

Abstract
The purpose of this study is to develop and validate an UPLC-MS/MS method to quantify different marker compounds from Xiao-Chai-Hu-Tang (XCHT, a Chinese traditional herbal) in biological samples and apply the method to pharmacokinetic study. A Waters BEH C18UPLC column was used with acetonitrile/0.1% formic acid mobile phases. The mass analysis was performed in a triple quadrupole mass spectrometer using multiple reaction monitoring (MRM) with positive scan mode. A one-step protein precipitation by methanol was used to extract the analytes from blood. Seventeen commercially available compounds from the different compositing herbals were selected as markers. The results revealed that all of the calibration curves showed good linear regression (r2 > 0.9918). The intra-day and inter-day precisions (RSD) of all of these markers at three different levels were less than 15.0% and the bias of the accuracies ranged from −13.5% to 16.6%. The extraction recoveries of all of these 17 markers were from 70.8% to 113.7% and the matrix effects ranged from 71.8% to 114.8%. The stabilities of these compounds in blood were evaluated by analyzing three replicates of QC samples at three different concentrations following storage at 25°C for 6 h, 4°C for 24 h, and −80°C for 30 days. All the samples displayed 85–15% precision and accuracy after various stability tests. The validated method was successfully applied to pharmacokinetic study in A/J mouse with oral administration of XCHT. All of these markers were detected and the pharmacokinetic parameters of 8 compounds were able to be calculated. This method is sensitive and reproducible that can be used for XCHT’s in vivo study.
Keywords: Xiao-Chai-Hu-Tang, UPLC-MS/MS, Pharmacokinetics
1. Introduction
Xiao Chai Hu Tang (XCHT), a well-known Chinese Traditional Medicine, is made from seven herbals including Bupleurum falcatum, Panax ginseng, Glycyrrhiza glabra, Zingiber officinale, Scutellaria baicalensis, Zizyphus jujube, and Pinellia ternate [1, 2]. This famous formula was initially recorded in the ancient medicinal book named Shanghanlun 2000 years ago [3]. XCHT is an approved drug by the China Food and Drug Administration (CFDA) primarily used for the treatment of liver diseases and is sold as different type of formulations (e.g., pills, pellets, oral liquid). In addition, XCHT, (Sho-saiko-to in Japanese), was introduced into Japan as an oriental classical medicine from China approximately 1500 years ago, and it is manufactured in Japan as an ethical drug on a modern industrial scale in which the quality of ingredients is standardized with Good Manufacturing Practices (GMP) regulation [4].
In vitro studies in cell lines and in vivo studies in animal models suggested that XCHT has multiple pharmacological functions including inhibition of hepatitis virus [5, 6], anti-inflammatory [7–10], immune-modulating [7, 11], liver protective effect [12, 13], anti-cancer [2, 14, 15], and renal protective effect [16]. Clinical trials demonstrated that XCHT improved liver pathology in patients with chronic viral hepatitis [2, 11, 17, 18]. It is reported that XCHT has been used to treat approximately one million patients with chronic viral liver diseases, liver dysfunction, liver fibrosis, and liver carcinogenesis [19].
However, it is also reported that XCHT has non-neglectable adverse effects. For example, Hsu et al reported that XCHT induced acute hepatitis in patients with chronic liver disorder [20]. Another example is that Itoh et al reported that XCHT induced liver injuries in clinical trials [12]. XCHT could also induce hypokalemia and hypertension after long term of treatments [21]. The mechanisms of these biological effects, including both therapeutic and adverse effects, are not well-studied. It is very important to quantify the in vivo concentrations of the phytochemical components of XCHT to help doctors to establish quality standards for proper clinical utility.
Different class of compounds have been reported from XCHT including flavonoids (e.g., wogonoside, baicalin), saponins, (e.g., ginsenosides, saikosaponin), phenol compounds (e.g, zingerone, 6-gingernol) [4]. In addition, there are a few publications reported the phytochemical and quality control studies of XCHT [22–24]. However, the components that are responsible for the efficacy have not been well identified. Multiple pharmacokinetic studies have been performed with administration of XCHT in human or animal models (e.g., rats, mice), only a couple of marker compounds, such as baicalin and wogonoside, were quantified in these studies [25, 26]. Since the active compounds have not been identified from XCHT, it is very important to monitor different types of compounds in the in vivo study for the purpose of good clinical practice. In the previously studies, quite a few efficacy experiments were performed using mice, but there is no analytical method available to quantify different components from XCHT in biological samples in mice blood samples. Therefore, in this paper, we establish a sensitive LC-MS method to quantify 17 components from XCHT and apply the method to a pharmacokinetic study in mice.
2. Experimental
2.1. Chemicals and reagents
The herbals were bought from Taihe Hospital (Shiyan, Hubei Province, China). Liquiritin, glycyrrhetinic acid, wogonoside, wogonin, baicalin, baicalein, saikosaponin a (SSa), saikosaponin d (SSd), zingerone, 6-gingernol, rutin and quecertin were purchased from the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). Ginsenoside Rd (Rd), ginsenoside Rg1 (Rg1), ginsenoside Rg3 (Rg3), ginsenoside Re (Re), formononetin, and daidzein were purchased from LKT Laboratories (St. Paul, MN). The chemical structures of these standard were shown in Fig. 1. The purity of all standards was 95.0% or above. Acetonitrile and methanol were purchased from Merck (Darmstadt, Germany). Other chemicals (analytical grade or better) were used as received.
Figure 1.
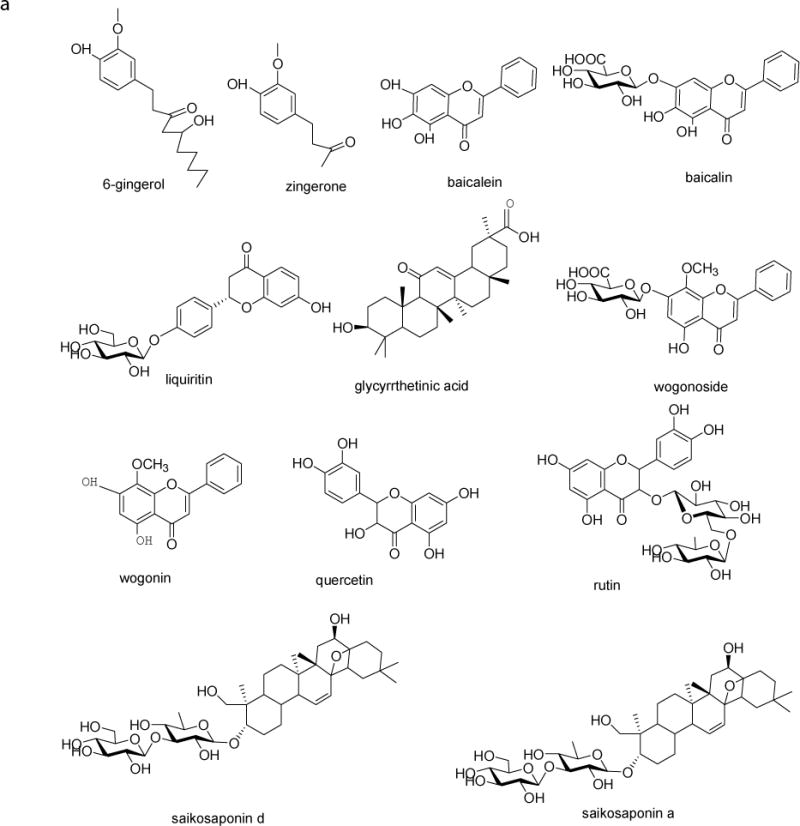
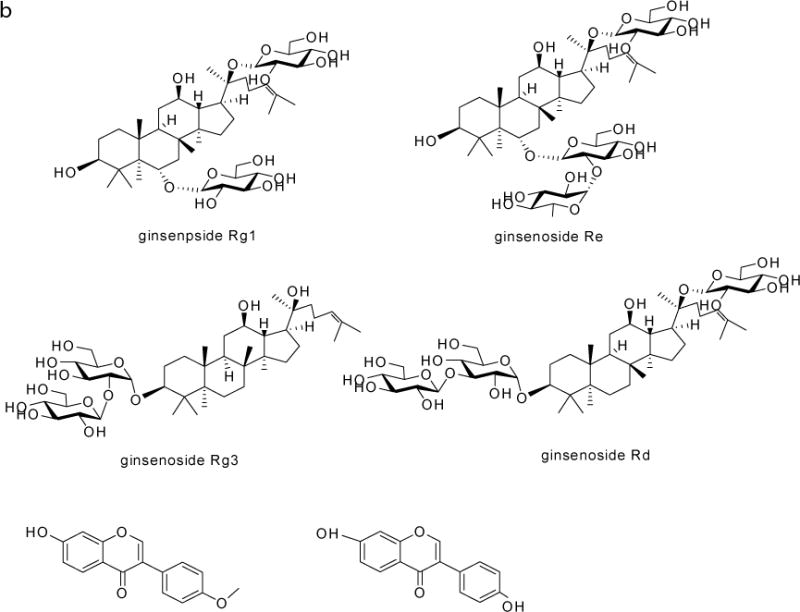
Chemical structures of the 17 marker compounds and daidzein (I.S.)
