

Abstract
Biodegradable hydrogels were synthesized by the click reaction of 4-arm azido-terminated PEG differing in molecular weight (2 100 and 8 800) and two alkyne-terminated peptides: [alkyne]-GFLGK-[alkyne] and ([alkyne]-GFLG)2K. The physical properties of in situ formed hydrogels were examined. The hydrogels were highly elastic as determined by rheological and micro-rheological studies. Swelling degree and enzymatic degradation by papain were dependent on the molecular weight of the PEG, but not the peptide. For PEG8800-based hydrogels, time-course analysis of degradation showed that the molecular weight of the soluble fraction quickly reached the PEG precursor value. These findings may guide future design of hydrogels with controllable mechanical properties and enzymatic degradability.
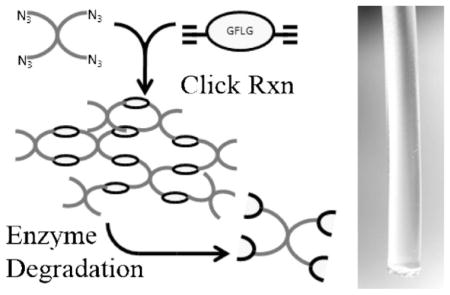
Keywords: biodegradable, click chemistry, hydrogels, peptide crosslinks, poly(ethylene glycol)
Introduction
Hydrogels were the first biomaterials designed for clinical use.[1] They have found applications in numerous areas, including tissue engineering, drug delivery, sensing, diagnostics, and microfluidics.[2,3]
Several factors need to be considered when designing biocompatible hydrogel biomaterials including controlled chemistry for the formation of reproducible, reversible 3D networks with precisely defined structures, and biodegradability. Living radical polymerization has been used for the control of the molecular weight distribution of the primary chains of the network;[4,5] copper catalyzed[6,7] or copper-free [8] Huisgen cycloaddition of azides and alkynes (click chemistry) and Michael addition[9] have been used to control crosslinking and/or attachment of biologically active molecules.
The combination of synthetic polymers with sequences/chains of natural polymers is a promising route for the design and synthesis of enzymatically degradable hybrid polymers/hydrogels.[2,10] To render a hybrid polymer/hydrogel degradable, the structure of the degradable sequences should match the active site of respective enzyme(s); oligopeptide sequences have been frequently used as degradable crosslinks in hydrogels. Hydrogels, containing oligopeptide crosslinks susceptible to chymotrypsin-catalyzed hydrolysis were synthesized by crosslinking N-(2-hydroxypropyl)methacrylamide (HPMA) copolymers containing reactive side-chains (terminated in p-nitrophenoxy groups) with oligopeptide-containing diamines.[10] The degradability of hydrogels was dependent on the length and detailed structure of the oligopeptide sequence and on the equilibrium degree of swelling (network density); the higher the degree of swelling, the faster the rate of degradation. The degree of swelling also has an impact on surface versus bulk degradation of the hydrogel. If the enzyme cannot diffuse into the hydrogel interior, only surface degradation takes place.[11] Similar HPMA-based hydrogels degradable by cathepsin B, a lysosomal thiol proteinase, have also been evaluated.[12] In further experiments, HPMA-based hydrogels with degradable crosslinks were shown to release FITC-dextran and daunomycin (covalently bound via oligopeptide spacer) during incubation with a mixture of lysosomal enzymes (Tritosomes) or chymotrypsin.[13] Recently, Plunkett et al. studied acrylamide-based hydrogels containing oligopeptides degradable by chymotrypsin.[14]
Poly(ethylene glycol) (PEG) is a hydrophilic polymer that has been used in several clinical applications. PEG-based hydrogels have been extensively studied. Hubbell’s laboratory used multiarm-PEG and Michael-type addition to synthesize extracellular matrix mimicking hydrogels degradable by cell-excreted matrix metalloproteinases.[9,15] PEG-based hydrogels containing Schiff base linkages were designed for low molecular weight (doxorubicin) drug delivery.[16] Azide-alkyne click chemistry was also used for the synthesis of PEG-based hydrogels.[6,17] Hawker and coworkers[6] synthesized hydrogels by reacting a tetraazide-modified tetraethylene glycol with telechelic alkyne terminated PEGs of varying molecular weight. Yang and coworkers employed 4-arm alkyne terminated PEG and crosslinked it with a series of RGD containing dialkyne-peptides. However, both alkyne groups were attached to the N-terminal lysine. Consequently, the side-chains were degradable, but the hydrogel crosslinked backbone remained stable.[17]
In this study we synthesized two 4-arm azide-terminated PEGs differing in molecular weight (2 100 and 8 800) and two alkyne-terminated peptides, [alkyne]-GFLGK-[alkyne] (GFLG1) and ([alkyne]-GFLG)2K (GFLG2). The hydrogels were synthesized by Huisgen cycloaddition (click chemistry) of PEG azides with peptide alkynes and contained enzymatically degradable crosslinks. The kinetics of the crosslinking reaction was monitored by dynamic rheology. The hydrogels were characterized by the equilibrium degree of swelling, their morphology, and bulk and micro-rheology. The relationship between the structure of the hydrogels and their degradability by papain, a thiol proteinase, was also evaluated.
