cholesterol-dependent manner. For example, Opper et al report that hypercholesterolemia is associated with increase in platelet reactivity.94 Since increased cholesterol-to-phospholipid ratio contributes to increased platelet activation, it is therefore probable that reducing the serum cholesterol level could result in decreased platelet reactivity. However, as such, we speculate whether excessively inhibiting platelet activity either by blocking Rho-GTPase signaling pathway or by decreasing serum cholesterol level would lead to increased risk of bleeding, and this merits further investigation.
Conclusions
In our brief review, we have described recent progress in understanding the roles Rho-GTPase plays in atherosclerosis and ASCVD, with an emphasis on aspects regarding the pleiotropic effects of statins therapy on endothelium protection, counter-proliferation of VSMC, anti-inflammation and anti-oxidation, and inhibition of platelet-activation that are clinically relevant (Figure2). In addition, we have detailed the potential additive benefits relevant to the combined therapy of statins plus ROCK antagonist that should be beneficial for reducing cardiovascular risk. Medications that reduce ROCK activity have been developed and are currently being tested. Hopefully, in the near future, these medications could be useful and helpful in the treatment of ASCVD.
Figure 2.
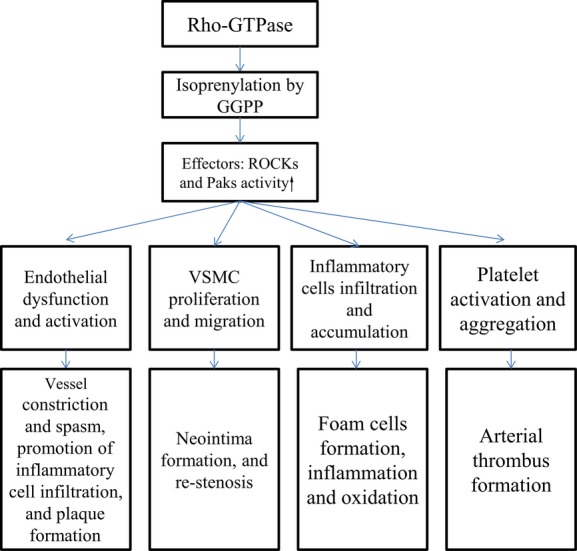
Rho-GTPase signaling pathway and its associated actions. GGPP indicates geranyl-geranylpyrophosphate; ROCK, Rho-kinase; VSMC, vascular smooth muscle cells.
Sources of Funding
This work was supported by grants from National Natural Science Foundation of China (81470571).
Disclosures
None.
References
- Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S) Lancet. 1994;344:1383–1389. [PubMed] [Google Scholar]
- Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels: the Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. N Engl J Med. 1998;339:1349–1357. doi: 10.1056/NEJM199811053391902. [DOI] [PubMed] [Google Scholar]
- Shepherd J, Cobbe SM, Ford I, Isles CG, Lorimer AR, MacFarlane PW, McKillop JH, Packard CJ. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia: West of Scotland Coronary Prevention Study Group. N Engl J Med. 1995;333:1301–1307. doi: 10.1056/NEJM199511163332001. [DOI] [PubMed] [Google Scholar]
- MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet. 2002;360:7–22. doi: 10.1016/S0140-6736(02)09327-3. [DOI] [PubMed] [Google Scholar]
- Sacks FM, Pfeffer MA, Moye LA, Rouleau JL, Rutherford JD, Cole TG, Brown L, Warnica JW, Arnold JM, Wun CC, Davis BR, Braunwald E. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels: Cholesterol and Recurrent Events Trial investigators. N Engl J Med. 1996;335:1001–1009. doi: 10.1056/NEJM199610033351401. [DOI] [PubMed] [Google Scholar]
- Cannon CP, Braunwald E, McCabe CH, Rader DJ, Rouleau JL, Belder R, Joyal SV, Hill KA, Pfeffer MA, Skene AM. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med. 2004;350:1495–1504. doi: 10.1056/NEJMoa040583. [DOI] [PubMed] [Google Scholar]
- Veillard NR, Braunersreuther V, Arnaud C, Burger F, Pelli G, Steffens S, Mach F. Simvastatin modulates chemokine and chemokine receptor expression by geranylgeranyl isoprenoid pathway in human endothelial cells and macrophages. Atherosclerosis. 2006;188:51–58. doi: 10.1016/j.atherosclerosis.2005.10.015. [DOI] [PubMed] [Google Scholar]
