

Summary
Somatic structural variants in tumor genomes can deregulate transcription through repositioning of enhancer elements. A new method, PEAR-ChIP, leverages paired-end H3K27ac ChIP-seq and current current computational methods to identify such events.
Cancer genomes often contain recurrent somatic structural variations (SVs) in the form of DNA amplifications, deletions, inversions, and translocations. Conventional approaches to pinpointing oncogenic drivers and tumor suppressor regions within SVs usually focused on putative oncogenes or tumor suppressor genes found within minimal common regions of alteration (1). This method has proven to be widely successful, leading to the detection of novel and recurrent oncogenes and tumor suppressor genes across several cancer types (2). In the case of DNA translocations, a similar approach has been used, where mapping of recurrent DNA breakpoints have revealed oncogenic gene fusion products that generate chimeric oncoproteins. These strategies prioritized candidate genes based on the rationale that the SVs should correspond with gene-level associated alteration such as increased DNA copy number or coding sequence change. However, by applying a gene-centric focus, SVs targeting non-coding regions of the genome have been largely unexplored.
Unclear in cancer genomic landscapes is the prevalence of SVs that lead to gene activation, independent of gene-disruption, such as rearrangement of DNA regulatory elements in noncoding regions of the genome. Identifying such events has been perhaps challenging in the past due to a limited capacity to detect complex rearrangements at high-resolution, and the ability to ascribe function to these alterations by determining the precise genes they regulate. These difficulties have been overcome, at least in part, by the increasing feasibility of whole-genome sequencing, allowing for more thorough characterization of cancer genomes. Furthermore, advancements in chromatin mapping using techniques such as chromatin immunoprecipitation combined with high-throughput sequencing (ChIP-seq) have unraveled the regulatory landscape of both normal and cancer epigenomes (3, 4). Histone modifications, specifically, indicate functional regions of the genome such as enhancers, marked by histone H3 lysine 4 mono-methylation (H3K4me1), and the potential activation status of those enhancers marked by histone H3 lysine 27 acetylation (H3K27ac).
Two recent studies leverage and integrate these genomic and epigenomic technologies, to identify highly recurrent SVs that reposition distal enhancer elements proximal to genetically intact oncogenes, termed ‘Enhancer Hijacking’ or ‘Enhancer Hitchhiking’ (5, 6). In aggressive subgroups of Medulloblastoma (Group 3 and 4), various classes of SVs such as tandem duplications, deletions, inversions, translocations, and other more complex rearrangements, converge to activate GFI1 or GFI1B oncogenic expression. This is accomplished by repositioning the intact GFI1B or GFI1B genes in close proximity with distal super enhancers (Highly active enhancer regions marked by extensive H3K27 acetylation) (6). Importantly, GFI1 and GFIB activation through ‘Enhancer Hijacking’, have not been reported in other cancers, and are the most prevalent driver events in Group 3 medulloblastoma. Similar observations have been observed in a type of acute myeloid leukemia (AML), characterized by chromosome 3q rearrangements (inv(3)/t(3;3)) that lead to aberrant expression of the stem-cell regulator EVI1 (5). The mechanism of EVI1 activation is caused by a chromosomal translocation, which relocates a GATA2 super enhancer proximal to the EVI1 oncogene. This single SV event not only activates EVI1 expression, but also removes an enhancer regulating GATA2, thereby leading to mono-allelic GATA2 expression and haplo-insufficiency. Examples in medulloblastoma and AML suggest that ‘Enhancer Hijacking’ events may be potentially common and driver alterations in other cancer types, and underscores the need for combined methodologies that leverage information from both genomic and epigenomic platforms.
In the current issue, Ryan and Drier et al., (2015) present a novel approach called PEAR-ChIP, which integrates H3K27ac ChIP-seq with paired-end sequencing (7) (Figure 1). Utilizing pre-existing computational tools to detect genomic rearrangements, PEAR-ChIP maps structural variations involving acetylated regulatory elements (PEAR-ChIP, Pinpointing Enhancer-Associated Rearrangements by Chromatin Immunoprecipitation and Paired-End Sequencing). They apply this methodology to investigate a cohort of 14 primary patient biopsies and 8 cell line models representing a diversity of B-cell lymphomas. Importantly, the authors validated several known SVs, and identified numerous types of novel chromosomal rearrangements that delineate various B-cell lymphoma subtypes.
Figure 1.