2.2. Preparation of XCHT
The powdered herbal materials, including Bupleuri Radix (12 g), Scutellariae Radix (9 g), Ginseng Radix (9 g), Glycyrrhizae Radix (6 g), Pinelliae Tuber (9 g), Zingiberis Rhizoma (9 g), and Jujubae Fructus (12 g), were extract twice with 8-fold volumes of water under reflux for 1 h. The combined filtrate was concentrated and abrown sticky extract (2 g/mL) was afforded. The sample for quality control was prepared by diluting the original extract for 1000-folds in 50% methanol. Before injection, the quality control samples were centrifuged for 15 min at 15,000 rpm, then, 100 μL of the supernatant was filtered through a 0.22 μm membrane and 20 μL of internal standard (daidzein 0.5 μM) was added for LC-MS analysis.
2.3. Instruments and conditions
2.3.1 UPLC
The UPLC conditions were: Waters Acquity™ with diode array detector (DAD); column, Acquity UPLC BEH C18 column (50 mm × 2.1 mm I.D., 1.7 μm, Waters, Milford, MA, USA); mobile phase A (MPA), 0.1% formic acid in water; mobile phase B (MPB), acetonitrile; gradient, 0–2 min, 5% B; 2.0–3.0 min, 10% B; 3.0–5.0 min, 10–15% B; 5.0–9.0 min, 15–20% B; 9.0–11.0 min, 20–40% B; 11.0–14.0 min, 40–45% B; 14.0–14.5 min, 45–60% B; 14.5–15.0 min, 60–100% B; 15.0–15.2 min, 100%; 15.2–16.0 min, 100–5% B; 16.0–17.0 5% B; column temperature, 45°C; sample temperature, 20°C; and injection volume, 10 μL.
2.3.2 MS
The MS analysis was performed on an API 5500 Qtrap triple quadrupole mass spectrometer (Applied Biosystem/MDS SCIEX, Foster City, CA, USA) equipped with a TurboIonSpray™ source. The compounds were determined by using MRM (multiple reaction monitoring) scan type in positive mode. The instrument dependent parameters for mass spectrum were set as follows: ion-spray voltage, 5.5 kV; ion source temperature, 500°C; nebulizer gas (gas 1), nitrogen, 40 psi; turbo gas (gas 2), nitrogen 40 psi; curtain gas, nitrogen 10 psi. Unit mass resolution was set in both mass-resolving quadruples Q1 and Q3. Compound-dependent parameters were listed in Table 1.
Table 1.
Compound-dependent parameters and Monitored ion pairs for 17 index compounds and daidzein (I.S.) in MRM mode for UPLC–MS/MS analysis.
| Analyte | Precursor ion | Product ion | Dwell | DP (V) | CE (V) | CXP (V) |
|---|---|---|---|---|---|---|
| (m/z) Q1 | (m/z) Q3 | Time (ms) | ||||
| Liquiritin | 419.3 | 257 | 100 | 29 | 14 | 12 |
| Rutin | 611 | 303 | 100 | 42 | 28 | 13 |
| Zingerone | 195 | 137 | 100 | 35 | 27 | 36 |
| Baicalin | 447 | 271 | 100 | 15 | 30 | 9 |
| Quercetin | 303 | 279 | 100 | 62 | 41 | 19 |
| Rg1 | 823 | 643.6 | 100 | 44 | 51 | 34 |
| Re | 969.7 | 789.6 | 100 | 23 | 59 | 13 |
| Wogonoside | 461 | 285 | 100 | 36 | 10 | 24 |
| Baicalein | 271 | 123 | 100 | 16 | 43 | 11 |
| Formononetin | 269 | 253 | 100 | 46 | 38 | 17 |
| Wogonin | 285 | 270 | 100 | 15 | 38 | 11 |
| (6)-Gingerol | 295 | 137 | 100 | 16 | 28 | 12 |
| SSa | 781.5 | 455 | 100 | 41 | 21 | 25 |
| Rd | 969.6 | 789.4 | 100 | 34 | 65 | 18 |
| SSd | 781.3 | 455 | 100 | 16 | 24 | 17 |
| Rg3 | 807.5 | 365 | 100 | 23 | 59 | 13 |
| GA | 471 | 137 | 100 | 24 | 43 | 13 |
| Daidzein | 255 | 153 | 100 | 26 | 42 | 13 |
DP: declustering potential; CE: collision energy; CXP: collision cell potential
2.4. Preparation of standard and quality control samples
The stock solutions of the 17 compounds were prepared in ethanol/DMSO (4:1) at a final concentration of 10 mM, respectively. To prepare standard curve samples in blood, the stock solution was serially diluted in 50% methanol to make a working solution at 0.6, 1.2, 2.4, 4.8, 9.77, 19.5, 39.1, 78.1, 156.0, 313.0, 625.0, 1,250.0, 2,500.0, 5,000.0, and 10,000.0 nM for baicalin, baicalein, wogonoside, formononetin, zingerone, 6-gingernol, rutin, quecertin, liquiritin, glycyrrhetinic acid, Rd, Rg1, Rg3, Re and 0.061, 0.122, 0.244, 0.488, 0.977, 1.95, 3.91, 7.81, 15.6,31.3, 62.5,125.0,250.0,500.0,1,000.0 nM for rutin, SSa, SSd. The working solution samples (10 μL) were then spiked into 10 μL bland mouse blood, add 2 μl of Vitamin C (20%), then the samples were extracted with 200 μL internal standard solution (0.5 μM daidzein in methanol) by vortex-mixing for 1 min. After centrifugation at 20,000 × g for 15 min, the supernatant was transferred to a new tube and evaporated to dryness under a stream of air. The residue was reconstituted in 80 μL of 50% acetonitrile and centrifuged at 20,000 × g for 15 min. IS working solution (0.5 μM) was prepared by diluting the stock solution in acetonitrile. The final concentrations of these analytes were0.006, 0.012, 0.024, 0.048, 0.098, 0.195, 0.391, 0.781, 1.563, 3.13, 6.25, 12.5, 25, 50, 100 nM for rutin, SSa, SSd; 0.061, 0.122, 0.244, 0.488, 0.977, 1.95, 3.91, 7.81, 15.6, 31.3, 62.5, 125.0, 250.0, 500.0, 1,000.0 nM for baicalin, baicalein, wogonoside, wogonin, formononetin, zingerone, 6-gingernol, rutin, quecertin, liquiritin, glycyrrhetinic acid, Rd, Rg1, Rg3, Re. The quality control (QC) samples were prepared at three different concentrations in the same way.
2.5. Method validation
2.5.1. Specificity and LLOD
The specificity of the method was determined by analyzing different blood samples for interference at the retention times of the analytes. Specificity was assessed by comparing the peak of an analyte in blank blood sample to that in a blank blood sample spiked with analyte at 0.024 nM for rutin, SSa, SSd and others at 0.24 nM.
