

Abstract
Multidrug ABC transporters can transport a wide range of drugs from the cell. Ongoing studies of the prototype mammalian multidrug resistance ATP-binding cassette transporter P-glycoprotein (ABCB1) have revealed many intriguing functional and biochemical features. However, a gap remains in our knowledge regarding the molecular basis of its broad specificity for structurally unrelated ligands. Recently, the first crystal structures of ligand-free and ligand-bound ABCB1 showed ligand binding in a cavity between its two membrane domains, and now previous observations on polyspecificity can be interpreted in a structural context. The new ABCB1 crystal structures also suggest a critical role for an axial rotation of transmembrane helices for high-affinity binding and low-affinity release of ligands during transmembrane transport.
Domain organisation
Multidrug ATP-binding cassette (ABC) transporters mediate the ATP-dependent extrusion of cytotoxic agents away from their intracellular targets1. They are pharmacologically important proteins in humans as they participate in the distribution and elimination of drugs in the body, and can confer drug resistance on cancer cells2-4. These transporters are also expressed in plants5 and in microbial pathogens associated with some of the most devastating diseases in our world; in this capacity they can impair antimicrobial chemotherapy6-8. Multidrug ABC transporters belong to the ABC superfamily that contains 48 representatives in the human genome alone9. In this superfamily, ABC exporters can be distinguished from ABC importers by the directionality of transport and distinct structural arrangements of the membrane domains (MDs). All ABC transporters contain two nucleotide-binding domains (NBDs), each carrying the namesake ABC motif, and two MDs, usually each containing 6 transmembrane helices (TMHs). In bacteria and archaea, ABC exporters are typically expressed as half-transporters, with one NBD and one MD on a single polypeptide chain. Two chains then assemble into a functional homo- or heterodimer. However, in eukarya ABC transporters are often expressed as a single polypeptide chain upon which the 4 domains are fused. The human multidrug resistance P-glycoprotein ABCB1, which was first described by Danø10, and Juliano and Ling11, and subsequently cloned as a full-length cDNA by Ueda and colleagues12, is a typical example of this architecture.
In general, the dimeric NBDs in multidrug ABC transporters act in concert to hydrolyse ATP and provide the free energy to drive directional transport against transmembrane concentration gradients for hydrophilic substrates and against the lipid-water partition coefficient for hydrophobic substrates. Although several models have been proposed to explain the energetic coupling between the NBDs and the transport by the MDs (reviewed in Ref. 13), these models will not be discussed. Instead, here we aim to provide the reader with an up-to-date view of drug–multidrug ABC transporter interactions. We will, in particular, address the structural features and mechanisms that allow the MDs of ABCB1 and bacterial homologs to bind and transport toxic ions and drugs (referred to as ligands). By comparing the recently published crystal structure of ligand-bound ABCB1 with available crystal structures of bacterial homologs in post-hydrolysis states, we suggest a role of helix rotation in ligand binding and release on opposite sides of the membrane.
Drug binding and transport
Our early knowledge about the ligand specificity of multidrug ABC transporters comes in large part from cell biological and biochemical experimentation. One recurrent theme that emerged from these studies is that hydrophobic ligands might interact with binding sites in ABCB1 which lie within the membrane. For example, the potency of inhibitors to modify ABCB1-mediated anthracycline transport is directly proportional to their ability to partition in the phospholipid bilayer14. Non-fluorescent acetoxymethyl precursors of Calcein (Calcein-AM) and 2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein (BCECF-AM) are extruded by ABCB1 and other systems before these precursors are converted into fluorescent Calcein and BCECF probes by cytoplasmic non-specific esterases15,16. The transport of the hydrophobic fluorescent dye 1-[4-(trimethylamino)phenyl]-6-phenylhexa-1,3,5-triene (TMA-DPH) occurs at a rate dependent on its concentration in the inner leaflet of the membrane16. Fluorescence energy transfer experiments on ABCB1 positioned a binding site for the dye Hoechst 33342 in the inner membrane leaflet17. In another setting, the ABCB1 homolog HlyB from Escherichia coli interacts with the signal sequence of α-hemolysin that forms an amphiphilic helix and binds to the cytoplasmic leaflet of the plasma membrane18. These examples support the notion that amphiphilic and hydrophobic ligands are ‘intercepted’ while they reside at the membrane, raising the suggestion that ABCB1 acts by a hydrophobic vacuum cleaner mechanism19,20. The physiological relevance of this mechanism for multidrug ABC transporters is related to the observed tendency of many (cationic) antimicrobial and anticancer chemotherapeutic agents to accumulate at the water–lipid interface of the membrane21, and for intracellular drugs at the inner leaflet16.