Experimental Part
Materials
Four-arm PEG terminated with hydroxy groups (PEG-4OH2 100 and PEG-4OH 8 800) was purchased from Polymer Source (Montreal, Canada). Side-chain protected Fmoc-amino acids, 2-chlorotrityl resin, and 2-(7-aza-1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU) were purchased from AAPPTec (Louisville, KY). Piperidine (PIP; 99.5+%, Biotech grade), triisopropylsilane (TIS; 99%), papain (EC 3.4.22.2), glutathione, 5-hexynoic acid, Bz-Phe-Val-Arg-NAp (chromogenic substrate), iodoacetic acid sodium salt, CuBr, L-ascorbic acid, and O-phthaladehyde (OPA)/3-mercaptopropionic acid (MPA) were purchased from Sigma–Aldrich (St. Louis, MO, USA). Ethyldiisopropylamine (DIPEA; 99%) was from Alfa Aesar (Ward Hill, MA, USA). Diethyl ether and dichloromethane (DCM) were purchased from Mallinckrodt Baker (Phillipsburg, NJ, USA). Trifluoroacetic acid (TFA; 99%) was purchased from Acros Organics (Morris Plains, NJ, USA). Surfactant free fluorescent yellow–green amidine modified polystyrene (PS) beads were from Interfacial Dynamics Corp. (Portland, OR).
Synthesis of Enzyme-Degradable Peptides with Dialkyne Modification
The functionalized peptides, flanked by alkyne groups at both ends, were synthesized using solid-phase methodology and manual Fmoc/tBu strategy on 2-chlorotrityl resin. As an example, the synthesis of GFLG1 is shown in Scheme 1. Fmoc-Lys(ivDde)–OH was employed as the first amino acid residue to introduce two free amine groups. The protecting groups (Fmoc- and ivDde-) were removed selectively; one was used to introduce alkyne group into the C-terminus of the peptide, whereas the other functioned in constructing the peptide sequence. Incorporation of alkyne at the N-terminus was accomplished by acylation of the peptide with 5-hexynoic acid using the standard coupling protocol (–COOH/DIPEA/HATU in DMF). The resin was then washed sequentially with 3 × DMF, 3 × DCM, and 3 × MeOH, and dried under vacuum. The resulting resin-bound peptides were cleaved from the resin, and side-chains were deprotected using TFA/H2O/TIS (95:2.5:2.5) cocktail. Crude peptides were purified by RP-HPLC (Agilent Technologies 1100, semipreparative Zorbax 300SB-C18 column, 250 mm × 9.4 mm, 300 Å pore size, 5 μm particle size, flow rate 2.5 mL · min−1) employing a gradient from 50 to 100% B over 30 min, where buffer A was 0.1% TFA in water and buffer B 0.1% TFA in 90:10 v/v methanol/water. The purity of peptides GFLG1 and GFLG2 was verified by analytical RP-HPLC (Figure S1 and S2 of Supporting Information). Peptide structures were ascertained by MALDI-TOF mass spectrometry (Voyager-DE STR Biospectrometry Workstation, Perseptive Biosystems) (Figure S3 and S4 of Supporting Information). For GFLG2, Fmoc-Lys(Fmoc)-OH was used as the first amino acid to incorporate a branched structure after removal of Fmoc-group. The end-modification was realized by acylation of the peptide with 5-hexynoic acid. The molecular structures of the two peptides are shown in Table 1.
Scheme 1.
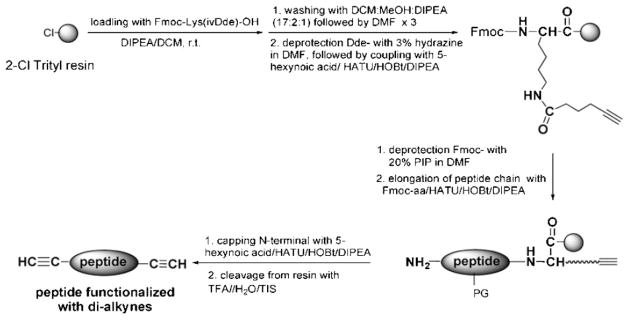
Synthesis of alkyne-modified peptide GFLG1.
Table 1.
Enzymatically degradable peptides with dialkyne modification.
| Peptide | Molecular structure | M+ |
|---|---|---|
| GFLG1 |
|
708.38 |
| GFLG2 |
|
1 082.58 |
Synthesis of Azido-Terminated Poly(ethylene glycol) (PEG)
The synthetic procedure consisted of two steps (Scheme 2A): 4-arm-PEG-OH (PEG-4OH) was first transformed into an active intermediate (mesylate-terminated PEG), followed by replacement with azido functional group. A typical procedure for synthesis of 4-arm PEG-N3 with M̄n 8 800 g · mol−1 (PEG-4N3 8 800) is briefly described. In a 25 mL dry round-bottom flask, PEG-4OH 8 800 (0.7 g, [OH] 0.32 mmol) was dissolved in 4.5 mL anhydrous DCM, then kept in an ice-ethanol bath (−10 to −15 °C). Triethylamine (0.88 mL, 6.4 mmol) was dropwise added to the flask under stirring before addition of 0.5 mL (6.4 mmol, 20×) methanesulfonyl chloride. The reaction was allowed to proceed at 0 °C for 3 h and then at room temperature for another 16 h. Precipitation was observed, and the color of the system gradually changed from colorless to yellow and then to a dark, tea-like color. At the end of reaction 20 mL DCM was added, and the precipitate was removed by filtration. The filtrate was washed sequentially with 1 N HCl, saturated NaCl solution, 1 N NaOH, and saturated NaCl solution. The organic phase was dried over MgSO4, and solvent was removed by evaporation. A yellowish powder (0.56 g) was obtained.
