

Abstract
Adoptive T cell therapy of cancer employs a large number of ex-vivo-propagated T cells which recognize their targets either by virtue of their endogenous T cell receptor (TCR) or via genetic reprogramming. However, both cell-extrinsic and intrinsic mechanisms often diminish the in-vivo potency of these therapeutic T cells, limiting their clinical efficacy and broader use. Direct activation of human T cells by Toll-like receptor (TLR) ligands induces T cell survival and proliferation, boosts the production of proinflammatory cytokines and augments resistance to regulatory T cell (Treg) suppression. Removal of the TLR ligand-binding region results in constitutive signalling triggered by the remaining cytosolic Toll/interleukin-1 receptor (TIR) domain. The use of such TIR domains therefore offers an ideal means for equipping anti-tumour T cells with the arsenal of functional attributes required for improving current clinical protocols. Here we show that constitutively active (ca)TLR-4 can be expressed efficiently in human T cells using mRNA electroporation. The mere expression of caTLR-4 mRNA in polyclonal CD8 and CD4 T cells induced the production of interferon (IFN)-γ, triggered the surface expression of CD25, CD69 and 4-1BB and up-regulated a panel of cytokines and chemokines. In tumour-infiltrating lymphocytes prepared from melanoma patients, caTLR-4 induced robust IFN-γ secretion in all samples tested. Furthermore, caTLR-4 enhanced the anti-melanoma cytolytic activity of tumour-infiltrating lymphocytes and augmented the secretion of IFN-γ, tumour necrosis factor (TNF)-α and granulocyte–macrophage colony-stimulating factor (GM-CSF) for at least 4 days post-transfection. Our results demonstrate that caTLR-4 is capable of exerting multiple T cell-enhancing effects and can potentially be used as a genetic adjuvant in adoptive cell therapy.
Keywords: CD8 T lymphocytes, constitutively active TLR-4, mRNA electroporation, tumour-infiltrating lymphocytes
Introduction
Different approaches to cancer treatment by adoptive cell therapy (ACT) are explored worldwide 1,2, and encouraging results are reported from a growing number of clinical trials. One approach exploits tumour-infiltrating lymphocytes (TILs) isolated from fresh tumour biopsies, which are capable of recognizing tumour antigens through their endogenous T cell receptor (TCR). TILs are reinfused to the patient either following an extended ex-vivo expansion and selection stage or after short-term culture in the absence of selection 3–6. Both procedures can achieve a high rate of objective response in patients with advanced refractory melanoma, including durable complete remission. With the recent demonstration that TILs against mutated peptides induce cancer regression in several tumours, autologous TILs are cloned and expanded, based on mutation recognition 7. In another approach, autologous or donor-derived polyclonal T cells are redirected genetically against tumour cells. This can be achieved with exogenous genes encoding carefully selected, often affinity-enhanced TCRs that redirect T cells against conventional major histocompatibility complex (MHC)-I epitopes 8,9. Alternatively, employing a strategy developed by us in the late 1980s 10, genes encoding chimeric antigen receptors (CARs) redirect T cells to recognize surface tumour antigens in an MHC-independent manner. CARs are currently examined in dozens of clinical trials, showing exceptionally high efficacy in B cell malignancies 11. New directions for applying ACT in cancer immunotherapy include the use of genetically modified donor T cells in allogeneic stem cell transplantation 12, the generation of off-the-shelf, universal T cells genetically edited to lack both TCR 13 and human leucocyte antigen (HLA) molecules 14 or autologous TCR gene therapy, which exploits the tumour-resident TCR repertoires for the development of personalized immunotherapy 15.
Although recent reports on enduring clinical responses stir much excitement, the field of ACT still faces major challenges, some posed by T cell-intrinsic and extrinsic factors, while others stem from the particular ACT protocol employed. Among the challenges that have to be overcome are the resilient microenvironment of the tumour, which exploits a variety of suppression and evasion mechanisms to avoid immunological attack (e.g. see 16); the compromised immune system of the patient, including a deteriorated T cell compartment, resulting from high tumour burden and previous chemo- or radiotherapy, functional T cell exhaustion following lengthy ex-vivo propagation and the acquisition of an unfavourable terminal effector T cell differentiation status, which impairs T cell function in vivo 17.
It is now well documented that T cells express different Toll-like receptors (TLRs) and are activated directly by TLR ligands. Engagement of these ligands has been shown to induce T cell survival and proliferation, lower the activation threshold, boost the production of interleukin (IL)-2 and interferon (IFN)-γ, render effector T cells refractory to suppression by regulatory T cells (Tregs) and promote differentiation into memory cells 18. Genetic removal of the TLR ligand-binding region results in constitutive signalling triggered by the remaining cytosolic Toll/interleukin-1 receptor (TIR) domain 19. A number of laboratories, including ours, exploited this characteristic and employed mRNA transfection for introducing constitutively active (ca)TLR-4 to ex-vivo-propagated dendritic cells for inducing their maturation and enhancing their T cell stimulatory capacity 20,21.
We hypothesized that truncated caTLRs expressed in anti-tumour T lymphocytes can augment their effector functions, alleviating, at least in part, some of the aforementioned hurdles confronted by therapeutic T cells in ACT. Here we describe the resulting phenotype and functional properties conferred on human T cells by the expression of caTLR-4 following gene delivery by mRNA electroporation.
Materials and methods
Antibodies
Phycoerythrin (PE)-CD25, allophycocyanin (APC)-CD8, fluorescein isothiocyanate (FITC)-CD69, FITC-CD8, FITC-CD4, PE-IFN-γ, FITC-CD107a, PE-cyanin 7 (Cy7)-CD8, Pacific blue CD107a, Pacific blue tumour necrosis factor (TNF)-α, APC-granulocyte–macrophage colony-stimulating factor (GM-CSF) and FITC-conjugated anti-HLA-A2 were purchased from Biolegend (San Diego, CA, USA).
Human tumour cell lines
Melanoma cell lines M171, M425 and M579 (all HLA-A2–) were established in the Sharett Institute of Oncology, Hadassah-Hebrew University Hospital laboratory, as described previously 22. The M579-A2 clone is a stable HLA-A2 transfectant of M579 cells, which expresses HLA-A2 following transfection with pcDNA3-HLA-A2 plasmid 22. All cell lines were cultured in RPMI-1640 supplemented with 10% heat-inactivated fetal calf serum (FCS), 2 mmol/l L-glutamine and combined antibiotics (all from Invitrogen Life Technologies, Carlsbad, CA, USA). M579-A2 cells were maintained in the same medium, supplemented with 1 mg/ml geneticin (Life Technologies).