Various studies aimed to locate the drug binding sites and key residues responsible for the interaction with ligands. In addition to ABCB14,22, the bacterial homologs LmrA23-25 from Lactococcus lactis and MsbA26-30 from E.coli are among the best-studied multidrug ABC transporters. The range of ligands that can be transported by these ABC systems overlaps, and indeed, LmrA can functionally substitute for ABCB1 in human lung fibroblast cells31. The MDs in ABCB1 and homodimeric LmrA share the common 6 + 6 helix arrangement32 that enables binding of ligands in the absence of the NBDs24,33. Studies of ligand–ligand interactions on ABCB1 revealed that some ligands interact with the transporter as single molecules, whereas others interact as pairs34. ABCB1 contains distinct sites for transport of rhodamine 123 (R-site) and Hoechst 33342 (H-site) in addition to a modulatory site for prazosin and progesterone35,36. The interaction of ligand at one of the transport competent sites enhances the ligand interaction at the other site35. LmrA is equally capable of binding and transporting two drug molecules in a positive cooperative manner23, further underlining the mechanistic similarities between mammalian and bacterial multidrug ABC transporters. Equilibrium binding measurements on ABCB1 provided evidence for three sites for transported ligands (vinblastine, pacitaxel, and Hoechst 33342), which can interact with ligand in the absence of exogenously added nucleotide, in addition to a modulatory site for nicardipine/GF120918 37. As a network of interactions between these kinetically distinguishable drug-binding sites exists, the possibility was raised that these sites are present on a common interacting surface.
Cross-linking experiments using photo-reactive drug analogs followed by peptide mapping38 provided a first glance at the locations of ligand binding in ABCB1. These insights were expanded by extensive cysteine scanning mutagenesis in which single mutants were tested for their ability to react with thiol-reactive substrates including dibromobimane39, methanethiosulfonate-verapamil (MTS-verapamil)40, and MTS-rhodamine B41. The non thiol-containing verapamil and rhodamine substrates were able to protect cysteines against labelling, thereby indicating that their MTS containing analogs bound ABCB1 at overlapping sites. The resulting data showed that residues on TMHs 6, 9 and 12 form the R-site pocket, whereas the H-site is created by residues located on TMHs 4, 6, 10, 11 and 12. Further studies examined the solvent accessibility of these regions. In LmrA, the highly soluble thiol-reactive compound fluorescein maleimide could access 11 out of 15 aromatic residues in the MD42. In similar experiments on ABCB1, the water soluble cysteine-reactive compounds sodium (2-sulfonatoethyl) methanethiosulfonate (MTS-ES) and [2-(trimethylammonium)ethyl)] methanethiosulfonate (MTS-ET) competed successfully with verapamil for site-specific labelling of single cysteine mutants43. As all of the experiments were carried out in the absence of ATP or non-hydrolysable nucleotide analogs, it was concluded that ABC multidrug transporters contain ligand-binding chambers that are exposed to the cytoplasm and are partially hydrated. A role of TMH 6 in drug binding was also found in MsbA, where Ser289 and Ser290 to Ala (SASA) substitutions in TMH 6 specifically inhibit the interaction of MsbA with ethidium and Taxol28.
The newly published inward-facing structures of ABCB144 exhibit structural features that give further credence to the previously discussed observations. The MDs indeed form a ligand-binding chamber that is exposed to the cytoplasm and contains a laterally open gap that is accessible to the inner leaflet of the membrane (Figure 1A). The V-shaped arrangement of the MDs in the membrane is also observed in the inward-facing structures of MsbA from E. coli and Vibrio cholera45 (Table 1). This conformation is supported by recent pulse double electron–electron resonance and fluorescence homo-transfer experiments using purified E. coli MsbA in detergent solution as well as when inserted in the phospholipid bilayer of liposomes46. In the outward-facing nucleotide-bound conformations of the ABCB1 homologs MsbA from Salmonella typhimurium45 and Sav1866 from Staphylococcus aureus47,48, and our ABCB1 models based on these templates (Figure 1B), the MDs also form a lateral gap that is exposed to the outer leaflet of the bilayer and the external environment. The inward-facing gap in ABCB1 (Figure 1A) and predicted outward-facing gap (Figure 1B) are connected by the ligand-binding surface at the centre of the membrane44 (Figure 1C). This ligand-binding surface might be preserved in the same position in the bacterial ABCB1 homologs. Residues Ser289 and Ser290 in E. coli MsbA that contribute to ligand specificity of the transporter28 are located at positions corresponding to those in TMH 6 and 12 of inward-facing ABCB1 at the top of the binding cavity for the co-crystallized Cyclic Pepide P-glycoprotein Inhibitors (CPPIs; QZ59-RRR and QZ59-SSS)44, close to the leaflet–leaflet interface (Figure 1C). The positioning of the binding cavity at the centre of the phospholipid bilayer lowers the activation energy for permeation of ligand through the acyl chain region, and grants alternating access to the cytosolic and extracellular facing leaflets of the membrane in a fashion that does not require significant movement of the ligand. Ligand binding at the leaflet–leaflet interface is also observed for members of the Major Facilitator Superfamily (MFS)49, which represents the largest group of secondary-active membrane transporters. The crystal structure of the lactose-H+ symporter LacY from E. coli shows the lactose-binding site in a hydrophobic environment at the centre of the membrane, consistent with the observation that the affinity of this MFS transporter for dansylated sugars increases with the hydrophobicity of the sugar derivatives50. Similarly, the ligand-binding site of the human MFS transporter Glut-1 (for glucose) can be labelled with lipophilic forskolin derivatives51. Hence, although sugars are not lipid-soluble substrates, their binding sites in sugar transporters can be accessible both from the aqueous phase and the phospholipid bilayer. EmrD, whose drug-binding site appears accessible from the lipid bilayer in a crystal structure52, transports uncouplers of oxidative phosphyorylation like meta-chloro carbonylcyanide phenylhydrazone (CCCP) directly from the cell membrane. Amphiphilic ligands, which insert with their hydrophobic moieties in the acyl chain region of one leaflet of the phospholipid bilayer and with their hydrophilic moieties in the polar head group region of that leaflet, likely invert during alternating access by ABCB1 by a mechanism that is not resolved experimentally. Thus, the “ON” and “OFF” sites originally proposed by Ambudkar and colleagues53 could reflect alternating access of the same binding cavity to the inner and outer membrane leaflets. The possibility exists, however, that the “ON” site in the central ligand binding cavity of ABCB1 is spatially linked to other binding regions located closer to or within the intracellular domains and/or NBDs30. This arrangement might provide access to more hydrophilic ligands from the cytoplasm (Figure 1A).
