

Abstract
Ion channels in the myocardial cellular membrane are responsible for allowing the cardiac action potential. Genetic abnormalities in these channels can predispose to life-threatening arrhythmias. We discuss the basic science of the cardiac action potential; outline the different clinical entities, including information regarding overlapping diagnoses, touching upon relevant genetics, new innovations in screening, diagnosis, risk stratification, and management. The special considerations of sudden unexplained death and sudden infant death syndrome are discussed. Scientists and clinicians continue to reconcile the rapidly growing body of knowledge regarding the molecular mechanisms and genetics while continuing to improve our understanding of the various clinical entities and their diagnosis and management in clinical setting. Two separate searches were run on the National Center for Biotechnology Information's website. The first using the term cardiac channelopathies was run on the PubMed database using filters for time (published in past 5 years) and age (birth-18 years), yielding 47 results. The second search using the medical subject headings (MeSH) database with the search terms “Long QT Syndrome” (MeSH) and “Short QT Syndrome” (MeSH) and “Brugada Syndrome” (MeSH) and “Catecholaminergic Polymorphic Ventricular Tachycardia” (MeSH), applying the same filters yielded 467 results. The abstracts of these articles were studied, and the articles were categorized and organized. Articles of relevance were read in full. As and where applicable, relevant references and citations from the primary articles where further explored and read in full.
Keywords: Brugada syndrome, cardiac channelopathy, catecholaminergic polymorphic ventricular tachycardia, long QT syndrome, short QT syndrome
INTRODUCTION
Our heart beats as the result of an elegant interplay of ions at the cellular level. Ion channels for sodium (Na+), potassium (K+), and calcium (Ca2+) in the myocardial cellular membrane are responsible for allowing this interplay across the membrane. When genetic abnormalities cause these channels to be dysfunctional, the resulting cardiac channelopathies can predispose to life-threatening arrhythmias.
This article is the first in a planned series, the purpose of which is to familiarize the reader with the relevant features of the major cardiac channelopathies-long QT syndrome (LQTS), short QT syndrome (SQTS), Brugada syndrome (BS), and catecholaminergic polymorphic ventricular tachycardia (CPVT). In this article we will discuss the basic science of the cardiac action potential, outline the different clinical entities (including some new information regarding overlapping diagnoses), specifically touching upon relevant genetics, new innovations in genetic screening, diagnosis, risk stratification, and management. The special considerations of sudden unexplained death (SUD) and sudden infant death syndrome (SIDS) are discussed. In future articles, we will go into further detail about CPVT, BS, and SQTS. LQTS has recently been comprehensively reviewed in this journal.[1]
THE ACTION POTENTIAL
The ventricular cardiac myocyte action potential is responsible for the propagation of the electrical impulse through the ventricles. The relevant features of the cardiac action potential are reviewed in Figure 1.
Figure 1.

Phases of the ventricular action potential with description of major events[1]
CLINICAL ENTITIES AND GENETIC CONSIDERATIONS
General considerations
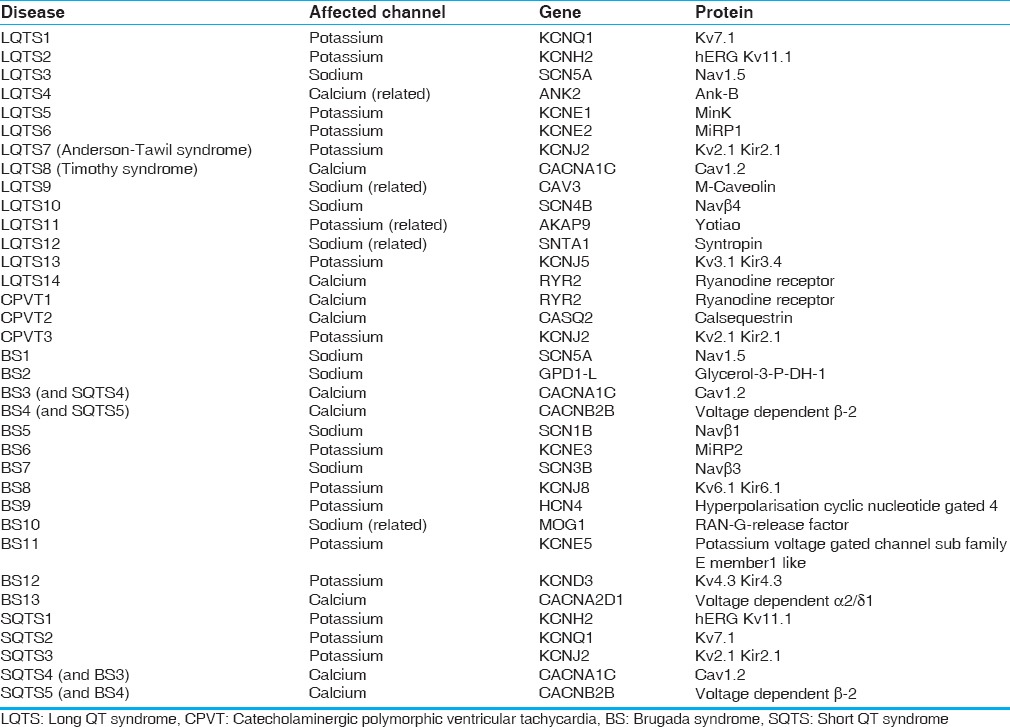
The cardiac channelopathies are caused by mutations affecting genes associated with various cardiac membrane channels (LQTS, SQTS, and BS) or cellular structures affecting Ca2+ availability (some forms of CPVT). The major genes and affected proteins are summarized in Table 1. The majority are autosomal dominant, with variable expressivity, although one form of LQTS is autosomal recessive (so-called Jervel and Lange-Nielson syndrome [JLNS]). Different triggers are classically associated with different syndromes, although these may vary within a given syndrome based on the specific mutation, as with LQTS. Initial symptoms include palpitations, syncope, near-syncope, seizures, and sudden cardiac death (SCD). Many channelopathies have typical findings on the electrocardiogram (EKG), but these findings may not always be present.[2]
Table 1.