Scheme 2.

(A) Synthesis of 4-arm azido-terminated PEG. (B) Synthesis of degradable hydrogels by click reaction of 4-arm PEG-N3 with dialkyne modified peptide.
Part of this intermediate (0.3 g, [OSO2CH3] 0.132 mmol) was dissolved in 5 mL DMF, and then NaN3 (0.43 g, 50×) was added. The reaction was undertaken at 80 °C for 20 h. DMF was removed by evaporation under reduced pressure. The resulting intermediate was redissolved in 30 mL DCM. Undissolved solid was removed by filtration. The filtrate was washed with saturated NaCl, dried over MgSO4, and then condensed and precipitated into cold diethyl ether. A fine powder was obtained. The yield was 82.5%. The product was confirmed by 13CNMR(CDCl3) and used without further purification.
PEG-4N3 with M̄n ≈2 100 g · mol−1 (PEG-4N3 2 100) was synthesized using a similar procedure.
Preparation of Hybrid Hydrogels via Click Reaction
All hydrogels were prepared by the reaction of 4-arm azide-terminated PEGs described above with dialkyne-flanked peptides in the presence of CuBr/L-ascorbic acid in DMF (Scheme 2B). For example, to prepare hybrid hydrogel PEG8800-GFLG1, PEG-4N3 8 800 (20 mg, [azide] 9 μmol), and GFLG1 (2.83 mg, [alkyne] 9 μmol) were dissolved in deoxygenated DMF (60 μL) in a 1 mL Eppendorf tube. L-Ascorbic acid solution (20 μL containing 0.35 mg L-ascorbic acid, 0.25× relative to alkyne and azide groups) was added to this tube, and the system was bubbled with nitrogen. CuBr solution (100 μL containing 0.56 mg CuBr, 0.5× relative to alkyne and azide groups) was added just prior to transfer of the mixture to the mold, and the reaction was carried out at room temperature. Gel formation was examined by vial tilting method.
Rheology
Gelation process was dynamically monitored by time sweep rheology using a TA-Instruments 550 Advanced Rheometer (New Castle, DE). Immediately after CuBr addition, sample was loaded on a Peltier plate underneath 20 mm diameter geometry with gap distance 103 μm, at 25 °C. Time sweeps were performed at fixed 1% strain amplitude and 6 rad · s−1 frequency until the curve achieved plateau, with data collection every 10–30 s. Following time sweeps, frequency sweep rheology, at fixed 1% strain amplitude over a frequency range from 0.1 to 100 rad · s−1, was performed to determine hydrogel rheological properties: storage, G′, and loss, G″, moduli.
Microrheology
Particle-tracking microrheological measurements were performed as previously described by us[18–20] and by others.[21,22] Surfactant free fluorescent yellow–green amidine modified PS beads (Interfacial Dynamics Corp., Portland, OR) (1 μm in diameter) were dispersed into the click reaction solution at 0.5% w/v final concentration. After gel formation (minimally 24 h after mixing of reagents), gel slices were mounted on slides underneath No. 1.5 coverslips and then visualized on a Nikon Eclipse E800 microscope equipped with a 100× oil-immersion objective. The samples were focused in a manner that avoided wall effects. The Brownian motion of the PS beads for each sample was analyzed by IDL software (Research Systems Inc., Boulder, CO) based on 3 000 consecutive images captured by a CCD camera (Dage-MTI, DC330) and recorded at intervals of 33 ms using StreamPix software (Norpix, Montreal, Canada). The mean square displacement (MSD) of tracer particles was determined using algorithms previously developed.[23]
Hydrogel Swelling
Hydrogels retrieved from the reaction molds were first immersed in DMF. To remove organic solvent and copper salts, hydrogel samples were incubated in a series of DMF/H2O solutions containing 10 mmol · L−1 EDTA (75:25, 50:50, 25:75, 0:100 v/v; each step lasted at least 12 h), and finally swollen inpureH2O until no weight changes were detectable. Hydrogels were then dried at room temperature until constant weight. Hydrogel equilibrium (gravimetric) swelling ratio was calculated by (Ww –Wd)/Wd, where Ww is the weight of fully swollen species and Wd is the weight of dry species. Kinetics of hydrogel swelling (volumetric) was performed in water and monitored under optical microscope (Nikon E800) using 2× objective. Gel radii were measured using ImagePro Plus software (Media Cybernetics, Bethesda, MD, USA). Assuming isotropic swelling, volume swelling ratio, Q, was calculated by (rt/r0)3, where rt is the radius of swelling gel at different time points, and r0 is the radius of the dried gel.
Scanning Electron Microscope (SEM)
Water-swollen hydrogel samples, prepared as described above, were shock-frozen by immersion into liquid nitrogen for 10 min, then lyophilized overnight. Freeze-dried samples were mounted on SEM aluminums tubs and sputtered with gold for 40 s to obtain a conductive surface. Observation of the hydrogel morphology was carried out on a SEM (Hitachi S-2460N SEM) at an accelerating voltage of 10 kV and appropriate magnifications.