Figure 1.

Model for ligand efflux by ABCB1. ABCB1 contains a central ligand-binding cavity close to the leaflet–leaflet interface of the membrane. During transport, this binding cavity is alternately exposed to the inside and outside surface of the membrane. The inward-facing structure of ABCB1 (A) and putative outward-facing structure (B) have a laterally open cleft in their MDs exposed to the lipid phase of the bilayer. In the inward-facing conformation, this “gap” could grant ligand (green) access to the binding site directly from the inner leaflet of the phospholipid bilayer and cytoplasm. When the transporter assumes its outward-facing conformation the ligand is expelled into the outer leaflet and/or aqueous extracellular medium. The outward-facing ABCB1 model in (B) is based on MsbA from S. thyphimurium45 and Sav1866 from S. aureus47. (C) Cross section through ABCB1 bound to two molecules of QZ59-SSS (black) reveals the central location of the ligand-binding site in relation to the phospholipid bilayer. The residues in ABCB1 corresponding to Ser289 and Ser290 in E. coli MsbA, which affect ligand specificity of this bacterial transporter28, are shown as brown spheres to highlight their juxtaposition to the bound ligand. The surface is rendered by hydrophobicity (orange) and hydrophilicity (blue). The model in (B) was generated using Modeller. Figure was generated in Chimera.
Table 1.
Available crystal structures of ABC multidrug exporters.
| Protein | Conformationa | Nucleotide | Drug | pdb code | Resolution (Å) | ref. | |
|---|---|---|---|---|---|---|---|
| 1 | Sav1866 | outward | ADPb | -- | 2HYD | 3.00 | 47 |
| 2 | Sav1866 | outward | AMP-PNP | -- | 2ONJ | 3.40 | 48 |
| 3 | MsbA | inward | -- | -- | 3B5W | 5.30 | 45 |
| 4 | MsbA | inward | -- | -- | 3B5X | 5.50 | 45 |
| 5 | MsbA | outward | AMP-PNP | -- | 3B5Y | 4.50 | 45 |
| 6 | MsbA | outward | ADP-OV | -- | 3B5Z | 4.20 | 45 |
| 7 | MsbA | outward | AMP-PNP | -- | 3B60 | 3.70 | 45 |
| 8 | Mouse ABCB1a |
inward | -- | -- | 3G5U | 3.80 | 44 |
| 9 | Mouse ABCB1a |
inward | -- | QZ59-RRR | 3G60 | 4.40 | 44 |
| 10 | Mouse ABCB1a |
inward | -- | 2 × QZ59- SSS |
3G61 | 4.35 | 44 |
Conformation refers to the inward or outward-facing orientations of the cavity formed by the MDs.
Abbreviations: ADP Adenosine-di-phosphate, AMP-PNP Adenosine 5′-(β,γ-imido)triphosphate, ADP-OV Adenosine-di-phosphate-orthovanadate, QZ59-RRR cyclic-tris-(R)-valineselenazole QZ59-SSS cyclic-tris-(S)-valineselenazole.
Based on the information inferred from the crystal structures and biochemical data, the transport mechanism can be broken down into at least 4 steps, which together describe alternating access of the ligand-binding cavity to either side of the membrane. (i) A ligand enters the binding cavity from the inner membrane leaflet through the lateral gaps between the two MDs or directly from the cytoplasm and binds the high-affinity ligand-binding site. (ii) The MDs undergo a conformational ‘switch’ upon binding and/or hydrolysis of ATP13,54,55, closing the binding pocket to the inner leaflet of the membrane and opening it to the outer leaflet. The binding site is thought to reduce its affinity for the ligand by decreasing favourable intermolecular contacts thereby increasing the off-rate. (iii) The ligand is released at the lateral gap into the outer leaflet of the membrane and/or external aqueous environment. (iv) After dissociation of the ligand the MDs reset to the inward-facing high-affinity state described under (i). It is important to note that although the alternating access model for ABCB1 often emphasizes a two-step oscillation between an inward-facing conformation and an outward-facing conformation (Figure 1), the conformational change pathways leading from inward-facing to outward-facing and from outward-facing back to inward-facing (collectively referred to as “transport cycle”) might be non-identical and most likely involve multiple intermediate states. The formation of a conformation with disengaged NBDs (and hence, low affinity for nucleotides), as obtained in the ABCB1 structure, could represent a compulsory step in this transport cycle.