Channelopathy associated genes[2]
Initial workup for these syndromes includes a thorough history and physical examination and 12 lead EKG. Treadmill exercise stress testing (LQTS and CPVT) and pharmacological testing (LQTS and BS) can be helpful in diagnosis.[2,3,4,5] Genetic testing can occasionally confirm the underlying mutation. Genetic testing can be helpful for risk stratification and also to guide genotype specific therapy, particularly for LQTS.[2] Risk stratification for some patients can be made a thorough assessment of their symptoms and clinical history, EKG abnormalities, and the location and nature of the underlying mutations.[6]
First line therapy usually involves medications, even sometimes for asymptomatic individuals discovered by ECG testing for other reasons or because other family members were diagnosed with the disease. Implantable cardioverter defibrillator (ICD) implantation has a clear role for secondary prevention for patients who have had a significant cardiac event (e.g., arrhythmia-related syncope or aborted sudden death [SD]) while on appropriate antiarrhythmic therapy. ICD implantation for primary prevention is more controversial.[7]
Left cardiac sympathetic denervation (LCSD) is a surgical intervention that has been shown to reduce the occurrence of ventricular arrhythmias in LQTS and CPVT.[8]
Long QT syndrome
As its name implies, LQTS is characterized by prolongation of the QT interval on ECGs. Figure 2 for an example of EKG findings in LQTS. However, abnormal QT interval prolongation is not always seen in patients with genetically confirmed LQTS and the QT interval prolongation can vary between ECGs. Individuals with LQTS are at risk for a polymorphic ventricular tachycardia called torsades de pointes (TdP) and this arrhythmia is responsible for cardiac events (e.g., syncope, aborted SD, and SD). Figure 3 shows an example of TdP. Approximately, 5% will present with fatal arrhythmia as the first event.[9]
Figure 2.
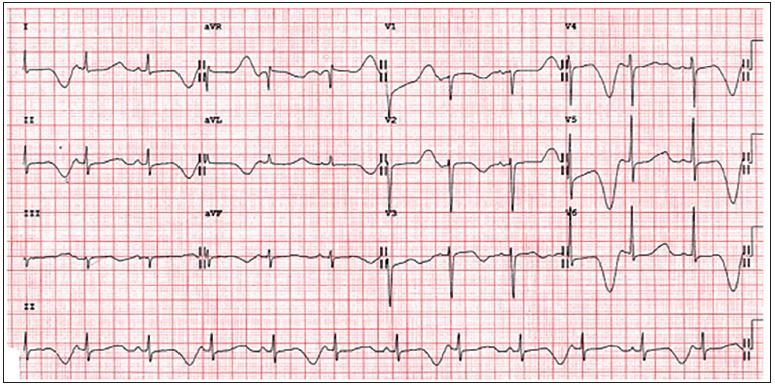
Long QT syndrome electrocardiogram with T-wave alternans
Figure 3.
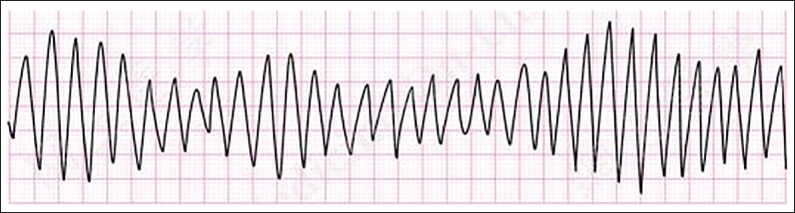
Torsades de pointes
LQTS is the most common cardiac channelopathy. LQTS is estimated to occur in about 1 in 2500 persons.[4,9] Initial classification of inheritable LQTS was as the autosomal dominant Romano-Ward syndrome, and the rarer autosomal recessive form associated with bilateral sensorineural deafness, the JLNS. We now understand that the majority of LQTS is autosomal dominant. There are currently 14 types of LQTS described based on the specific gene involved, location of a mutation along the gene, and associated noncardiac findings. Subtypes are associated with different triggering events such as exercise, swimming, or loud sounds such as alarms.[9]
The first genes responsible for LQTS were elucidated in 1995. The majority of cases are ascribed to three major genes causing three major syndromes - KCNQ1 causing LQTS1 (35%), KCNH2 causing LQTS2 (30%) and SCN5A causing LQTS3 (10%).[4,9] Mutations in KCNQ1 and KCNH2 result in loss of function mutations of K+ repolarization currents, while SCN5A mutations result in gain of function mutations in the Na+ channel.[4] JLNS is thought to be a homozygous form of LQTS1.
Torsades in individuals with LQTS1 is more typically triggered by exercise, especially swimming or diving.[4,10] Cardiac events in individuals with LQTS2 can be triggered by sudden loud noises or being startled. LQTS3 patients tend to have cardiac events while at rest or asleep[10] because the Na+ current is more prolonged at slower stimulus frequency.[11] However, there is considerable overlap in triggers for different LQTS types.
The Schwartz and Crotti score, proposed in 1993 and revised in 2011[12] can be used to diagnose LQTS when the situation is unclear [Table 2]. The Goldenburg criteria can be used to risk stratify clinically [Table 3].[4] Holter monitoring has not shown to be efficacious in the evaluation of LQTS.[13]
Table 2.
Updated Schwartz score[3]

Table 3.