Enzyme-Catalyzed Degradation of Hybrid Hydrogels
Hydrogel degradation was performed in McIlvaine’s buffer (50 mmol · L−1 citrate/0.1 M phosphate) at pH 6.0, 37 °C, using papain as model enzyme. Papain, at concentration of 0.05 mg · mL−1 (determined by UV at 280 nm, with absorbance of 25 at 1%[24]), was reduced with 5 × 10−3 M glutathione for 5 min. The enzyme activity was confirmed using Bz-PheValArg-NAp as chromogenic substrate. A series of dried hydrogels (4–10 mg) were pre-swollen in the above buffer before addition of enzyme solution (3 mg · hydrogel · mL−1 enzyme solution). At predetermined time points after addition of enzyme, hydrogel samples were removed from the medium and re-immersed in a large volume of water to remove salts before wet mass weighing and drying. The medium was treated with enzyme inhibitor solution (3 × 10−3 M iodoacetic acid sodium salt in McIlvaine’s buffer, 10 μL) to stop the hydrogel degradation. Then, the medium was analyzed with size-exclusion chromatography on an AKTA FPLC system (Pharmacia) equipped with miniDAWN TREOS and OptilabEX detectors (Wyatt Technology, Santa Babara, CA, USA) using a Superose12 HR/10/30 column with PBS (pH 7) as mobile phase. The average molecular weight of the degraded products was determined by light scattering, UV, RI and calculated by ASTRA software. The peptide content in remaining hydrogels was determined by amino acid analysis (see below). The weight loss (WL) was calculated as
where Wt is the dry weight at selected time point and W0 is the dry weight of original gel.
Amino Acid Analysis
Hydrogel samples (2–3 mg)at selected time points were hydrolyzed with 0.5 mL of 6 N HCl in sealed ampoules at 160 °C for 3 h. Then, the samples were dried under vacuum (with the protection of NaOH pellet). The hydrolysates were dissolved in 1 mL of distilled water. Pre-column OPA/MPA derivatization followed by RP-HPLC analysis (Agilent Technologies 1100, Eclipse XDB-C8 column, 4.6 mm × 150 mm, with Ex 229 nm and Em 450 nm) was used. The glycine (G) and L-phenylalanine (F) contents in the hydrogels were determined.
Results and Discussion
Synthesis of 4-Arm Azido-Terminated Poly(ethylene glycol)s (PEGs) and Peptides with Dialkyne Modification
The synthesis of tetraazido-functionalized PEG (PEG-4N3) followed a published procedure[25] with slight modifications. As shown in Scheme 2A, the activation of 4-arm hydroxy-terminated PEG(M̄n = 2 100 and 8 800) with mesyl chloride, followed by nucleophilic substitution with sodium azide produced PEG-4N3. The presence of azido groups was evidenced by the methylene resonance in 13C NMR spectrum (Figure 1). The presence of the peak at δ 50.9 ppm (CH2N3) and the absence of the peak at δ 61.7 ppm (CH2OH) in the product demonstrated that the conversion to azido groups was nearly 100%.
Figure 1.
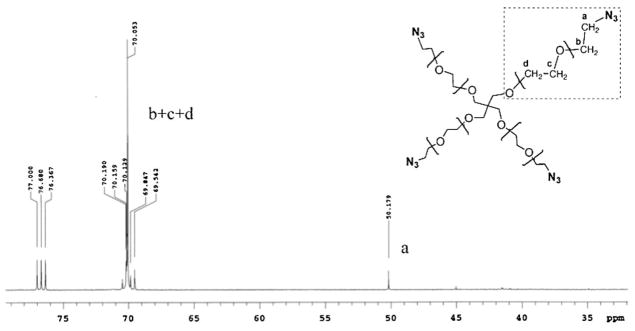
13C NMR spectrum (CDCl3) of PEG-4N3 8 800.
The inclusion of alkyne groups at both ends of degradable peptides, GFLG1 and GFLG2, was achieved via standard Fmoc/tBu solid-phase peptide synthesis, and by incorporation of lysine (K) residue that created a branched structure for the introduction of either an alkyne group (for GFLG1) or a second copy of the degradable sequence (GFLG2). The identities of the peptides were ascertained by MALDI-TOF mass spectra, indicating that the alkyne groups were stable under peptide synthesis/cleavage conditions. The yields of both peptides were in the range of 70–90%. Thus, the synthetic approach provided an efficient method for the insertion of alkyne groups into peptide sequences.
Hydrogel Formation
The degradable PEG hydrogels were formed by click reaction between 4-arm azido-terminated PEG and dialkyne-modified peptides, GFLG1 or GFLG2. The reaction conditions, such as the molar ratio of azide/alkyne and the catalyst system, were selected based on preliminary experiments (results not shown). The gelation was performed at equimolar azide/alkyne ratio at room temperature, using DMF as solvent and 0.5 equivalent of copper bromide/0.25 equivalent of L-ascorbic acid as the catalyst system. We observed that the PEG concentration significantly affects the gelation process. To optimize gelation conditions, three different PEG concentrations (from 2.5 to 10 wt.-%) were tested (Table 2). No hydrogel formation was observed at the lowest polymer concentration of 2.5%. As expected, higher concentration of polymer in the reaction mixture shortened the time needed for gel formation and increased the reaction yield (Table 2). Consequently, in further experiments, a polymer concentration of 10 wt.-% was used in agreement with similar reactions described in the literature.[8,17]. Moreover, the occurrence of gelation was determined more accurately from rheological curves as the crossover of storage modulus, G′, and loss modulus, G″.