2.5.2. Linearity and LLOD
Calibration curves were prepared the same way as described in section 2.4. The linearity of each calibration curves were determined by plotting the peak area ratio of the 17 analytes to I.S. in mice blood. Least-squares linear regression method (1/x2 weight) was used to determine the slope, intercept and correlation coefficient of linear regression equation. The lower limit of detection (LLOD) was defined based on a signal-to-noise ratio of 10:1.
2.5.3. Precision and accuracy
The intra/inter-day precision and accuracy were determined by injecting three different concentration of QC samples on the same day or on three different days. The precision was evaluated by relative standard deviation (RSD), and accuracy was expressed as relative error (RE).
2.5.4. Extraction recovery and matrix effect
The extraction recoveries of the 17 analytes, together with the I.S., were evaluated by comparing the relative peak areas obtained from blank blood spiked with analytes and those obtained from water spiked with the same amount of analytes. The matrix effect was determined by comparing the relative peak areas obtained from blank blood extract spiked with these analytes to those from mobile phase extract spiked with the same amount of the analytes. These evaluations were performed according to the recommended validation procedures reported by Matuszewski [27].
2.5.5. Stability
The stabilities of these compounds were tested by analyzing 3 replicate QC samples at three different concentrations. The freeze and thaw stability were determined after three freeze-thaw cycles (from −20°C to 25°C on consecutive days. Long-term stability was studied by storing QC samples at −80°C for 30 days. Short-term stability was assessed by analyzing QC samples kept at room temperature for 6 h. The post-preparation stability was tested by determining the extracted QC samples kept in the auto-sampler at 4°C for 24 h.
2.6. Application in pharmacokinetic study
2.6.1. Animals
The animal protocol used in this study was approved by the University of Houston’s Institutional Animal Care and Uses Committee. Male A/J mice (22–25 g, 8–10 weeks old) were from Harlan Laboratory (Indianapolis, IN) and kept in an environmentally controlled room (temperature: 25±2°C, humidity: 50±5%, 12 h dark-light cycle) for at least 1 week before the experiments.
2.6.2. Experimental design
Mice were fasted for 12 h with free access of water prior to the pharmacokinetic experiment. The crude XCHT extract was suspended in the oral suspension vehicle and administrated by oral gavage at a dose of 500 mg/kg. Blood samples (about 20 μL) were collected in heparinized tubes at 0, 5 min, 15 min, 30 min, 1, 2, 3, 4, 6, 8, 12, and 24 h by snipping the tail and stored at −20°C until analysis.
2.6.3. Sample preparation
The blood (10 μL) was spiked with 10 μl of50% methanol, add 2 μL Vitamin C (20%). The sample other prepared steps the same way as described in section 2.4.
2.6.4 Data Analysis
WinNonlin 3.3 (Pharsight, Mountain View, CA) was used for the pharmacokinetic data analysis and the non-compartmental model was applied.
2.6.5 Statistical Analysis
The data in this study were presented as means±S.D., if not specified otherwise. Significance differences were assessed by using Student’s t test or one-way analysis of variance. A p<0.05 was considered asstatistically significant.
3. Results and discussion
3.1. Method development
The chromatographic conditions were optimized to improve the peak shape, sensitivity and through-put. Different mobile phases including different concentration of formic acid (pH from 2, 2.5, 3 and 4), ammonium acetate (2.5 mM, strong ammonia adjusted pH 6.5, 7.4, 8.0 and 9.0), methanol, and acetonitrile were tested as the mobile phase. The 0.1% of formic acid and acetonitrile were selected as mobile phases. A representative chromatogram is shown in Fig. 2.
Figure 2.
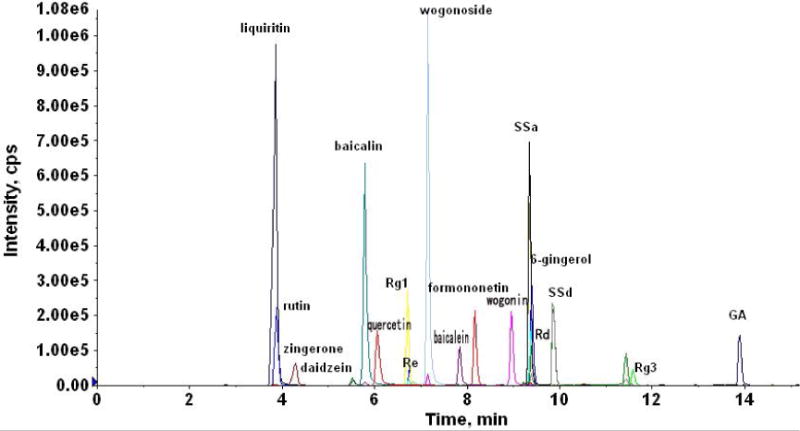
A representative MRM chromatogram of the 17 marker compounds and daidzein (I.S.).
To improve the septicity, MRM (multiple reaction monitoring) scan type was used in this analysis. To improve the sensitivity, the compound dependent parameters and the instrument dependent parameters were optimized by tuning the 17 standard analytes with infusion. Both negative and positive mode were tested. The optimized MS/MS transitions and the compound dependent parameters of all the analyte sand IS were showed in Table 1.
3.2. Method validation
3.2.1. Specificity
There is no significant interference with the 17 analytes in the chromatogram. The retention times of liquiritin, rutin, zingerone, baicalin, quecertin, Rg1, Re, wogonoside, baicalein, formononetin, wogonin, 6-gingerol, SSa, Rd, SSd, Rg3, GA were 3.85, 3.88, 4.28, 5.78, 6.05, 6.69, 6.73, 7.13, 7.81, 8.15, 8.93, 9.34, 9.36, 9.36, 9.84, 11.55, and 13.87, respectively. A representative MRM chromatograms of blank plasma; blank plasma spiked with the analytes at LLOQs, and plasma samples after oral administration of XCHT extract for 0.25 h (for GA at 4 h) was shown in Fig 4.
Figure 4.