Polyspecificity of the drug-binding cavity
The two reported structures of ABCB1 in complex with one QZ59-RRR molecule or two QZ59-SSS molecules (Figure 2) are consistent with previous observations that ABCB1 and LmrA can bind more than one ligand simultaneously23,34,56. The cavity formed by ABCB1 encloses a volume of approximately 6000 cubic-Å44 thus providing ample space for two drug molecules. The ABCB1–CPPI interactions are dominated by the hydrophobic side chains of nonpolar Phe and polar Tyr residues that reside in the apex of the ligand-binding cavity in TMH 1, 6, 7, and 12 (Table 2). This data agrees with previous suggestions regarding the importance of aromatic residues in ABCB1–ligand binding57,58. Phe and Tyr residues account for ~ 40 % and ~ 60 % of the ABCB1–CPPI interaction as measured by buried surface area in the case of ABCB1 bound to a single CPPI and two CPPI molecules, respectively. The binding of the structurally different CPPIs is imparted by the utilization of different combinations of Phe and Tyr side chains and by the flexibility of these side chains, allowing them to form rotamers with different orientations in the two non-identical drug-bound states (Figure 2). In addition to aromatic side chains, aliphatic side chains and less abundant polar hydrogen-bond donor side chains (Ser, Thr, Gln, Asn)59 are found, the latter mostly in the walls of the cavity leading to the apex. Hence, structurally diverse ligands can be accommodated in the binding cavity by stacking and cation-π interactions and by hydrophobic and hydrogen bonding interactions, offering a structural explanation for the polyspecificity of this transporter. The chemical nature of the ABCB1–CPPI interactions is comparable to that observed in structures of other protein–hydrophobic ligand complexes. A well-known example is provided by soluble multidrug binding transcriptional regulators, which have a similar multidrug specificity as the multidrug transporters they regulate. In the transcriptional regulator QacR from S. aureus, the plasticity of the ligand-binding site is similarly affected by the flexibility and redundancy of ligand-binding residues (including Tyr and Phe side chains)60,61 as observed in ABCB1. In addition, at least two compounds (the bivalent aromatic diamidines DB75 and DB359) can bind QacR in more than one orientation62.
Figure 2.

ABCB1 can use different combinations of flexible side chains in its ligand-binding cavity to create binding sites for different ligands. Structure of ABCB1 complexed with one molecule of QZ59-RRR (red) or two molecules of QZ59-SSS (black). Superimposition of the ligand-binding sites: residues interacting with QZ59-RRR are rendered in red whereas residues interacting with QZ59-SSS are rendered in black. The magnified stereo view (inset) reveals alternative side chain rotamers in the two different drug bound crystal structures. Up to 60 % of the ABCB1–CPPI interactions in the binding cavity is based on interactions with Phe and Tyr side chains. The use of different sets of Phe and Tyr residues in binding of QZ-RRR and QZ59-SSS, and flexibility of these aromatic side chains contribute to the specificity of the ligand-binding cavity for different CPPIs. The N-terminal half of ABCB1 is rendered in green, the C-terminal half in blue. Figure was generated in Pymol.
Table 2.
Summary of interactions between ABCB1 and ligandsa
The ligand-binding residues in ABCB1 that lie in proximity to the CPPI ligands overlap extensively with those previously identified in biochemical studies on ABCB163,64, LmrA42 and MsbA28 suggesting that these bacterial transporter also bind ligands in the apex of the binding cavity in the inward-facing conformation. However, a direct comparison of the apex regions in the crystal structures of mouse ABCB1 and MsbA, and homology models of human ABCB1, the human hepatic ABCB1 homolog ABCB465, Sav1866 and LmrA reveals that many hydrophobic residues are large (Tyr, Phe, Trp) in ABCB1 (mouse and human) and ABCB4, but smaller (Gly, Ala, Leu, Ile, Val, Met) in MsbA, Sav1866 and LmrA (Figure 3). Hence, the overlapping ligand specificities of these multidrug ABC transporters26,31,65,66 are not based on sequence conservations, but instead, rely on functional conservations of residues. The size changes in non-polar residues in the bacterial transporters and loss of the aromaticity of these residues creates steric changes that alter the affinity for cationic aromatic ligands. ABCB4 interacts with zwitterionic phosphatidylcholine65, MsbA interacts with the net anionic lipid A anchor of lipopolysaccharides67, and more recently, dimeric LmrA was found to specifically bind anionic cardiolipin68; these steric changes in the binding cavity might relate to differences in lipid specificity of these ABC multidrug transporters.
Figure 3.
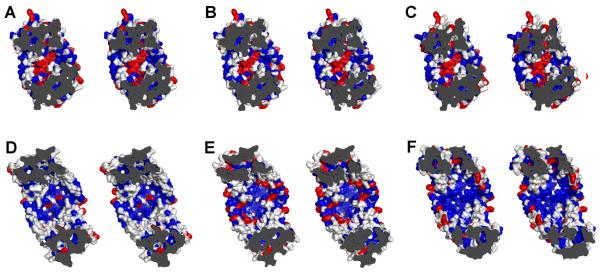
Stereo views of inward-facing ligand-binding pockets. Views are shown for (A) ABCB1a (mouse), (B) ABCB1 (human), (C) ABCB4 (human) (D) MsbA (E. coli), (E) Sav1866 (S. aureus), and (F) LmrA (L. lactis). Aromatic residues (Tyr, Phe, Trp) are colored red, hydrophobic residues (Gly, Ala, Leu, Ile, Val, Met) are blue and all other residues are colored white. The bacterial models (Sav1866 and LmrA) were based on a full model of MsbA (E. coli) and the mammalian models (ABCB1 and ABCB4) were based on the crystal structure of ABCB1a. The models were created using Swiss Model.
Helix rotation drives affinity change?