- Baba TT, Ohara-Nemoto Y, Miyazaki T, Nemoto TK. Involvement of geranylgeranylation of Rho and Rac GTPases in adipogenic and RANKL expression, which was inhibited by simvastatin. Cell Biochem Funct. 2013;31:652–659. doi: 10.1002/cbf.2951. [DOI] [PubMed] [Google Scholar]
- Loirand G, Sauzeau V, Pacaud P. Small G proteins in the cardiovascular system: physiological and pathological aspects. Physiol Rev. 2013;93:1659–1720. doi: 10.1152/physrev.00021.2012. [DOI] [PubMed] [Google Scholar]
- Satoh K, Fukumoto Y, Shimokawa H. Rho-kinase: important new therapeutic target in cardiovascular diseases. Am J Physiol Heart Circ Physiol. 2011;301:H287–H296. doi: 10.1152/ajpheart.00327.2011. [DOI] [PubMed] [Google Scholar]
- Shimokawa H, Takeshita A. Rho-kinase is an important therapeutic target in cardiovascular medicine. Arterioscler Thromb Vasc Biol. 2005;25:1767–1775. doi: 10.1161/01.ATV.0000176193.83629.c8. [DOI] [PubMed] [Google Scholar]
- Tousoulis D, Oikonomou E, Siasos G, Stefanadis C. Statins in heart failure—with preserved and reduced ejection fraction: an update. Pharmacol Ther. 2014;141:79–91. doi: 10.1016/j.pharmthera.2013.09.001. [DOI] [PubMed] [Google Scholar]
- Qin YW, Ye P, He JQ, Sheng L, Wang LY, Du J. Simvastatin inhibited cardiac hypertrophy and fibrosis in apolipoprotein E-deficient mice fed a “Western-style diet” by increasing PPAR alpha and gamma expression and reducing TC, MMP-9, and Cat S levels. Acta Pharmacol Sin. 2010;31:1350–1358. doi: 10.1038/aps.2010.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brault M, Ray J, Gomez YH, Mantzoros CS, Daskalopoulou SS. Statin treatment and new-onset diabetes: a review of proposed mechanisms. Metabolism. 2014;63:735–745. doi: 10.1016/j.metabol.2014.02.014. [DOI] [PubMed] [Google Scholar]
- Kaarbo M, Crane DI, Murrell WG. RhoA is highly up-regulated in the process of early heart development of the chick and important for normal embryogenesis. Dev Dyn. 2003;227:35–47. doi: 10.1002/dvdy.10283. [DOI] [PubMed] [Google Scholar]
- Weis M, Heeschen C, Glassford AJ, Cooke JP. Statins have biphasic effects on angiogenesis. Circulation. 2002;105:739–745. doi: 10.1161/hc0602.103393. [DOI] [PubMed] [Google Scholar]
- Sata M, Nishimatsu H, Suzuki E, Sugiura S, Yoshizumi M, Ouchi Y, Hirata Y, Nagai R. Endothelial nitric oxide synthase is essential for the HMG-CoA reductase inhibitor cerivastatin to promote collateral growth in response to ischemia. FASEB J. 2001;15:2530–2532. doi: 10.1096/fj.01-0415fje. [DOI] [PubMed] [Google Scholar]
- Lee DH, Shi J, Jeoung NH, Kim MS, Zabolotny JM, Lee SW, White MF, Wei L, Kim YB. Targeted disruption of ROCK1 causes insulin resistance in vivo. J Biol Chem. 2009;284:11776–11780. doi: 10.1074/jbc.C900014200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mita S, Kobayashi N, Yoshida K, Nakano S, Matsuoka H. Cardioprotective mechanisms of Rho-kinase inhibition associated with eNOS and oxidative stress-LOX-1 pathway in Dahl salt-sensitive hypertensive rats. J Hypertens. 2005;23:87–96. doi: 10.1097/00004872-200501000-00017. [DOI] [PubMed] [Google Scholar]
- Alberts AW, Chen J, Kuron G, Hunt V, Huff J, Hoffman C, Rothrock J, Lopez M, Joshua H, Harris E, Patchett A, Monaghan R, Currie S, Stapley E, Albers-Schonberg G, Hensens O, Hirshfield J, Hoogsteen K, Liesch J, Springer J. Mevinolin: a highly potent competitive inhibitor of hydroxymethylglutaryl-coenzyme A reductase and a cholesterol-lowering agent. Proc Natl Acad Sci USA. 1980;77:3957–3961. doi: 10.1073/pnas.77.7.3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsini A, Maggi FM, Catapano AL. Pharmacology of competitive inhibitors of HMG-CoA reductase. Pharmacol Res. 1995;31:9–27. doi: 10.1016/1043-6618(95)80042-5. [DOI] [PubMed] [Google Scholar]
