

Abstract
The objective of this study was to explore the involvement of collectin liver 1 (CL-L1) and collectin kidney 1 (CL-K1) and other pattern recognition molecules (PRMs) of the lectin pathway of the complement system in a cross-sectional cohort of systemic lupus erythematosus (SLE) patients. Concentrations in plasma of CL-L1, CL-K1, mannan-binding lectin (MBL), M-ficolin, H-ficolin and L-ficolin were determined in 58 patients with SLE and 65 healthy controls using time-resolved immunoflourometric assays. The SLE patients' demographic, diagnostic, clinical and biochemical data and collection of plasma samples were performed prospectively during 4 months. CL-L1, CL-K1 and M-ficolin plasma concentrations were lower in SLE patients than healthy controls (P-values < 0·001, 0·033 and < 0·001, respectively). H-ficolin concentration was higher in SLE patients (P < 0·0001). CL-L1 and CL-K1 plasma concentrations in the individuals correlated in both patients and controls. Patients with low complement component 3 (C3) demonstrated a negative correlation between C3 and CL-L1 and CL-K1 (P = 0·022 and 0.031, respectively). Patients positive for anti-dsDNA antibodies had lower levels of MBL in plasma than patients negative for anti-dsDNA antibodies (P = 0·02). In a cross-sectional cohort of SLE patients, we found differences in the plasma concentrations of CL-L1, CL-K1, M-ficolin and H-ficolin compared to a group of healthy controls. Alterations in plasma concentrations of the PRMs of the lectin pathway in SLE patients and associations to key elements of the disease support the hypothesis that the lectin pathway plays a role in the pathogenesis of SLE.
Keywords: complement system, lectin pathway, pattern recognition molecules, systemic lupus erythematosus
Introduction
Systemic lupus erythematosus (SLE) is a complex autoimmune disease, which affects primarily women of childbearing age, and disease progression involves both the innate and the adaptive immune system. It is a severe systemic disease potentially affecting multiple organ systems, but the pathogenesis remains unresolved 1. However, several studies indicate that the innate immune system, and complement in particular, plays a key role. In the daily clinic, measurements of complement proteins (e.g. C3 and C4) are used in monitoring SLE disease activity 2 and complement component 1q (C1q) plays a role in the pathogenesis, as deficiency is associated strongly with SLE 3–5.
The complement system is part of the innate immune defence. It comprises three initiating pathways: the classical, alternative and lectin pathways 6,7. The latter pathway activates the complement system when one of six pattern recognition molecules (PRMs) – mannan-binding lectin (MBL), M-ficolin, L-ficolin, H-ficolin, collectin liver 1 (CL-L1) or collectin kidney 1 (CL-K1), in complex with the MBL-associated serine proteases (MASPs) – bind to a surface recognized by the PRMs. The MASPs then become enzymatically active and initiate the common pathway of the complement system 8. This activation results in opsonization of the target, chemotaxis of leucocytes and lysis of the recognized cell 6.
CL-L1 and CL-K1 are the last members of the PRMs of the lectin pathway to be discovered 9,10. Both PRMs are found in circulation with MASPs 11,12, and it was revealed recently that most of the CL-K1 exists in serum in the form of heteromeric complexes with CL-L1, making up a CL–LK heterocomplex. The heteromeric complex, CL–LK, interacts avidly with MASPs and mediates complement activation potently in comparison with, e.g. CL-K1 alone 11,12. In the present work we have chosen to detect both of the two different polypeptide chains (CL-L1 and CL-K1 polypeptide chains) of the complex and make statistical analyses separately for the concentrations of each of them. The preceding literature describes examinations of the protein as if there are two different molecules, and CL-K1 was shown to bind carbohydrate moieties such as fucose and mannose and CL-L1 to bind mannose, N-acetylglucosamine, galactose and fucose 9,10,12,13; more recently, the recognition domain of the CL-K1 polypeptide chain was described in complex with a di-mannose structure 14. As is also the case for the other soluble pattern recognition molecules mentioned above, a very low affinity between the single recognition domain and, for example, monosaccharides is seen. Only when clustering of the domains through oligomerization happens will there be a high avidity binding towards patterns of carbohydrate structures. Using the CL-K1 polypeptide chain alone, it was also found that an activation of complement could be observed 15.
CL-K1 binds DNA-coated surfaces, which leads to C4b-deposition via MASPs, and could thus play a role in response to particles and surfaces presenting extracellular DNA, such as apopototic cells, neutrophil extracellular traps and biofilms 16. The concentration of the two polypeptide chains, CL-L1 and CL-K1, is found in closely associated concentrations, again indicating that they exist as one complex, CL–LK 17.