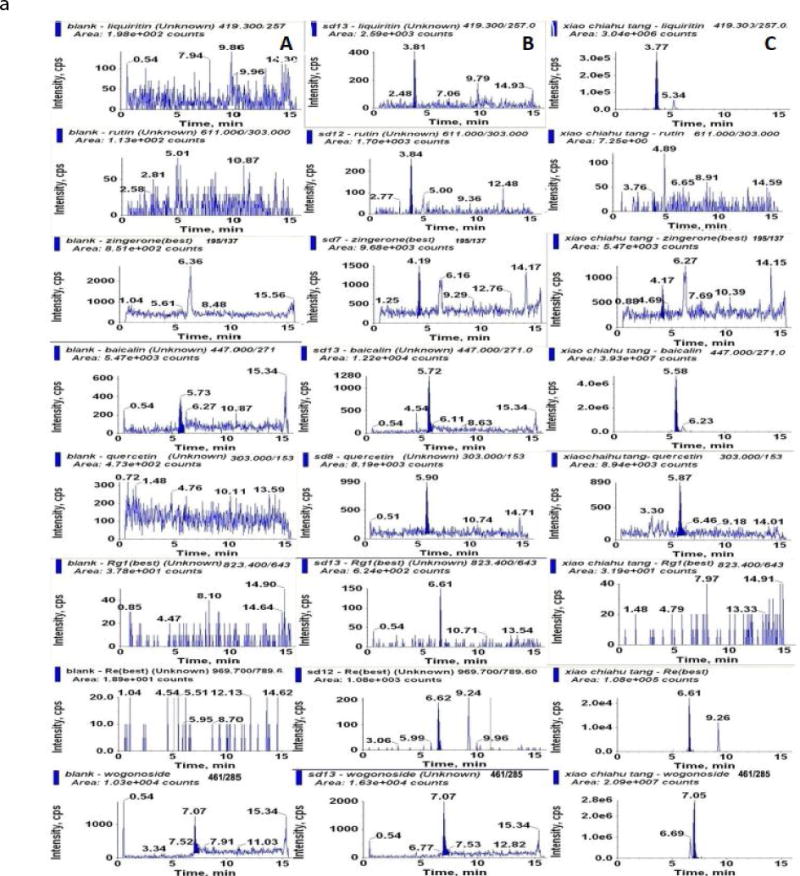
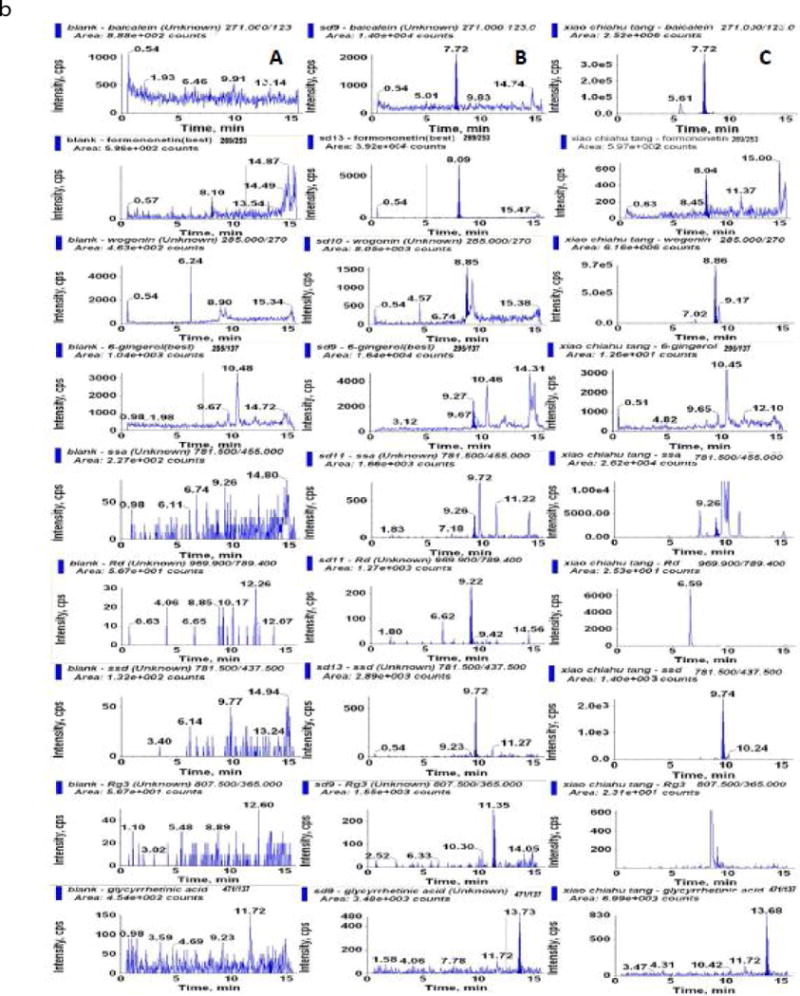
Representative MRM chromatograms of (A) blank plasma; (B) blank plasma spiked with the analytes at LLOQs; (C) plasma samples after oral administration of XCHT extract for 0.25 h (for GA at 4 h).
3.2.2. Linearity of calibration curves and LLOQs (low limit of quantification)
The calibration curves and LLOQs of the seventeen analytes were summarized in Table 2. All calibration curves exhibited good linearity with correlation coefficient (r) within the range of 0.9918–0.9984. The LLOQs were appropriate for quantitative detection of analytes in the pharmacokinetic studies.
Table 2.
The regression equations, linear range, LLOD and LLOQs of the seventeen analytes in blood
| Components | Range (nM) | Calibration curves | Correlation coefficient (r) | LLOQ (nM) | LLOD (nM) |
|---|---|---|---|---|---|
| Liquiritin | 0.122~1000 | Y=0.125X+0.00005 | 0.9984 | 0.122 | 0.061 |
| Rutin | 0.0488~100 | Y=0.183X+0.00054 | 0.9937 | 0.0488 | 0.0488 |
| Zingerone | 1.95~1000 | Y=0.0063X+0.0113 | 0.9942 | 1.95 | 0.244 |
| Baicalin | 1.95~1000 | Y=0.00242X+0.00390 | 0.9918 | 1.95 | 0.122 |
| Quercetin | 0.244~1000 | Y=0.0151X+0.004 | 0.9951 | 0.244 | 0.244 |
| Rg1 | 0.122~1000 | Y=0.0327X+0.00262 | 0.9946 | 0.122 | 0.061 |
| Re | 0.488~1000 | Y=0.00561X+0.0003 | 0.9977 | 0.488 | 0.488 |
| Wogonoside | 0.977~1000 | Y=0.519X+0.87 | 0.9939 | 0.977 | 0.244 |
| Baicalein | 0.488~1000 | Y=0.114X+0.688 | 0.9933 | 0.488 | 0.061 |
| Formononetin | 0.977~1000 | Y=0.129X+0.968 | 0.9935 | 0.977 | 0.061 |
| Wogonin | 0.488~1000 | Y=0.601X+1.43 | 0.9947 | 0.488 | 0.061 |
| (6)-Gingerol | 1.95~1000 | Y=0.0508X+2.04 | 0.9960 | 1.95 | 0.977 |
| SSa | 0.0488~100 | Y=0.0851X+0.0003 | 0.9930 | 0.0488 | 0.0488 |
| Rd | 0.977~1000 | Y=0.0157X+0.003 | 0.9943 | 0.977 | 0.122 |
| SSd | 0.0488~100 | Y=0.133X+0.0066 | 0.9966 | 0.0488 | 0.0244 |
| Rg3 | 0.488~1000 | Y=0.00549X+0.00115 | 0.9937 | 0.488 | 0.244 |
| GA | 0.488~1000 | Y=0.0552X+0.0275 | 0.9965 | 0.488 | 0.244 |
3.2.3. Precision and accuracy
The intra-day and inter-day precisions (RSD) at three different levels were both less than 15.0% and the accuracies (RE) ranged from −13.7% to 14.8% (Table 3)
Table 3.