Having explored how ligands can bind in the ABCB1 ligand-binding cavity, we will investigate how the ligand-binding cavity can switch from high-affinity in the inward-facing state to low-affinity in the outward-facing state, allowing dissociation of the ligand. Fortunately, the inward-facing structures of ABCB1 are complemented by the previously published outward-facing structures of the homologous bacterial ABC transporters MsbA45 and Sav186647,48. Both were crystallised in nucleotide-bound form, with the drug-binding cavity exposed to the extracellular side. In our outward-facing ABCB1 models that were based on these structures, the TMHs that carry the majority of residues lining the ligand-binding cavity in the inward-facing ABCB1 structure have rotated along the length of their axis by up to 90°. The CPPI binding residues situated along TMHs 1, 6, 7 and 12 are forced to follow this rotation (Figure 4). As a result, the amino acid side chains that previously reached into the ligand-binding cavity are now partially or completely buried in the MDs, or are even facing the lipid bilayer. Thus, when the transporter assumes its outward-facing state in an efflux reaction coupled to ATP-binding and hydrolysis, the ligand-binding surface is deprived of the residues that support the strongest interactions with the ligand. As a result, the ligand dissociates and diffuses away. A similar conclusion on helix rotation was reached in a study in which published amino acid substitutions in ABCB1 that are associated with a change-in-specificity phenotype69 were superimposed on the outward-facing structure of MsbA from Salmonella typhimurium28. As observed for Ser289 and Ser290, these residues are partially occluded or faced away from the drug-binding surface in this outward-facing conformation28. Biochemical evidence for helix rotation was obtained by Loo and Clarke70, who observed that thiol-cross-linking between TMH 6 and 12 in ABCB1 was altered by ATP hydrolysis in a fashion requiring rotation of one or both helices. In considering the structural similarity between ABCB1, LmrA, Sav1866 and MsbA, it is possible that the mechanism involving helix rotations is conserved among multidrug ABC transporters.
Figure 4.

Conformational switch in ABCB1 from the inward to the outward facing state is accompanied by rotation of the transmembrane helices. Helix rotations are evident from a comparison of positions of atoms that comprise helices and bulky amino acid side chains. During the rotation, side chains important for ligand binding to the inward-facing conformation move away from the binding cavity. As favourable protein–ligand interactions are disrupted, the reduced binding affinity in the outward-facing conformation allows dissociation of the ligand from the binding cavity. Inward-facing and outward-facing conformations of the ABCB1 MDs are seen from the outside of the cell. Loops and the NBD are omitted for clarity. Residues involved in ligand binding are shown as sticks. Residues residing on TMHs 1, 6, 7 and 12 are shown in red. Residues contributed from other helices are rendered in green. Arrows refer to helix rotation from the inward to the outward state. To generate the outward-facing view, the residues from ABCB1 were superimposed onto the outward-facing structure of S. thyphimurium MsbA using Modeller. Figure was generated in Pymol.
Concluding remarks
This review summarizes that the insights gained by the first structures of ABCB1 are complementary with the established biochemical understanding of this transporter and its bacterial homologs. Yet these structures also allow us to ask more questions. The CPPI ligands QZ59-RRR and QZ59-SSS clearly inhibit ABCB1-ATPase activity in the detergent solubilised form as well as ABCB1-mediated transport and drug resistance in intact cells44. However, the CPPI inhibitors are bound to a ligand-binding cavity in ABCB1 that can also bind ligands that do get transported. What distinguishes the inhibitor from a transported ligand? Do non-transported inhibitors and transported ligands bind different sets of residues? If the V-shaped ABCB1 conformation is part of the transport cycle, could CPPI inhibitors prevent the tight dimerisation of half-transporters necessary for propagation of this cycle? Could these inhibitors sterically hinder the helix rotations required for their release at the opposite side of the membrane? The answers to these questions might hold important clues to the future development of modulators of multidrug ABC transporters, which would have an enormous impact on the efficacy of chemotherapeutic treatment of infectious diseases and cancer. Progress will require further structural and biochemical characterizations of a variety of transport cycle intermediates of mouse and human ABCB1 and their bacterial homologs that provide useful and accessible models.
Acknowledgments
We apologize to all colleagues whose work was not cited in this brief review. We thank James Smith and Alan Senior for discussions. Our work was supported the Biotechnology and Biological Sciences Research Council (BBSRC) (to HWvV), and the Jasper L. and Jack Denton Wilson Foundation (to IU), and by grants from the US Army (W81XWH-05-1-0316), NIH (GM61905), and the Skaggs Chemical Biology Foundation (to GC).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure Statement
The authors have declared that no competing interests exist.