Five different mutations have been identified in the gene encoding CL-K1 (COLEC11) associated with the severe developmental 3MC syndrome 18. A recent clinical study observed higher plasma concentrations of CL-K1 in patients with disseminated intravascular coagulation 19.
There is considerable evidence that defects in the removal of apoptotic cells is associated with autoimmune disease 20,21. PRMs of the lectin pathway appear to assist in disposing of dying host cells by binding apoptotic cells, promoting macropinocytosis of the debris 22–24. Accordingly, MBL-deficient mice also clear apoptotic cells poorly 25.
MBL have been implicated in the pathogenesis of SLE with conflicting results 21,26. However, the polymorphism in the MBL2 gene at codon 54, which gives rise to an amino acid change resulting in low MBL concentrations in plasma, has been associated with SLE susceptibility 27. A more pronounced association of MBL deficiency and SLE has also been seen in patients with accompanying complement deficiency 28. Further, anti-MBL antibodies are present in sera from SLE patients and influence the functional activity of MBL 29. There have been only few reports addressing the ficolins in relation to SLE. High serum concentrations of H-ficolin and low L-ficolin concentrations have been measured in serum from SLE patients 30,31, but only the high H-ficolin levels were confirmed in a recent report 32. To our knowledge, there are no publications on CL-K1 or CL-L1 in SLE.
The aim of this pilot study was to assess the plasma concentrations of CL-K1, CL-L1 and the remaining PRMs of the lectin pathway in patients diagnosed with SLE and in age- and gender-matched healthy controls. Further, we analysed for associations between the PRMs of the lectin pathway and disease manifestations in SLE.
Materials and methods
Patients
From a cross-sectional cohort of 58 SLE patients, we collected prospectively plasma samples, demographic, biochemical and clinical data including American College of Rheumatology (ACR) classification criteria 33, SLE disease activity index score (SLEDAI) 2 and organ damage using the Systemic Lupus International Collaborating Clinics/American College of Rheumatology Damage Index (SDI) 34. The inclusion criteria were: fulfilment of the 1997 revised ACR classification criteria for SLE 33, female, aged more than 18 years and exclusion of incapacitated patients. For comparison, plasma samples were collected from 65 age- and gender-matched healthy blood donors (Table1). Only females were included, as it would not be possible to ensure anonymity for males in a group of 58 SLE patients.
Table 1.
Demographics and clinical characteristics of systemic lupus erythematosus (SLE) patients and demographics of healthy controls
| Characteristics | Patients (n = 58) |
|---|---|
| M/F | 0/58 |
| Mean age at diagnosis ± s.d. (range) | 32 ± 2·3 (14–64) |
| Mean age at inclusion ± s.d. (range) | 46·1 ± 13·1 (24–69) |
| Ethnic Danes (%) | 58 (100) |
| Hydroxychloroquine treatment n (%) | 45 (78) |
| Prednisolone treatment n (%) | 22 (38) |
| Mycophenolate mofetil n (%) | 6 (10) |
| Azathioprine n (%) | 10 (17) |
| No treatment n (%) | 6 (10) |
| ACR criteria | |
| Malar rash (ACR-1) n (%) | 27 (46·6) |
| Discoid rash (ACR-2) n (%) | 11(19·0) |
| Photosensitivity (ACR-3) n (%) | 28 (48·3) |
| Oronasal ulcers (ACR-4) n (%) | 13 (22·4) |
| Arthritis (ACR-5) n (%) | 46 (79·3) |
| Serositis (ACR-6) n (%) | 14 (24·1) |
| Nephritis (ACR-7) n (%) | 7 (12·1) |
| CNS (ACR-8) n (%) | 4 (6·89) |
| Haematological (ACR-9) n (%) | 33 (57·0) |
| Immunological (ACR-10) n (%) | 46 (79·3) |
| ANA (ACR-11) n (%) | 57 (98·3) |
| Present anti-ds-DNA level (*103 IU/l) ± s.d. | 33·3 ± 15·7 |
| Disease activity/organ damage | |
| SLEDAI mean ± s.d. | 6·48 ± 4·03 |
| SDI mean ± s.d. | 1·72 ± 1·56 |
| Characteristics healthy controls | |
| M/F | 0/65 |
| Mean age ± s.d. (range) | 44·03 ± 12·2 (21–64) |
ANA = anti-nuclear antibody; CNS = central nervous system; SDI = Systemic Lupus International Collaborating Clinics/American College of Rheumatology Damage Index; SLEDAI = SLE disease activity index; ACR = American College of Rheumatology; M/F = male/female; s.d. = standard deviation.