Intra-day and inter-day precisions and accuracies for the determination of seventeen compounds from the assay samples (mean±SD, n = 3)
| components | C (nM) | Inter-day | Intra-day | ||||
|---|---|---|---|---|---|---|---|
| Observed C (nM) | Precise (RSD, %) | Accuracy (Re, %) | Observed C (nM) | Precise (RSD, %) | Accuracy (Re, %) | ||
| Liquiritin | 0.488 | 0.571±0.079 | 13.9 | 11 | 0.5±0.027 | 5. 4 | 2.5 |
| 62.5 | 57.93±0.42 | 4.1 | −7.3 | 62.4±6.44 | 10.3 | −0.1 | |
| 500 | 470.3±34.9 | 7.4 | −5.9 | 521.7±19.3 | 3.7 | 4.3 | |
| rutin | 0.0488 | 0.0536±0.006 | 11.2 | 13 | 0.0485±0.004 | 8.7 | −0.5 |
| 6.25 | 6.29±0.609 | 9.7 | 0.6 | 6.25±0.63 | 10.1 | 0 | |
| 50 | 45.6±3.3 | 7.2 | −8.7 | 49.1±6.9 | 14.1 | −1.8 | |
| zingerone | 3.9 | 3.34±0.452 | 13.47 | 12.7 | 3.37±0.46 | 13.6 | −13.5 |
| 62.5 | 56.03±6.5 | 11.6 | −10.3 | 62±6.4 | 10.3 | −0.8 | |
| 500 | 489±12.4 | 12.4 | −2.2 | 522.33±56.72 | 10.9 | 4.5 | |
| baicalin | 3.9 | 0.369±0.0313 | 8.46 | 8.46 | 3.87±0.47 | 12.1 | −0.8 |
| 62.5 | 1.793±0.298 | 16.6 | 16.6 | 54.5±4.9 | 8.9 | −12.8 | |
| 500 | 52.56±1.401 | 2.67 | 2.67 | 513±67 | 13.1 | 2.6 | |
| quercetin | 3.9 | 4.13±0.298 | 7.22 | 7.22 | 4.36±0.395 | 9.1 | 11.7 |
| 62.5 | 60.2±7.3 | 12.2 | −3.6 | 60.4±7.4 | 12.3 | −3.3 | |
| 500 | 438.7±21.22 | 4.8 | −12.3 | 470±44.9 | 9.6 | −6 | |
| Rg1 | 0.488 | 0.466±0.065 | 13.9 | −4.5 | 0.448±0.017 | 3.7 | −8.2 |
| 62.5 | 59.13±7.22 | 12.2 | −5.4 | 71.7±5.76 | 8 | 14.8 | |
| 500 | 468±22.91 | 4.9 | −6.4 | 474±13.5 | 2.9 | −5.2 | |
| Re | 3.9 | 3.63±0.467 | 12.9 | −6.9 | 3.52±0.274 | 7.8 | −9.7 |
| 62.5 | 66.6±3.86 | 5.8 | 6.6 | 68.7±3.1 | 4.5 | 9.9 | |
| 500 | 497.33±6.65 | 1.3 | −0.5 | 472±27.1 | 5.7 | −5.6 | |
| wogonoside | 0.488 | 0.463±0.03 | 6.5 | −5.1 | 0.469±0.001 | 0.86 | −4 |
| 62.5 | 58.7±4.1 | 7 | −4.1 | 63.8±0.69 | 1.1 | 2.1 | |
| 500 | 472.3±28.3 | 6 | −5.5 | 488±41.7 | 8.5 | −2.4 | |
| baicalein | 3.9 | 3.84±0.344 | 8.95 | −0.03 | 3.90±0.21 | 5.5 | 0.2 |
| 62.5 | 62.1±7.1 | 11.4 | −0.6 | 64.97±4.5 | 7 | 3.9 | |
| 500 | 423.3±20.5 | 4.8 | −15.3 | 453.3±11.84 | 2.6 | −9.3 | |
| Formononetin | 3.9 | 4.1±0.28 | 4.8 | 5.1 | 3.47±0.129 | 3.7 | −10.9 |
| 62.5 | 63.56±2.9 | 4.6 | 1.7 | 72.4±6.05 | 8.3 | 15.9 | |
| 500 | 491±13.2 | 2.7 | −1.8 | 475.3±26.9 | 5.7 | −4.9 | |
| wogonin | 0.488 | 0.461±0.014 | 3 | −6.5 | 0.421±0.0456 | 10.9 | −13.7 |
| 62.5 | 57.3±3.38 | 5.9 | −8.4 | 62.9±3.91 | 6.2 | 0.6 | |
| 500 | 471±36.3 | 7.7 | −5.8 | 486.3±23.5 | 4.8 | −2.7 | |
| (6)-Gingerol | 3.9 | 3.93±0.19 | 4.83 | 0 | 4.08±0.145 | 3.6 | 4.7 |
| 62.5 | 54.06±6.38 | 11.8 | −12.6 | 65.6±6.4 | 9.7 | 5 | |
| 500 | 443.7±67.7 | 15.3 | −11.3 | 514.3±33.5 | 6.5 | 2.9 | |
| SSa | 0.39 | 0.367±0.05 | 13.8 | −5.8 | 0.043±0.006 | 12.1 | 0.7 |
| 6.25 | 5.84±0.517 | 8.9 | −6.5 | 6.04±0.262 | 4.3 | −3.4 | |
| 50 | 48.96±6.14 | 12.5 | −2.1 | 52.6±1.97 | 3.7 | 5.2 | |
| Rd | 0.488 | 0.504±0.071 | 14.16 | 3.3 | 0.479±0.047 | 9.9 | −1.7 |
| 62.5 | 63.5±7.0 | 11 | 1.6 | 60.86±3.9 | 6.5 | −2.6 | |
| 500 | 527±33.45 | 6.3 | 5.4 | 477.7±23.54 | 4.9 | −4.5 | |
| SSd | 0.0488 | 0.050±0.006 | 12 | 1 | 0.0398±0.0 | 0 | −18 |
| 6.25 | 6.16±0.44 | 7.2 | −1.4 | 6.59±0.113 | 1.7 | 5.4 | |
| 50 | 49±3.16 | 6.4 | −1.9 | 50.16±2.35 | 4.7 | 0.3 | |
| Rg3 | 0.488 | 0.482±0.074 | 15.1 | −1.1 | 0.466±0.0056 | 1.2 | −4.5 |
| 62.5 | 59.7±1.31 | 2.2 | −4.5 | 63.5±2.54 | 4 | 1.7 | |
| 500 | 461.7±36.5 | 7.9 | −7.7 | 494.3±27.31 | 5.5 | −1.1 | |
| GA | 0.488 | 0.462±0.038 | 10.8 | −5.4 | 0.489±0.0356 | 7.3 | 0.3 |
| 62.5 | 58.7±6.17 | 10.5 | −6.1 | 63.46±5.61 | 8.8 | 1.5 | |
| 500 | 485.3±33 | 6.8 | −2.9 | 529.33±25 | 4.7 | 5.9 | |
3.2.4. Recovery and matrix effect
The recoveries of these 17 analytes ranged from 70.8 to 115% (Table 4). The matrix effects were between 71.8% and 114.6% suggesting that there was no significant ion suppression in this method.
Table 4.
The Matrix effects, extraction recoveries and stability of seventeen analytes in mice plasma under different storage conditions
| Component | Spiked concentration |
Matrix effect (%, mean± SD, n = 3) |
Recovery (%, mean± SD, n = 3) |
25°C for 6 h | 4°C for 24 h | freeze–thaw | Frozen for 30 days | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Precision (RSD, %) |
Accuracy (RE, %) |
Precision (RSD, %) |
Accuracy (RE, %) |
Precision (RSD, %) |
Accuracy (RE, %) |
Precision (RSD, %) |
Accuracy | ||||
| (RE, %) | |||||||||||
| Liquiritin | 0.488 | 103±16.7 | 87.7±14.8 | 14.5 | 7.3 | 9.4 | 14.4 | 14.4 | 13.6 | 9.4 | 13.9 |
| 62.5 | 97.8±5.8 | 99.1±1.6 | 14.4 | 5.5 | 3.8 | 15 | 4.0 | 12.5 | 3.3 | 13.8 | |
| 500 | 103.3±4.6 | 92.8±6.6 | 4.9 | 12.7 | 4.4 | 12.8 | 5.6 | 14.0 | 1.6 | 10.9 | |