Table 2.
Synthesis of PEG-based hybrid hydrogels by click reactions.a)
| No. | PEG-4N3
M̄n
|
Dialkyne peptide | PEG Conc, w/v
|
Gelation time | Yieldb) |
|---|---|---|---|---|---|
| g · mol−1 | % | % | |||
| 1 | 8 800 | GFLG1 | 2.5 | no gel | – |
| 2 | 8 800 | GFLG1 | 5 | h | 66.9 |
| 3 | 8 800 | GFLG1 | 10 | 12 minc) | 94.6 |
| 4 | 8 800 | GFLG2 | 10 | 9 minc) | 58.1 |
| 5 | 2 100 | GFLG1 | 10 | 9 minc) | 56.8 |
| 6 | 2 100 | GFLG2 | 10 | ≈l0 minc) | 45.1 |
Experimental conditions: [azide]0/[alkyne]0/[CuBr]0/[L-ascorbic acid]0 = 1:1:0.5:0.25; rt in DMF;
Yield = (weight of dried gel)/(initial weight of PEG-4N3 + initial weight of dialkyne peptide);
Gelation time was obtained from dynamic time-sweep rheology experiments.
The effect of peptide structure on the gelation process was also examined. There were no significant differences in gelation time for peptides GFLG1 and GFLG2, but the yield of hydrogel was slightly lower when the longer sequence (GFLG2) was used (Table 2, gels 4 and 6). As described by Liu,[17] the lengths of the peptide crosslinkers may influence the gelation process. Alkyne groups in GFLG1 and GFLG2 possess the same reactivity; thus, there were no differences in initial gelation time. However, generally, peptides have a high persistence length;[26] once the viscosity of the reaction mixture increases, the longer GFLG2 has a lower mobility thus leading to incomplete reaction compared to the shorter GFLG1.
Poly(ethylene glycol) (PEG) length has an opposite effect compared to peptide length. The yield of the hydrogel for the PEG2100 was lower than that for PEG8800 (Table 2). One could hypothesize that the intramolecular reaction of peptide alkynes with the same PEG molecule is more probable with PEG2100 due to the closer vicinity of the polymer arm termini.
Physical Characterization of Hydrogels
Hydrogel formation was monitored by bulk dynamic rheology experiments. Under constant strain and frequency, G′ and G″ were measured immediately after addition of catalyst CuBr (Figure 2A–C). Onset of gelation was observed at ≈10 min (insets in Figure 2A–C) and a plateau was reached in a range of 70–150 min.
Figure 2.

Hydrogel formation. (A–C) Time-sweep rheology of gel formation, as monitored by increase in G′, storage modulus. Gel points were identified when G′ was equal to G″, the loss modulus, as shown in the insets. A, PEG8800GFLG1; B, PEG8800GFLG2; C, PEG2100GFLG1. (D–F) Frequency sweep rheology of hydrogels. D, PEG8800GFLG1; E, PEG8800GFLG2; F, PEG2100GFLG1. (A–F) Rheology studies were performed ≥3 times; open diamond represents G′, closed diamond G″. (G) Microrheology of hydrogels, monitored by MSD versus lag time. (H) Top, hydrogel obtained following rheological characterization; bottom, hydrogel molded in plastic syringe, and used for enzymatic degradation studies.
Following time-sweep experiments of hydrogel formation, frequency-sweeps were performed to more thoroughly characterize the rheological properties of the hydrogels (Figure 2D–F). Hydrogels showed highly elastic or gel-like character, with G′ values 2–3 orders of magnitude greater than G″. Inour study, G′ values were around 6 000 Pa when using PEG8800 and a 1:1 azide/alkyne ratio at 10% w/v, which mirrored values in click reaction work from Anseth’s group, where they observed mean final elastic (storage) modulus of 5 160 Pa when using PEG10000 and a 1:1 azide/acetylene ratio at 13.5% w/v.[27]
A significant increase in G′ was observed when PEG2100 (Figure 2C and F) was used compared to PEG8800 (Figure 2A, B, D, and E). This finding suggests that the PEG chain length strongly affects the mechanical properties of hydrogels, whereas the peptide composition (GFLG1 or GFLG2) had a minor effect (GFLG2 did reach plateau faster than the GFLG1 peptide: 70 min with GFLG2, compared to 120 or 150 min with GFLG1).
In addition to classical bulk rheology, we also performed particle-tracking microrheology. We have used this technique successfully to analyze rheological properties — mainly degree of gelation — in numerous soft materials, ranging from primarily viscous to primarily elastic[18–20] Briefly, particle-tracking microrheology is based on the Brownian motion of beads embedded within materials; these motions can be tracked to calculate MSDs, which relate to the material’s viscoelastic properties. Some advantages of microrheology techniques include wide frequency range, local environmental probing, very small volumes, and cost efficiency,[28] due to few requirements of fluorescent microscope and specific software (see Experimental Part). On a log–log MSD plot (MSD vs. lag time), a primarily viscous liquid yields a slope of 1, viscoelastic between 0 and 1, and elastic of 0.[18,29] We obtained MSD plots (Figure 2G) with slopes near 0, which indicated elastic hydrogels and supported our bulk rheology findings.