Methods
CPT™ tubes (BD Vacutainers®; BD Diagnostics, Franklin Lakes, NJ, USA) were used to collect peripheral venous blood samples. The time from collection to processing the blood did not exceed 1 h. Blood samples were centrifuged at 1800 g for a minimum of 30 min at room temperature in a horizontal rotor, aliquoted and then stored at −80°C.
Plasma concentrations of CL-L1 35, CL-K1 36, MBL 37, M-ficolin 38 and H-ficolin 39 were determined using time-resolved immunofluorometric assays (TRIFMA). The assays and specific antibodies for CL-K1, CL-L1, MBL, M-ficolin and H-ficolin were produced in-house, and for each protein a detailed description for the assays can be found within the references given above 35–39. We estimated L-ficolin plasma concentrations using an enzyme-linked immunosorbent assay (ELISA) (Hycult Biotech®, Uden, the Netherlands), according to the manufacturer's instructions.
In brief, samples were thawed at 4°C overnight and prediluted fourfold in Tris-buffered saline (10 mM Tris, 145 mM NaCl, 15 nM NaN2 pH 7·4) and thereafter freeze–thaw cycles were kept at a minimum. The TRIFMA assays are traditional sandwich immunoassays using a combination of two monoclonal antibodies; one is used to coat the microtitre plate, the other antibody is biotinylated and used to detect the antigen bound to the coating antibody. Europium-labelled streptavidin, which binds biotin, is added, and the europium is detected after the addition of enhancement solution on a fluorometer (Victor X5®; PerkinElmer, Waltham, MA, USA). All samples were added in duplicate and the analysis was repeated if the coefficient of variation (%CV) was greater than 20% between the two wells. For all assays, internal controls were used to ensure reproducibility and plates were repeated if the quality controls varied with %CV > 15 compared with our laboratory standard values. Regarding the concentrations given for CL-L1, for the present report we determined the concentration of CL-L1 in standard serum using a new and better-characterized preparation of recombinant CL-L1, resulting in levels approximately three times lower than given in our previous report 35.
Statistics
The data were checked for normality by Q–Q plots and histograms, and Gaussian distribution could not be assumed. Therefore, non-parametric tests were used for the statistical analysis. The Mann–Whitney U-test was used for comparison of plasma levels of the proteins in patients and controls and correlation analysis was performed calculating Spearman's rank correlation coefficient. P-values < 0·05 were considered statistically significant. Stata® version 12 and GraphPad Prism® software package (version 6·0) were used for data management and statistical calculations.
Ethics statement
The Regional Committee on Health Research Ethics (case no. 1-10-72-214-13) and the Danish data protection agency approved the study. The project was performed in pursuance of the Helsinki Declaration.
Results
The study cohort was comparable with other European SLE cohorts, except for a lower number of patients with kidney infection (Table1).
The concentrations of CL-L1, CL-K1 and M-ficolin in plasma were found to be lower in patients than in healthy controls (P-values < 0·0001, < 0·0334 and < 0·0001, respectively). The median concentration of H-ficolin in patients was significantly higher than in healthy controls. We observed no statistically significant difference between median concentration of MBL and L-ficolin in SLE patients and healthy controls (Table2 and Supporting information, Fig. S1).
Table 2.
Plasma concentrations of the pattern recognition molecules of the lectin complement pathway in systemic lupus erythematosus (SLE) patients and healthy controls
| Results | SLE patients (n = 58) Median plasma conc. μg/ml (range) | Healthy controls (n = 65) Median plasma conc. μg/ml (range) | Mann–Whitney P-value |
|---|---|---|---|
| CL-L1 | 0·25 (0·16–0·37) | 0·31 (0·20–0·51) | <0·001 |
| CL-K1 | 0·33 (0·22–0·45) | 0·34 (0·25–0·50) | 0·033 |
| MBL | 0·75 (0·00–3,25) | 0·70 (4–4,203) | 0·520 |
| M-ficolin | 0·30 (0·01–1·01) | 0·49 (0·16–1·63) | <0·001 |
| L-ficolin | 2·50 (1·09–6·11) | 2·40 (0·32–6·44) | 0·612 |
| H-ficolin | 21·5 (6·64–53·9) | 16·0 (5·17–25·3) | <0·001 |
CL-L1 = collectin liver 1; CL-K1 = collectin kidney 1; MBL = mannan-binding lectin.
A positive correlation (r = 0·576, P < 0·0001) was found between the concentrations of CL-K1 and CL-L1 in plasma in both patients and in healthy controls (Fig. 1).
Figure 1.