- Bischoff KM, Rodwell VW. Biosynthesis and characterization of (S)-and (R)-3-hydroxy-3-methylglutaryl coenzyme A. Biochem Med Metab Biol. 1992;48:149–158. doi: 10.1016/0885-4505(92)90060-c. [DOI] [PubMed] [Google Scholar]
- Corsini A, Bellosta S, Baetta R, Fumagalli R, Paoletti R, Bernini F. New insights into the pharmacodynamic and pharmacokinetic properties of statins. Pharmacol Ther. 1999;84:413–428. doi: 10.1016/s0163-7258(99)00045-5. [DOI] [PubMed] [Google Scholar]
- Cannon CP, Steinberg BA, Murphy SA, Mega JL, Braunwald E. Meta-analysis of cardiovascular outcomes trials comparing intensive versus moderate statin therapy. J Am Coll Cardiol. 2006;48:438–445. doi: 10.1016/j.jacc.2006.04.070. [DOI] [PubMed] [Google Scholar]
- Ray KK, Seshasai SR, Erqou S, Sever P, Jukema JW, Ford I, Sattar N. Statins and all-cause mortality in high-risk primary prevention: a meta-analysis of 11 randomized controlled trials involving 65,229 participants. Arch Intern Med. 2010;170:1024–1031. doi: 10.1001/archinternmed.2010.182. [DOI] [PubMed] [Google Scholar]
- Waters DD, Schwartz GG, Olsson AG, Zeiher A, Oliver MF, Ganz P, Ezekowitz M, Chaitman BR, Leslie SJ, Stern T. Effects of atorvastatin on stroke in patients with unstable angina or non-Q-wave myocardial infarction: a Myocardial Ischemia Reduction with Aggressive Cholesterol Lowering (MIRACL) substudy. Circulation. 2002;106:1690–1695. doi: 10.1161/01.cir.0000031568.40630.1c. [DOI] [PubMed] [Google Scholar]
- Schwartz GG, Olsson AG, Ezekowitz MD, Ganz P, Oliver MF, Waters D, Zeiher A, Chaitman BR, Leslie S, Stern T. Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes: the MIRACL study: a randomized controlled trial. JAMA. 2001;285:1711–1718. doi: 10.1001/jama.285.13.1711. [DOI] [PubMed] [Google Scholar]
- Dong M, Yan BP, Liao JK, Lam YY, Yip GW, Yu CM. Rho-kinase inhibition: a novel therapeutic target for the treatment of cardiovascular diseases. Drug Discov Today. 2010;15:622–629. doi: 10.1016/j.drudis.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimokawa H, Rashid M. Development of Rho-kinase inhibitors for cardiovascular medicine. Trends Pharmacol Sci. 2007;28:296–302. doi: 10.1016/j.tips.2007.04.006. [DOI] [PubMed] [Google Scholar]
- Nakagawa O, Fujisawa K, Ishizaki T, Saito Y, Nakao K, Narumiya S. ROCK-I and ROCK-II, two isoforms of Rho-associated coiled-coil forming protein serine/threonine kinase in mice. FEBS Lett. 1996;392:189–193. doi: 10.1016/0014-5793(96)00811-3. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Liao JK. Rho kinase: an important mediator of atherosclerosis and vascular disease. Curr Pharm Des. 2009;15:3108–3115. doi: 10.2174/138161209789057986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rikitake Y, Oyama N, Wang CY, Noma K, Satoh M, Kim HH, Liao JK. Decreased perivascular fibrosis but not cardiac hypertrophy in ROCK1+/− haploinsufficient mice. Circulation. 2005;112:2959–2965. doi: 10.1161/CIRCULATIONAHA.105.584623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto R, Li Y, Noma K, Hiroi Y, Liu PY, Taniguchi M, Ito M, Liao JK. FHL2 prevents cardiac hypertrophy in mice with cardiac-specific deletion of ROCK2. FASEB J. 2013;27:1439–1449. doi: 10.1096/fj.12-217018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noma K, Rikitake Y, Oyama N, Yan G, Alcaide P, Liu PY, Wang H, Ahl D, Sawada N, Okamoto R, Hiroi Y, Shimizu K, Luscinskas FW, Sun J, Liao JK. ROCK1 mediates leukocyte recruitment and neointima formation following vascular injury. J Clin Invest. 2008;118:1632–1644. doi: 10.1172/JCI29226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laufs U, Liao JK. Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. J Biol Chem. 1998;273:24266–24271. doi: 10.1074/jbc.273.37.24266. [DOI] [PubMed] [Google Scholar]