References
- 1.Higgins CF. Multiple molecular mechanisms for multidrug resistance transporters. Nature. 2007;446:749–757. doi: 10.1038/nature05630. [DOI] [PubMed] [Google Scholar]
- 2.Borst P, Oude Elferink R. Mammalian ABC transporters in health and disease. Annu. Rev. Biochem. 2002;71:537–592. doi: 10.1146/annurev.biochem.71.102301.093055. [DOI] [PubMed] [Google Scholar]
- 3.Gottesman MM, et al. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat. Rev. Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- 4.Holland IB, et al., editors. ABC Proteins: From Bacteria to Man. Academic Press; 2003. [Google Scholar]
- 5.Verrier PJ, et al. Plant ABC proteins - a unified nomenclature and updated inventory. Trends Plant Sci. 2008;13:151–159. doi: 10.1016/j.tplants.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 6.Ouellette M, et al. Multidrug resistance and ABC transporters in parasitic protozoa. J. Mol. Microbiol. Biotechnol. 2001;3:201–206. [PubMed] [Google Scholar]
- 7.Sipos G, Kuchler K. Fungal ATP-binding cassette (ABC) transporters in drug resistance and detoxification. Curr. Drug Targets. 2006;7:471–481. doi: 10.2174/138945006776359403. [DOI] [PubMed] [Google Scholar]
- 8.Nikaido H. Multidrug resistance in bacteria. Annu. Rev. Biochem. 2009;78:119–146. doi: 10.1146/annurev.biochem.78.082907.145923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dean M, et al. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 2001;11:1156–1166. doi: 10.1101/gr.184901. [DOI] [PubMed] [Google Scholar]
- 10.Dano K. Active outward transport of daunomycin in resistant Ehrlich ascites tumor cells. Biochim. Biophys. Acta. 1973;323:466–493. doi: 10.1016/0005-2736(73)90191-0. [DOI] [PubMed] [Google Scholar]
- 11.Juliano RL, Ling V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim. Biophys. Acta. 1976;455:152–162. doi: 10.1016/0005-2736(76)90160-7. [DOI] [PubMed] [Google Scholar]
- 12.Ueda K, et al. Expression of a full-length cDNA for the human MDR1 gene confers resistance to colchicine, doxorubicin, and vinblastine. Proc. Natl. Acad. Sci. USA. 1997;94:3004–3009. doi: 10.1073/pnas.84.9.3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seeger MA, van Veen HW. Molecular basis of multidrug transport by ABC transporters. Biochim. Biophys. Acta. 2009;1794:725–737. doi: 10.1016/j.bbapap.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 14.Friche E, et al. Effect of anthracycline analogs on photolabelling of P-glycoprotein by [125I]iodomycin and [3H]azidopine: relation to lipophilicity and inhibition of daunorubicin transport in multidrug resistant cells. Br. J. Cancer. 1993;67:226–231. doi: 10.1038/bjc.1993.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Homolya L, et al. Fluorescent cellular indicators are extruded by the multidrug resistance protein. J. Biol. Chem. 1993;268:22493–22496. [PubMed] [Google Scholar]
- 16.Bolhuis H, et al. Multidrug resistance in Lactococcus lactis: evidence for ATP-dependent drug extrusion from the inner leaflet of the cytoplasmic membrane. EMBO J. 1996;15:4239–4245. [PMC free article] [PubMed] [Google Scholar]
- 17.Qu Q, Sharom FJ. Proximity of bound Hoechst 33342 to the ATPase catalytic sites places the drug binding site of P-glycoprotein within the cytoplasmic membrane leaflet. Biochemistry. 2002;41:4744–4752. doi: 10.1021/bi0120897. [DOI] [PubMed] [Google Scholar]
- 18.Sheps JA, et al. Hemolysin transport in Escherichia coli. Point mutants in HlyB compensate for a deletion in the predicted amphiphilic helix region of the HlyA signal. J. Biol. Chem. 1995;270:14829–14834. doi: 10.1074/jbc.270.24.14829. [DOI] [PubMed] [Google Scholar]
- 19.Higgins CF, Gottesman MM. Is the multidrug transporter a flippase? Trends Biochem. Sci. 1992;17:18–22. doi: 10.1016/0968-0004(92)90419-a. [DOI] [PubMed] [Google Scholar]
- 20.Gottesman MM, Pastan I. Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu. Rev. Biochem. 1993;62:385–427. doi: 10.1146/annurev.bi.62.070193.002125. [DOI] [PubMed] [Google Scholar]
- 21.Siarheyeva A, et al. Localization of multidrug transporter substrates within model membranes. Biochemistry. 2006;45:6203–6221. doi: 10.1021/bi0524870. [DOI] [PubMed] [Google Scholar]
- 22.Ambudkar SV, Gottesman MM, editors. Meth. Enzymol. Vol. 292. Elsevier; 1998. ABC Transporters: Biochemical, Cellular, and Molecular Aspects. [Google Scholar]
- 23.van Veen HW, et al. The homodimeric ATP-binding cassette transporter LmrA mediates multidrug transport by an alternating two-site (two-cylinder engine) mechanism. EMBO J. 2000;19:2503–2514. doi: 10.1093/emboj/19.11.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Venter H, et al. An ABC transporter with a secondary-active multidrug translocator domain. Nature. 2003;426:866–870. doi: 10.1038/nature02173. [DOI] [PubMed] [Google Scholar]
- 25.Shilling R, et al. A critical role of a carboxylate in proton conduction by the ATP-binding cassette multidrug transporter LmrA. FASEB J. 2005;19:1698–1700. doi: 10.1096/fj.04-3558fje. [DOI] [PubMed] [Google Scholar]