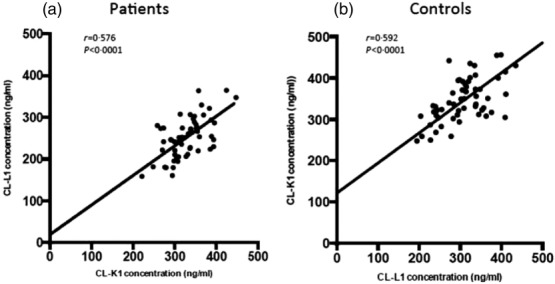
Correlation of collectin liver 1 (CL-L1) and collectin kidney 1 (CL-K1) plasma concentration in systemic lupus erythematosus (SLE) patients (a) and healthy controls (b).
For patients with discoid rash we observed higher plasma concentrations of CL-L1, M-ficolin and H-ficolin compared to patients without this manifestation (Table3). In patients with mucosal ulcers or lymphopenia, we also observed higher H-ficolin concentrations in plasma compared to patients who did not display these symptoms. In the patients with lymphopenia, there was a positive correlation between lymphocyte count and H-ficolin concentration, although it did not reach statistical significance (Table4).
Table 3.
Differences in plasma concentrations of the pattern recognition molecules in patients with and without characteristic manifestations of systemic lupus erythematosus (SLE)
| Patients with manifestations | Patients without manifestations | ||||
|---|---|---|---|---|---|
| Manifestation | n | n | P | ||
| CL-L1 median (range) | CL-L1 median (range) | ||||
| Discoid rash | 11 | 0·281 (0·161–0·330) | 47 | 0·247 (0·159–0·365) | 0·036 |
| M-ficolin median (range) | M-ficolin median (range) | ||||
| Discoid rash | 11 | 0·444 (0·098–0·878) | 47 | 0·228 (0·010–1·013) | 0·028 |
| H-ficolin median (range) | H-ficolin median (range) | ||||
| Discoid rash | 11 | 31·3 (15·8–42·8) | 47 | 20·5 (6·64–54·0) | 0·012 |
| Lymphopenia | 30 | 24·7 (12·8–54·0) | 28 | 20·3 (6·64–40·7) | 0·022 |
| Mucosal ulcers | 13 | 31·3 (13·3–54·0) | 45 | 20·5 (6·64–51·1) | 0·037 |
| MBL median (range) | MBL median (range) | ||||
| Anti-dsDNA-positive | 34 | 0·311 (0·003–2·265) | 24 | 0·970 (0·010–3·249) | 0·020 |
Lymphopenia was determined as blood counts < 1·30 × 109/l. Measurement of anti-ds-DNA were considered positive >10 × 103 IU/l. CL-L1 = collectin liver 1.
Table 4.
Spearman's rank correlation between key laboratory findings in the systemic lupus erythematosus (SLE) patients and plasma concentrations of the pattern recognition molecules
| Concentration | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CL-L1 | CL-K1 | MBL | M-ficolin | L-ficolin | H-ficolin | |||||||
| rho P | rho P | rho P | rho P | rho P | rho P | |||||||
| Lymphopenia (n = 30) | −0·128 | 0·500 | −0·203 | 0·281 | −0·298 | 0·109 | 0·116 | 0·542 | 0·406 | 0·055 | 0·353 | 0·056 |
| Anti-ds-DNA-positive (n = 34) | 0·281 | 0·120 | 0·303 | 0·091 | 0·125 | 0·496 | −0·275 | 0·127 | −0·097 | 0·624 | 0·018 | 0·923 |
| Low C3 (n = 25) | −0·464 | 0·022 | −0·442 | 0·031 | −0·014 | 0·941 | 0·002 | 0·992 | 0·199 | 0·386 | 0·067 | 0·750 |
Lymphopenia was determined as blood counts < 1·30 × 109/l. Measurement of anti-ds-DNA was considered positive > 10 × 103 IU/l. Low C3 was determined as plasma concentrations <0·90 g/l. CL-L1 = collectin liver 1; CL-K1 = collectin kidney 1; MBL = mannan-binding lectin.
Patients who were positive for anti-dsDNA antibodies showed lower levels of MBL than anti-dsDNA antibody-negative patients (P = 0·020). A negative correlation was found between concentrations of both CL-L1 and CL-K1 and C3 concentration in patients with low C3 concentrations (Table4 and Fig. 2).
Figure 2.