Goldenburg criteria for risk stratification in long QT[4]

Initial management of LQTS involves medication-beta blockers are the mainstay of therapy for LQTS 1 and LQTS2,[4] and are emerging to be beneficial in LQTS3 also.[6] The efficacy of beta blockers for LQTS3 may be mutation specific.[11] Beta-blockers are clearly effective in LQTS1 and LQTS2 though it appears females with LQTS2 are less protected. Beta-blocker therapy for LQTS3 is more efficacious than previously assumed.[10] Na+ channel blockers such as mexiletine, flecainide, and ranolazine have been used in LQTS3.[11] The response to Na+ channel blockade also appears to be mutation specific in LQTS3 and indicates a need for close EKG monitoring while testing the QT shortening effect of the Na+ channel blocker.[10] LCSD has also been shown to be beneficial in patients who are symptomatic despite beta blocker therapy.[6,8] Avoidance of drugs which can prolong the QT interval or trigger arrhythmias is important.[6] (An updated list of medications to avoid in patients with LQTS can be found at the website https://www.crediblemeds.org). Exercise restrictions are also important depending on the type of LQTS.[4]
Catecholaminergic polymorphic ventricular tachycardia
Individuals with CPVT generally have normal resting EKGs with a structurally and functionally normal heart. These patients are at risk for exercise-induced syncope or SD secondary to catecholamine dependent polymorphic ventricular tachycardia (often a bidirectional ventricular tachycardia). Cardiac events occur most commonly in children and adolescents, predominantly young males.[4,9] It is a significant cause of SUD which is discussed below.[4]
Exercise stress testing, Holter monitor recording, looping event monitors and implantable loop recorders can reveal ventricular ectopy or tachycardia during exercise or stress to help make the diagnosis.[4] Bidirectional ventricular tachycardia during such events is specific but insensitive.[10]
Autosomal dominant mutations in the RYR2 gene which encodes the ryanodine receptor/Ca2+ release channel, account for 50-65% of CPVT. This receptor regulates the release of Ca2+ from the sarcoplasmic reticulum during the plateau of the action potential. Mutations result in uncontrolled release of Ca2+ during catecholaminergic stimulation.[4,9] A rarer subtype, CPVT2 is caused by autosomal recessive mutations in CASQ2 which encodes calsequestrin, the major Ca2+ binding protein in the sarcoplasmic reticulum. This has more severe features and earlier onset.[4,9] A third syndrome, CPVT3 occurs with mutations in the KCNJ2 K+ channel gene, and can be misdiagnosed as LQTS7 (Anderson-Taweil syndrome).[4]
There is less data available to help risk stratification and prognostication based on mutation or affected domain.[10] However, we do know that CPVT3 may be more responsive to primary therapy with flecainide or mexiletine and that LCSD has lesser effect in CPVT3.[10]
Untreated CPVT has mortality as high as 30-50%, with earlier episodes associated with poor prognosis.[4] First line therapy is with beta-blockers, however there is higher breakthrough rates during therapy.[10] In a retrospective study from Mayo clinic, 8 years rate of arrhythmic events for patients with CPVT treated with beta-blockers was 37%, near-fatal events was 15%, and fatal events was 6%.[8] Addition of Ca2+ channel blockers improves protection.[4] Flecainide has been shown to be beneficial in CPVT patients.[14] LCSD has been shown to be effective in patients who were symptomatic despite beta-blocker therapy.[8] ICD placement is indicated in cases of aborted SCD, episodes during exercise, and with poor control on medication,[4] but should be reserved as a last resort in CPVT.[10] Due to the nature of CPVT, the fear of ICD discharge may result in more catecholaminergic discharge, and may lead to a vicious cycle of tachycardia and shocks, known as ICD storm.[8]
Brugada syndrome
BS accounts for 4-12% of SCD.[4] BS appears related to mutations affecting Na+ channels. There are currently more than 300 mutations described, mostly autosomal dominant, affecting the SCN5A gene, leading to loss of Na+ channel function in a variety of ways (Interestingly, mutations in other parts of this gene lead to LQTS3). Other genes affecting the Na+ channel are also implicated in BS are summarized in Table 1.[4,6,9]
BS is typified by classical finding of coved ST-segment elevation in anterior precordial leads, as demonstrated in Figure 4.[6] Cardiac events secondary to ventricular tachycardia typically occur in young adults but have been described in children and infants.[4] Individuals with BS develop a monomorphic ventricular tachycardia; often precipitated during sleep or rest, and especially fever.[6] It is thought that some SCN5A mutations alter the Na+ channel in a temperature dependent manner. Males have arrhythmic events more frequently, and there is thought to be a gender effect on ion channel expression.[11]
Figure 4.
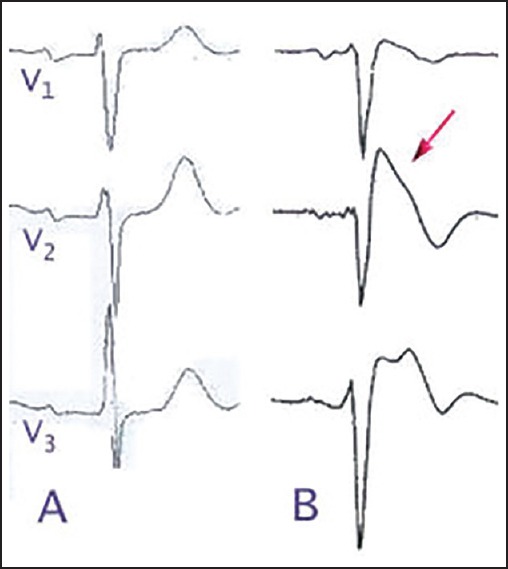
Brugada syndrome electrocardiogram findings. (a) Normal, (b) Brugada syndrome
The EKG findings are often concealed, but drug challenge with Na+ channel blockers such as flecainide and ajmaline can be used to elicit these EKG findings.[11] The role of electrophysiological testing is controversial.[6]
Data regarding gene specific prognostication and stratification is sparse. The presence of SCN5A does not seem to affect the clinical outcome.[10]
Quinidine and beta blockers have been used to treat BS.[6] It seems that in children, clinical picture is frequently dominated by tachycardia induced conduction disturbances, such as fever related arrhythmias.[6] Fever management is important in BS.[11] It is important to avoid certain medications. An updated list of medications to avoid in patients with BS can be found at the website (www.brugada-drugs.org).