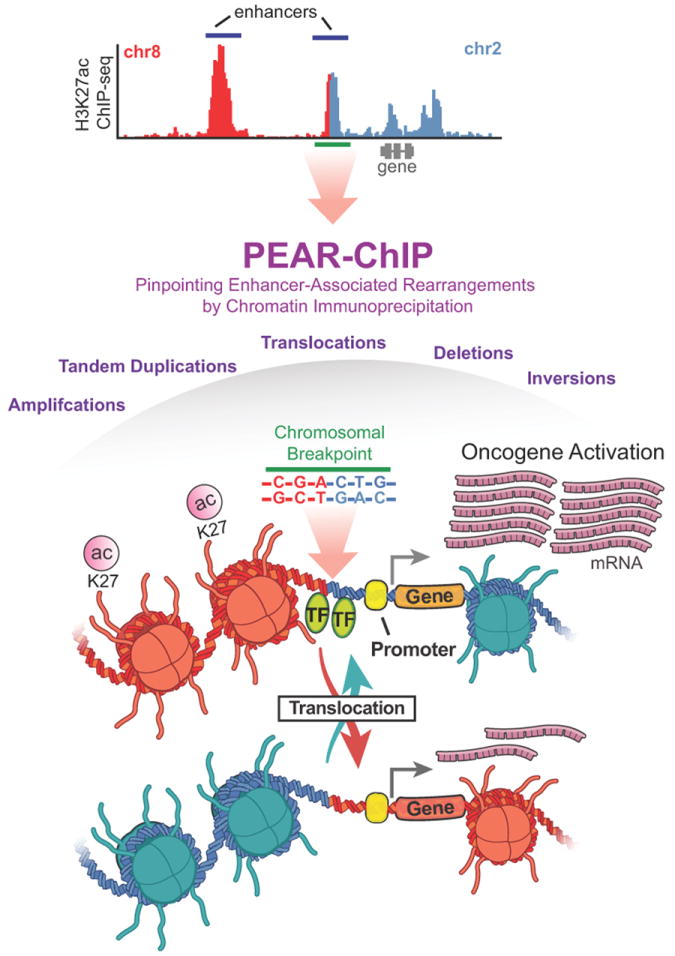
An illustration of the PEAR-ChIP approach used to identify genomic rearrangements within regions of H3K27 acetylation. The top panel is a representative H3K27ac ChIP-seq profile with chromosome 8 reads in red, and chromosome 2 in blue. The breakpoint region is shown as a green bar, and this local region is expanded in a schematic in the lower panel.
This approach is first validated in mantle cell lymphoma (MCL) primary tissue and cell lines, all of which harbor reciprocal translocations between the IGH J recombination region (Chromosome 14) and a gene-desert region located > 300 kb from CCND1 (Chromosome 11). In all samples, H3K27ac enrichment was observed, and extended from the IGH μ intronic enhancer and overlapping the J recombination region. PEAR-ChIP identified the precise t(11;14) breakpoints, all of which contained peaks of acetylation signal. This methodology was then applied to a heterogeneous cohort of primary high-grade B-cell lymphomas (HGBs), cell lines, and lymph node biopsies from patients with chronic lymphocytic leukemia/small lymphocytic leukemia (CLL/SLL). PEAR-ChIP identified several known rearrangements linking an IGH enhancer exclusively to the 5’ side of MYC revealing potentially significant and specific mechanisms of cis-regulation. This was in contrast to a collection of non-IGH rearrangement breakpoints that occurred on the 3’ side of MYC.
In cases where HGB cell lines were also profiled by whole-genome sequencing, PEAR-ChIP detected all six inter-chromosomal or large-scale inversions with improved sensitivity (2-17 times more supporting reads) despite lower sequencing coverage (10-31 fold lower). Further, PEAR-ChIP was sensitive for the detection of other large-scale rearrangements containing several known translocation targets, including BCL2, CIITA, and PDCD1LG2, and novel translocation partners including PDCD1LG2-NCOA3 and CIITA-IL4R. In addition to translocation events, PEAR-ChIP identified several other classes of structural variations that repositioned candidate enhancer elements of oncogenic significance, including small-scale intra-chromosomal deletions and inversions. In several examples, kilobase-scale tandem duplications were detected targeting acetylated putative enhancers up-stream of the rho GTPase-activating gene TAGAP, and duplication of regions harboring an interferon-responsive enhancer upstream of the inducible nitric oxide synthase gene NOS2. These events represent potentially novel mechanisms of oncogenic regulation currently unexplored in HGB biology.