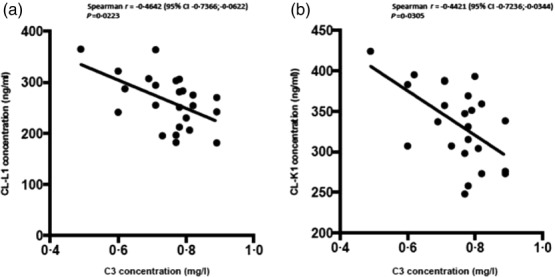
Correlation between C3-plasma concentrations and collectin liver 1 (CL-L1) (a) and collectin kidney 1 (CL-K1) (b) in systemic lupus erythematosus (SLE) patients with low C3-count (low = C3 < 0·90 mg/l).
No associations were found between disease activity (SLEDAI) or accumulated organ damage (SDI score) with concentrations in plasma of the PRM (Supporting information, Table S1).
Discussion
Adaptive immunity has been the main focus of research in SLE, particularly in the attempts to develop new therapies for the disease. Intuitively, this makes sense, as autoantibodies and immune complexes, which attack the patient's own tissue, characterize the disease 40. However, B cell-depleting therapy, targeting the antibody-producing cells, have had disappointing clinical efficacy in active SLE 41–43. These observations have led to a renewed interest in the potential role of the innate immune system in the pathogenesis of SLE.
In the innate immune system, deficiencies in the complement system have already been associated with SLE 1, and this makes pattern recognition within the lectin pathway interesting.
In this cross-sectional cohort of SLE-patients, we found plasma concentrations of several PRMs of the lectin pathway to be altered significantly, compared to healthy controls with low levels in plasma of CL-L1, CL-K1 and M-ficolin and high levels of H-ficolin in SLE patients.
The clear correlation between CL-L1 and CL-K1 substantiates the findings by Henriksen et al. 16 that the two proteins form a heteromeric complex (CL–LK) and thereby exist in more or less equal amounts in the circulation. It is noteworthy that, in the present report, the levels of CL-L1 for the healthy individuals are lower than we reported originally for blood donors 35, as we have now managed to develop a better quantification of the CL-L1 content in our standard serum. Further, both proteins are lower in SLE patients than healthy controls, and there is a negative correlation between the plasma concentrations of both CL-L1 and CL-K1 and C3 in patients with low C3. Whether the low levels of CL-L1 and CL-K1 in SLE patients in our study are caused by polymorphisms in the genes encoding the proteins 17, due to consumption because of increased activation of the lectin pathway, antibodies against the proteins or faster clearance of the proteins due to binding to apoptotic cells, remains to be investigated.
It would seem paradoxical that we observe lower concentrations of CL-K1 and CL-L1 in SLE patients compared to healthy controls. However, when the analysis is confined to the subgroup of patients with low C3, in general the patients with the highest disease activity, they have higher levels of CL-L1 and CL-K1. The same complexity is seen when viewing the classical pathway of the complement system. The immune complexes that deposit in the kidney effectively activate the complement system via the classical pathway 44. However, deficiency of complement C1q, which is necessary for classical pathway activation, leads to SLE in more than 90% of cases 3. This seems to be a paradox, as complement is thought to contribute to the damage seen in SLE 45. However, it is possible that complement activation acts as a double-edged sword, being highly important in preventing SLE and exacerbating it once the disease has been established 44.
For MBL, we did not see a difference in plasma concentrations between patients and healthy controls. We did, however, observe a very clear difference in MBL concentrations between SLE patients who were positive for anti-dsDNA antibodies and those who were not, indicating a potential pathogenic role of MBL in the subset of SLE patients who are positive for anti-dsDNA antibodies. The genetic association of low MBL with SLE 27 would probably have been stronger if only patients positive for anti-dsDNA antibodies were examined.
High plasma concentrations of H-ficolin in SLE patients and an association to lymphopenia have been described previously 30,32, and Ucieklak et al. described an association between high levels of H-ficolin and SLE glomerulonephritis. We could not confirm the association between glomerulonephritis and H-ficolin, but this could be explained by the relatively small number of patients in the subgroups. In our SLE cohort, 12% of patients had kidney involvement. Approximately 18% of approximately 200 SLE patients in our out-patient clinic are treated for nephritis. This could possibly represent a selection bias, or could be due to chance.
We found that SLE patients with discoid skin manifestations had higher plasma concentrations of CL-L1, M-ficolin and H-ficolin than patients without discoid lesions. Recently, Wallim et al. 46 described MBL depositions in lesional skin of SLE patients, but not in non-lesional skin or in discoid skin lesions. Our findings support a different pathogenic mechanism in the discoid skin manifestation of SLE and indicate an association with the lectin pathway proteins.