Human lymphocytes
Human peripheral blood lymphocytes (PBL) were obtained from healthy donors by Ficoll Hypaque (Amersham, Uppsala, Sweden) gradient centrifugation, cultured in complete medium (CM) consisting of RPMI-1640 supplemented with 10% heat-inactivated human AB serum, 300 and 6000 IU/ml recombinant human IL-2 for PBLs and TILs, respectively (rhIL-2; Chiron, Amsterdam, the Netherlands), 2 mmol/l L-glutamine, 1 mmol/l sodium pyruvate, 1% non-essential amino acids, 25 mmol/l HEPES (pH 7·4), 50 μmol/l β-mercaptoethanol and combined antibiotics (Invitrogen Life Technologies). All TIL and blood samples were obtained under an institutional review body (IRB) approval (Hadassah Hebrew University Hospital).
Generating bulk TIL populations and cloning peptide-specific TILs
TIL microcultures were initiated and expanded from tumour specimens taken from resected metastases of melanoma patients, under IRB approval (no. 383-23.12.05), as described previously 23. On day 14 of TIL initiation, the lymphocytes were washed with phosphate-buffered saline (PBS), resuspended in PBS supplemented with 0.5% bovine serum albumin (BSA) and stained with FITC-conjugated HLA-A*0201/MART-126–35 dextramer (Immudex, Copenhagen, Denmark) for 30 min at 4°C. The lymphocytes were then incubated with APC-conjugated mouse anti-human CD8 (eBioscience, San Diego, CA, USA) for an additional 30 min at 4°C. CD8+ lymphocytes, positively stained by the dextramer (CD8+/dextramer+ cells), were sorted by a BD FACSAria (BD Biosciences, San Jose, CA, USA) and directly cloned at one or two cells per well in 96-well plates in the presence of anti-CD3 (30 ng/ml; eBioscience), rhIL-2 (6000 IU/ml) and 4 Gy-irradiated 5 × 104 allogeneic PBMCs as feeder cells. Five days later, rhIL-2 (6000 IU/ml) was added and renewed every 2 days thereafter. On day 14, the clones were assayed for IFN-γ secretion in a peptide-specific manner following their co-incubation with MART-126–35–pulsed T2 cells (peptides were synthesized commercially and purified [>95%) by reverse-phase high-performance liquid chromatography (HPLC) by Biomer Technology, Pleasanton, CA, USA] using commercially available enzyme-linked immunosorbent assay (ELISA) reagents (R&D Systems, Minneapolis, MN, USA). The MART-126–35–reactive clones used in this study (clones 1C9 and 1H8) were expanded further with anti-CD3 (30 ng/ml), rhIL-2 and 50-fold excess irradiated feeder cells. For specific TIL cultures explored in this study see Table1. TIL cultures explored in this study were prepared from resected metastases of melanoma patients under IRB approval (Hadassah Hebrew University Hospital).
Table 1.
Anti-melanoma tumour infiltrating lymphocytes (TILs) explored in this study
| Designation | T cell composition | Specificity |
|---|---|---|
| TIL425 | ∼100% CD8 | – |
| TIL433 | ∼70% CD8, 30% CD4 | – |
| TIL2045 | ∼70% CD8, 30% CD4 | – |
| 1C9 | CD8 T cell clone | MART-126–35/A2 |
| 1H8 | CD8 T cell clone | MART-126–35/A2 |
Cloning human β2-microglobulin (hβ2m)-anchor constructs
For cloning hβ2m-TLR-4, the transmembrane and cytoplasmic (TC) portion of human TLR-4 (hTLR-4) seq (GenBank NM 138554) was cloned by reverse transcriptase polymerase chain reaction (RT–PCR) from mRNA of THP-I cells with the forward primer 5′-CCC TCG AGC ACC TGT CAG ATG AAT AAG ACC-3′ and the reverse primer 5′-CGC GCG GCC GCT GGG CAA GAA ATG CCT CAG GAG GT-3′. For constructing the negative control hβ2m-HLA-A2 gene, the TC portions of HLA-A2 were also cloned by RT–PCR from THP-1 mRNA using the forward primer 5′-GCC TCG AGC CAG CCC ACC ATC CCC ATC-3′ and the reverse primer 5′-GCG CGG CCG CTC ACA CTT TAC AAG CTG TGA G-3′. The full-length hβ2m gene was cloned previously by us from Jurkat cells mRNA by RT–PCR. All products were cloned in a single step into pGEM4Z/A64.
Production of in-vitro-transcribed mRNA
Template DNA for in-vitro transcription was cloned into pGEM4Z/GFP/A64 vector 24, a kind gift from Dr E. Gilboa, University of Miami, following removal of the green fluorescent protein (GFP) insert to create pGEM4Z/A64. Appropriate plasmids were linearized using SpeI restriction enzyme (NEB, Ipswitch, MA, USA) and were used as DNA templates for the in-vitro transcription reaction. Transcription was conducted in a final 20-μl reaction mix at 37°C using AmpliCap-MaxTM T7 High Yield Message Maker Kit (Epicentre Biotechnologies, Madison, WI, USA) to generate 5′-capped in-vitro-transcribed mRNA. Purification of mRNA was performed by DNase-I digestion, followed by LiCl precipitation, according to the manufacturer's instructions. mRNA quality was checked by agarose gel electrophoresis and concentration was determined by spectrophotometric analysis at optical diameter (OD) 260. RNA was stored at −80°C in small aliquots.
Isolation and stimulation of T cells
Thawed or fresh PBLs from healthy donors or TILs from melanoma patients were cultured in growth medium in the presence of 300 or 6000 U/ml IL-2, respectively, for 24 h. Cells were then separated to CD4 and CD8 subsets by positive selection (BD Biosciences) and activated for 72 h in the presence of plate-bound anti-CD3 and 0·5 µg/ml soluble anti-CD28 monoclonal antibodies (mAb) and 100 U/ml IL-2. This step is necessary, as high-yield mRNA transfection of T cells requires an activated state. Stimulated T cells were then cultured for another 24 h in complete medium without antibodies and cytokines to stop stimulation.
mRNA electroporation
Stimulated PBLs and TILs were washed twice and resuspended in OptiMEM medium to a final concentration of 3 × 107/ml (Gibco BRL, Grand Island, NY, USA). Cells and cuvettes were kept on ice for 5 min before electroporation. A volume of 0·1–0·4 ml cells (at 4ºC) was mixed with RNA at 10 µg/0·1 ml and electroporated in a cooled 2-mm cuvette, using an ECM830 Electro Square Wave Porator (Harvard Apparatus BTX, Holliston, MA, USA) at LV mode, 500 V, 1 ms, one pulse. After electroporation, the cells were transferred immediately to fresh culture medium.