| rutin | 0.049 | 71.8±10.4 | 98.7±6.39 | 10.9 | 13.2 | 12.3 | 9.3 | 14.7 | 11.7 | 12.3 | 8.3 |
| 6.25 | 101.6±2.5 | 82.5±3.3 | 7.2 | 3.1 | 1.1 | 12.7 | 13.6 | 11.9 | 1.1 | 8.5 | |
| 50 | 94.8±3.2 | 79.9±5.3 | 9.3 | 6.3 | 9.9 | 12.1 | 11.1 | 16.7 | 9.9 | 7.9 | |
| zingerone | 3.9 | 75.9±12.9 | 70.8±12.9 | 5.3 | 7.5 | 13.8 | 4.6 | 8.9 | 5.8 | 11.8 | −5.9 |
| 62.5 | 93.1±10.4 | 101.2±15.4 | 8.7 | −8.6 | 9.2 | −1 | 0.5 | −13.2 | 9 | −0.9 | |
| 500 | 105.7±2.6 | 86.7±8.4 | 8.1 | −14.6 | 11.5 | 11.4 | 14.1 | 7.9 | 5.1 | 12.7 | |
| baicalin | 3.9 | 72.2±6.7 | 75.4±1.1 | 6.7 | −5.7 | 15.9 | 3.8 | 15.0 | 14.7 | 14.9 | 6.7 |
| 62.5 | 80.3±8.3 | 76.8±1.1 | 10.8 | −5.3 | 6 | 14.8 | 12.5 | −1.1 | 6 | 14.1 | |
| 500 | 91.7±6.9 | 72.8±4.2 | 14.3 | 1.1 | 7.9 | 5.2 | 4.7 | 9.6 | 7.9 | 4.9 | |
| quercetin | 3.9 | 116.7±16.7 | 85.3±8.1 | 12.9 | −4.8 | 12.8 | −1.3 | 8.7 | −10.5 | 8.2 | −2.6 |
| 62.5 | 101.7±12.4 | 85.4±3.5 | 11.5 | −12.9 | 11.1 | 5.12 | 13.1 | −13.0 | 11.1 | 4.8 | |
| 500 | 96.5±5.2 | 86.2±6.1 | 3.3 | 10.1 | 3.3 | 14.72 | 6.2 | 12.9 | 3.3 | 14.7 | |
| Rg1 | 0.488 | 114.6±15.3 | 110.6±15.6 | 12.4 | 14.8 | 7.1 | 4.7 | 9.3 | −6.9 | 10.5 | 7.2 |
| 62.5 | 107±13.2 | 109.1±9.0 | 6.3 | 7.4 | 2.1 | 10.1 | 0.9 | 10.2 | 2.1 | 9.2 | |
| 500 | 104±7.8 | 104.5±12.4 | 11.2 | 8.5 | 0.9 | 12.12 | 12.3 | 12.1 | 0.9 | 12.5 | |
| Re | 3.9 | 99.9±17.4 | 106.4±5.4 | 14.6 | 10.1 | 11.82 | −5.4 | 10.3 | 7.1 | 8 | 10.4 |
| 62.5 | 101±12.8 | 112.1±7.9 | 9.9 | 8.8 | 2.4 | 13.76 | 9.2 | 14.3 | 2.4 | 9.7 | |
| 500 | 106.5±5.5 | 105.3±9.6 | 14.3 | −0.3 | 6.8 | 9.7 | 13.3 | 4.5 | 6.8 | 8.9 | |
| wogonoside | 0.488 | 110.2±12.9 | 81.2±6.2 | 10.2 | 14.1 | 0.86 | 2.3 | −14.1 | −12.7 | 13.7 | −10.9 |
| 62.5 | 111.8±9.6 | 83.4±1.7 | 9.4 | 8.5 | 12.7 | 15.1 | 13.0 | 11.9 | 12.7 | 10.1 | |
| 500 | 99.2±3.2 | 87.8±7 | 6.8 | 1.4 | 14.9 | 12.6 | 13.7 | 12.4 | 14.9 | 11.2 | |
| baicalein | 3.9 | 89.5±14.9 | 84±13.6 | 14.0 | 10.3 | 10.1 | 12.3 | 10.3 | −8.9 | 8.4 | −11.4 |
| 62.5 | 88.9±12 | 82.3±2.6 | 7.3 | 9.9 | 5 | −14 | 10.3 | −13.7 | 5 | −14.5 | |
| 500 | 96.6±0.9 | 83.6±8.4 | 8.3 | 0.7 | 10.3 | 13.1 | 16.1 | 3.5 | 10.3 | 9 | |
| Formononetin | 3.9 | 112.7±14.5 | 95.2±12.2 | 10.7 | 8.0 | 14.7 | −8.9 | 8.4 | 8.6 | 14.7 | 13.5 |
| 62.5 | 115±17 | 97±10.6 | 10.7 | 11.8 | 8.6 | 13.6 | 5.1 | 15.3 | 8.6 | 13 | |
| 500 | 93.3±10.2 | 93.5±9.1 | 11.5 | −10.9 | 6.7 | 5 | 10.9 | 1.4 | 6.7 | 4.9 | |
| wogonin | 3.9 | 100.9±10.3 | 91.6±6.3 | 9.3 | 11.4 | 15.2 | 9.2 | 7.3 | 13.1 | 15.2 | 8.4 |
| 62.5 | 96.7±11.6 | 96±13.5 | 13.6 | 9.5 | 11 | 14.8 | 15.2 | 13.4 | 11 | 15 | |
| 500 | 105.7±9.8 | 89.7±6.1 | 12.8 | 3.1 | 9.9 | 13.5 | 14.8 | 14.0 | 9.9 | 10.3 | |
| (6)-Gingerol | 3.9 | 111.7±10.3 | 109.1±11.7 | 9.9 | 2.8 | 10.2 | 2.6 | 10.9 | 3.7 | 10.8 | 4.4 |
| 62.5 | 86.8±8.6 | 81.1±10.9 | 6.0 | −2.3 | 9.8 | −8.4 | 5.6 | −3.8 | 9.2 | −12.1 | |
| 500 | 99.4±6.6 | 81.7±6.7 | 10.9 | −1.0 | 11.5 | 2 | 12.2 | 2.4 | 11.5 | 1.2 | |
| SSa | 0.39 | 105.1±15.4 | 91±8.5 | 10.7 | 0.7 | 12 | −7.4 | 10.4 | 0.8 | 12 | −6.7 |
| 6.25 | 101.8±12.1 | 100.4±9.6 | 12.4 | −1.5 | 8.3 | 12 | 13.2 | 8.2 | 8.3 | 10.8 | |
| 50 | 97.6±5.1 | 103±8.5 | 14.9 | 7.4 | 7.5 | 10.6 | 7.9 | 16.9 | 7.5 | 12.1 | |
| Rd | 0.488 | 85.3±17.2 | 98.2±11.2 | 9.3 | 13.3 | 13.8 | −4 | 8.9 | 9.5 | 11.9 | 11.6 |
| 62.5 | 113.3±12.4 | 107±12.9 | 14.1 | −3.7 | 2.7 | 12.8 | 10.6 | 4.5 | 2.7 | 11.4 | |
| 500 | 102±4.5 | 98.8±8.9 | 12.6 | −7.1 | 10.9 | 12.3 | 16.0 | 9.6 | 10.9 | 12.2 | |
| SSd | 0.049 | 108.3±15.2 | 98.06±16.6 | 2.7 | 6.8 | 10.4 | −19 | 3.4 | 8.6 | 10.4 | 8.7 |
| 6.25 | 86.7±13.6 | 115±10.3 | 9.2 | −13.0 | 1.7 | 13.1 | 12.3 | 12.7 | 1.7 | 12.5 | |
| 50 | 105.9±11.9 | 102.6±13.1 | 12.4 | 2.4 | 6.6 | 14.5 | 11.5 | 12.3 | 6.6 | 15.0 | |
| Rg3 | 0.488 | 112.7±3.6 | 113.7±14.9 | 12.8 | 9.2 | 5.5 | 1.57 | 5.7 | 7.4 | 5.5 | 8.0 |
| 6.25 | 114.8±13.6 | 104±3.6 | 6.2 | 9.2 | 2.4 | 8.64 | 3.3 | 7.4 | 2.4 | 8.0 | |
| 500 | 103.9±4.3 | 99.2±9.2 | 9.3 | −4.3 | 7.3 | 0.4 | 5.0 | 3.3 | 7.3 | 0.4 | |
| GA | 3.9 | 113±8.9 | 92.8±10 | 9.4 | 2.1 | 11.8 | 5.4 | 7.8 | 0.7 | 3.7 | 6.9 |
| 62.5 | 106.4±5.7 | 92.2±9.4 | 11.1 | 1.6 | 2.6 | 12 | 12.6 | 6.4 | 2.6 | 14.0 | |
| 500 | 92.6±5.4 | 89.9±9.1 | 5.8 | 8.1 | 6.2 | 14.6 | 9.2 | 11.9 | 6.2 | 13.5 | |
3.2.5. Stability
The results of the stability studies showed that all of the 17 analytes were stable in mouse blood samples for 1 month at −80degree (RE: −12.6% to 14.8%, RSD <14.5%), within three freeze-thaw cycles (RE: −13.7% to 14.7%, RSD < 15%), 4°C for 24 h (RE: −14% to 14.8%, RSD < 15%) and for 6 h at room temperature (RE: −14.6% to 14.8%, RSD < 14.9%).
