

Abstract
The control of motion of one element with respect to others in an interlocked architecture allows for different co-conformational states of a molecule. This can result in variations of physical or chemical properties. The increase of knowledge in the field of molecular interactions led to the design, the synthesis, and the study of various systems of molecular machinery in a wide range of interlocked architectures. In this field, the discovery of new molecular stations for macrocycles is an attractive way to conceive original molecular machines. In the very recent past, the triazolium moiety proved to interact with crown ethers in interlocked molecules, so that it could be used as an ideal molecular station. It also served as a molecular barrier in order to lock interlaced structures or to compartmentalize interlocked molecular machines. This review describes the recently reported examples of pH-sensitive triazolium-containing molecular machines and their peculiar features.
Keywords: lasso structures, macrocycles, molecular machines, molecular muscles, rotaxane, triazolium
Introduction
Since the conformational state of a molecule is closely related to its intrinsic properties, interlocked molecular architectures, or more precisely, interlocked molecular machines,1 for which a component can be mechanically moved with control relative to another, can be considered of high interest. Among interlocked molecules, rotaxanes occupy an important place. They comprise a macrocycle which surrounds a molecular axle that is usually ended by bulky stoppers in order to avoid any disassembly of the structure.
If the chemical system is designed with the aim of acting as a molecular machine, different sites of interactions (i.e. molecular stations) must be present in the threaded axle so that each of them can bind the macrocycle with different affinities. Decreasing the interactions of the best molecular station for the macrocycle or increasing those of the poorest molecular station can result in the controlled shuttling of the macrocycle along the molecular thread. More exactly, this change excites the molecular system out of equilibrium, before it relaxes to the energetically favored co-conformation via the so-called Brownian motion. In these singular chemical species, not only the presence of the macrocycle around the threaded axle but also its controlled variable localization can give rise to the modulation of the chemical and physical properties of the molecule. Hence, novel molecular stations for various macrocycles have been the subject of various research endeavors in the last two decades.
In order to reversibly tune the affinity of a molecular site of interaction for the macrocycle, different kinds of stimuli have been used to trigger the shuttling of the macrocycle along the thread: pH,2 temperature3 or solvent variation,4 and photo-,5 electro-,6 or chemical7 reactions. Among the plethora of systems that have been described until now, we would like to focus in this review on the pH-dependent molecular machines that are exclusively based on the interactions of crown ethers with ammonium and triazolium8 molecular stations. For a long time, the ammonium motif has been known to interact quite strongly with crown ethers such as 24-crown-8,9 so that it can serve as a template for driving the rotaxane formation. On the other hand, the triazolium moiety was only reported in 2008 as a weak molecular station for dibenzo-24-crown-8 (DB24C8).13,39 Therefore, systems containing ammonium and triazolium molecular stations for crown ethers can be quite efficiently used for designing new molecular machines. More precisely, because of their quite different affinities for crown ethers, the ammonium moiety can act as the best site of interactions, whereas the triazolium becomes the predominant site of localization of the macrocycle in basic medium after deprotonation of the ammonium, because no more interaction remains between the revealed amine and the crown ether.
The easy access to triazolium compounds makes the synthetic route to such interlocked molecular machines straightforward and versatile. A particular asset of the system is the fact that triazolium can be obtained from azide and alkyne fragments, which allows any kind of chemical variations on the triazole moiety depending on the precursors used. When making the triazolium molecular station through nucleophilic substitution, it is interesting to note the possibility to graft on the triazole nitrogen atom any moiety which can play different roles. Thus, so far, different triazolium-containing molecular architectures, from rotaxane to the more sophisticated double-lasso rotamacrocycle via molecular muscles, have been synthesized and studied.
Some of these molecular machines have only been prepared to overcome a synthetic challenge or for a purely “chemical design” aim, whereas others have already found utility as switchable catalysts for organic reactions, switchable fluorescent probes, or extendable materials. In all the reported structures, different molecular motions arising from the interlocked architecture have been accurately studied. Some of them directly concern the translation of the macrocycle in a [2]rotaxane, which can be accompanied in some cases by a rotary motion, whereas others relate the tightening/loosening of lasso compounds, or even a molecular “jump rope” motion in a double-lasso structure. Triazolium can also be used as a multipurpose molecular station that can act as a kinetic barrier in order to trap the macrocycle in a compartment of the threaded axle, making the system unbalanced and not at equilibrium.