Analysis of mRNA caTLR-4 expression and duration
Integrity of caTLR-4 protein following mRNA transfection of TILs was determined by Western blot analysis using polyclonal rabbit anti-hβ2m antibodies, followed by horseradish peroxidase (HRP)-conjugated goat antibodies against anti-rabbit IgG (Dako, Carpinteria, CA, USA). Stably-transfected K562 cells expressing hβ2m-TLR-4 served as a positive control. Persistence of caTLR-4 mRNA was determined by RT–PCR using the following primers: forward primer: 5′-GAG ATG GGA GCC CTC GAG-3′; reverse primer: 5′-AGC ACA CTG AGG ACC GAC AC-3′.
Flow cytometry
For direct staining, cells were washed with cold fluorescence activated cell sorter (FACS) buffer (PBS with 1% FCS and 0·1% sodium azide) and incubated for 30 min at 4°C in the dark with the relevant antibodies (at the concentrations recommended by the manufacturer). Intracellular cytokine staining was performed according to the eBioscience protocol. Cells were washed once using 4 ml FACS buffer, resuspended in 0·3 ml PBS with 0·1% sodium azide and analysed by flow cytometry. Data were analysed by LSRII (BD Biosciences) and FCS Express (De Novo Software, Thornhill, Ontario, Canada).
Monitoring cytokine and chemokine production
Growth media were monitored 48 h post-transfection for IFN-γ levels by ELISA, and for relative levels of 36 chemokines and cytokines using the Proteome Profiler Cytokine Array Kit (both from R&D Systems), according to the manufacturer's protocol. For assaying the anti-melanoma response, 24 h post-electroporation TILs at 1 × 106 cells/well were washed and co-cultured in CM with either autologous melanoma, an HLA-I mismatched melanoma or with no target cells, at an effector-to-target ratio of 1 : 1 at 37°C, for 24 h. At 1-day intervals the cells were washed and fixed in 1% formaldehyde. T cell reactivity was evaluated by flow cytometry analysis for CD107a and intracellular staining for IFN-γ, tumour necrosis factor (TNF)-α and GM-CSF.
Results
Expression of caTLR-4 in human T cells following mRNA electroporation
Transfection efficiency of human T cells using electroporation was optimized using enhanced green fluorescent protein (EGFP)-transfected CD4 and CD8 T cell subsets from healthy donors’ PBLs and TILs obtained from a melanoma patient biopsy (Fig. 1a). Transfection levels were uniform, reaching 94% in PBLs and 92% in TILs with no apparent reduction in cell viability.
Figure 1.
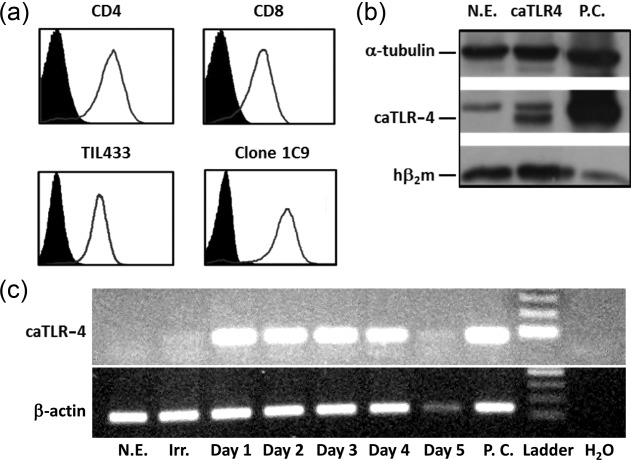
mRNA transfection of human T cells. (a) Transfection efficiency of enhanced green fluorescent protein (EGFP)-electroporated T cell populations by flow cytometry. CD4 (upper left) and CD8 (upper right) T cells from a healthy donor's peripheral blood lymphocytes (PBLs) and bulk tumour-infiltrating lymphocytes (TILs) (TIL433, lower left) or a TIL-derived clone (1C9, lower panel) activated with anti-CD3/anti-CD28 monoclonal antibodies (mAbs). (b) Detection of the hβ2m-Toll-like receptor 4 (TLR-4) protein product in mRNA-transfected human PBLs by Western blot analysis, using anti-human hβ2m antibody. α-tubulin serves as a reference. (c) Reverse transcription–polymerase chain reaction (RT–PCR) analysis for stability of the electroporated hβ2m-TLR-4 mRNA performed at 1-day intervals for 5 days post-transfection using hβ2m-forward and TLR-4-reverse primers. RT–PCR performed on the same RNA samples with primers specific to the human β-actin gene served as reference. N.E. = no electroporation; P.C. = positive control (stable K562 transfectants expressing hβ2m-TLR-4); Irr. = irrelevant mRNA.
We then verified that, following transfection, caTLR-4 mRNA is translated into a protein which can be recognized by a specific antibody. To allow both detection of the protein product and reduce its spontaneous internalization 21, we fused the full-size hβ2m genetically to the N-terminus of the truncated TLR-4 as a molecular tag. Figure 1b shows Western blot analysis of human T cells transfected with mRNA encoding hβ2m-TLR-4. As expected, two bands were visible: a lower molecular weight band consistent with the endogenously expressed β2m, and a higher band, corresponding to the β2m-TLR-4 construct protein product. PCR analysis of the transfected cells over time (Fig. 1c) revealed that the introduced mRNA is visible for as long as 4 days post-transfection. Stably transfected K562 cells expressing hβ2m-TLR-4 served as positive control in both assays.
caTLR-4 mRNA-transfected lymphocytes acquire an activated phenotype
To determine the phenotypical changes induced by caTLR-4 mRNA transfection in human T cells, the surface expression of CD25, CD69 and CD137, three markers of T cell activation, was analysed by flow cytometry 24 h post-transfection. In CD4 T cells elevation was evident for CD25 and, to a lesser extent, for CD69 and CD137, whereas in CD8 T cells CD25 and CD137 expression was augmented markedly but no change in CD69 could be detected (Fig. 2a). Expression of these markers was then evaluated in anti-melanoma TILs daily for 3 days post-electroporation. The results, presented in Fig. 2b, show that CD25 and CD69 remained elevated for at least 3 days, whereas increase in CD137 could be observed for 48 h and then declined. These effects are indicative of long-term functional activation achieved as a result of caTLR-4 mRNA transfection. Neither in this series of experiments nor in the following experiments could we detect TLR-4 on the surface of cultured TILs or polyclonal CD8 T cells (not shown).