- 26.Reuter G, et al. The ATP binding cassette multidrug transporter LmrA and lipid transporter MsbA have overlapping substrate specificities. J. Biol. Chem. 2003;278:35193–35198. doi: 10.1074/jbc.M306226200. [DOI] [PubMed] [Google Scholar]
- 27.Woebking B, et al. Drug-lipid A interactions on the Escherichia coli ABC transporter MsbA. J. Bacteriol. 2005;187:6363–6369. doi: 10.1128/JB.187.18.6363-6369.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Woebking B, et al. Functional role of transmembrane helix 6 in drug binding and transport by the ABC transporter MsbA. Biochemistry. 2008;47:10904–10914. doi: 10.1021/bi800778d. [DOI] [PubMed] [Google Scholar]
- 29.Siarheyeva A, Sharom FJ. The ABC transporter MsbA interacts with lipid A and amphipathic drugs at different sites. Biochem. J. 2009;419:317–328. doi: 10.1042/BJ20081364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smriti PZ, et al. Mapping daunorubicin-binding sites in the ATP-binding cassette transporter MsbA using site-specific quenching by spin labels. J. Biol. Chem. 2009;284:13904–13913. doi: 10.1074/jbc.M900837200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Veen HW, et al. A bacterial antibiotic-resistance gene that complements the human multidrug-resistance P-glycoprotein gene. Nature. 1998;391:291–295. doi: 10.1038/34669. [DOI] [PubMed] [Google Scholar]
- 32.Grimard V, et al. Structure and dynamics of the membrane-embedded domain of LmrA investigated by coupling polarized ATR-FTIR spectroscopy and (1)H/(2)H exchange. Biochemistry. 2001;40:11876–11886. doi: 10.1021/bi010017+. [DOI] [PubMed] [Google Scholar]
- 33.Loo TW, Clarke DM. The transmembrane domains of the human multidrug resistance P-glycoprotein are sufficient to mediate drug binding and trafficking to the cell surface. J. Biol. Chem. 1999;274:24759–24765. doi: 10.1074/jbc.274.35.24759. [DOI] [PubMed] [Google Scholar]
- 34.Ayesh S, et al. Co-operative, competitive and non-competitive interactions between modulators of P-glycoprotein. Biochim. Biophys. Acta. 1996;1316:8–18. doi: 10.1016/0925-4439(96)00008-7. [DOI] [PubMed] [Google Scholar]
- 35.Shapiro AB, Ling V. Positively cooperative sites for drug transport by P-glycoprotein with distinct drug specificities. Eur. J. Biochem. 1997;250:130–137. doi: 10.1111/j.1432-1033.1997.00130.x. [DOI] [PubMed] [Google Scholar]
- 36.Shapiro AB, et al. Stimulation of P-glycoprotein-mediated drug transport by prazosin and progesterone. Evidence for a third drug-binding site. Eur. J. Biochem. 1999;259:841–850. doi: 10.1046/j.1432-1327.1999.00098.x. [DOI] [PubMed] [Google Scholar]
- 37.Martin C, et al. Communication between multiple drug binding sites on P-glycoprotein. Mol. Pharmacol. 2000;58:624–632. doi: 10.1124/mol.58.3.624. [DOI] [PubMed] [Google Scholar]
- 38.Peer M, et al. Photoaffinity labeling of P-glycoprotein. Mini Rev. Med. Chem. 2005;5:165–172. doi: 10.2174/1389557053402738. [DOI] [PubMed] [Google Scholar]
- 39.Loo TW, Clarke DM. Identification of residues within the drug-binding domain of the human multidrug resistance P-glycoprotein by cysteine-scanning mutagenesis and reaction with dibromobimane. J. Biol. Chem. 2000;275:39272–39278. doi: 10.1074/jbc.M007741200. [DOI] [PubMed] [Google Scholar]
- 40.Loo TW, Clarke DM. Defining the drug-binding site in the human multidrug resistance P-glycoprotein using a methanethiosulfonate analog of verapamil, MTS-verapamil. J. Biol. Chem. 2001;276:14972–14979. doi: 10.1074/jbc.M100407200. [DOI] [PubMed] [Google Scholar]
- 41.Loo TW, Clarke DM. Location of the rhodamine-binding site in the human multidrug resistance P-glycoprotein. J. Biol. Chem. 2002;277:44332–44338. doi: 10.1074/jbc.M208433200. [DOI] [PubMed] [Google Scholar]
- 42.Poelarends GJ, Konings WN. The transmembrane domains of the ABC multidrug transporter LmrA form a cytoplasmic exposed, aqueous chamber within the membrane. J. Biol. Chem. 2002;277:42891–42898. doi: 10.1074/jbc.M206508200. [DOI] [PubMed] [Google Scholar]
- 43.Loo TW, et al. The drug-binding pocket of the human multidrug resistance P-glycoprotein is accessible to the aqueous medium. Biochemistry. 2004;43:12081–12089. doi: 10.1021/bi049045t. [DOI] [PubMed] [Google Scholar]
- 44.Aller SG, et al. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science. 2009;323:1718–1722. doi: 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ward A, et al. Flexibility in the ABC transporter MsbA: Alternating access with a twist. Proc. Natl. Acad. Sci. USA. 2007;104:19005–19010. doi: 10.1073/pnas.0709388104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Borbat PP, et al. Conformational motion of the ABC transporter MsbA induced by ATP hydrolysis. PLoS Biol. 2007;5:e271. doi: 10.1371/journal.pbio.0050271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dawson RJ, Locher KP. Structure of a bacterial multidrug ABC transporter. Nature. 2006;443:180–185. doi: 10.1038/nature05155. [DOI] [PubMed] [Google Scholar]
