

Aging is a multifactorial process that is characterized by the accumulation of damaged cellular components and that leads progressively to deterioration of biological functions.1 Recently, increased cellular production of reactive oxygen species (ROS) and decreased removal of oxidized proteins have been implicated in the aging process.2 During aging, ROS generation is amplified, leading to the accumulation of oxidative damage to cells. Such enhancements can result from the lack of proportional regulation of antioxidant systems in the body.
Mitochondria are a central source of ROS under stressful energetic and redox conditions: They produce 85% to 90% of cellular ROS.3 Mitochondria are the energy-generating organelles that play crucial roles in the fundamental cellular processes, including supply of metabolic intermediates, calcium homeostasis, and apoptotic signaling. During mitochondrial respiration, redox reactions occur, but a small percentage of electrons do not complete the series of passages through the electron transport chain and thus generate ROS mainly at complexes I and III of the electron transport chain. Because of the high reliance of cardiomyocytes on mitochondrial energy metabolism, the heart is always challenged by a considerable burden of ROS throughout the lifespan. Morphological and functional alterations of mitochondria have been observed in the aged myocardium, including mitochondrial swelling, matrix derangement, and loss of cristae.4 Dysfunctional mitochondria produce less ATP, release greater amounts of ROS, and have a higher propensity to trigger apoptosis during cardiac aging.5 These observations emphasize the importance of understanding the mechanism that links mitochondrial dysfunction and age-related heart diseases.
The mitochondrial thioredoxin system is the major H2O2 scavenger providing a primary defense against ROS produced by the mitochondrial respiratory chain. The mitochondrial thioredoxin system is composed of thioredoxin 2, peroxiredoxin 3 (thioredoxin peroxidase 2), and thioredoxin reductase 2 (Txnrd2). Thioredoxin 2 is localized to the mitochondrial matrix and found at the highest levels in metabolically active tissues such as the heart.6 Peroxiredoxin 3 contains a mitochondrial localization sequence, is found exclusively in the mitochondrion, and uses thioredoxin 2 as the electron donor to detoxify H2O2 generated by the mitochondrial metabolism. Txnrd2 is a selenocysteine-containing homodimeric flavoenzyme necessary for maintaining thioredoxin 2 in its reduced state using electrons from NADPH. Txnrd2 contains a typical mitochondrial import sequence that keeps the protein within the mitochondria.7 Consequently, Txnrd2 governs the reducing activity of thioredoxin 2 in mitochondria, thereby controlling cellular redox balance through peroxiredoxin 3 in the mammalian heart.8 It has become more clear that aging is associated with significant downregulation of Trxrd2 expressions in the mammalian heart9; however, an important question is whether Trxrd2 represents an active protective response in the aging heart.
In this issue of Journal of the American Heart Association (JAHA), Kiermayer et al provided remarkably important insights regarding a crucial role of Txnrd2 in the aging heart.10 The important functions of Txnrd2 in the aging heart are not totally surprising considering the indispensability of Txnrd2 for embryonic cardiac development11; however, elegant work by Kiermayer et al further detailed how Txnrd2 preserves the mitochondrial integrity, redox homeostasis, and cardiac function in the aging heart. With their studies, they demonstrated that inducible cardiomyocyte-specific deletion of Txnrd2 caused left ventricular hypertrophy and dysfunction with reduced systemic blood pressure in aged mice but not in younger mice. These changes in the aged Txnrd2 knockout hearts appear to be attributed to severe mitochondrial degeneration (eg, loss of cristae), leading to progressive heart failure with consequent bioenergic insufficiency during aging.
Several important molecular findings emerged from this study (Figure1). First, deletion of Txnrd2 in the heart induced protein stabilization of the α subunit of hypoxia-inducible factor 1 (HIF-1α). HIF-1 is a master transcription factor that controls cellular metabolism for adaptation to low oxygen availability.12 Under normoxia, HIF-1α undergoes rapid proteasomal degradation. When oxygen is low, HIF-1α is stabilized and translocated to the nucleus, where it stimulates transcription of genes encoding glucose transporters and enzymes of glycolysis and negatively regulates tricarboxylic acid cycle enzymes.13 It has been known that even under normoxic conditions, ROS can inhibit HIF-1α degradation and increase HIF-1α expression.14 In this study, by using an integrated approach, the authors demonstrated that a loss of Txnrd2 induced HIF-1–mediated adaptive and protective metabolic responses under normoxia. These responses included transcriptional changes of HIF-1 target genes such as glucose transporter 1 (GLUT1) and a significant decrease in fatty acid catabolism. Mitochondria from the aged Txnrd2 knockout mice consumed less oxygen, suggesting reduced oxidative phsophorylation. Interestingly, despite reduced oxidative phosphorylation, the ATP content in the whole myocardium did not change in Txnrd2 knockout hearts. Given the fact that HIF-1 is activated, it is tempting to speculate that the HIF-1–induced glycolytic pathway compensates for the limited energy supply from the tricarboxylic acid cycle. A metabolic shift from oxidative phosphorylation to glycolysis makes sense under severe oxidative stress in mitochondria because the glycolytic pathway could provide key intermediates for de novo synthesis of nucleotides, amino acids, and lipid, in which the oxidative pentose phosphate pathway produces NADPH to counter oxidative stress.15
Figure 1.