Figure 2.
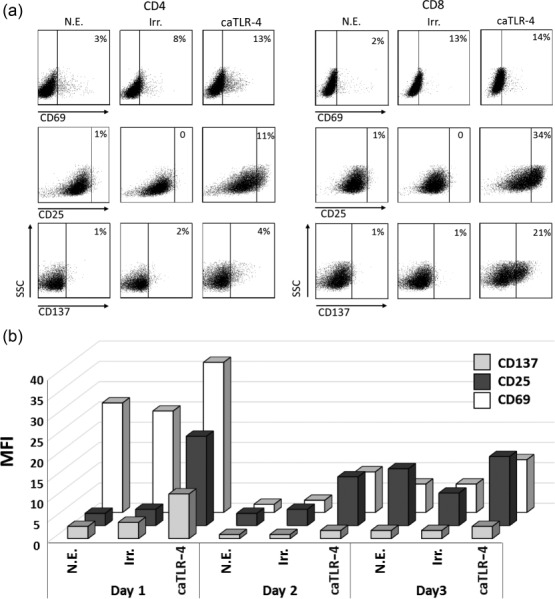
Constitutively active Toll-like receptor 4 (caTLR-4) mRNA transfection induces prolonged expression of T cell activation markers in human CD8 T cells. (a) Flow cytometry analysis for the activation markers CD137, CD69 and CD25 on CD4 and CD8 T cells from a healthy donor's peripheral blood lymphocytes (PBL) transfected with hβ2m-TLR-4 mRNA, performed 24 h post-transfection. (b) Flow cytometry analysis of anti-melanoma bulk tumour-infiltrating lymphocytes (TILs) (TIL433) transfected with hβ2m-TLR-4 mRNA, sampled at three 1-day intervals starting 24 h post-transfection. Mean fluorescence intensity (MFI) for the indicated activation markers is presented. N.E. = no electroporation; Irr. = irrelevant mRNA.
Increased spontaneous IFN-γ secretion in caTLR-4-modified T lymphocytes
In order to determine the impact of caTLR-4 modification on the effector function of T lymphocytes, production of IFN-γ, a reliable marker of T cell activation, was determined. Following caTLR-4 transfection of human anti-tumour effector T cells (TILs and CD8 T cell clones), spontaneously increased secretion of IFN-γ was detected consistently, even in the absence of cognate peptide antigen stimulation. As shown in Fig. 3a, IFN-γ secretion by CD4 and CD8 cells transfected with as little as 1 µg mRNA increased five- and threefold, respectively, in comparison to irrelevant control (mRNA encoding hβ2m-HLA-A2). This effect was enhanced further following transfection with 5 µg mRNA. As lipopolysaccharide (LPS), the natural ligand of TLR-4, was reported to induce secretion of IFN-γ by human T cells 25, we examined the effect of LPS on T cells. We found that no IFN-γ was secreted in response to the addition of LPS, in agreement with other works 18,26. Consequently, LPS could not serve as a positive control for caTLR-4-mediated T cell activation. Next, we examined the effect of TLR-4 mRNA transfection on a TIL-derived CD8 T cell clone and on bulk TILs. Figure 3b reveals a substantial increase in IFN-γ secretion in all three TIL-derived T cell populations (clone 1C9 and TILs 433 and 2045) following transfection, and a smaller increase in a fourth one (TIL 425).
Figure 3.
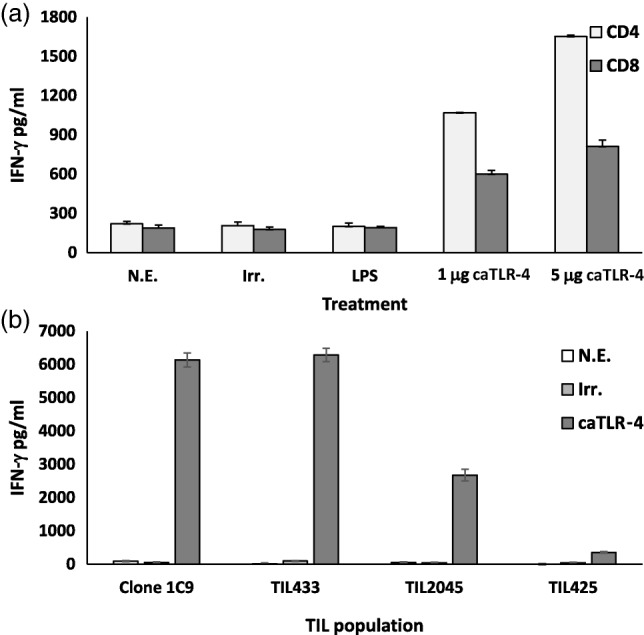
Spontaneous interferon (IFN)-γ secretion following hβ2m-Toll-like receptor 4 (TLR-4) transfection. (a) CD4 and CD8 T cells isolated from a healthy donor's peripheral blood lymphocytes (PBL) and (b) the indicated tumour-infiltrating lymphocyte (TIL)-derived T cell populations were activated and transfected with hβ2m-TLR-4 mRNA. IFN-γ levels in the culture medium were measured 24 h post-transfection by enzyme-linked immunosornent assay (ELISA). Lipopolysaccharide (LPS)-treated, untransfected (no electrop.) and irrelevant mRNA-transfected cells served as controls.
caTLR-4 mRNA-transfected CD8 T cells secrete an array of cytokines and chemokines
We screened the growth medium of mRNA-transfected CD8+ TIL clone 1H8 for the presence of additional cytokines, chemokines and growth factors using a proteome profiler kit (Fig. 4). The levels of regulated upon activation normal T cell expressed and secreted (RANTES) (CCL5), macrophage migration inhibitory factor (MIF), soluble intercellular adhesion molecule-1 (sICAM1), IFN-γ, IL-8, plasminogen activator inhibitor-1 (PAI-1) and macrophage inflammatory protein (MIP)-1α (CCL3) were all increased compared to the same T cell populations transfected with irrelevant mRNA and/or activated by CD3 triggering.
Figure 4.