In this review, we propose to give the current state of art on triazolium-based crown ether molecular machines. We will first describe the general synthetic accesses to triazolium-based interlocked molecules. We will then successively highlight examples that have been reported with a pure chemical design aim, and those designed for a defined application. A following section will be devoted to triazolium-based molecular muscles and their use as building blocks in polymers. The relative affinity of the DB24C8 for triazolium and pyridinium amide molecular stations will then be exemplified. This will lead us to discuss about the compartmentalization of three-station-containing molecular machines by using triazolium as both a molecular station and a barrier for the macrocycle. The final sections will relate on more sophisticated mono- and double-lasso architectures with their relative motions.
Synthetic Accesses to Triazolium-Based Interlocked Compounds and 1H NMR Characterization
General synthetic access to triazolium-containing rotaxane molecular machines
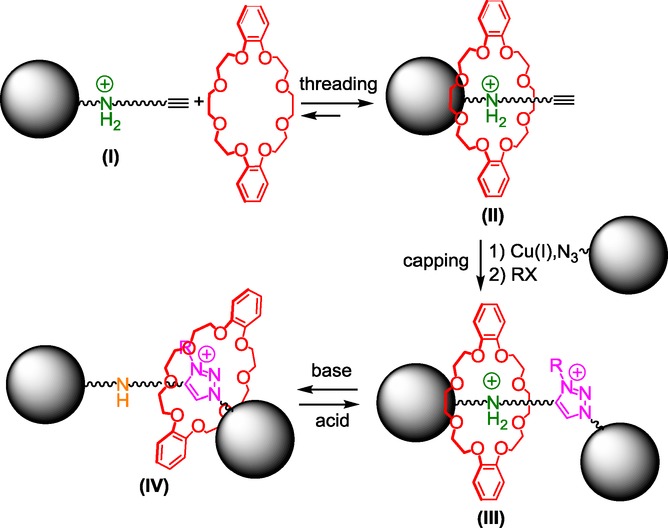
Triazolium-based rotaxane molecular machines are usually obtained through a threading-capping strategy using a two-step sequence beginning with a copper(I)-catalyzed Huisgen[10] alkyne–azide 1,3-dipolar cycloaddition,11 also called “CuAAC click chemistry”, then by the subsequent alkylation of the triazole (Figure 1).
Figure 1.
General chemical route to triazolium-containing molecular machines.
The classical straightforward chemical route to triazolium-containing molecular machines begins from a molecular axle I that bears a secondary ammonium moiety as the template to thermodynamically drive the formation of the semi-rotaxane II and an alkyne function at one extremity of the thread for further chemical connection. The dibenzo-24-crown-8 interacts quite well with the ammonium template, so that the conversion to the semi-rotaxane II can be generally quantitative. The CuAAC click reaction between the alkyne-containing semi-rotaxane II and the azido “stopper” groups then allows the capping of the threaded axle and affords the locked [2]rotaxane architecture III. The inverse was also found in the literature, that is to say an alkyne stopper group with an azido-containing semi-rotaxane. Subsequent regioselective N-alkylation of the triazole provides the triazolium as the second molecular station. This latter has a much poorer affinity for the crown ether than the ammonium. As a consequence, in the two-station-based [2]rotaxane III, the macrocycle mainly resides around the ammonium site. However, deprotonation of the ammonium molecular station triggers the translational motion of the crown ether toward the triazolium station (in IV). The process can be reversed by adding an acid. Interestingly, although triazolium interacts well with crown ether in a locked rotaxane molecular architecture, this moiety cannot be used as a template to prepare rotaxane from disassembled elements, because the intermolecular interactions between crown ether and triazolium are much too weak. Indeed, when attempting to thread a crown ether with a triazolium-containing axle, one may assume that the enthalpic energy (this latter being directly related to the weak noncovalent interactions between the elements) cannot overcome the opposite change in entropic energy. On contrary, in a mechanically interlocked [2]rotaxane, the change in entropies between the deprotonated and the protonated rotaxanes seems much lower. In that case, the enthalpic factor which is driven by interactions between triazolium and the crown ether become predominant, which corroborates the fact that triazolium is a good molecular station for the macrocycle in such a locked structure. Taking into account such considerations, we recently imagined a diverted strategy to prepare a wide range of triazolium-based rotaxanes that are devoid of any other template.