3.3 Selection of the marker compounds
Totally 17 commercially available compounds were selected as markers in the analysis. These markers are identified from different herbs: SSd, SSa, rutin, and quercetin are from Bupleurum falcatum, Rd, Re, Rg1, and Rg3 are from Panax ginseng, liquiritin, and GA arefrom glycyrrhiza glabra, zingerone, and 6-gingerolare from zingiber officinale, baicalin, quecertin, wogonoside, baicalein, formononetin, and wogonin are from scutellaria baicalensis. We selected different compounds from the five herbs in this method to quantify these components in the pharmacokinetic study.
3.4 Contents of the 17 marker compounds in the XCHT
The contents of the 17 marker compounds in XCHT extract were determined by LC-MS method. The results showed that the contents per gram of XCHT extract were liquiritin 17.6 mg, rutin, 0.30 mg, zingerone, 0.13 mg, baicalin, 58.9 mg, quecertin, 0.02 mg, Rg1, 1.29 mg, Re, 30.9 mg, wogonoside, 15.8 mg, baicalein, 8.8 mg, formononetin, 0.05 mg, wogonin, 13.5 mg, 6-gingerol, 0.96 mg, SSa, 0.39 mg, Rd, 2.35 mg, SSd, 8.96 mg, Rg3, 0.60 mg, and GA, 0.03 mg.
3.5 Pharmacokinetic study
The validated method was applied to determine the blood concentrations of 17 components in mice after oral administration of XCHT extract at a dose of 0.5 g/kg (equivalent to 8.8 mg/kg of liquiritin, 0.15 mg/kg of rutin, 0.07 mg/kg of zingerone, 29.5 mg/kg ofbaicalin, 0.01 mg/kg of quecertin, 0.65 mg/kg of Rg1, 15.5 mg/kg of Re, 7.9 mg/kg of wogonoside, 4.4 mg/kg of baicalein, 0.03 mg/kg of formononetin, 6.8 mg/kg of wogonin, 0.48 mg/kg of 6-gingerol, 0.19 mg/kg of SSa, 1.17 mg/kg of Rd, 4.48 mg/kg of SSd, 0.30 mg/kg of Rg3, 0.02 mg/kg of GA). The result showed that at this dose, only liquiritin, baicalin, Re, wogonoside, baicalein, wogonin, SSd, and GA were detected in the blood. The other compounds were not detected due to low dose. The mean blood concentration-time profiles of these detected compounds are shown in Fig. 3. The estimated pharmacokinetic parameters are listed in Table 5.
Figure 3.
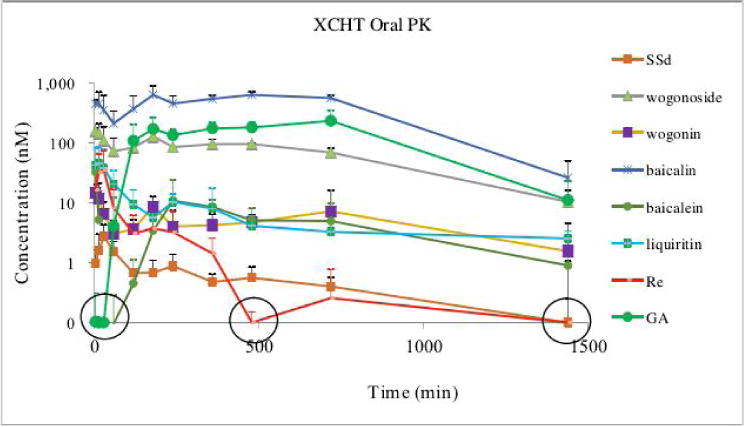
Plasma concentrations of the detected compounds after p.o. administration of XCHT in A/J mice. Blood sample (10 μL) was spiked with 10 μl of 50% methanol, add 2 μL Vitamin C (20%), which was further extracted by 200 μL internal standard solution (0.5 μM daizein in methanol) by vortex-mixing for 1 min. After centrifugation at 20,000 × g for 15 min, the supernatant was transferred to a new tube and evaporated to dryness under a stream of air. The residue was reconstituted in 80 μL of 50% acetonitrile and centrifuged at 20,000 × g for 15 min for LC-MS injection. The circled concentrations were out of the linear range.
Table 5.
Pharmacokinetic parameters of the 8 analytes in male A/J mouse after oral administration of XCHT (mean±SD, n=6); Tmax:median (min~max)
| Compounds | Cmax (nmol/L) | Tmax (h) | t1/2 (h) | AUC0-t (nmol·h/L) | AUC0-∞ (nmol·h/L) |
|---|---|---|---|---|---|
| Liquiritin | 48.68±38.17 | 0.026 (0.083~0.5) | 9.33±3.42 | 137.47±41.73 | 137.47±41.73 |
| Baicalin | 787.0±123.16 | 2.57 (0.5~3) | 3.11±0.95 | 8957.38±2077.4 | 9711.88±709.27 |
| Re | 50.17±40.07 | 0.25 (0.083~0.5) | 2.72±2.15 | 42.29±26.5 | 45.01±28.9 |
| Wogonoside | 174.25±42.12 | 1.625 (0.083~3) | 4.77±1.24 | 1446.54±330.91 | 1555.74±120.91 |
| Baicalein | 32.73±15.02 | 0.083 (0.083~0.083) | 6.85±4.32 | 85.36±25.98 | 119.10±33.82 |
| Wogonin | 18.78±6.27 | 0.167 (0.083~0.25) | 6.16±4.85 | 109.97±87.80 | 116.49±82.20 |
| SSd | 3.42±0.74 | 0.50 (0.125~0.5) | 12.18±5.32 | 25.84±18.5 | 25.84±18.5 |
| GA | 263.25±87.31 | 3.00 (2~12) | 4.77±1.93 | 3297.32±1178.49 | 3297.32±1178.49 |
These detected compounds belong to two classes: saponins and flavonoid. Re, SSd, GA are saponins, while baicalin, wogonoside, baicalein, and wogoninare flavonoids. The Tmax of baicalein, and wogonin were 0.083±0 h (5 min), and 0.17±0.09 h (10 min) respectively suggesting that these two compounds were absorbed rapidly after administration. The Tmaxof baicalin and wogonoside, which is glucuronide of baicalein or wogonin (Fig. 1), is significantly slower (2.13 ±1.18 hour for baicalin, 1.58±1.63 hour for wogonoside) than that of baicalein, and wogonin. This observation is understandable because usually the absorption of glucuronide is slower than its aglycone [28]. The content of GA in the XCHT extract is 0.03 mg/g, however, the Cmax of GA is 263.25±87.31, which is higher than the other compounds. Moreover, the Tmax of GA is 7.25±5.50, which is significantly slower than the other compounds. One of the possible reason for this observation is that GA is a metabolites of other saponins in glycyrrhiza Radix by microflora [29]. More experiment is needed to explain the PK profiles of these components in XCHT.
In the PK study, we successfully detected most of these compounds in the blood using a dose of 0.5 g/kg, which is translated from the dose used for human study. We calculated the PK parameters for 8 compounds (table 5) as these compounds were detected at all of the time points. However, we couldn’t get the PK parameters for the other compounds as these compounds can be only detected at certain time point(s) (table 6). Since the contents of the semarker compounds are highly different in XCHT, for example, baicalin 58.9 mg/g vs formononetin 0.05 mg/gram, a super-high dose may be needed in order to calculate the PK parameters for all of these 17 markers. The purpose of this paper is to report an LC-MS method to quantify different class of markers from XCHT in biological samples, PK studies at different dose is actually out of the scope of this study.
Table 6.