In cases of survived arrest, only ICD implantation has been shown to be of proven efficacy. Placement of ICD in symptomatic children is not without potential problems and is not completely protective against arrhythmic death.[15] Inappropriate shocks and other complications related to the presence of an ICD may occur at a higher rate for children, and may be compounded by the likely need for an ICD (with multiple revisions) for many decades.[15,16] The use of electrophysiology testing (EPT) for risk stratification and determining who should get an ICD for primary prevention remains controversial, as studies have found different rates of subsequent clinical events in patients with positive electrophysiology studies, only partly explained by differences in protocols used.[15] Additionally, there are nontrivial false positive and false negative rates with EPT, as well as poor reproducibility.[17]
Short QT syndrome
SQTS was first described in 2000. It is characterized by shortening of the QT interval. It is the severest form of the major channelopathies, with cardiac arrest/SCD being the most common presentation. Events occur during rest, sleep or exertion.[4]
Gain of function mutations in three K+ channel genes have been associated with SQTS, causing three syndromes-SQTS1 caused by mutations in hERG, SQTS2 caused by mutations in KCNQ1 and SQTS3 caused by mutations in KCNJ2.[4]
ICD placement is the only proven therapy in SQTS and is the first line of therapy given the severe nature of the syndrome.[4]
Other channelopathies
Other channelopathies involving mutations affecting the Na+ channel include cardiac conduction disease, dilated cardiomyopathy, sick sinus syndrome, and familial atrial fibrillation.[6]
SUDDEN UNEXPLAINED DEATH AND CHANNELOPATHIES
Sudden unexplained death
The SD of young persons is a tragic event. On autopsy examination, many different causes of SD can be identified.[18] The most common cause of SD is SCD, defined as death from a cardiac cause occurring shortly after the onset of symptoms.[6] Many cases of SCD have identifiable abnormalities such as hypertrophic cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy, coronary artery anomalies or myocarditis.[18] However, a significant proportion of SD (3-53%) has no identifiable cause on autopsy examination, and these are labeled SUD.[18] SUD occurs at about 33/million people/year.[19] SUD of young persons is a rare occurrence, best studied in the developed world, with an incidence of 1.3-8.5/100,000 patient years.[18]
Cardiac channelopathies account for approximately one-third of SUD cases.[6] SD can be the first presentation of an underlying channelopathy. Evaluations of patients with SUD has included molecular autopsy (genetic assessment of the deceased individual, looking for underlying pathogenic mutations which may explain the SUD) and cascade screening (clinical and/or genetic investigation of immediate relatives of the deceased person), in an attempt to find individuals at risk for potentially treatable life-threatening arrhythmias.[18]
Clinical and genetic evaluation
In clinical assessments of surviving family members after SUD, the diagnostic yield for inheritable cardiac arrhythmias has been 22-53%, with a higher likelihood when the decedent was younger (1-10 years). The limitation of clinical assessment alone is that there are incomplete penetrance and variable expressivity of channelopathy causing mutations, resulting in underdiagnosis.[18]
“Next Generation” genetic sequencing panels geared toward the detection of the major channelopathy causing mutations are becoming more readily available, cheaper, faster, and more accurate. Next Generation sequencing for LQTS testing has a sensitivity of 72%.[20]
Molecular autopsy series have revealed mutations associated with channelopathies in approximately 25-35%.[18,21] The largest molecular autopsy series, from the Mayo Clinic, revealed that 26% of SUD cases were associated with a LQTS or CPVT associated mutation, and another 16% had polymorphisms previously associated with inherited arrhythmias. Females were more likely to have a mutation, especially in adolescents. Mutation positive females tended to have LQTS associated mutations, and males more often had CPVT associated mutations.[18,22]
Standardized guidelines for autopsy practices in SUD in the young have been put forward by the trans-Tasman response against SD in the young in 2008, and an expert consensus statement was published by the Heart Rhythm Society and the European Heart Rhythm Association in 2011.[18,22]
Currently, there are no consensus guidelines for the extent of clinical evaluation of survivors, but it is reasonable to include thorough history and physical examination, EKG, stress testing (if age appropriate), Holter monitor recording (especially if stress testing cannot be done), and echocardiogram.[18] An algorithm for clinical decision making for testing of family members is outlined [Table 4].[23]
Table 4.
Decision making algorithm[5]
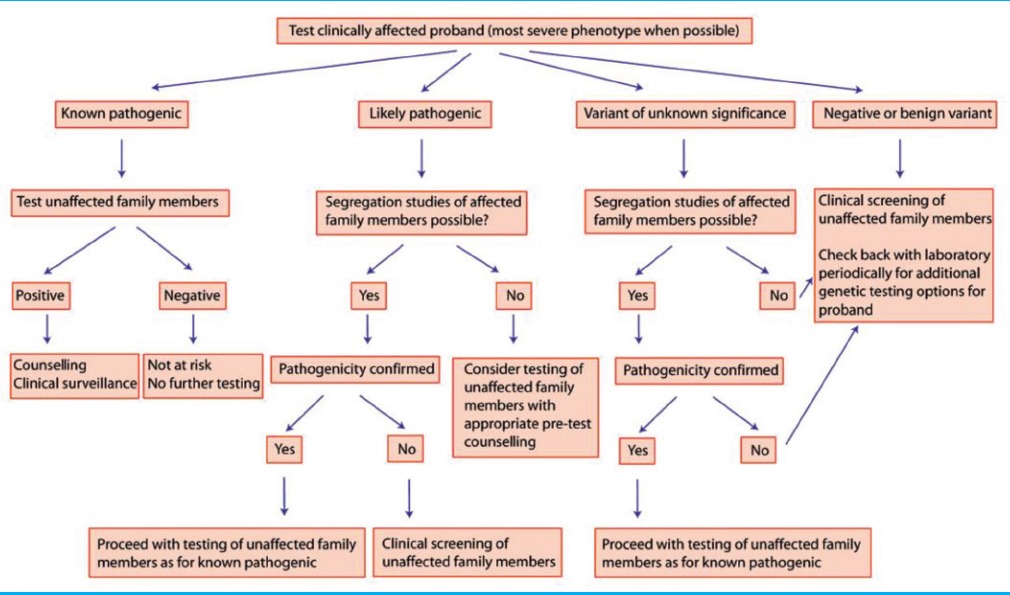
Although both clinical and molecular screening methods appear to reveal a roughly similar rate of cardiac channelopathies, it is unclear which method will be more cost effective. In their elegant review paper, Tester et al. discussed that this will depend on multiple factors: The type of SD (SIDS expected to be less revealing than SUD), the extent of the clinical workup, the size of the family, and with regard to molecular autopsy, whether to perform all-inclusive screening or tiered approach.[9]
It is reasonable to follow and consider subsequent re-testing of family members in cases of SUD where there is reasonable clinical suspicion of a cardiac channelopathy and a disease associated mutation is not initially found, since current testing is not 100% sensitive, and it is likely that the fidelity of tests and the spectrum of relevant mutations will advance with time.[23]
Team approach
Genetic counselors also form an important part of the management team, helping to guide testing of the index patient and family members, and aiding clinicians and patients in interpreting test results.[23,24]
Special considerations in resuscitated cardiac arrest
Approximately 28% of survivors of cardiac arrest have cardiac channelopathies.[25] Based on available data it appears that about two-third of survivors have an inherited cardiac disease, but cardiac arrest due to channelopathies is survived less frequently than structural inheritable cardiac pathology. Proposed clinical investigation pathway includes a thorough history and physical examination, EKG, echocardiogram, and possibly a treadmill exercise stress test. If the echocardiogram does not reveal the likely etiology of SD, clinical investigation of the family is important (including family medical history and EKGs of available family members). Genetic testing may be beneficial in confirming the presence of a channelopathy. In the absence of diagnosis, provocative tests such as ajmaline or flecainide administration for BS, or epinephrine or isoproterenol for CPVT may be helpful.[19]