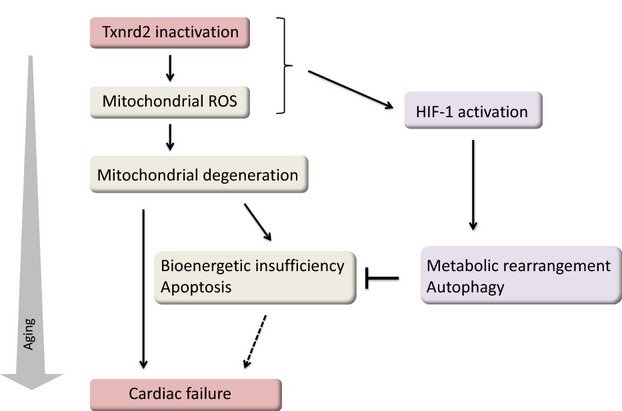
A possible regulatory mechanism by which Txnrd2 inactivation leads to the progression of age-related heart failure. A prominent hypothesis of Txnrd2 inactivation postulates that accumulating mitochondrial damage by increased generation of ROS results in progressive mitochondrial degeneration with consequent bioenergic insufficiency and cardiomyocyte death during aging. Txnrd2 inactivation induces stabilization of the α subunit of HIF-1 and transcriptional activation of HIF-1. The HIF-1–driven metabolic adaptation and increased autophagy may be necessary for resistance to mitochondrial oxidative damage and an energy crisis in the aged cardiomyocytes. These pathways suggest integrated regulation of the mitochondrial thioredoxin system that confers resistance to oxidative stress in cardiac senescence. HIF-1 indicates hypoxia-inducible factor 1; ROS, reactive oxygen species; Txnrd2, thioredoxin reductase 2.
Second, the authors demonstrated that the autophagic pathway was activated in Txnrd2-deficient mitochondria, whereas cardiomyocyte apoptosis was not significantly induced by deletion of Txnrd2 in the aging heart. Autophagy or mitophagy has been shown to degrade dysfunctional mitochondria, limiting ROS production and eliminating oxidatively damaged organelles and protein aggregates.16 Under conditions of oxidative stress, autophagy can be activated to promote the release of recycled nutrients into the microenvironment, and these nutrients may be used by adjacent cells to support their survival.17 During aging, continuous constitutive autophagy is necessary for maintaining intact cardiac structure and function.18 Based on these pieces of evidence, it is reasonable to hypothesize that the housekeeping property of autophagy plays a protective role as a compensatory mechanism in the aged Txnrd2 knockout cardiomyocytes to antagonize cell death. Although not addressed in this paper, the induction of autophagy might be dependent on the stress response of HIF-1, which is specifically stabilized by deletion of Txnrd2. This is supported by the observation that HIF-1–dependent activation of autophagy and glycolysis greatly enhances anabolic growth in cancer cells,19 linking activation of autophagic and glycolytic pathways with cell survival.
Finally, further mechanical questions remain to be addressed to fully understand the role of Txnrd2 in age-related heart failure. Is the function of Txnrd2, for example, mediated solely through mitochondrial thioredoxin 2 activity in the aging heart or is another pathway involved? It seems that both thioredoxin 2 and Txnrd2 are essential for endogenous cardioprotection in a similar way.20 Thioredoxin 2 cardiac knockout mice share many specific phenotypes with the aged Txnrd2 cardiac knockout mice, including a progressive loss of mitochondrial integrity and function20; however, thioredoxin reductase is a promiscuous enzyme capable of interacting with many other endogenous proteins and substrates including lipoic acid, lipid hydroperoxides, the cytotoxic peptide NK-lysin, vitamin K3, dehydroascorbic acid, the ascorbyl free radical, and tumor suppressor p53.21 Because Txnrd2 can reduce bulky proteins to small molecules, mechanisms may include multiple signaling pathways to control mitochondrial metabolism and cardiac function.
Kiermayer et al described a previously unrecognized metabolic response to mitochondrial ROS in the aging heart. Their findings revealed significant redox adaptations to cardiac aging and uncovered a novel role of Txnrd2 as a mechanism to control morphological and functional integrity of mitochondria for proper metabolic arrangements. Although these data warrant further research on the role of Txnrd2, the work by Kiermayer et al provides an important perspective regarding the relationship between evolutionarily conserved mechanisms of mitochondrial metabolism and cardiac senescence.
Sources of Funding
This work was supported by an American Heart Association Award 13GRNT16870007 (to Yoshioka).
Disclosures
None.