Guided by information gained from chromosome conformation capture (3C) experiments, the authors utilized the PEAR-ChIP approach to investigate the complex dynamics of BCL6 and MYC oncogenic regulation in HGB. 3C studies revealed that MCL and SLL were delineated by 5’ interacting enhancers with the MYC promoter, while HGBs were distinctly characterized by 3’ enhancers. In the case of BCL6, a gene known to frequently rearrange in HGB, multiple 3’ super enhancers were detected and demonstrated to form contact loops with the BCL6 promoter. Furthermore, PEAR-ChIP was successful in detecting a tandem duplication spanning one of the super enhancers, in addition to breakpoints within acetylated regions proximal to BCL6. Understanding the native enhancer-promoter regulation of BCL6 and MYC in HGB, allowed the authors to explore the product of translocations between the BCL6 and MYC loci. In one sample, harboring a t(3;8)(q27;q24) rearrangement, PEAR-ChIP identified a breakpoint that swapped the BCL6 super enhancer region described above, with the 3’ MYC enhancer regions prevalent in HGB lines, classified as an ‘Enhancer Swap’. The authors report that, in independent rearrangements, the BCL6 locus is capable of acting as a ‘donor’ or ‘recipient’ of enhancer elements, thus revealing novel mechanisms in cancer gene de-regulation.
These findings emphasize that rearrangement of enhancer elements by structural variation are potentially a general mechanism of oncogenic activation across cancer. PEAR-ChIP represents both a sensitive and cost effective approach to identify such alterations. One important consideration is the requirement for breakpoints to be located within acetylated regions in order to be detected by PEAR-ChIP. Breakpoints occurred within acetylated loci in many of the B-cell lymphomas in this study, and Group 3 and 4 Medulloblastomas, but this may not be the case in other cancers. The authors suggest that in such scenarios, one complementary approach would be to pair the high-resolution breakpoint detection of PEAR-ChIP with a low-resolution, genome-wide platform, such as long-insert mate pair sequencing. While ‘Enhancer Hijacking’ events represent novel modes of oncogenic regulation that are independent of gene disruption, care should be taken to validate that the effects upon gene expression are indeed direct mechanisms. As shown in this study, and EVI1 re-arranged AML (5), chromosome conformation capture experiments can complement such analyses providing insights about enhancers in their native loci, and the consequence of their repositioning. Beyond structural variations, recurrent somatic mutations have recently been identified in regulatory regions across different cancer types (8-10). With increasing understanding of cancer epigenomes, armed with integrative tools such as PEAR-ChIP, revisiting cancer genome studies may be warranted, and may reveal novel mechanisms of oncogenic regulation.
Acknowledgments
Financial Support:
This work was supported by The Banting Fellowship (SCM), and NIH grants: R01CA160356 (PCS), CA154130 (JNR), R01 CA169117 (JNR), R01 CA171652 (JNR), R01 NS087913 (JNR), R01 NS089272 (JNR).
Footnotes
Conflict of Interest:
The authors of this manuscript have no conflicts to disclose.
References
- 1.Santarius T, Shipley J, Brewer D, Stratton MR, Cooper CS. A census of amplified and overexpressed human cancer genes. Nature reviews Cancer. 2010;10:59–64. doi: 10.1038/nrc2771. [DOI] [PubMed] [Google Scholar]
- 2.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Akhtar-Zaidi B, Cowper-Sal-lari R, Corradin O, Saiakhova A, Bartels CF, Balasubramanian D, et al. Epigenomic enhancer profiling defines a signature of colon cancer. Science. 2012;336:736–9. doi: 10.1126/science.1217277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, et al. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–47. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Groschel S, Sanders MA, Hoogenboezem R, de Wit E, Bouwman BA, Erpelinck C, et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell. 2014;157:369–81. doi: 10.1016/j.cell.2014.02.019. [DOI] [PubMed] [Google Scholar]
- 6.Northcott PA, Lee C, Zichner T, Stutz AM, Erkek S, Kawauchi D, et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature. 2014;511:428–34. doi: 10.1038/nature13379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ryan RJH, Drier Y, Whitton H, Cotton MJ, Kaur J, Issner R, et al. Detection of Enhancer-Associated Rearrangements Reveals Mechanisms of Oncogene Dysregulation in B-cell Lymphoma. Cancer Discovery. 2015 doi: 10.1158/2159-8290.CD-15-0370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mansour MR, Abraham BJ, Anders L, Berezovskaya A, Gutierrez A, Durbin AD, et al. Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science. 2014;346:1373–7. doi: 10.1126/science.1259037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Katainen R, Dave K, Pitkanen E, Palin K. CTCF/cohesin-binding sites are frequently mutated in cancer. Nature genetics. 2015;47:818–21. doi: 10.1038/ng.3335. [DOI] [PubMed] [Google Scholar]
- 10.Kivioja T, Valimaki N, Gylfe AE, Ristolainen H, Hanninen UA, Cajuso T, et al. Recurrent somatic mutations in regulatory regions of human cancer genomes. Nature genetics. 2015;47:710–6. doi: 10.1038/ng.3332. [DOI] [PMC free article] [PubMed] [Google Scholar]