Cardiac channelopathies and sudden infant death syndrome
SIDS is defined as SD of seemingly healthy children of age <10-year. The incidence of SIDS varies across the world.[26] About 10-20% of SIDS cases are attributed to genetic mutations associated with channelopathies.[6,26,27]
Mutations associated with LQTS1 and LQTS3 have been related to SIDS.[26] Mutations in the genes encoding the beta sub-units of Na channels have also been implicated.[28] Interestingly, a loss of function mutation in the K+ channel encoding gene KCNJ8 has also been associated with SIDS. It is hypothesized that this mutation causes maladaptation to stress such as endotoxemia.[27] A Japanese study looking more broadly at the characteristics of all infantile LQTS, found that 84% of all cases were diagnosed in the fetal or neonatal period. LQTS1 was associated with most risk of a first cardiac event, but LQTS2 and LQTS3 more exclusively caused VT or TdP.[29] QT intervals were found to be longest around 2 months of age.[30] Fetal magnetocardiography and echocardiography have been used to assess fetal LQTS. Sinus bradycardia is a common finding. Transplacental magnesium and lidocaine, and prenatal beta-blocker therapies have been used for management.[31]
While less commonly studied or identified, mutations associated with CPVT, SQTS, and BS have been linked to SIDS.[26]
There is an ongoing debate about the utility of ECG screening in newborns. Studies from Italy and Japan demonstrate similar detection rates of LQTS from EKG screening of neonates (1:2500), and argue for the cost-effectiveness and efficacy of this approach.[30] A high number of false positives and the high number needed to treat are considerations against this.[26] Another approach would be to screen newborns in the setting of prior SIDS in first-degree relatives.[26] One must be cautious in the interpretation of neonatal ECGs for LQTS. For unknown reasons, many newborns have a prolonged QT interval without electrolyte abnormalities or LQTS, especially in the first few days of life.
Overlapping diagnoses
A number of syndromes and symptoms are associated in the literature with channelopathies.
JLNS has been associated with about 4% of patients with the bilateral sensorineural loss.[32] However this association may have been overestimated in the era before genetic testing, and newer studies seem to reflect the similar rate of LQTS causing mutations in deaf children, as in the general population.[33]
There is overlap between seizure disorders and cardiac channelopathies. Sudden unexpected death in epilepsy (SUDEP) has an incidence of 6-9/1000 person years in epilepsy surgery programs. Channelopathy associated mutations have been identified in 13% of patients with SUDEP.[34] Seizures triggered by exercise, emotion, sudden stimuli, seizures unresponsive to anti-seizure medications, and seizures in the setting of family history of SD, syncope, or obvious EKG abnormalities, should all be viewed with suspicion for underlying channelopathy.[35]
In patients with BS, fever is a well-known arrhythmogenic trigger because SCN5A mutations alter the temperature sensitivity of fast inactivation of the Na+ channel. This may cause “Apparent Life-Threatening Events (ALTEs)” and even SUD or SIDS in susceptible infants in the setting of febrile illnesses.[3]
As many as 30% of victims of drowning-related deaths have been found to have cardiac channelopathies.[36,37]
Patients with SCN5A mutations have been found to have irritable bowel syndrome (IBS). In a recent study, 2% of patients with IBS were found to have SCN5A mutations, and in one case, mexiletine administration even caused normalization of bowel habits. It is hypothesized that channelopathies are involved in the pathogenesis of some forms of IBS.[38]
There is also a co-existence of iron-deficiency anemia, hypergastrinemia and gastric hyperplasia associated with LQT1. This suggests both a role for the gene KCNQ1 in gastric secretion, but also a role for gastrin as a marker of arrhythmia severity.[39,40]
Interestingly, in a study from the USA, 36% of patients with drug-induced LQTS possessed known arrhythmia associated mutations.[41]
New innovations in diagnosis and management
Since the elucidation of the genetic basis of LQTS in the mid-1990's, there has been tremendous progress made in the discovery of putative mutations and genes responsible for different channelopathies. With this ever-enlarging fund of known pathogenic mutations, comes the growing number of variants of unknown significance (VUS). Researchers continue to work on better understanding how to stratify the risk of life-threatening arrhythmia based on the genotype and phenotype of the individual.
The delta T50 is a measure of the variability of ventricular repolarization (at 50% of the T-wave downslope). It has been used to identify patients with LQTS in combination with QT interval cut-offs, as well as identifying patients at higher risk for cardiac events.[42]
Rest and exercise QT interval measurements have been used to create a validated algorithm for diagnosing LQTS.[43] End recovery QT interval measurements have also been used, and in combination with clinical history and mutation-specific information, can aid in understanding the pathogenicity of VUS.[44]
A recent study assessed the combination of age and sex associated risk factors with mutation-specific information for stratifying LQTS, and found that during childhood age range, male risk of SD is higher, whereas post-adolescence the risk is similar. The presence of mutations affecting the cytoplasmic loop of the K+ channel (known to increase the risk of SD), has a greater effect on females than males.[45] It is possible that similar effects may be discovered in other channelopathies. Patients with mutations in the KCNQ1 gene affecting a specific portion of the K channel (C-loop missense mutations) demonstrate a higher risk for life-threatening events and have a greater benefit from beta-blocker therapy.[46] However, some studies have not reflected the same increase in QTc on exercise, nor improvement with beta blockade.[47]
Copy number variations (CNV) are a form of genetic abnormality which may explain the genetic basis of channelopathies in cases where there is no identifiable point mutation.[48] It is conceivable that in the future, CNV may be added to genetic screens.