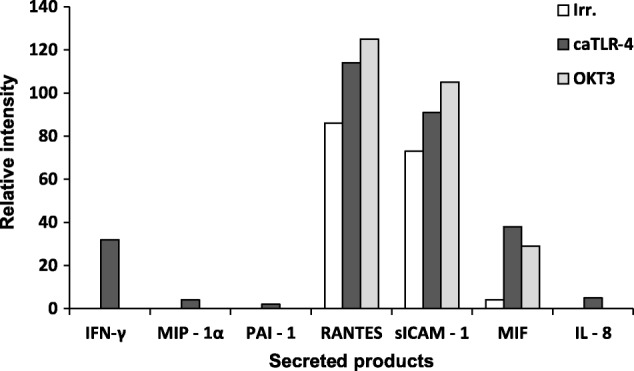
Constitutively active Toll-like receptor 4 (caTLR-4) mRNA induces the secretion of a broad range of cytokines and chemokines. Semi-quantitative analysis for selected factors secreted from caTLR-4 mRNA-transfected CD8 tumour-infiltrating lymphocytes (TILs) (clone 1H8). Cells transfected with an irrelevant mRNA served as negative controls and OKT3-activated T cells served as a positive control. Relative level (percentage of positive control) of the indicated factors was determined 48 h post-transfection using the Proteome Profiler Human Cytokine Array Kit (R&D Systems).
Melanoma-specific T cell responses are improved by caTLR-4
In order to determine the effect of caTLR-4 mRNA transfection on the activation of T lymphocytes in response to tumour cells, we incubated TILs at a 1 : 1 ratio with HLA-A2-matched melanoma (M579-A2) or with mismatched melanoma controls (M171 and M579) 24 h post-transfection and determined the level of secreted proinflammatory cytokines and the expression of CD107a, a degranulation marker attesting for cytotoxic activity. The results showed an increase in the percentage of CD8+ T cells producing TNF-α and IFN-γ in caTLR-4 mRNA-transfected TILs compared to irrelevant mRNA transfection (Fig. 5a). The expression of the CD107a degranulation marker increased robustly on caTLR-4-transfected CD8+ cells compared to untransfected or irrelevant mRNA-transfected TILs (66, 13 and 28%, respectively, Fig. 5b). These results demonstrate that, in addition to improving basal lymphocyte activation state, caTLR-4 transfection leads to enhanced specific anti-melanoma response of T cells.
Figure 5.
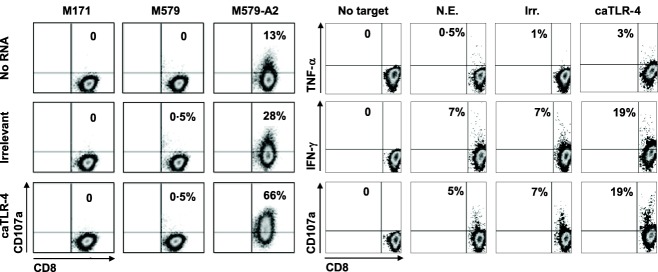
Constitutively active Toll-like receptor 4 (caTLR-4) mRNA transfection enhances melanoma-specific activity of tumour-infiltrating lymphocytes (TILs). (a) Melanoma-specific CD107a mobilization. caTLR-4 mRNA or irrelevant mRNA transfected TILs (clone 1C9) were co-cultured with human leucocyte antigen (HLA)-A2 matched (M579-A2) and mismatched (M171 and M579) melanoma cells for 3 h. CD107a expression on CD8+ T cells was determined by flow cytometry. (b) Proinflammatory cytokine production. caTLR-4 mRNA, irrelevant mRNA transfected or untransfected TILs (bulk TIL425) were co-cultured with M579-A2 melanoma cells for 4 h. Intracellular interferon (IFN)-γ and tumour necrosis factor (TNF)-α production and CD107a mobilization in CD8+ T cells was determined by flow cytometry.
Because the duration of the anti-tumour response is critical to potential clinical efficacy, we performed a series of experiments to monitor the anti-melanoma response of transfected TILs over several days. Production of IFN-γ, TNF-α and GM-CSF and expression of CD107a were determined at 4 successive days starting 24 h post-electroporation. The results (Fig. 6) clearly show an enhanced response to melanoma in caTLR-4-transfected cells, reflected in increased production of cytokines and higher CD107a surface mobilization. Even at day 4, a slight increase was still noticeable on the transfected cells. Remarkably, although a surge of IFN-γ production was observed 24 h post-transfection (Fig. 5) and no cytokines were secreted afterwards (not shown), enhanced cytokine secretion by TLR-4-transfected cells reappeared in response to incubation with autologous melanoma target cells (Fig. 6). These results indicate that the increased cytokine secretion was due to response to melanoma cells and not a spontaneous occurrence.
Figure 6.
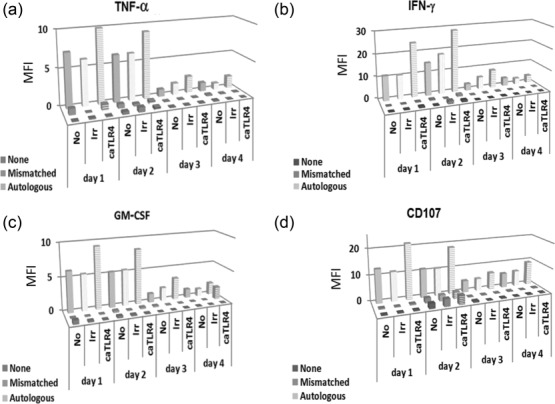
Constitutively active Toll-like receptor 4 (caTLR-4) mRNA enhances prolonged expression of key activation markers by human tumour-infiltrating lymphocytes (TILs) in response to melanoma cells. Anti-melanoma TILs (TIL425), either non-transfected (No), or transfected with caTLR-4 or irrelevant (Irr) mRNA were incubated with autologous melanoma cells, HLA-mismatched melanoma or no target cells (None). Cells were subjected to intracellular staining and flow cytometry analysis every 24 h for 4 days.
Discussion
ACT is a treatment for cancer whereby patients are transfused with a large number of ex-vivo activated and expanded tumour-reactive T cells. These T cells are either derived from the pre-existing lymphocyte pool or reprogrammed genetically to recognize tumour antigens. A growing number of ACT trials reveal an unprecedented high rate of clinical response, including complete remission in patients which are refractory to all other treatments 1,27. However, in different ACT protocols T cell exhaustion, which is manifested in down-regulation of effector mechanisms, is a critical drawback, limiting therapeutic efficacy and wider use of ACT 28
Attempts to enhance the curative potential of tumour-reactive T cells by genetic modification have been focusing largely on the delivery of cytokine genes. These include, for example, IL-2 29,30, IL-12 (either expressed constantly 31,32 or induced by T cell activation via a nuclear factor of activated T cells (NFAT)-responsive promoter 33–35, IL-15 36 or TNF-α 37). Here we present a novel genetic approach designed to alleviate T cell exhaustion and improve anti-tumour activity, which is based on the recently documented ability of TLR ligands to act on T cells in a direct manner. We showed that the delivery of a single mRNA encoding caTLR-4 into T cell populations derived from blood CD8 and CD4 T cells or anti-melanoma TILs up-regulated key activation markers, triggered the secretion of a panel of proinflammatory cytokines and enhanced antigen-specific T cell response. The induction of such a wide range of T cell-enhancing effects via the mere expression of a single gene, in the complete absence of any additional ligands or cytokines, offers a new T cell-enhancing modality for use in cancer ACT.