- Takemoto M, Sun J, Hiroki J, Shimokawa H, Liao JK. Rho-kinase mediates hypoxia-induced downregulation of endothelial nitric oxide synthase. Circulation. 2002;106:57–62. doi: 10.1161/01.cir.0000020682.73694.ab. [DOI] [PubMed] [Google Scholar]
- Nohria A, Grunert ME, Rikitake Y, Noma K, Prsic A, Ganz P, Liao JK, Creager MA. Rho kinase inhibition improves endothelial function in human subjects with coronary artery disease. Circ Res. 2006;99:1426–1432. doi: 10.1161/01.RES.0000251668.39526.c7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto M, Nakayama M, Goto TM, Amano M, Komori K, Kaibuchi K. Rho-kinase phosphorylates eNOS at threonine 495 in endothelial cells. Biochem Biophys Res Commun. 2007;361:462–467. doi: 10.1016/j.bbrc.2007.07.030. [DOI] [PubMed] [Google Scholar]
- Hordijk PL. Regulation of NADPH oxidases: the role of Rac proteins. Circ Res. 2006;98:453–462. doi: 10.1161/01.RES.0000204727.46710.5e. [DOI] [PubMed] [Google Scholar]
- Yeh LH, Park YJ, Hansalia RJ, Ahmed IS, Deshpande SS, Goldschmidt-Clermont PJ, Irani K, Alevriadou BR. Shear-induced tyrosine phosphorylation in endothelial cells requires Rac1-dependent production of ROS. Am J Physiol. 1999;276:C838–C847. doi: 10.1152/ajpcell.1999.276.4.C838. [DOI] [PubMed] [Google Scholar]
- Sohn HY, Keller M, Gloe T, Morawietz H, Rueckschloss U, Pohl U. The small G-protein Rac mediates depolarization-induced superoxide formation in human endothelial cells. J Biol Chem. 2000;275:18745–18750. doi: 10.1074/jbc.M000026200. [DOI] [PubMed] [Google Scholar]
- Laufs U, La Fata V, Plutzky J, Liao JK. Upregulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitors. Circulation. 1998;97:1129–1135. doi: 10.1161/01.cir.97.12.1129. [DOI] [PubMed] [Google Scholar]
- Rawlings R, Nohria A, Liu PY, Donnelly J, Creager MA, Ganz P, Selwyn A, Liao JK. Comparison of effects of rosuvastatin (10 mg) versus atorvastatin (40 mg) on rho kinase activity in Caucasian men with a previous atherosclerotic event. Am J Cardiol. 2009;103:437–441. doi: 10.1016/j.amjcard.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu PY, Liu YW, Lin LJ, Chen JH, Liao JK. Evidence for statin pleiotropy in humans: differential effects of statins and ezetimibe on rho-associated coiled-coil containing protein kinase activity, endothelial function, and inflammation. Circulation. 2009;119:131–138. doi: 10.1161/CIRCULATIONAHA.108.813311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SP, Bolli R. The ubiquitous role of nitric oxide in cardioprotection. J Mol Cell Cardiol. 2006;40:16–23. doi: 10.1016/j.yjmcc.2005.09.011. [DOI] [PubMed] [Google Scholar]
- Wagner AH, Schwabe O, Hecker M. Atorvastatin inhibition of cytokine-inducible nitric oxide synthase expression in native endothelial cells in situ. Br J Pharmacol. 2002;136:143–149. doi: 10.1038/sj.bjp.0704678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong X, Ma Y, Ruan Y, Fu G, Wu S. Long-term atorvastatin improves age-related endothelial dysfunction by ameliorating oxidative stress and normalizing eNOS/iNOS imbalance in rat aorta. Exp Gerontol. 2014;52:9–17. doi: 10.1016/j.exger.2014.01.015. [DOI] [PubMed] [Google Scholar]