A diverted strategy to yield triazolium rotaxane devoid of any other template
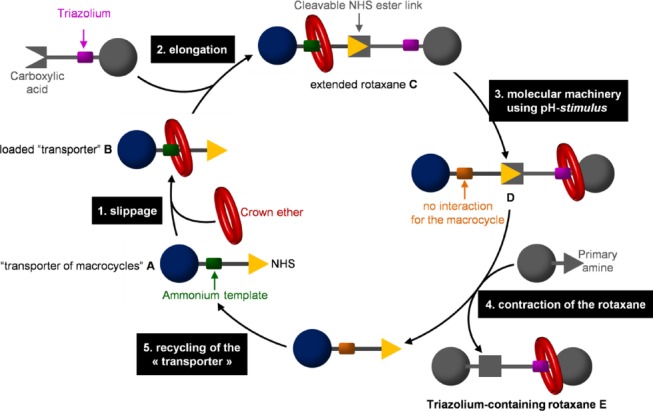
Since the triazolium moiety cannot be utilized as an efficient template for the formation of crown-ether-based rotaxane, we recently reported a general diverted strategy to yield triazolium-based rotaxanes (Figure 2).12
Figure 2.
A diverted route to triazolium-containing rotaxanes.
The strategy relies on the use of a so-called macrocycle “transporter” A which contains an ammonium template in order to efficiently bind a crown ether. Compound A has also been designed to be further extended at one of its extremities with a triazolium-containing axle through a N-hydroxysuccinimide (NHS) ester bond. Once connected, the extended ammonium- and triazolium-based rotaxane C can undergo co-conformational changes through pH variation. Indeed, deprotonation of the ammonium station triggers the shuttling of the macrocycle from its initial ammonium location to the triazolium D. The NHS ester moiety, which has been chosen as a linker between the two initial axle fragments, allows for the contraction of the axle when reacting with an amine. This last reaction (step 4) provides triazolium-based rotaxane E, while the macrocycle “transporter” is recycled at the same time (step 5). One of the advantages of this synthesis remains in the fact that the loaded macrocycle transporter B can be purified and stored over time. Moreover, the possibility to recycle the transporter is one more asset that makes this strategy very attractive and quite direct from B.
Because it is very recent, this second strategy has not yet been widely used. On contrary, the strategy reported in 2008 has now been used by several groups to prepare triazolium-based molecular machines which are described hereafter.