A typical method to produce TILs for clinical administration relies upon the selection of tumour-reactive T cells based on IFN-γ secretion, followed by an intensive expansion phase. However, at the end of expansion, T cells often fail to perform their expected effector functions due to activation-induced exhaustion 38–41. To shorten the expansion phase and minimize T cell exhaustion, the use of short-term cultured ‘young’ TILs is based on a simplified protocol, which avoids screening for specific tumour recognition 42,43. The use of caTLR-4 offers another way to rescue exhausted T cells from loss of their potential effector functions. Furthermore, the enhancement of T cell activity could potentially obviate the need for large cell numbers, thus significantly facilitating ACT. As shown in Fig. 6, no significant cytokine release could already be observed in the absence of autologous target cells 24 h post-transfection. However, a clear increase, typically between 1.5- and twofold, could be observed in the fraction of cells responding specifically to the autologous melanoma cells at least 4 days post-transfection for all four activation markers tested. These results suggest that caTLR-4 may have driven functional de-differentiation of otherwise terminally differentiated, non-responsive yet melanoma-specific T cells in the TIL sample.
Equally important with regard to potential application of caTLR-4 is the rapid decline in the spontaneous induction of proinflammatory cytokines and chemokines observed in the absence of cognate target cells (Fig. 6). Among the secreted products shown to be up-regulated by caTLR-4 (Figs 6), IFN-γ, TNF-α, GM-SCF, IL-8 and MIP-1α are associated commonly with cytokine release syndrome. This potentially life-threatening phenomenon is reported frequently in clinical trials evaluating CAR T cell therapy and is attributed to massive T cell activation 44. The results presented in Fig. 6 reveal that although caTLR-4 was functional at least 4 days post-electroporation, the induced, antigen-independent cytokine secretion was only short-lived, expiring within 24 h after mRNA delivery.
Overall, the cytokines secreted by the transfected T cells point towards an improved functional phenotype. In vivo, secretion of IFN-γ, which was elevated in the different experimental settings examined in this study, inhibits tumour cell proliferation, sensitizes tumour cells to apoptosis, up-regulates MHC classes I and II expression and stimulates anti-tumour immune activity 45. GM-CSF is known to enhance the anti-tumour response, as shown by its relevance in active immunization (e.g. 46) and ACT 47. CD137 (4-1BB) receptor signalling has emerged as a powerful co-stimulatory pathway to maintain the survival of transferred T cells in vivo 48. In recent landmark clinical trials using anti-CD19 CAR T cells in B cell malignancies, inclusion of a CD137 signalling endo-domain was found to be critical to maintain T cell persistence and anti-tumour activity 49,50. Up-regulation of CD107a indicates degranulation, which is a prerequisite for perforin-mediated effector CD8 T cell cytotoxicity 51.
Our study supports the growing appreciation of the use of mRNA electroporation for delivering therapeutic genes to human T cells 52–54. The use of mRNA is entirely safe, as it obviates the risk of insertional mutagenesis which is associated with commonly used viral vectors. Electroporation is efficient, drives high and uniform expression, preserves maximal cell viability, allows gene function several days post-transfection and facilitates the co-expression of a predefined gene mixture (Weinstein-Marom et al., manuscript in preparation). Indeed, mRNA is currently evaluated clinically with CARs targeting mesothelin in malignant pleural mesothelioma and pancreatic cancer (NCT01355965, NCT01897415) 55 CD19 in Hodgkin's lymphoma (NCT02277522) and c-MET in breast cancer (NCT01837602). Further studies report constantly on progress made in the improvement of mRNA-based protocols and their adaptation to the clinical setting (e.g. 56,57). The caTLR-4 gene product exerts its multiple effects via continuous signalling, so that stable expression is expected to maintain autonomous activation circuits operational, resulting in perpetual T cell activation and systemic toxicity. A solution such as the use of transient, yet powerful, expression driven by mRNA is therefore mandatory. Importantly, transfection of a non-relevant mRNA which served as negative control in our experiments rarely caused non-specific T cell activation compared to non-transfected cells. Thus, the enhanced activation observed following caTLR-4 transfection could not be attributed to mRNA electroporation per se, which entails the engagement of TLR-7 with single-stranded RNA in the endosomal compartment 58.
Taken together, the functional properties conferred to effector T cells by direct caTLR-4-mediated activation may reverse the major suppressive mechanisms operating at the tumour site and enable T cells to maximize their anti-tumour activity. These consequences may bear important implications on the efficacy of ACT. Studies evaluating the effects of caTLR-4 on the function of tumour-specific T cells in vivo and the improvement of the anti-tumour response are under way.
Acknowledgments
We wish to acknowledge the devoted technical work of Anna Kuznetz, Inna Ben David and Yael Gelfand. This study was supported by a research grants from Dr Miriam and Sheldon G. Adelson Medical Research Foundation (AMRF), a Project Grant from the Israel Cancer Research Fund, the Chief Scientist of the Ministry of Industry, Trade and Labor (Israel), Israel Cancer Association, Deutsche Forschungsgemeinschaft (DFG) and the Perlstein Family Fund.
Author contributions
A. P. and G. E. performed the experiments; G. G., A. M., M. L. and T. P. designed the study and A. P., S. F., M. L. and G. G. wrote the paper.
Disclosure
There are no financial or commercial conflicts of interest.