Figure 2H qualitatively demonstrates hydrogel formation. Top image shows a gel that was formed during the time-sweep rheology; here, the hydrogel was removed from the plate immediately after rheology. Bottom image shows a hydrogel formed using a plastic syringe mold. This particular gel was used for enzymatic degradation studies (see discussion in later section).
The swelling degree of hydrogels was measured both gravimetrically and volumetrically, and consistent data were obtained. Figure 3A shows the equilibrium swelling ratios that were calculated by comparing the weights of the dry and water-swollen hydrogels; time-course studies of volumetric swelling ratios were performed to assess the kinetics of hydrogel swelling (Figure 3B). In both experiments, PEG8800GFLG1 showed significantly higher degree of swelling than PEG2100GFLG1. Equilibrium swollen state was reached in more than 4 h with PEG8800GFLG1, whereas this state was reached in about 1 h with PEG2100GFLG1. Both hydrogels, PEG8800GFLG1 and PEG2100GFLG1, were prepared under same weight concentration (10 wt.-%). In the same volume, the number of functional groups in PEG2100GFLG1 is 4.5 times that of PEG8800GFLG1, which leads to a higher crosslinking density, consequently resulting in higher G′. This hypothesis was supported by SEM images (Figure 4) in which condensed pore sizes were observed in PEG2100GFLG1 compared to PEG8800GFLG1. Similar observations of the relationship between polymer length and swelling degree were also reported by Lutolf and Hubbell.[26]
Figure 3.
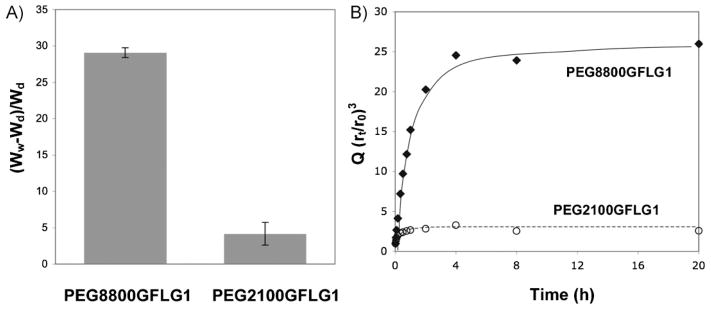
Hydrogel swelling. (A) Equilibrium degree of swelling (in water), which was measured by (Ww – Wd)/Wd, where Ww is the weight of fully swollen species and Wd is the weight of dry species. Standard deviation bars are indicated (n = 5). (B) Kinetics of swelling (in water), as measured by Q, which was calculated by (rt/r0)3, where rt is the radius of gel at time point and r0 is the radius of the dried gel.
Figure 4.

Scanning electron microscope (SEM) imaging of hydrogels. Freeze-dried hydrogels were imaged at 200× and 800× magnification.
The difference in the polymer–water interaction parameter, χ, between PEG8800 and PEG2100 based hydrogels also contributes to the lower swelling degree of hydrogel PEG2100GFLG1. As PEG is the only water-soluble component, the higher the PEG content, the higher the swelling degree. The weight ratio of PEG:peptide changes from 7 in PEG8800GFLG1 to 1.5 in PEG2100GFLG1, leading to an increase in χ with concomitant decrease in swelling ability.
From these studies, we confirmed that the structure of the polymer component (PEG 2 100 or 8 800) played an important role in determining the physical properties of the hydrogel.
Hydrogel Degradation
Hydrogels crosslinked by GFLG1 and GFLG2 were incubated with papain for enzymatic degradation. The time-course of hydrogel degradation was characterized by three parameters: (i) hydrogel WL, (ii) size-exclusion chromatography of the soluble fraction, and (iii) amino acid analysis of the remaining hydrogel samples.
Papain (EC 3.4.22.2) is a thiol protease of wide specificity. It has a large active site which extends over about 25 Å and can be divided into seven subsites, each accommodating one amino acid residue.[30,31] The subsite S2 specifically interacts with phenylalanine (F) (nomenclature from[30]); in substrates containing –F–X–Y–, the peptide bond between amino acid residues X and Y is cleaved. Other factors important for the formation of enzyme–(polymer) substrate complexes are the steric hindrance of the polymeric chain and the length of the oligopeptide sequence.[10] In a three-dimensional hydrogel structure, there is transport (diffusion) limitation for the enzyme to reach the peptide substrate. However, peptides bound to PEG chain termini accommodate into the active site of chymotrypsin more easily than peptides bound to synthetic polymer side chains.[32] Consequently, the PEG chain may accommodate relatively easily into the distant subsites of papain’s active site.
The WL of hydrogels during degradation is shown in Figure 5. PEG8800 hydrogels degraded considerably faster than those based on PEG2100, indicating that PEG chain length highly affected the rate of degradation. This is related to the differences in hydrogel properties: degree of swelling and elastic modulus. PEG8800 hydrogels showed higher degree of swelling and lower elastic moduli compared to PEG2100 hydrogels. This finding is in good agreement with published data on the chymotrypsin-catalyzed surface degradation of HPMA-based hydrogels containing oligopeptide crosslinks.[11] However, the structure of the crosslinks, GFLG1 versus GFLG2, did not have an impacton the rate of degradation. Apparently, the enzyme–substrate interactions are controlled by diffusion, and the presence of two enzyme attachment modes for GFLG2 when compared to GFLG1 did not result in differences in the rate of degradation.
Figure 5.