Since the first report in 2006, bench researchers have made use of “induced pluripotent stem cell” systems to study the electrophysiological and pharmacological characteristics of cardiomyocyte cells that are specific to an individual patient and his/her mutation and channelopathy. This technology has huge potential to aid our understanding of individual channelopathies and further steer the management of channelopathies in an individualistic, genotype-specific manner in the future.[49,50,51]
One of the potential areas of debate is the feasibility and efficacy of population screening in some form, to identify channelopathies and potentially prevent lethal arrhythmias. School based EKG screening programs have reflected similar rates and clinical characteristics as larger population studies.[52] In a population study from New Zealand, cardiac/genetic registries in combination with the family screening program, have been shown to be a feasible alternative to widespread EKG screening.[53]
CONCLUSION
Inherited cardiac channelopathies are a very important cause of arrhythmia-related cardiac events, including SUD. The past two decades have seen rapid strides in our understanding of the disease burden, epidemiology, genetic basis and clinical correlation of the channelopathies. Scientists and clinicians continue to reconcile the rapidly growing body of knowledge regarding the molecular mechanisms and genetics, with risk stratification and management strategies for the clinical entities. As genetic testing becomes more rapidly available, we hope that it will be more easily available across the globe. In the meanwhile, we continue to improve our understanding of the various clinical entities and their diagnosis and management in clinical setting even without gene testing. The future promises exciting new developments in our ability to use the genetic and molecular information to better screen, recognize, and tailor management of these potentially lethal cardiac channelopathies.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Shah M, Carter C. Long QT syndrome: A therapeutic challenge. Ann Pediatr Cardiol. 2008;1:18–26. doi: 10.4103/0974-2069.41051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beckmann BM, Pfeufer A, Kääb S. Inherited cardiac arrhythmias: Diagnosis, treatment, and prevention. Dtsch Arztebl Int. 2011;108:623–33. doi: 10.3238/arztebl.2011.0623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chockalingam P, Rammeloo LA, Postema PG, Hruda J, Clur SA, Blom NA, et al. Fever-induced life-threatening arrhythmias in children harboring an SCN5A mutation. Pediatrics. 2011;127:e239–44. doi: 10.1542/peds.2010-1688. [DOI] [PubMed] [Google Scholar]
- 4.Sarquella-Brugada G, Campuzano O, Iglesias A, Sánchez-Malagón J, Guerra-Balic M, Brugada J, et al. Genetics of sudden cardiac death in children and young athletes. Cardiol Young. 2013;23:159–73. doi: 10.1017/S1047951112001138. [DOI] [PubMed] [Google Scholar]
- 5.Schwartz PJ, Crotti L, Insolia R. Long-QT syndrome: From genetics to management. Circ Arrhythm Electrophysiol. 2012;5:868–77. doi: 10.1161/CIRCEP.111.962019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chockalingam P, Wilde A. The multifaceted cardiac sodium channel and its clinical implications. Heart. 2012;98:1318–24. doi: 10.1136/heartjnl-2012-301784. [DOI] [PubMed] [Google Scholar]
- 7.Olde Nordkamp LR, Wilde AA, Tijssen JG, Knops RE, van Dessel PF, de Groot JR. The ICD for primary prevention in patients with inherited cardiac diseases: Indications, use, and outcome: A comparison with secondary prevention. Circ Arrhythm Electrophysiol. 2013;6:91–100. doi: 10.1161/CIRCEP.112.975268. [DOI] [PubMed] [Google Scholar]
- 8.Coleman MA, Bos JM, Johnson JN, Owen HJ, Deschamps C, Moir C, et al. Videoscopic left cardiac sympathetic denervation for patients with recurrent ventricular fibrillation/malignant ventricular arrhythmia syndromes besides congenital long-QT syndrome. Circ Arrhythm Electrophysiol. 2012;5:782–8. doi: 10.1161/CIRCEP.112.971754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tester DJ, Ackerman MJ. The molecular autopsy: Should the evaluation continue after the funeral? Pediatr Cardiol. 2012;33:461–70. doi: 10.1007/s00246-012-0160-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schwartz PJ, Ackerman MJ, George AL, Jr, Wilde AA. Impact of genetics on the clinical management of channelopathies. J Am Coll Cardiol. 2013;62:169–80. doi: 10.1016/j.jacc.2013.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Amin AS, Asghari-Roodsari A, Tan HL. Cardiac sodium channelopathies. Pflugers Arch. 2010;460:223–37. doi: 10.1007/s00424-009-0761-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schwartz PJ, Crotti L. QTc behavior during exercise and genetic testing for the long-QT syndrome. Circulation. 2011;124:2181–4. doi: 10.1161/CIRCULATIONAHA.111.062182. [DOI] [PubMed] [Google Scholar]
- 13.Mauriello DA, Johnson JN, Ackerman MJ. Holter monitoring in the evaluation of congenital long QT syndrome. Pacing Clin Electrophysiol. 2011;34:1100–4. doi: 10.1111/j.1540-8159.2011.03102.x. [DOI] [PubMed] [Google Scholar]
- 14.Khoury A, Marai I, Suleiman M, Blich M, Lorber A, Gepstein L, et al. Flecainide therapy suppresses exercise-induced ventricular arrhythmias in patients with CASQ2-associated catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2013;10:1671–5. doi: 10.1016/j.hrthm.2013.08.011. [DOI] [PubMed] [Google Scholar]