Deciphering the complex interaction of the lectin pathway proteins in the pathological mechanisms of SLE is a challenging task; the limitations posed by the small patient subgroups in the present study are obvious, and call for additional investigations in large SLE cohorts. However, it appears that there are significant changes in the PRMs of the lectin pathway in SLE patients and that the associations with key manifestations of the disease found in our study indicate a pathogenic role in SLE.
In conclusion, we observed significant concentration changes in plasma of the PRMs of the lectin pathway in SLE patients compared to healthy controls and found significant associations with central manifestations of SLE. Our findings support a pathogenic role of the lectin pathway in SLE.
Acknowledgments
The authors would like to acknowledge the Danish Rheumatism Association, who supported the project.
Author contributions
A. T. and L. J. performed the laboratory experiments; A. T., B. D. and K. S. was in charge of collecting clinical data and blood samples; S. H., S. T. and J. C. J. developed the assays used in the project and supervised laboratory procedures; M. J. L. handled the blood samples after they were drawn and collected and handled the control material. A. T., S. T. and K. S. wrote the manuscript and all authors participated in the editing of the article.
Disclosure
The authors declare no conflicts of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Fig. S1. Scatterplots of the plasma concentration of all the pattern recognition molecules of the lectin pathway in patients and healthy controls.
Table S1. Spearman's rank correlation between disease activity score, organ damage score and plasma concentrations of the pattern recognition molecules
References
- Bryan AR, Wu EY. Complement deficiencies in systemic lupus erythematosus. Curr Allergy Asthma Rep. 2014;14:448. doi: 10.1007/s11882-014-0448-2. [DOI] [PubMed] [Google Scholar]
- Bombardier C, Gladman DD, Urowitz MB, Caron D, Chang CH. Derivation of the SLEDAI. A disease activity index for lupus patients. The Committee on Prognosis Studies in SLE. Arthritis Rheum. 1992;35:630–40. doi: 10.1002/art.1780350606. [DOI] [PubMed] [Google Scholar]
- Walport MJ, Davies KA, Botto M. C1q and systemic lupus erythematosus. Immunobiology. 1998;199:265–85. doi: 10.1016/S0171-2985(98)80032-6. [DOI] [PubMed] [Google Scholar]
- Botto M, Dell'Agnola C, Bygrave AE, et al. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat Genet. 1998;19:56–9. doi: 10.1038/ng0598-56. [DOI] [PubMed] [Google Scholar]
- Belot A, Cimaz R. Monogenic forms of systemic lupus erythematosus: new insights into SLE pathogenesis. Pediatr Rheumatol Online J. 2012;10:21. doi: 10.1186/1546-0096-10-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walport MJ. Complement. First of two parts. N Engl J Med. 2001;344:1058–66. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- Thiel S, Vorup-Jensen T, Stover CM, et al. A second serine protease associated with mannan-binding lectin that activates complement. Nature. 1997;386:506–10. doi: 10.1038/386506a0. [DOI] [PubMed] [Google Scholar]
- Kjaer TR, Thiel S, Andersen GR. Toward a structure-based comprehension of the lectin pathway of complement. Mol Immunol. 2013;56:413–22. doi: 10.1016/j.molimm.2013.05.007. [DOI] [PubMed] [Google Scholar]
- Ohtani K, Suzuki Y, Eda S, et al. Molecular cloning of a novel human collectin from liver (CL-L1) J Biol Chem. 1999;274:13681–9. doi: 10.1074/jbc.274.19.13681. [DOI] [PubMed] [Google Scholar]
- Keshi H, Sakamoto T, Kawai T, et al. Identification and characterization of a novel human collectin CL-K1. Microbiol Immunol. 2006;50:1001–13. doi: 10.1111/j.1348-0421.2006.tb03868.x. [DOI] [PubMed] [Google Scholar]
- Henriksen ML, Brandt J, Andrieu JP, et al. Heteromeric complexes of native collectin kidney 1 and collectin liver 1 are found in the circulation with MASPs and activate the complement system. J Immunol. 2013;191:6117–27. doi: 10.4049/jimmunol.1302121. [DOI] [PubMed] [Google Scholar]
- Hansen S, Selman L, Palaniyar N, et al. Collectin 11 (CL-11, CL-K1) is a MASP-1/3-associated plasma collectin with microbial-binding activity. J Immunol. 2010;185:6096–104. doi: 10.4049/jimmunol.1002185. [DOI] [PubMed] [Google Scholar]