Usual 1H NMR characterization of ammonium and N-methyltriazolium in interactions with DB24C8 in a pHsensitive [2]rotaxane molecular machine
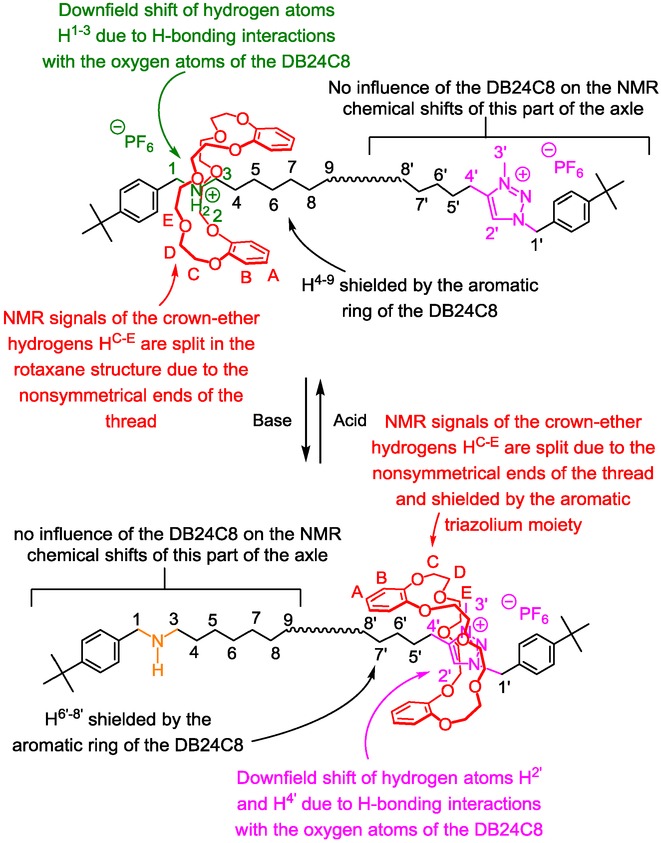
The weak interactions between the DB24C8 for the ammonium and the triazolium moieties induce typical displacements in 1H NMR chemical shifts with respect to the free elements (Figure 3).
Figure 3.
Usual main influence of the DB24C8 on the 1H NMR chemical shifts of the ammonium and N-methyltriazolium stations.
In all the interlocked structures, the NMR signals of the hydrogen atoms of the crown ether are split because they are facing the two nonsymmetrical ends of the threaded axle. Moreover, the hydrogen atoms HE, and to a lesser extent HD, are usually shielded because they are located in the shielding cavity of the benzylammonium moiety. There are multiple influences of the crown ether on the thread. At the protonated state, the DB24C8 mainly interacts with the ammonium station through hydrogen bonds and ion–dipole interactions. In this co-conformation, hydrogen atoms H1–3, which belong to the ammonium site, are all dramatically shifted downfield, due to their hydrogen-bonding interactions with the oxygen atoms of the DB24C8. At the same time, methylene hydrogens H4–9, which are next to the ammonium, experience more or less the shielding effect of the aromatic rings of the DB24C8. No other chemical changes are observed for the other part of the threaded molecular axle, corroborating the localization of the macrocycle. At the deprotonated state, the DB24C8 interacts with the triazolium site. This new localization can be evidenced by the usual main 1H NMR variations. With respect to the free molecular axle, H2’ and in a lesser extent H4’ are both shifted downfield in the rotaxane, due to hydrogen-bonding interactions with the oxygen atoms of the DB24C8. At the same time, hydrogens H6′–8’ are shifted upfield due to their localization in the shielding cavity of the aromatic rings of the DB24C8.
In other triazolium-containing rotaxanes, some variations of the exposed rules can be observed for hydrogens H1’ and H4’ especially if the triazolium is located further to the bulky end. In that less constrained case, one may instead observe a shielding effect for these hydrogens.
Molecular Design in the [2]Rotaxane Series
A pH-sensitive mannosyl [2]rotaxane molecular machine
We reported the first example of a pH-sensitive [2]rotaxane molecular machine possessing a triazolium molecular station for the DB24C8 in 2008 (Scheme 1).13 Its synthesis appeared quite straightforward and efficient, and the molecular machinery easy to drive and characterize. The targeted DB24C8-based [2]rotaxane 4 consists of an encircled axle which contains an anilinium and a N-methyltriazolium molecular stations. Two bulky extremities have been chosen for the thread in order to prevent the macrocycle from any unthreading. One extremity is a mannose derivative, whereas the other one is a di-tert-butyl anilinium moiety. The anilinium was chosen as the pH-dependent station and as the template to drive the rotaxane formation. The triazolium serves as the second station of much poorer affinity for the DB24C8. The preparation of the [2]rotaxane molecular architecture was achieved according to the threading and end-capping strategy described in Section 2.1. The anilinium alkyne 1 was able to thread quite instantaneously through the DB24C8 because of the hydrogen-bonding and ion–dipole interactions between the oxygen atoms of the crown ether and both the ammonium charge and the hydrogen atoms H14 and H15. The obtained semi-rotaxane 2 was then capped at its alkyne extremity using CuAAC click chemistry with the mannosyl azide derivative. The subsequent methylation of the triazole revealed the triazolium station and afforded the pH-sensitive [2]rotaxane molecular machine 4. After deprotonation of the anilinium station, the DB24C8 has no reason to sit anymore over the aniline because it has no more interaction with it. Therefore, the DB24C8 moves toward the N-methyltriazolium and interacts with it. Protonation of 5 inverts the translational process of the DB24C8 toward the anilinium.