- 48.Dawson RJ, Locher KP. Structure of the multidrug ABC transporter Sav1866 from Staphylococcus aureus in complex with AMP-PNP. FEBS Lett. 2007;581:935–938. doi: 10.1016/j.febslet.2007.01.073. [DOI] [PubMed] [Google Scholar]
- 49.Law CJ, et al. Ins and outs of Major Facilitator Superfamily antiporters. Annu. Rev. Microbiol. 2008;62:289–305. doi: 10.1146/annurev.micro.61.080706.093329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schuldiner S, et al. Microenvironment of the binding site in the lac carrier protein. Proc. Natl. Acad. Sci. USA. 1977;74:1851–1854. doi: 10.1073/pnas.74.5.1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wadzinski BE, et al. Localization of the forskolin photolabelling site within the monosaccharide transporter of human erythrocytes. Biochem. J. 1990;272:151–158. doi: 10.1042/bj2720151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yin Y, et al. Structure of the multidrug transporter EmrD from Escherichia coli. Science. 2006;312:741–744. doi: 10.1126/science.1125629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ambudkar SV, et al. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu. Rev. Pharmacol. Toxicol. 1999;39:361–398. doi: 10.1146/annurev.pharmtox.39.1.361. [DOI] [PubMed] [Google Scholar]
- 54.Higgins CF, Linton KJ. The ATP switch model for ABC transporters. Nat. Struct. Mol. Biol. 2004;11:918–926. doi: 10.1038/nsmb836. [DOI] [PubMed] [Google Scholar]
- 55.Sauna ZE, Ambudkar SV. About a switch: how P-glycoprotein (ABCB1) harnesses the energy of ATP binding and hydrolysis to do mechanical work. Mol. Cancer Ther. 2007;6:13–23. doi: 10.1158/1535-7163.MCT-06-0155. [DOI] [PubMed] [Google Scholar]
- 56.Loo TW, et al. Simultaneous binding of two different drugs in the binding pocket of the human multidrug resistance P-glycoprotein. J. Biol. Chem. 2003;278:39706–39710. doi: 10.1074/jbc.M308559200. [DOI] [PubMed] [Google Scholar]
- 57.Pawagi AB, et al. Transmembrane aromatic amino acid distribution in P-glycoprotein. A functional role in broad substrate specificity. J. Mol. Biol. 1994;235:554–564. doi: 10.1006/jmbi.1994.1013. [DOI] [PubMed] [Google Scholar]
- 58.Ambudkar SV, et al. P-glycoprotein: From genomics to mechanism. Oncogene. 2003;22:7468–7485. doi: 10.1038/sj.onc.1206948. [DOI] [PubMed] [Google Scholar]
- 59.Seelig A, et al. Substrate recognition by P-glycoprotein and the multidrug resistance-associated protein MRP1: a comparison. Int. J. Clin. Pharmacol. Ther. 2000;38:111–122. doi: 10.5414/cpp38111. [DOI] [PubMed] [Google Scholar]
- 60.Schumacher MA, et al. Structural mechanism of the simultaneous binding of two drugs to a multidrug-binding protein. EMBO J. 2004;23:2923–2930. doi: 10.1038/sj.emboj.7600288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Peters KM, et al. QacR-cation recognition is mediated by a redundancy of residues capable of charge neutralization. Biochemistry. 2008;47:8122–8129. doi: 10.1021/bi8008246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brooks BE, et al. Multidrug-binding transcription factor QacR binds the bivalent aromatic diamidines DB75 and DB359 in multiple positions. J. Am. Chem. Soc. 2007;129:8389–8395. doi: 10.1021/ja072576v. [DOI] [PubMed] [Google Scholar]
- 63.Loo TW, et al. Transmembrane segment 7 of human P-glycoprotein forms part of the drug-binding pocket. Biochem. J. 2006;399:351–359. doi: 10.1042/BJ20060715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Loo TW, et al. Transmembrane segment 1 of human P-glycoprotein contributes to the drug-binding pocket. Biochem. J. 2006;396:537–545. doi: 10.1042/BJ20060012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Smith AJ, et al. MDR3 P-glycoprotein, a phosphatidylcholine translocase, transports several cytotoxic drugs and directly interacts with drugs as judged by interference with nucleotide trapping. J. Biol. Chem. 2000;275:23530–23539. doi: 10.1074/jbc.M909002199. [DOI] [PubMed] [Google Scholar]
- 66.Velamakanni S, et al. Multidrug transport by the ABC transporter Sav1866 from Staphylococcus aureus. Biochemistry. 2009;47:9300–9308. doi: 10.1021/bi8006737. [DOI] [PubMed] [Google Scholar]
- 67.Doerrler WT, et al. An Escherichia coli mutant defective in lipid export. J. Biol. Chem. 2001;276:11461–11464. doi: 10.1074/jbc.C100091200. [DOI] [PubMed] [Google Scholar]
- 68.Velamakanni S, et al. A multidrug ABC transporter with a taste for salt. PLoS One. 2009;4:e6137. doi: 10.1371/journal.pone.0006137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shilling RA, et al. New light on multidrug binding by an ATP-binding-cassette transporter. Trends Pharmacol. Sci. 2006;27:195–203. doi: 10.1016/j.tips.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 70.Loo TW, Clarke DM. Cross-linking of human multidrug resistance P-glycoprotein by the substrate, tris-(2-maleimidoethyl)amine, is altered by ATP hydrolysis. Evidence for rotation of a transmembrane helix. J. Biol. Chem. 2001;276:31800–31805. doi: 10.1074/jbc.M103498200. [DOI] [PubMed] [Google Scholar]