- Selman L, Hansen S. Structure and function of collectin liver 1 (CL-L1) and collectin 11 (CL-11, CL-K1) Immunobiology. 2012;217:851–63. doi: 10.1016/j.imbio.2011.12.008. [DOI] [PubMed] [Google Scholar]
- Venkatraman Girija U, Furze CM, Gingras AR, et al. Molecular basis of sugar recognition by collectin-K1 and the effects of mutations associated with 3MC syndrome. BMC Biol. 2015;13:27. doi: 10.1186/s12915-015-0136-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma YJ, Skjoedt MO, Garred P. Collectin-11/MASP complex formation triggers activation of the lectin complement pathway–the fifth lectin pathway initiation complex. J Innate Immun. 2013;5:3242–50. doi: 10.1159/000345356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriksen ML, Brandt J, Iyer SS, Thielens NM, Hansen S. Characterization of the interaction between collectin 11 (CL-11, CL-K1) and nucleic acids. Mol Immunol. 2013;56:757–67. doi: 10.1016/j.molimm.2013.07.011. [DOI] [PubMed] [Google Scholar]
- Bayarri-Olmos R, Hansen S, Henriksen ML, et al. Genetic variation of COLEC10 and COLEC11 and association with serum levels of collectin liver 1 (CL-L1) and collectin kidney 1 (CL-K1) PLOS ONE. 2015;10:e0114883. doi: 10.1371/journal.pone.0114883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooryck C, Diaz-Font A, Osborn DP, et al. Mutations in lectin complement pathway genes COLEC11 and MASP1 cause 3MC syndrome. Nat Genet. 2011;43:197–203. doi: 10.1038/ng.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Ohtani K, Larvie M, et al. Elevated plasma CL-K1 level is associated with a risk of developing disseminated intravascular coagulation (DIC) J Thromb Thrombolysis. 2014;38:331–8. doi: 10.1007/s11239-013-1042-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walport MJ. Complement. Second of two parts. N Engl J Med. 2001;344:1140–4. doi: 10.1056/NEJM200104123441506. [DOI] [PubMed] [Google Scholar]
- Manderson AP, Botto M, Walport MJ. The role of complement in the development of systemic lupus erythematosus. Annu Rev Immunol. 2004;22:431–56. doi: 10.1146/annurev.immunol.22.012703.104549. [DOI] [PubMed] [Google Scholar]
- Runza VL, Schwaeble W, Mannel DN. Ficolins: novel pattern recognition molecules of the innate immune response. Immunobiology. 2008;213:297–306. doi: 10.1016/j.imbio.2007.10.009. [DOI] [PubMed] [Google Scholar]
- Honore C, Hummelshoj T, Hansen BE, Madsen HO, Eggleton P, Garred P. The innate immune component ficolin 3 (Hakata antigen) mediates the clearance of late apoptotic cells. Arthritis Rheum. 2007;56:1598–607. doi: 10.1002/art.22564. [DOI] [PubMed] [Google Scholar]
- Jensen ML, Honore C, Hummelshoj T, Hansen BE, Madsen HO, Garred P. Ficolin-2 recognizes DNA and participates in the clearance of dying host cells. Mol Immunol. 2007;44:856–65. doi: 10.1016/j.molimm.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Stuart LM, Takahashi K, Shi L, Savill J, Ezekowitz RA. Mannose-binding lectin-deficient mice display defective apoptotic cell clearance but no autoimmune phenotype. J Immunol. 2005;174:3220–6. doi: 10.4049/jimmunol.174.6.3220. [DOI] [PubMed] [Google Scholar]
- Monticielo OA, Mucenic T, Xavier RM, Brenol JC, Chies JA. The role of mannose-binding lectin in systemic lupus erythematosus. Clin Rheumatol. 2008;27:413–9. doi: 10.1007/s10067-008-0838-8. [DOI] [PubMed] [Google Scholar]
- Lee YH, Lee HS, Choi SJ, Ji JD, Song GG. The association between the mannose-binding lectin codon 54 polymorphism and systemic lupus erythematosus: a meta-analysis update. Mol Biol Rep. 2012;39:5569–74. doi: 10.1007/s11033-011-1361-6. [DOI] [PubMed] [Google Scholar]
- Saevarsdottir S, Kristjansdottir H, Grondal G, Vikingsdottir T, Steinsson K, Valdimarsson H. Mannan-binding lectin and complement C4A in Icelandic multicase families with systemic lupus erythematosus. Ann Rheum Dis. 2006;65:1462–7. doi: 10.1136/ard.2005.046086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seelen MA, Trouw LA, van der Hoorn JW, et al. Autoantibodies against mannose-binding lectin in systemic lupus erythematosus. Clin Exp Immunol. 2003;134:335–43. doi: 10.1046/j.1365-2249.2003.02274.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen T, Munthe-Fog L, Garred P, Jacobsen S. Serum levels of ficolin-3 (Hakata antigen) in patients with systemic lupus erythematosus. J Rheumatol. 2009;36:757–9. doi: 10.3899/jrheum.080361. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Saito R, Asano T, et al. Serum L-ficolin levels in patients with systemic lupus erythematosus. Mod Rheumatol. 2012;22:899–902. doi: 10.1007/s10165-012-0616-y. [DOI] [PubMed] [Google Scholar]