References
- Hinrichs CS, Rosenberg SA. Exploiting the curative potential of adoptive T-cell therapy for cancer. Immunol Rev. 2014;257:56–71. doi: 10.1111/imr.12132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee C. The use of endogenous T cells for adoptive transfer. Immunol Rev. 2014;257:250–63. doi: 10.1111/imr.12134. [DOI] [PubMed] [Google Scholar]
- Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol. 2012;12:269–81. doi: 10.1038/nri3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu R, Forget M, Chacon J, et al. Adoptive T-cell therapy using autologous tumor-infiltrating lymphocytes for metastatic melanoma: current status and future outlook. Cancer J. 2012;18:160–75. doi: 10.1097/PPO.0b013e31824d4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itzhaki O, Levy D, Zikich D, et al. Adoptive T-cell transfer in melanoma. Immunotherapy. 2013;5:79–90. doi: 10.2217/imt.12.143. [DOI] [PubMed] [Google Scholar]
- Phan GQ, Rosenberg SA. Adoptive cell transfer for patients with metastatic melanoma: the potential and promise of cancer immunotherapy. Cancer Control. 2013;20:289–97. doi: 10.1177/107327481302000406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran E, Turcotte S, Gros A, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. 2014;344:641–5. doi: 10.1126/science.1251102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merhavi-Shoham E, Haga-Friedman A, Cohen CJ. Genetically modulating T-cell function to target cancer. Semin Cancer Biol. 2012;22:14–22. doi: 10.1016/j.semcancer.2011.12.006. [DOI] [PubMed] [Google Scholar]
- Zhang L, Morgan RA. Genetic engineering with T cell receptors. Adv Drug Deliv Rev. 2012;64:756–62. doi: 10.1016/j.addr.2011.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshhar Z, Waks T, Gross G. The emergence of T-bodies/CAR T cells. Cancer J. 2014;20:123–6. doi: 10.1097/PPO.0000000000000027. [DOI] [PubMed] [Google Scholar]
- Gill S, June CH. Going viral: chimeric antigen receptor T-cell therapy for hematological malignancies. Immunol Rev. 2015;263:68–89. doi: 10.1111/imr.12243. [DOI] [PubMed] [Google Scholar]
- Cieri N, Mastaglio S, Oliveira G, Casucci M, Bondanza A, Bonini C. Adoptive immunotherapy with genetically modified lymphocytes in allogeneic stem cell transplantation. Immunol Rev. 2014;257:165–80. doi: 10.1111/imr.12130. [DOI] [PubMed] [Google Scholar]
- Torikai H, Reik A, Liu P, et al. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood. 2012;119:5697–705. doi: 10.1182/blood-2012-01-405365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torikai H, Reik A, Soldner F, et al. Toward eliminating HLA class I expression to generate universal cells from allogeneic donors. Blood. 2013;122:1341–9. doi: 10.1182/blood-2013-03-478255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnemann C, Mezzadra R, Schumacher TNM. TCR repertoires of intratumoral T-cell subsets. Immunol Rev. 2014;257:72–82. doi: 10.1111/imr.12140. [DOI] [PubMed] [Google Scholar]
- Gajewski TF, Woo S, Zha Y, et al. Cancer immunotherapy strategies based on overcoming barriers within the tumor microenvironment. Curr Opin Immunol. 2013;25:268–76. doi: 10.1016/j.coi.2013.02.009. [DOI] [PubMed] [Google Scholar]
- Wu RC, Liu S, Chacon JA, et al. Detection and characterization of a novel subset of CD8 +CD57 + T cells in metastatic melanoma with an incompletely differentiated phenotype. Clin Cancer Res. 2012;18:2465–77. doi: 10.1158/1078-0432.CCR-11-2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds JM, Dong C. Toll-like receptor regulation of effector T lymphocyte function. Trends Immunol. 2013;34:511–9. doi: 10.1016/j.it.2013.06.003. [DOI] [PubMed] [Google Scholar]
- Ozinsky A, Underhill DM, Fontenot JD, et al. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between toll-like receptors. Proc Natl Acad Sci USA. 2000;97:13766–71. doi: 10.1073/pnas.250476497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilgenhof S, Van Nuffel AMT, Corthals J, et al. Therapeutic vaccination with an autologous mRNA electroporated dendritic cell vaccine in patients with advanced melanoma. J Immunother. 2011;34:448–56. doi: 10.1097/CJI.0b013e31821dcb31. [DOI] [PubMed] [Google Scholar]
- Cafri G, Amram E, Rinott G, et al. Coupling presentation of MHC class I peptides to constitutive activation of antigen-presenting cells through the product of a single gene. Int Immunol. 2011;23:453–61. doi: 10.1093/intimm/dxr033. [DOI] [PubMed] [Google Scholar]
- Machlenkin A, Uzana R, Frankenburg S, et al. Capture of tumor cell membranes by trogocytosis facilitates detection and isolation of tumor-specific functional CTLs. Cancer Res. 2008;68:2006–13. doi: 10.1158/0008-5472.CAN-07-3119. [DOI] [PubMed] [Google Scholar]
- Dudley ME, Wunderlich JR, Shelton TE, Even J, Rosenberg SA. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J Immunother. 2003;26:332–42. doi: 10.1097/00002371-200307000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boczkowski D, Nair SK, Nam JH, Lyerly HK, Gilboa E. Induction of tumor immunity and cytotoxic T lymphocyte responses using dendritic cells transfected with messenger RNA amplified from tumor cells. Cancer Res. 2000;60:1028–34. [PubMed] [Google Scholar]
- Komai-Koma M, Gilchrist DS, Xu D. Direct recognition of LPS by human but not murine CD8+ T cells via TLR4 complex. Eur J Immunol. 2009;39:1564–72. doi: 10.1002/eji.200838866. [DOI] [PubMed] [Google Scholar]
- Komai-Koma M, Jones L, Ogg GS, Xu D, Liew FY. TLR2 is expressed on activated T cells as a costimulatory receptor. Proc Natl Acad Sci USA. 2004;101:3029–34. doi: 10.1073/pnas.0400171101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett DM, Singh N, Porter DL, Grupp SA, June CH. Chimeric antigen receptor therapy for cancer. Annu Rev Med. 2014;65:333–47. doi: 10.1146/annurev-med-060512-150254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmadzadeh M, Johnson LA, Heemskerk B, et al. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood. 2009;114:1537–44. doi: 10.1182/blood-2008-12-195792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treisman J, Hwu P, Minamoto S, et al. Interleukin-2-transduced lymphocytes grow in an autocrine fashion and remain responsive to antigen. Blood. 1995;85:139–45. [PubMed] [Google Scholar]