- 15.Conte G, Sieira J, Ciconte G, de Asmundis C, Chierchia GB, Baltogiannis G, et al. Implantable cardioverter-defibrillator therapy in Brugada syndrome: A 20-year single-center experience. J Am Coll Cardiol. 2015;65:879–88. doi: 10.1016/j.jacc.2014.12.031. [DOI] [PubMed] [Google Scholar]
- 16.Conte G, Dewals W, Sieira J, de Asmundis C, Ciconte G, Chierchia GB, et al. Drug-induced Brugada syndrome in children: Clinical features, device-based management, and long-term follow-up. J Am Coll Cardiol. 2014;63:2272–9. doi: 10.1016/j.jacc.2014.02.574. [DOI] [PubMed] [Google Scholar]
- 17.Priori SG, Gasparini M, Napolitano C, Della Bella P, Ottonelli AG, Sassone B, et al. Risk stratification in Brugada syndrome: Results of the PRELUDE (PRogrammed ELectrical stimUlation preDictive valuE) registry. J Am Coll Cardiol. 2012;59:37–45. doi: 10.1016/j.jacc.2011.08.064. [DOI] [PubMed] [Google Scholar]
- 18.Boczek NJ, Tester DJ, Ackerman MJ. The molecular autopsy: An indispensable step following sudden cardiac death in the young? Herzschrittmacherther Elektrophysiol. 2012;23:167–73. doi: 10.1007/s00399-012-0222-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Skinner JR. Investigation following resuscitated cardiac arrest. Arch Dis Child. 2013;98:66–71. doi: 10.1136/archdischild-2011-301515. [DOI] [PubMed] [Google Scholar]
- 20.Lieve KV, Williams L, Daly A, Richard G, Bale S, Macaya D, et al. Results of genetic testing in 855 consecutive unrelated patients referred for long QT syndrome in a clinical laboratory. Genet Test Mol Biomarkers. 2013;17:553–61. doi: 10.1089/gtmb.2012.0118. [DOI] [PubMed] [Google Scholar]
- 21.Basso C, Carturan E, Pilichou K, Rizzo S, Corrado D, Thiene G. Sudden cardiac death with normal heart: Molecular autopsy. Cardiovasc Pathol. 2010;19:321–5. doi: 10.1016/j.carpath.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 22.Tester DJ, Medeiros-Domingo A, Will ML, Haglund CM, Ackerman MJ. Cardiac channel molecular autopsy: Insights from 173 consecutive cases of autopsy-negative sudden unexplained death referred for postmortem genetic testing. Mayo Clin Proc. 2012;87:524–39. doi: 10.1016/j.mayocp.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Caleshu C, Day S, Rehm HL, Baxter S. Use and interpretation of genetic tests in cardiovascular genetics. Heart. 2010;96:1669–75. doi: 10.1136/hrt.2009.190090. [DOI] [PubMed] [Google Scholar]
- 24.Kauferstein S, Kiehne N, Jenewein T, Biel S, Kopp M, König R, et al. Genetic analysis of sudden unexplained death: A multidisciplinary approach. Forensic Sci Int. 2013;229:122–7. doi: 10.1016/j.forsciint.2013.03.050. [DOI] [PubMed] [Google Scholar]
- 25.van der Werf C, Hofman N, Tan HL, van Dessel PF, Alders M, van der Wal AC, et al. Diagnostic yield in sudden unexplained death and aborted cardiac arrest in the young: The experience of a tertiary referral center in The Netherlands. Heart Rhythm. 2010;7:1383–9. doi: 10.1016/j.hrthm.2010.05.036. [DOI] [PubMed] [Google Scholar]
- 26.Tfelt-Hansen J, Winkel BG, Grunnet M, Jespersen T. Cardiac channelopathies and sudden infant death syndrome. Cardiology. 2011;119:21–33. doi: 10.1159/000329047. [DOI] [PubMed] [Google Scholar]
- 27.Tester DJ, Tan BH, Medeiros-Domingo A, Song C, Makielski JC, Ackerman MJ. Loss-of-function mutations in the KCNJ8-encoded Kir6.1 K(ATP) channel and sudden infant death syndrome. Circ Cardiovasc Genet. 2011;4:510–5. doi: 10.1161/CIRCGENETICS.111.960195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tan BH, Pundi KN, Van Norstrand DW, Valdivia CR, Tester DJ, Medeiros-Domingo A, et al. Sudden infant death syndrome-associated mutations in the sodium channel beta subunits. Heart Rhythm. 2010;7:771–8. doi: 10.1016/j.hrthm.2010.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Horigome H, Nagashima M, Sumitomo N, Yoshinaga M, Ushinohama H, Iwamoto M, et al. Clinical characteristics and genetic background of congenital long-QT syndrome diagnosed in fetal, neonatal, and infantile life: A nationwide questionnaire survey in Japan. Circ Arrhythm Electrophysiol. 2010;3:10–7. doi: 10.1161/CIRCEP.109.882159. [DOI] [PubMed] [Google Scholar]
- 30.Yoshinaga M, Ushinohama H, Sato S, Tauchi N, Horigome H, Takahashi H, et al. Electrocardiographic screening of 1-month-old infants for identifying prolonged QT intervals. Circ Arrhythm Electrophysiol. 2013;6:932–8. doi: 10.1161/CIRCEP.113.000619. [DOI] [PubMed] [Google Scholar]
- 31.Anuwutnavin S, Wanitpongpan P, Chungsomprasong P, Soongswang J, Srisantiroj N, Wataganara T. Fetal long QT syndrome manifested as atrioventricular block and ventricular tachycardia: A case report and a review of the literature. Pediatr Cardiol. 2013;34:1955–62. doi: 10.1007/s00246-012-0507-1. [DOI] [PubMed] [Google Scholar]
- 32.Niaz A, Rizvi SF, Khurram D. Prevalence of long QT syndrome and other cardiac defects in deaf-mute children. J Ayub Med Coll Abbottabad. 2011;23:5–8. [PubMed] [Google Scholar]
- 33.Chang RK, Lan YT, Silka MJ, Morrow H, Kwong A, Smith-Lang J, et al. Genetic variants for long QT syndrome among infants and children from a statewide newborn hearing screening program cohort. J Pediatr. 2014;164(590):5.e1–3. doi: 10.1016/j.jpeds.2013.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Partemi S, Cestèle S, Pezzella M, Campuzano O, Paravidino R, Pascali VL, et al. Loss-of-function KCNH2 mutation in a family with long QT syndrome, epilepsy, and sudden death. Epilepsia. 2013;54:e112–6. doi: 10.1111/epi.12259. [DOI] [PubMed] [Google Scholar]