Hydrogel WL. During degradation catalyzed by 0.05 mg · mL−1 papain in citrate-phosphate buffer (pH 6.0) at 37 °C, the hydrogel WL was determined from WL = Wt/W0 × 100% where Wt is dry weight at selected time point and W0 is dry weight of original gel.
The analysis of cleavage products may contribute to our understanding of hydrogel degradation. Size-exclusion chromatography was used to detect changes in the soluble fraction of papain-catalyzed PEG8800GFLG1 hydrogel degradation, at different time points (Figure 6). With the exception of the initial time points, where higher molecular weight fractions were detected (at elution volume ≈11 mL, indicated by arrows), the molecular weight of the soluble fraction quickly reached the initial value of the PEG precursor.
Figure 6.

Size-exclusion chromatography of soluble fractions. Soluble fractions were obtained during degradation of hydrogel PEG8800GFLG1 with 0.05 mg · mL−1 papain in citrate-phosphate buffer (pH 6.0) at 37 °C. Inset shows initial molecular weight of PEG-4N3 8 800 polymer, for comparison.
The importance of F in position P2 of the substrate and the significance of at least four amino acid residues in the oligopeptide substrate (to achieve high rate of degradation) have been previously demonstrated to be important in the papain-catalyzed cleavage of branched HPMA copolymer with oligopeptide crosslinks.[33]
Apparently, the crosslinks in PEG hydrogels might preferentially align with F in the S2 subsite; consequently, the first cleavage would occur between L and G. However, the wide specificity of papain suggests that other sites of initial cleavage are also possible. The changes in peptide content in the hydrogel PEG2100GFLG1 during papain degradation were analyzed by amino acid analysis. The G/F ratio in the hydrogel fraction at 0, 3, and 16 h of degradation was 2.1, 1.8, and 1.3, respectively (Figure 7). This indicates that the peptide cleavage occurs at more than one site.
Figure 7.

Amino acid analysis of hydrogels. G/F ratios were determined from PEG2100GFLG1 hydrogels degraded by 0.05 mg · mL−1 papain in citrate-phosphate buffer (pH 6.0) at 37 °C, at selected time points.
Conclusion
PEG-based peptide-containing hydrogels were synthesized by click chemistry and shown to have elastic rheological properties, along with relatively high degrees of network swelling. Hydrogels based on PEG8800 showed lower elastic moduli, higher degree of swelling, and larger pore sizes compared to PEG2100, which is likely a result of the higher hydrophobicity of the PEG2100 hydrogel, and due to the higher peptide:PEG ratio and higher crosslinking density. Using a papain enzyme degradation assay, we observed significant differences in hydrogel degradability between PEG 2 100 and 8 800-based hydrogels. Time-course analysis of degradation showed that the molecular weight of the soluble fraction quickly reached that of the PEG precursor. Finally, we have introduced in this work a “click hydrogel” system and shown how specific changes in composition can tune the hydrogel’s mechanical properties and sensitivity to enzymatic degradation.
Supplementary Material
Acknowledgments
This research was supported in part by NIH grant EB 005288 and the University of Utah Research Foundation TCP Program. We thank Dr. Pavla Kopečková for valuable discussion and the Dr. Patrick Kiser laboratory for assistance and use of Rheometer.
Footnotes
Supporting information for this article is available at the bottom of the article’s abstract page, which can be accessed from the journal’s homepage at http://www.mbs-journal.de, or from the author.
Contributor Information
Jiyuan Yang, Departments of Pharmaceutics and Pharmaceutical Chemistry/CCCD, 20 S 2030 E, BPRB 205B, University of Utah, Salt Lake City, Utah 84112, USA Fax: (+801) 581 7848.
Michael T. Jacobsen, Departments of Pharmaceutics and Pharmaceutical Chemistry/CCCD, 20 S 2030 E, BPRB 205B, University of Utah, Salt Lake City, Utah 84112, USA Fax: (+801) 581 7848
Huaizhong Pan, Departments of Pharmaceutics and Pharmaceutical Chemistry/CCCD, 20 S 2030 E, BPRB 205B, University of Utah, Salt Lake City, Utah 84112, USA Fax: (+801) 581 7848.
Jindřich Kopeček, Email: jindrich.kopecek@utah.edu, Departments of Pharmaceutics and Pharmaceutical Chemistry/CCCD, 20 S 2030 E, BPRB 205B, University of Utah, Salt Lake City, Utah 84112, USA Fax: (+801) 581 7848. Department of Bioengineering, University of Utah, Salt Lake City, Utah 84112-9452, USA.