- Heemskerk B, Liu K, Dudley ME, et al. Adoptive cell therapy for patients with melanoma, using tumor-infiltrating lymphocytes genetically engineered to secrete interleukin-2. Hum Gene Ther. 2008;19:496–510. doi: 10.1089/hum.2007.0171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerkar SP, Muranski P, Kaiser A, et al. Tumor-specific CD8+ T cells expressing interleukin-12 eradicate established cancers in lymphodepleted hosts. Cancer Res. 2010;70:6725–34. doi: 10.1158/0008-5472.CAN-10-0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegram HJ, Lee JC, Hayman EG, et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. 2012;119:4133–41. doi: 10.1182/blood-2011-12-400044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chmielewski M, Kopecky C, Hombach AA, Abken H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011;71:5697–706. doi: 10.1158/0008-5472.CAN-11-0103. [DOI] [PubMed] [Google Scholar]
- Zhang L, Kerkar SP, Yu Z, et al. Improving adoptive T cell therapy by targeting and controlling IL-12 expression to the tumor environment. Mol Ther. 2011;19:751–9. doi: 10.1038/mt.2010.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnasamy D, Yu Z, Kerkar SP, et al. Local delivery of interleukin-12 using T cells targeting VEGF receptor-2 eradicates multiple vascularized tumors in mice. Clin Cancer Res. 2012;18:1672–83. doi: 10.1158/1078-0432.CCR-11-3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowley J, Monie A, Hung CF, Wu TC. Expression of IL-15RA or an IL-15/IL-15RA fusion on CD8+ T cells modifies adoptively transferred T-cell function in cis. Eur J Immunol. 2009;39:491–506. doi: 10.1002/eji.200838594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwu P, Yannelli J, Kriegler M, et al. Functional and molecular characterization of tumor-infiltrating lymphocytes transduced with tumor necrosis factor-α cDNA for the gene therapy of cancer in humans. J Immunol. 1993;150:4104–15. [PubMed] [Google Scholar]
- Zhou J, Shen X, Huang J, Hodes RJ, Rosenberg SA, Robbins PF. Telomere length of transferred lymphocytes correlates with in vivo persistence and tumor regression in melanoma patients receiving cell transfer therapy. J Immunol. 2005;175:7046–52. doi: 10.4049/jimmunol.175.10.7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Khong HT, Dudley ME, et al. Survival, persistence, and progressive differentiation of adoptively transferred tumor-reactive T cells associated with tumor regression. J Immunother. 2005;28:258–67. doi: 10.1097/01.cji.0000158855.92792.7a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell JDJ, Dudley ME, Robbins PF, Rosenberg SA. Transition of late-stage effector T cells to CD27+ CD28+ tumor-reactive effector memory T cells in humans after adoptive cell transfer therapy. Blood. 2005;105:241–50. doi: 10.1182/blood-2004-06-2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Zhou J, Hathcock KS, et al. Persistence of tumor infiltrating lymphocytes in adoptive immunotherapy correlates with telomere length. J Immunother. 2007;30:123–9. doi: 10.1097/01.cji.0000211321.07654.b8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran KQ, Zhou J, Durflinger KH, et al. Minimally cultured tumor-infiltrating lymphocytes display optimal characteristics for adoptive cell therapy. J Immunother. 2008;31:742–51. doi: 10.1097/CJI.0b013e31818403d5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besser MJ, Shapira-Frommer R, Treves AJ, et al. Minimally cultured or selected autologous tumor-infiltrating lymphocytes after a lympho-depleting chemotherapy regimen in metastatic melanoma patients. J Immunother. 2009;32:415–23. doi: 10.1097/CJI.0b013e31819c8bda. [DOI] [PubMed] [Google Scholar]
- Xu X, Tang Y. Cytokine release syndrome in cancer immunotherapy with chimeric antigen receptor engineered T cells. Cancer Lett. 2014;343:172–8. doi: 10.1016/j.canlet.2013.10.004. [DOI] [PubMed] [Google Scholar]
- Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat Rev Immunol. 2006;6:836–48. doi: 10.1038/nri1961. [DOI] [PubMed] [Google Scholar]
- Cheever MA, Higano CS. PROVENGE (sipuleucel-T) in prostate cancer: the first FDA-approved therapeutic cancer vaccine. Clin Cancer Res. 2011;17:3520–6. doi: 10.1158/1078-0432.CCR-10-3126. [DOI] [PubMed] [Google Scholar]
- Spear P, Barber A, Rynda-Apple A, Sentman CL. Chimeric antigen receptor T cells shape myeloid cell function within the tumor microenvironment through IFN-γ and GM-CSF. J Immunol. 2012;188:6389–98. doi: 10.4049/jimmunol.1103019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Chacon JA, Li Y, Wu RC, et al. Costimulation through the CD137/4-1BB pathway protects human melanoma tumor-infiltrating lymphocytes from activation-induced cell death and enhances antitumor effector function. J Immunother. 2011;34:236–50. doi: 10.1097/CJI.0b013e318209e7ec. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalos M, Levine BL, Porter DL, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3 doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–17. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betts MR, Brenchley JM, Price DA, et al. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods. 2003;281:65–78. doi: 10.1016/s0022-1759(03)00265-5. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Zheng Z, Cohen CJ, et al. High-efficiency transfection of primary human and mouse T lymphocytes using RNA electroporation. Mol Ther. 2006;13:151–9. doi: 10.1016/j.ymthe.2005.07.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkholz K, Hombach A, Krug C, et al. Transfer of mRNA encoding recombinant immunoreceptors reprograms CD4+ and CD8+ T cells for use in the adoptive immunotherapy of cancer. Gene Ther. 2009;16:596–604. doi: 10.1038/gt.2008.189. [DOI] [PubMed] [Google Scholar]
- Thomas S, Klobuch S, Besold K, et al. Strong and sustained effector function of memory- versus naive-derived T cells upon T-cell receptor RNA transfer: implications for cellular therapy. Eur J Immunol. 2012;42:3442–53. doi: 10.1002/eji.201242666. [DOI] [PubMed] [Google Scholar]
- Beatty GL, Haas AR, Maus MV, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce antitumor activity in solid malignancies. Cancer Immunol Res. 2014;2:112–20. doi: 10.1158/2326-6066.CIR-13-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett DM, Liu X, Jiang S, June CH, Grupp SA, Zhao Y. Regimen-specific effects of RNA-modified chimeric antigen receptor T cells in mice with advanced leukemia. Hum Gene Ther. 2013;24:717–27. doi: 10.1089/hum.2013.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug C, Wiesinger M, Abken H, et al. A GMP-compliant protocol to expand and transfect cancer patient T cells with mRNA encoding a tumor-specific chimeric antigen receptor. Cancer Immunol. Immunother. 2014;63:999–1008. doi: 10.1007/s00262-014-1572-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobold S, Wiedemann G, Rothenfußer S, Endres S. Modes of action of TLR7 agonists in cancer therapy. Immunotherapy. 2014;6:1085–95. doi: 10.2217/imt.14.75. [DOI] [PubMed] [Google Scholar]