- Kureishi Y, Luo Z, Shiojima I, Bialik A, Fulton D, Lefer DJ, Sessa WC, Walsh K. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat Med. 2000;6:1004–1010. doi: 10.1038/79510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu R, Cai A, Dong Y, Zhou Y, Yu D, Huang Y, Zheng D, Rao S, Feng Y, Mai W. SDF-1alpha upregulation by atorvastatin in rats with acute myocardial infarction via nitric oxide production confers anti-inflammatory and anti-apoptotic effects. J Biomed Sci. 2012;19:99. doi: 10.1186/1423-0127-19-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Cheng M, Liao YH, Hu Y, Wu M, Wang Q, Qin B, Wang H, Zhu Y, Gao XM, Goukassian D, Zhao TC, Tang YL, Kishore R, Qin G. Rosuvastatin enhances angiogenesis via eNOS-dependent mobilization of endothelial progenitor cells. PLoS One. 2013;8:e63126. doi: 10.1371/journal.pone.0063126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao XM, Wu CF, Lu GP. Atorvastatin inhibits homocysteine-induced dysfunction and apoptosis in endothelial progenitor cells. Acta Pharmacol Sin. 2010;31:476–484. doi: 10.1038/aps.2010.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenpeck KL, Gauthier TW, Lefer AM. Inhibition of endothelial-derived nitric oxide promotes P-selectin expression and actions in the rat microcirculation. Gastroenterology. 1994;107:1050–1058. doi: 10.1016/0016-5085(94)90229-1. [DOI] [PubMed] [Google Scholar]
- De Caterina R, Libby P, Peng HB, Thannickal VJ, Rajavashisth TB, Gimbrone MA, Jr, Shin WS, Liao JK. Nitric oxide decreases cytokine-induced endothelial activation: nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J Clin Invest. 1995;96:60–68. doi: 10.1172/JCI118074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seye CI, Yu N, Gonzalez FA, Erb L, Weisman GA. The P2Y2 nucleotide receptor mediates vascular cell adhesion molecule-1 expression through interaction with VEGF receptor-2 (KDR/Flk-1) J Biol Chem. 2004;279:35679–35686. doi: 10.1074/jbc.M401799200. [DOI] [PubMed] [Google Scholar]
- Chen XL, Zhang Q, Zhao R, Ding X, Tummala PE, Medford RM. Rac1 and superoxide are required for the expression of cell adhesion molecules induced by tumor necrosis factor-alpha in endothelial cells. J Pharmacol Exp Ther. 2003;305:573–580. doi: 10.1124/jpet.102.047894. [DOI] [PubMed] [Google Scholar]
- Cai A, Zheng D, Dong Y, Qiu R, Huang Y, Song Y, Jiang Z, Rao S, Liao X, Kuang J, Dai G, Mai W. Efficacy of atorvastatin combined with adipose-derived mesenchymal stem cell transplantation on cardiac function in rats with acute myocardial infarction. Acta Biochim Biophys Sin (Shanghai) 2011;43:857–866. doi: 10.1093/abbs/gmr087. [DOI] [PubMed] [Google Scholar]
- Braun-Dullaeus RC, Mann MJ, Dzau VJ. Cell cycle progression: new therapeutic target for vascular proliferative disease. Circulation. 1998;98:82–89. doi: 10.1161/01.cir.98.1.82. [DOI] [PubMed] [Google Scholar]
- Chandrasekar B, Mummidi S, Mahimainathan L, Patel DN, Bailey SR, Imam SZ, Greene WC, Valente AJ. Interleukin-18-induced human coronary artery smooth muscle cell migration is dependent on NF-kappaB- and AP-1-mediated matrix metalloproteinase-9 expression and is inhibited by atorvastatin. J Biol Chem. 2006;281:15099–15109. doi: 10.1074/jbc.M600200200. [DOI] [PubMed] [Google Scholar]