- 35.Hazle MA, Shellhaas RA, Bradley DJ, Dick M nd, Lapage MJ. Arrhythmogenic channelopathy syndromes presenting as refractory epilepsy. Pediatr Neurol. 2013;49:134–7. doi: 10.1016/j.pediatrneurol.2013.03.017. [DOI] [PubMed] [Google Scholar]
- 36.Kenny D, Martin R. Drowning and sudden cardiac death. Arch Dis Child. 2011;96:5–8. doi: 10.1136/adc.2010.185215. [DOI] [PubMed] [Google Scholar]
- 37.Tester DJ, Medeiros-Domingo A, Will ML, Ackerman MJ. Unexplained drownings and the cardiac channelopathies: A molecular autopsy series. Mayo Clin Proc. 2011;86:941–7. doi: 10.4065/mcp.2011.0373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Beyder A, Mazzone A, Strege PR, Tester DJ, Saito YA, Bernard CE, et al. Loss-of-function of the voltage-gated sodium channel NaV1.5 (channelopathies) in patients with irritable bowel syndrome. Gastroenterology. 2014;146:1659–68. doi: 10.1053/j.gastro.2014.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Diamant UB, Jensen SM, Winbo A, Stattin EL, Rydberg A. Vectorcardiographic recordings of the Q-T interval in a pediatric long Q-T syndrome population. Pediatr Cardiol. 2013;34:245–9. doi: 10.1007/s00246-012-0425-2. [DOI] [PubMed] [Google Scholar]
- 40.Rice KS, Dickson G, Lane M, Crawford J, Chung SK, Rees MI, et al. Elevated serum gastrin levels in Jervell and Lange-Nielsen syndrome: A marker of severe KCNQ1 dysfunction? Heart Rhythm. 2011;8:551–4. doi: 10.1016/j.hrthm.2010.11.039. [DOI] [PubMed] [Google Scholar]
- 41.Ramirez AH, Shaffer CM, Delaney JT, Sexton DP, Levy SE, Rieder MJ, et al. Novel rare variants in congenital cardiac arrhythmia genes are frequent in drug-induced torsades de pointes. Pharmacogenomics J. 2013;13:325–9. doi: 10.1038/tpj.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Abrahamsson C, Dota C, Skallefell B, Carlsson L, Frison L, Berggren A, et al. Assessment of ventricular repolarization variability with the DeltaT50 method improves identification of patients with congenital long QT syndromes. Ann Noninvasive Electrocardiol. 2013;18:240–50. doi: 10.1111/anec.12016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sy RW, van der Werf C, Chattha IS, Chockalingam P, Adler A, Healey JS, et al. Derivation and validation of a simple exercise-based algorithm for prediction of genetic testing in relatives of LQTS probands. Circulation. 2011;124:2187–94. doi: 10.1161/CIRCULATIONAHA.111.028258. [DOI] [PubMed] [Google Scholar]
- 44.Obeyesekere MN, Sy RW, Klein GJ, Gula LJ, Modi S, Conacher S, et al. End-recovery QTc: A useful metric for assessing genetic variants of unknown significance in long-QT syndrome. J Cardiovasc Electrophysiol. 2012;23:637–42. doi: 10.1111/j.1540-8167.2011.02265.x. [DOI] [PubMed] [Google Scholar]
- 45.Costa J, Lopes CM, Barsheshet A, Moss AJ, Migdalovich D, Ouellet G, et al. Combined assessment of sex-and mutation-specific information for risk stratification in type 1 long QT syndrome. Heart Rhythm. 2012;9:892–8. doi: 10.1016/j.hrthm.2012.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barsheshet A, Goldenberg I, O-Uchi J, Moss AJ, Jons C, Shimizu W, et al. Mutations in cytoplasmic loops of the KCNQ1 channel and the risk of life-threatening events: Implications for mutation-specific response to ß-blocker therapy in type 1 long-QT syndrome. Circulation. 2012;125:1988–96. doi: 10.1161/CIRCULATIONAHA.111.048041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Laksman ZW, Hamilton RM, Chockalingam P, Ballantyne E, Stephenson EA, Gross GJ, et al. Mutation location effect on severity of phenotype during exercise testing in type 1 long-QT syndrome: Impact of transmembrane and C-loop location. J Cardiovasc Electrophysiol. 2013;24:1015–20. doi: 10.1111/jce.12172. [DOI] [PubMed] [Google Scholar]
- 48.Barc J, Briec F, Schmitt S, Kyndt F, Le Cunff M, Baron E, et al. Screening for copy number variation in genes associated with the long QT syndrome: Clinical relevance. J Am Coll Cardiol. 2011;57:40–7. doi: 10.1016/j.jacc.2010.08.621. [DOI] [PubMed] [Google Scholar]
- 49.Rosenzweig A. Illuminating the potential of pluripotent stem cells. N Engl J Med. 2010;363:1471–2. doi: 10.1056/NEJMe1007902. [DOI] [PubMed] [Google Scholar]
- 50.Egashira T, Yuasa S, Suzuki T, Aizawa Y, Yamakawa H, Matsuhashi T, et al. Disease characterization using LQTS-specific induced pluripotent stem cells. Cardiovasc Res. 2012;95:419–29. doi: 10.1093/cvr/cvs206. [DOI] [PubMed] [Google Scholar]
- 51.Terrenoire C, Wang K, Tung KW, Chung WK, Pass RH, Lu JT, et al. Induced pluripotent stem cells used to reveal drug actions in a long QT syndrome family with complex genetics. J Gen Physiol. 2013;141:61–72. doi: 10.1085/jgp.201210899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yoshinaga M, Kucho Y, Sarantuya J, Ninomiya Y, Horigome H, Ushinohama H, et al. Genetic characteristics of children and adolescents with long-QT syndrome diagnosed by school-based electrocardiographic screening programs. Circ Arrhythm Electrophysiol. 2014;7:107–12. doi: 10.1161/CIRCEP.113.000426. [DOI] [PubMed] [Google Scholar]
- 53.Earle N, Crawford J, Smith W, Hayes I, Shelling A, Hood M, et al. Community detection of long QT syndrome with a clinical registry: An alternative to ECG screening programs? Heart Rhythm. 2013;10:233–8. doi: 10.1016/j.hrthm.2012.10.043. [DOI] [PubMed] [Google Scholar]