References
- 1.Wichterle O, Lím D. Nature. 1960;185:117. [Google Scholar]
- 2.Kopeček J, Yang J. Polym Int. 2007;56:1078. [Google Scholar]
- 3.Kopeček J. J Polym Sci, Part A: Polym Chem. 2009;47:5928. doi: 10.1002/pola.23607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bencherif SA, Gao H, Srinivasan A, Siegwart DJ, Hollinger JO, Washburn NR, Matyjaszewski K. Biomacromolecules. 2009;10:1795. doi: 10.1021/bm900213u. [DOI] [PubMed] [Google Scholar]
- 5.Kirkland SE, Hensarling RM, McConaughy SD, Guo Y, Jarrett WL, McCormick CL. Biomacromolecules. 2008;9:481. doi: 10.1021/bm700968t. [DOI] [PubMed] [Google Scholar]
- 6.Malkoch M, Vestberg R, Gupta N, Mespouille L, Dubois P, Mason AF, Hedrick JL, Liao Q, Frank CW, Kingsbury K, Hawker CJ. Chem Commun. 2006:2774. doi: 10.1039/b603438a. [DOI] [PubMed] [Google Scholar]
- 7.Ossipov DA, Hilborn J. Macromolecules. 2006;39:1709. [Google Scholar]
- 8.DeForest CA, Polizzotti BD, Anseth KS. Nat Mater. 2009;8:659. doi: 10.1038/nmat2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rizzi SC, Ehrbar M, Halstenberg S, Raeber GP, Schmoekel HG, Hagenmüller H, Müller R, Weber FE, Hubbell JA. Biomacromolecules. 2006;7:3019. doi: 10.1021/bm060504a. [DOI] [PubMed] [Google Scholar]
- 10.Kopeček J, Rejmanová P. Enzymatically Degradable Bonds in Synthetic Polymers. In: Bruck SD, editor. Controlled Drug Delivery. I. CRC Press; Boca Raton, FL: 1983. p. 81. [Google Scholar]
- 11.Ulbrich K, Strohalm J, Kopeček J. Biomaterials. 1982;3:150. doi: 10.1016/0142-9612(82)90004-7. [DOI] [PubMed] [Google Scholar]
- 12.Rejmanová P, Pohl J, Baudyš M, Kostka V, Kopeček J. Makromol Chem. 1983;184:2009. [Google Scholar]
- 13.Šubr V, Duncan R, Kopeček J. J Biomater Sci, Polym Ed. 1990;1:261. doi: 10.1163/156856289x00145. [DOI] [PubMed] [Google Scholar]
- 14.Plunkett KN, Berkowski KL, Moore JS. Biomacromolecules. 2005;6:632. doi: 10.1021/bm049349v. [DOI] [PubMed] [Google Scholar]
- 15.West JL, Hubbell JA. Macromolecules. 1999;32:241. [Google Scholar]
- 16.Saito H, Hoffman AS, Ogawa HI. J Bioact Compatible Polym. 2007;22:589. [Google Scholar]
- 17.Liu SQ, Ee PLR, Ke CY, Hedrick JL, Yang YY. Biomaterials. 2009;30:1453. doi: 10.1016/j.biomaterials.2008.11.023. [DOI] [PubMed] [Google Scholar]
- 18.Xu C, Breedveld V, Kopeček J. Biomacromolecules. 2005;6:1739. doi: 10.1021/bm050017f. [DOI] [PubMed] [Google Scholar]
- 19.Yang J, Xu C, Kopečková P, Kopeček J. Macromol Biosci. 2006;6:201. doi: 10.1002/mabi.200500208. [DOI] [PubMed] [Google Scholar]
- 20.Radu-Wu LC, Yang J, Wu K, Kopeček J. Biomacromolecules. 2009;10:2319. doi: 10.1021/bm9005084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Banwell EF, Abelardo ES, Adams DJ, Birchall MA, Corrigan A, Donald AM, Kirkland M, Serpell LC, Butler MF, Woolfson DN. Nat Mater. 2009;8:596. doi: 10.1038/nmat2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Larsen TH, Furst EM. Phys Rev Lett. 2008;100:146001. doi: 10.1103/PhysRevLett.100.146001. [DOI] [PubMed] [Google Scholar]
- 23.Crocker JC, Valentine MT, Weeks ER, Gisler T, Kaplan PD, Yodh AG, Weitz DA. Phys Rev Lett. 2000;85:888. doi: 10.1103/PhysRevLett.85.888. [DOI] [PubMed] [Google Scholar]
- 24.Glazer AN, Smith EL. J Biol Chem. 1961;236:2948. [PubMed] [Google Scholar]
- 25.Gao H, Matyjaszewski K. J Am Chem Soc. 2007;129:6633. doi: 10.1021/ja0711617. [DOI] [PubMed] [Google Scholar]
- 26.Lutolf MP, Hubbell JA. Biomacromolecules. 2003;4:713. doi: 10.1021/bm025744e. [DOI] [PubMed] [Google Scholar]
- 27.Polizzotti BD, Fairbanks BD, Anseth KS. Biomacromolecules. 2008;9:1084. doi: 10.1021/bm7012636. [DOI] [PubMed] [Google Scholar]
- 28.Cicuta P, Donald AM. Soft Matter. 2007;3:1449. doi: 10.1039/b706004c. [DOI] [PubMed] [Google Scholar]
- 29.Mason TG, Weitz DA. Phys Rev Lett. 1995;74:1250. doi: 10.1103/PhysRevLett.74.1250. [DOI] [PubMed] [Google Scholar]
- 30.Schechter I, Berger A. Biochem Biophys Res Commun. 1967;27:157. doi: 10.1016/s0006-291x(67)80055-x. [DOI] [PubMed] [Google Scholar]
- 31.Schechter I, Berger A. Biochem Biophys Res Commun. 1968;32:898. doi: 10.1016/0006-291x(68)90326-4. [DOI] [PubMed] [Google Scholar]
- 32.Ulbrich K, Strohalm J, Kopeček J. Makromol Chem. 1986;187:1131. [Google Scholar]
- 33.Ulbrich K, Zacharieva EI, Obereigner B, Kopeček J. Biomaterials. 1980;1:199. doi: 10.1016/0142-9612(80)90017-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.