References
- Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petropoulos I, Friguet B. Maintenance of proteins and aging: the role of oxidized protein repair. Free Radic Res. 2006;40:1269–1276. doi: 10.1080/10715760600917144. [DOI] [PubMed] [Google Scholar]
- Aon MA, Stanley BA, Sivakumaran V, Kembro JM, O’Rourke B, Paolocci N, Cortassa S. Glutathione/thioredoxin systems modulate mitochondrial H2O2 emission: an experimental-computational study. J Gen Physiol. 2012;139:479–491. doi: 10.1085/jgp.201210772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs HG, Colgan JA, Lazarus ML. Ultrastructure of the aging myocardium: a morphometric approach. Am J Anat. 1977;150:63–71. doi: 10.1002/aja.1001500105. [DOI] [PubMed] [Google Scholar]
- Dutta D, Calvani R, Bernabei R, Leeuwenburgh C, Marzetti E. Contribution of impaired mitochondrial autophagy to cardiac aging: mechanisms and therapeutic opportunities. Circ Res. 2012;110:1125–1138. doi: 10.1161/CIRCRESAHA.111.246108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spyrou G, Enmark E, Miranda-Vizuete A, Gustafsson J. Cloning and expression of a novel mammalian thioredoxin. J Biol Chem. 1997;272:2936–2941. doi: 10.1074/jbc.272.5.2936. [DOI] [PubMed] [Google Scholar]
- Miranda-Vizuete A, Damdimopoulos AE, Pedrajas JR, Gustafsson JA, Spyrou G. Human mitochondrial thioredoxin reductase cDNA cloning, expression and genomic organization. Eur J Biochem. 1999;261:405–412. doi: 10.1046/j.1432-1327.1999.00286.x. [DOI] [PubMed] [Google Scholar]
- Stanley BA, Sivakumaran V, Shi S, McDonald I, Lloyd D, Watson WH, Aon MA, Paolocci N. Thioredoxin reductase-2 is essential for keeping low levels of H(2)O(2) emission from isolated heart mitochondria. J Biol Chem. 2011;286:33669–33677. doi: 10.1074/jbc.M111.284612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrbach S, Gruenler S, Teschner M, Holtz J. The thioredoxin system in aging muscle: key role of mitochondrial thioredoxin reductase in the protective effects of caloric restriction? Am J Physiol Regul Integr Comp Physiol. 2006;291:R927–R935. doi: 10.1152/ajpregu.00890.2005. [DOI] [PubMed] [Google Scholar]
- Kiermayer C, Northrup E, Schrewe A, Walch A, Hrabe de Angelis M, Schoensiegel F, Zischka H, Prehn C, Adamski J, Bekeredjian R, Ivandic B, Kupatt C, Brielmeier M. Heart-specific knockout of the mitochondrial thioredoxin reductase (Txnrd2) induces metabolic and contractile dysfunction in the aging myocardium. J Am Heart Assoc. 2015;4:e002153. doi: 10.1161/JAHA.115.002153. doi: 10.1161/JAHA.115.002153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad M, Jakupoglu C, Moreno SG, Lippl S, Banjac A, Schneider M, Beck H, Hatzopoulos AK, Just U, Sinowatz F, Schmahl W, Chien KR, Wurst W, Bornkamm GW, Brielmeier M. Essential role for mitochondrial thioredoxin reductase in hematopoiesis, heart development, and heart function. Mol Cell Biol. 2004;24:9414–9423. doi: 10.1128/MCB.24.21.9414-9423.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med. 2003;9:677–684. doi: 10.1038/nm0603-677. [DOI] [PubMed] [Google Scholar]
- Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11:325–337. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- Finkel T. Signal transduction by mitochondrial oxidants. J Biol Chem. 2012;287:4434–4440. doi: 10.1074/jbc.R111.271999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441–464. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- Linton PJ, Gurney M, Sengstock D, Mentzer RM, Jr, Gottlieb RA. This old heart: cardiac aging and autophagy. J Mol Cell Cardiol. 2015;83:44–54. doi: 10.1016/j.yjmcc.2014.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–995. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi O, Otsu K. Role of autophagy in aging. J Cardiovasc Pharmacol. 2012;60:242–247. doi: 10.1097/FJC.0b013e31824cc31c. [DOI] [PubMed] [Google Scholar]
- Capparelli C, Whitaker-Menezes D, Guido C, Balliet R, Pestell TG, Howell A, Sneddon S, Pestell RG, Martinez-Outschoorn U, Lisanti MP, Sotgia F. CTGF drives autophagy, glycolysis and senescence in cancer-associated fibroblasts via HIF1 activation, metabolically promoting tumor growth. Cell Cycle. 2012;11:2272–2284. doi: 10.4161/cc.20717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q, Zhou HJ, Zhang H, Huang Y, Hinojosa-Kirschenbaum F, Fan P, Yao L, Belardinelli L, Tellides G, Giordano FJ, Budas GR, Min W. Thioredoxin-2 inhibits mitochondrial reactive oxygen species generation and apoptosis stress kinase-1 activity to maintain cardiac function. Circulation. 2015;131:1082–1097. doi: 10.1161/CIRCULATIONAHA.114.012725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustacich D, Powis G. Thioredoxin reductase. Biochem J. 2000;346(Pt 1):1–8. [PMC free article] [PubMed] [Google Scholar]