The concentrations of those compounds only detected at certain time points in the PK study.
| Compounds | ||||||||
|---|---|---|---|---|---|---|---|---|
| Time (min) | rutin | zingerone | quecertin | Rg1 | 6-gingerol | SSa | Rd | Rg3 |
| 5 | ND | ND | ND | ND | ND | 0.69±0.26 | ND | 1.33±0.48 |
| 15 | ND | ND | ND | 0.80±0.50 | ND | 1.17±0.88 | ND | ND |
| 30 | ND | ND | ND | ND | ND | 1.02±0.75 | ND | 3.74±3.73 |
| 60 | ND | ND | ND | ND | ND | 1.15±0.77 | ND | ND |
| 120 | ND | ND | ND | ND | ND | 0.85±0.51 | ND | ND |
| 360 | ND | ND | ND | ND | ND | ND | 5.07±6.67 | 1.61±0.53 |
| 480 | ND | ND | ND | 1.16±0.29 | ND | ND | ND | ND |
| 720 | ND | ND | ND | ND | ND | ND | 6.23±7.96 | ND |
| 1440 | ND | ND | ND | ND | ND | ND | 5.20±7.64 | ND |
ND not detected
4. Conclusion
A rapid, specific, and sensitive LC-MS method to quantify 17 components in XCHT was developed and validated. The main advantages of this method are (1) only 10 μL of blood is needed; (2) 17 marker compounds belong to different compound class in XCHT are simultaneously quantified; (3) the sample preparation procedure is simple; (4) recovery is good and matrix effect is minor. This method was successfully used in the pharmacokinetics study. This method is also valuable for human clinical study because it should allow even higher sensitivity than reported here since a large blood volume is usually available and thereby may be used to concentrate the analyte before analysis.
Highlights.
An UPLC-MS/MS method to quantify 17 compounds in Xiao-Chai-Hu-Tangin blood was developed
The sensitivity and robust method was validated.
The sensitivity and robust method was applied for pharmacokinetic study in mice.
Acknowledgments
This work was supported by a grant from National Institute of Health GM070737 to MH. MZ was supported by a training grant from Taihe Hospital, and RS was also supported by a training grant from Hubei University of Medicine.
Abbreviations
- UPLC
ultra-performance liquid chromatography
- I.S
internal standard
- DP
declustering potential
- CE
collision energy
- CXP
collision cell exit potential
- AUC
area under the curve
- QC
quality control
- LLOD
lower limit of detection
- LLOQ
lower limit of quantification
- XCHT
Xiao-Chai-Hu-Tang
- MPA
mobile phase A
- MPB
mobile phase B
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fujiwara K, Mochida S, Nagoshi S, et al. J Ethnopharmacol. 1995;46:107–114. doi: 10.1016/0378-8741(95)01235-6. [DOI] [PubMed] [Google Scholar]
- 2.Zheng N, Dai J, Cao H, et al. Evid Based Complement Alternat Med. 2013;2013:529458. doi: 10.1155/2013/529458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Qin XK, Li P, Han M, et al. Zhong Xi Yi Jie He Xue Bao. 2010;8:312–320. doi: 10.3736/jcim20100403. [DOI] [PubMed] [Google Scholar]
- 4.Ohtake N, Nakai Y, Yamamoto M, et al. J Chromatogr B Analyt Technol Biomed Life Sci. 2004;812:135–148. doi: 10.1016/j.jchromb.2004.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang JS, Wang KC, Liu HW, et al. Am J Chin Med. 2007;35:341–351. doi: 10.1142/S0192415X07004862. [DOI] [PubMed] [Google Scholar]
- 6.Cheng PW, Ng LT, Lin CC. Int Immunopharmacol. 2006;6:1003–1012. doi: 10.1016/j.intimp.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 7.Horie Y, Kajihara M, Yamagishi Y, et al. J Gastroenterol Hepatol. 2001;16:1260–1266. doi: 10.1046/j.1440-1746.2001.02622.x. [DOI] [PubMed] [Google Scholar]
- 8.Miyahara M, Tatsumi Y. Yakugaku Zasshi. 1990;110:407–413. doi: 10.1248/yakushi1947.110.6_407. [DOI] [PubMed] [Google Scholar]
- 9.Yoshida K, Mizukawa H, Honmura A, et al. Am J Chin Med. 1993;21:171–177. doi: 10.1142/S0192415X93000200. [DOI] [PubMed] [Google Scholar]
- 10.Yoshida K, Mizukawa H, Honmura A, et al. Am J Chin Med. 1994;22:183–189. doi: 10.1142/S0192415X9400022X. [DOI] [PubMed] [Google Scholar]
- 11.Deng G, Kurtz RC, Vickers A, et al. J Ethnopharmacol. 2011;136:83–87. doi: 10.1016/j.jep.2011.04.008. [DOI] [PubMed] [Google Scholar]
- 12.Itoh S, Marutani K, Nishijima T, et al. Dig Dis Sci. 1995;40:1845–1848. doi: 10.1007/BF02212712. [DOI] [PubMed] [Google Scholar]
- 13.Amagaya S, Hayakawa M, Ogihara Y, et al. J Ethnopharmacol. 1989;25:181–187. doi: 10.1016/0378-8741(89)90020-2. [DOI] [PubMed] [Google Scholar]
- 14.Huang Y, Marumo K, Murai M. Keio J Med. 1997;46:132–137. doi: 10.2302/kjm.46.132. [DOI] [PubMed] [Google Scholar]
- 15.Ito H, Shimura K. Jpn J Pharmacol. 1986;41:307–314. doi: 10.1254/jjp.41.307. [DOI] [PubMed] [Google Scholar]
- 16.Lin CC, Lin LT, Yen MH, et al. Evid Based Complement Alternat Med. 2012;2012:984024. doi: 10.1155/2012/984024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kakumu S, Yoshioka K, Wakita T, et al. Int J Immunopharmacol. 1991;13:141–146. doi: 10.1016/0192-0561(91)90091-k. [DOI] [PubMed] [Google Scholar]
- 18.Yamashiki M, Nishimura A, Huang XX, et al. Dev Immunol. 1999;7:17–22. doi: 10.1155/1999/62564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamashiki M, Nishimura A, Suzuki H, et al. Hepatology. 1997;25:1390–1397. doi: 10.1002/hep.510250615. [DOI] [PubMed] [Google Scholar]
- 20.Hsu LM, Huang YS, Tsay SH, et al. J Chin Med Assoc. 2006;69:86–88. doi: 10.1016/S1726-4901(09)70119-4. [DOI] [PubMed] [Google Scholar]
- 21.Homma M, Ishihara M, Qian W, et al. Yakugaku Zasshi. 2006;126:973–978. doi: 10.1248/yakushi.126.973. [DOI] [PubMed] [Google Scholar]
- 22.Tsuji H, Osaka S, Kiwada H. Chem Pharm Bull (Tokyo) 1991;39:1004–1008. doi: 10.1248/cpb.39.1004. [DOI] [PubMed] [Google Scholar]
- 23.Shimaoka A, Seo S, Minato H. J Chem Soc Perkin. 1975;1:2043–2048. doi: 10.1039/p19750002043. [DOI] [PubMed] [Google Scholar]
- 24.Yen MH, Lin CC, Chuang CH, et al. J Ethnopharmacol. 1991;34:155–165. doi: 10.1016/0378-8741(91)90033-a. [DOI] [PubMed] [Google Scholar]
- 25.Zhu Z, Zhao L, Liu X, et al. J Chromatogr B Analyt Technol Biomed Life Sci. 2010;878:2184–2190. doi: 10.1016/j.jchromb.2010.06.021. [DOI] [PubMed] [Google Scholar]
- 26.Li C, Homma M, Oka K. J Chromatogr B Biomed Sci Appl. 1997;693:191–198. doi: 10.1016/s0378-4347(96)00514-2. [DOI] [PubMed] [Google Scholar]
- 27.Matuszewski BK, Constanzer ML, Chavez-Eng CM. Anal Chem. 2003;75:3019–3030. doi: 10.1021/ac020361s. [DOI] [PubMed] [Google Scholar]
- 28.Gao S, Hu M. Mini Rev Med Chem. 2010;10:550–567. doi: 10.2174/138955710791384081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Montoro P, Maldini M, Russo M, et al. J Pharm Biomed Anal. 2011;54:535–544. doi: 10.1016/j.jpba.2010.10.004. [DOI] [PubMed] [Google Scholar]