- Hein E, Nielsen LA, Nielsen CT, et al. Ficolins and the lectin pathway of complement in patients with systemic lupus erythematosus. Mol Immunol. 2015;63:209–14. doi: 10.1016/j.molimm.2014.07.003. [DOI] [PubMed] [Google Scholar]
- Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725. doi: 10.1002/art.1780400928. [DOI] [PubMed] [Google Scholar]
- Gladman D, Ginzler E, Goldsmith C, et al. The development and initial validation of the systemic lupus international collaborating clinics/American College of Rheumatology damage index for systemic lupus erythematosus. Arthritis Rheum. 1996;39:363–9. doi: 10.1002/art.1780390303. [DOI] [PubMed] [Google Scholar]
- Axelgaard E, Jensen L, Dyrlund TF, et al. Investigations on collectin liver 1. J Biol Chem. 2013;288:23407–20. doi: 10.1074/jbc.M113.492603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selman L, Henriksen ML, Brandt J, et al. An enzyme-linked immunosorbent assay (ELISA) for quantification of human collectin 11 (CL-11, CL-K1) J Immunol Methods. 2012;375:182–8. doi: 10.1016/j.jim.2011.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel S, Moller-Kristensen M, Jensen L, Jensenius JC. Assays for the functional activity of the mannan-binding lectin pathway of complement activation. Immunobiology. 2002;205:446–54. doi: 10.1078/0171-2985-00145. [DOI] [PubMed] [Google Scholar]
- Wittenborn T, Thiel S, Jensen L, Nielsen HJ, Jensenius JC. Characteristics and biological variations of M-ficolin, a pattern recognition molecule, in plasma. J Innate Immun. 2010;2:167–80. doi: 10.1159/000218324. [DOI] [PubMed] [Google Scholar]
- Krarup A, Sorensen UB, Matsushita M, Jensenius JC, Thiel S. Effect of capsulation of opportunistic pathogenic bacteria on binding of the pattern recognition molecules mannan-binding lectin, L-ficolin, and H-ficolin. Infect Immun. 2005;73:1052–60. doi: 10.1128/IAI.73.2.1052-1060.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsokos GC. Systemic lupus erythematosus. N Engl J Med. 2011;365:2110–21. doi: 10.1056/NEJMra1100359. [DOI] [PubMed] [Google Scholar]
- Merrill J, Buyon J, Furie R, et al. Assessment of flares in lupus patients enrolled in a phase II/III study of rituximab (EXPLORER) Lupus. 2011;20:709–16. doi: 10.1177/0961203310395802. [DOI] [PubMed] [Google Scholar]
- Rovin BH, Furie R, Latinis K, et al. Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: the Lupus Nephritis Assessment with Rituximab study. Arthritis Rheum. 2012;64:1215–26. doi: 10.1002/art.34359. [DOI] [PubMed] [Google Scholar]
- Furie R, Petri M, Zamani O, et al. A phase III, randomized, placebo-controlled study of belimumab, a monoclonal antibody that inhibits B lymphocyte stimulator, in patients with systemic lupus erythematosus. Arthritis Rheum. 2011;63:3918–30. doi: 10.1002/art.30613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjoberg AP, Trouw LA, Blom AM. Complement activation and inhibition: a delicate balance. Trends Immunol. 2009;30:83–90. doi: 10.1016/j.it.2008.11.003. [DOI] [PubMed] [Google Scholar]
- Reid KB, Porter RR. The proteolytic activation systems of complement. Annu Rev Biochem. 1981;50:433–64. doi: 10.1146/annurev.bi.50.070181.002245. [DOI] [PubMed] [Google Scholar]
- Wallim LR, Nisihara R, Skare T, Mocelin V, Messias-Reason IJ. Mannose binding lectin deposition in skin of lupus erythematosus patients: a case series. Hum Immunol. 2014;75:629–32. doi: 10.1016/j.humimm.2014.04.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Scatterplots of the plasma concentration of all the pattern recognition molecules of the lectin pathway in patients and healthy controls.
Table S1. Spearman's rank correlation between disease activity score, organ damage score and plasma concentrations of the pattern recognition molecules