- Shimizu K, Aikawa M, Takayama K, Libby P, Mitchell RN. Direct anti-inflammatory mechanisms contribute to attenuation of experimental allograft arteriosclerosis by statins. Circulation. 2003;108:2113–2120. doi: 10.1161/01.CIR.0000092949.67153.74. [DOI] [PubMed] [Google Scholar]
- Laufs U, Marra D, Node K, Liao JK. 3-Hydroxy-3-methylglutaryl-CoA reductase inhibitors attenuate vascular smooth muscle proliferation by preventing rho GTPase-induced down-regulation of p27(Kip1) J Biol Chem. 1999;274:21926–21931. doi: 10.1074/jbc.274.31.21926. [DOI] [PubMed] [Google Scholar]
- Sawada N, Itoh H, Ueyama K, Yamashita J, Doi K, Chun TH, Inoue M, Masatsugu K, Saito T, Fukunaga Y, Sakaguchi S, Arai H, Ohno N, Komeda M, Nakao K. Inhibition of rho-associated kinase results in suppression of neointimal formation of balloon-injured arteries. Circulation. 2000;101:2030–2033. doi: 10.1161/01.cir.101.17.2030. [DOI] [PubMed] [Google Scholar]
- Maruhashi T, Noma K, Iwamoto Y, Iwamoto A, Oda N, Kajikawa M, Matsumoto T, Hidaka T, Kihara Y, Chayama K, Nakashima A, Goto C, Liao JK, Higashi Y. Critical role of exogenous nitric oxide in ROCK activity in vascular smooth muscle cells. PLoS One. 2014;9:e109017. doi: 10.1371/journal.pone.0109017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Fukutomi T, Zago AC, Ehlers R, Detmers PA, Wright SD, Rogers C, Simon DI. Simvastatin reduces neointimal thickening in low-density lipoprotein receptor-deficient mice after experimental angioplasty without changing plasma lipids. Circulation. 2002;106:20–23. doi: 10.1161/01.cir.0000022843.76104.01. [DOI] [PubMed] [Google Scholar]
- Eto Y, Shimokawa H, Hiroki J, Morishige K, Kandabashi T, Matsumoto Y, Amano M, Hoshijima M, Kaibuchi K, Takeshita A. Gene transfer of dominant negative Rho kinase suppresses neointimal formation after balloon injury in pigs. Am J Physiol Heart Circ Physiol. 2000;278:H1744–H1750. doi: 10.1152/ajpheart.2000.278.6.H1744. [DOI] [PubMed] [Google Scholar]
- Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, Tamakawa H, Yamagami K, Inui J, Maekawa M, Narumiya S. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature. 1997;389:990–994. doi: 10.1038/40187. [DOI] [PubMed] [Google Scholar]
- Liu HW, Halayko AJ, Fernandes DJ, Harmon GS, McCauley JA, Kocieniewski P, McConville J, Fu Y, Forsythe SM, Kogut P, Bellam S, Dowell M, Churchill J, Lesso H, Kassiri K, Mitchell RW, Hershenson MB, Camoretti-Mercado B, Solway J. The RhoA/Rho kinase pathway regulates nuclear localization of serum response factor. Am J Respir Cell Mol Biol. 2003;29:39–47. doi: 10.1165/rcmb.2002-0206OC. [DOI] [PubMed] [Google Scholar]
- Wassmann S, Laufs U, Baumer AT, Muller K, Konkol C, Sauer H, Bohm M, Nickenig G. Inhibition of geranylgeranylation reduces angiotensin II-mediated free radical production in vascular smooth muscle cells: involvement of angiotensin AT1 receptor expression and Rac1 GTPase. Mol Pharmacol. 2001;59:646–654. doi: 10.1124/mol.59.3.646. [DOI] [PubMed] [Google Scholar]
- Kumai T, Takeba Y, Matsumoto N, Nakaya S, Tsuzuki Y, Yanagida Y, Hayashi M, Kobayashi S. Fasudil attenuates sympathetic nervous activity in the adrenal medulla of spontaneously hypertensive rats. Life Sci. 2007;81:1193–1198. doi: 10.1016/j.lfs.2007.08.008. [DOI] [PubMed] [Google Scholar]
- Guilluy C, Bregeon J, Toumaniantz G, Rolli-Derkinderen M, Retailleau K, Loufrani L, Henrion D, Scalbert E, Bril A, Torres RM, Offermanns S, Pacaud P, Loirand G. The Rho exchange factor Arhgef1 mediates the effects of angiotensin II on vascular tone and blood pressure. Nat Med. 2010;16:183–190. doi: 10.1038/nm.2079. [DOI] [PubMed] [Google Scholar]
- Wang Y, Zheng XR, Riddick N, Bryden M, Baur W, Zhang X, Surks HK. ROCK isoform regulation of myosin phosphatase and contractility in vascular smooth muscle cells. Circ Res. 2009;104:531–540. doi: 10.1161/CIRCRESAHA.108.188524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Jiang J, Yin H, Wang L, Tian R, Li H, Wang Z, Li D, Wang Y, Gui Y, Walsh MP, Zheng XL. Atorvastatin inhibits myocardin expression in vascular smooth muscle cells. Hypertension. 2012;60:145–153. doi: 10.1161/HYPERTENSIONAHA.112.195644. [DOI] [PubMed] [Google Scholar]
- Wirth A, Schroeter M, Kock-Hauser C, Manser E, Chalovich JM, De Lanerolle P, Pfitzer G. Inhibition of contraction and myosin light chain phosphorylation in guinea-pig smooth muscle by p21-activated kinase 1. J Physiol. 2003;549:489–500. doi: 10.1113/jphysiol.2002.033167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster DB, Shen LH, Kelly J, Thibault P, Van Eyk JE, Mak AS. Phosphorylation of caldesmon by p21-activated kinase: Implications for the Ca(2+) sensitivity of smooth muscle contraction. J Biol Chem. 2000;275:1959–1965. doi: 10.1074/jbc.275.3.1959. [DOI] [PubMed] [Google Scholar]
- Mack CP, Somlyo AV, Hautmann M, Somlyo AP, Owens GK. Smooth muscle differentiation marker gene expression is regulated by RhoA-mediated actin polymerization. J Biol Chem. 2001;276:341–347. doi: 10.1074/jbc.M005505200. [DOI] [PubMed] [Google Scholar]
- Weissberg PL, Bennett MR. Atherosclerosis–an inflammatory disease. N Engl J Med. 1999;340:1928–1929. [PubMed] [Google Scholar]
- Wojciak-Stothard B, Entwistle A, Garg R, Ridley AJ. Regulation of TNF-alpha-induced reorganization of the actin cytoskeleton and cell-cell junctions by Rho, Rac, and Cdc42 in human endothelial cells. J Cell Physiol. 1998;176:150–165. doi: 10.1002/(SICI)1097-4652(199807)176:1<150::AID-JCP17>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Li S, Chen BP, Azuma N, Hu YL, Wu SZ, Sumpio BE, Shyy JY, Chien S. Distinct roles for the small GTPases Cdc42 and Rho in endothelial responses to shear stress. J Clin Invest. 1999;103:1141–1150. doi: 10.1172/JCI5367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojciak-Stothard B, Williams L, Ridley AJ. Monocyte adhesion and spreading on human endothelial cells is dependent on Rho-regulated receptor clustering. J Cell Biol. 1999;145:1293–1307. doi: 10.1083/jcb.145.6.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzima E, Del PMA, Kiosses WB, Mohamed SA, Li S, Chien S, Schwartz MA. Activation of Rac1 by shear stress in endothelial cells mediates both cytoskeletal reorganization and effects on gene expression. EMBO J. 2002;21:6791–6800. doi: 10.1093/emboj/cdf688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng C, Azcutia V, Aikawa E, Figueiredo JL, Croce K, Sonoki H, Sacks FM, Luscinskas FW, Aikawa M. Statins suppress apolipoprotein CIII-induced vascular endothelial cell activation and monocyte adhesion. Eur Heart J. 2013;34:615–624. doi: 10.1093/eurheartj/ehs271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallat Z, Gojova A, Sauzeau V, Brun V, Silvestre JS, Esposito B, Merval R, Groux H, Loirand G, Tedgui A. Rho-associated protein kinase contributes to early atherosclerotic lesion formation in mice. Circ Res. 2003;93:884–888. doi: 10.1161/01.RES.0000099062.55042.9A. [DOI] [PubMed] [Google Scholar]
- Wang HW, Liu PY, Oyama N, Rikitake Y, Kitamoto S, Gitlin J, Liao JK, Boisvert WA. Deficiency of ROCK1 in bone marrow-derived cells protects against atherosclerosis in LDLR−/− mice. FASEB J. 2008;22:3561–3570. doi: 10.1096/fj.08-108829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen WE, Zicha D, Ridley AJ, Jones GE. A role for Cdc42 in macrophage chemotaxis. J Cell Biol. 1998;141:1147–1157. doi: 10.1083/jcb.141.5.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley AJ. Rho proteins, PI 3-kinases, and monocyte/macrophage motility. FEBS Lett. 2001;498:168–171. doi: 10.1016/s0014-5793(01)02481-4. [DOI] [PubMed] [Google Scholar]
- Smith SD, Jaffer ZM, Chernoff J, Ridley AJ. PAK1-mediated activation of ERK1/2 regulates lamellipodial dynamics. J Cell Sci. 2008;121:3729–3736. doi: 10.1242/jcs.027680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak B, Mulhaupt F, Myit S, Mach F. Statins as a newly recognized type of immunomodulator. Nat Med. 2000;6:1399–1402. doi: 10.1038/82219. [DOI] [PubMed] [Google Scholar]
- Senokuchi T, Matsumura T, Sakai M, Yano M, Taguchi T, Matsuo T, Sonoda K, Kukidome D, Imoto K, Nishikawa T, Kim-Mitsuyama S, Takuwa Y, Araki E. Statins suppress oxidized low density lipoprotein-induced macrophage proliferation by inactivation of the small G protein-p38 MAPK pathway. J Biol Chem. 2005;280:6627–6633. doi: 10.1074/jbc.M412531200. [DOI] [PubMed] [Google Scholar]
- Argmann CA, Edwards JY, Sawyez CG, O’Neil CH, Hegele RA, Pickering JG, Huff MW. Regulation of macrophage cholesterol efflux through hydroxymethylglutaryl-CoA reductase inhibition: a role for RhoA in ABCA1-mediated cholesterol efflux. J Biol Chem. 2005;280:22212–22221. doi: 10.1074/jbc.M502761200. [DOI] [PubMed] [Google Scholar]
- Aslan JE, McCarty OJ. Rho GTPases in platelet function. J Thromb Haemost. 2013;11:35–46. doi: 10.1111/jth.12051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goggs R, Williams CM, Mellor H, Poole AW. Platelet Rho GTPases—a focus on novel players, roles and relationships. Biochem J. 2015;466:431–442. doi: 10.1042/BJ20141404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneider NC, Egger P, Dunzendorfer S, Wiedermann CJ. Rho-GTPase-dependent platelet-neutrophil interaction affected by HMG-CoA reductase inhibition with altered adenosine nucleotide release and function. Arterioscler Thromb Vasc Biol. 2002;22:1029–1035. doi: 10.1161/01.atv.0000018306.68268.86. [DOI] [PubMed] [Google Scholar]
- Aslan JE, Itakura A, Haley KM, Tormoen GW, Loren CP, Baker SM, Pang J, Chernoff J, McCarty OJ. p21 activated kinase signaling coordinates glycoprotein receptor VI-mediated platelet aggregation, lamellipodia formation, and aggregate stability under shear. Arterioscler Thromb Vasc Biol. 2013;33:1544–1551. doi: 10.1161/ATVBAHA.112.301165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iida Y, Doi T, Tokuda H, Matsushima-Nishiwaki R, Tsujimoto M, Kuroyanagi G, Yamamoto N, Enomoto Y, Tanabe K, Otsuka T, Iwama T, Ogura S, Kozawa O, Iida H. Rho-kinase regulates human platelet activation induced by thromboxane A2 independently of p38 MAP kinase. Prostaglandins Leukot Essent Fatty Acids. 2015;94:73–81. doi: 10.1016/j.plefa.2014.11.006. [DOI] [PubMed] [Google Scholar]
- Opper C, Clement C, Schwarz H, Krappe J, Steinmetz A, Schneider J, Wesemann W. Increased number of high sensitive platelets in hypercholesterolemia, cardiovascular diseases, and after incubation with cholesterol. Atherosclerosis. 1995;113:211–217. doi: 10.1016/0021-9150(94)05448-r. [DOI] [PubMed] [Google Scholar]