Scheme 1.
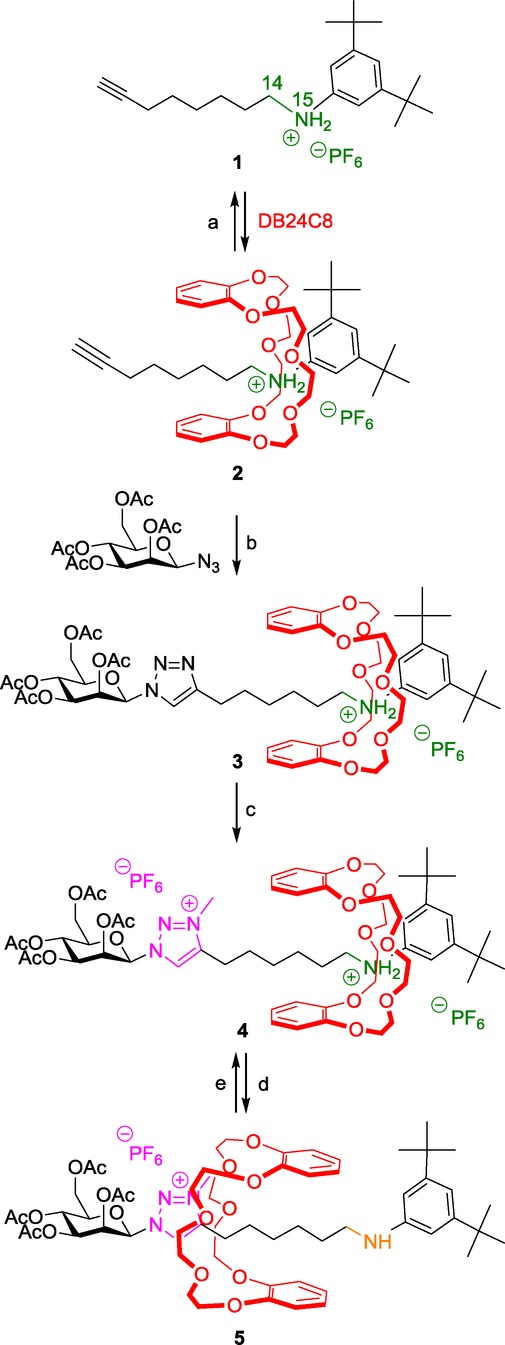
Preparation of a mannosyl pH-sensitive [2]rotaxane molecular machine. Reagents and conditions: a) DB24C8 (2 equiv), CH2Cl2, RT; b) 2,6-lutidine, Cu(MeCN)4PF6, CH2Cl2, RT, 24 h, 72 %; c) 1) CH3I, 2) NH4PF6, quantitative; d) DIEA; e) 1) HCl/Et2O, 2) NH4PF6.
This system appeared very appealing, first because of its quite easy synthetic access, second because of the efficiency of the molecular machinery, and third because of the easy characterization of the molecular machinery by 1H NMR. Numerous other molecules based on this system have been reported since then. As a closely related example, Chen et al. proposed in 2010 the synthesis of a [3]rotaxane molecular machine, in which two triptycene derivatives are linked together thanks to two DB24C8 spacers, the latter being each threaded by two distinct axles.14 Other aesthetic examples are highlighted below.
A pH-sensitive molecular elevator
Using a similar approach (i.e. CuAAC click reaction followed by methylation of the triazole) although with different precursors, Liu et al. reported in 2013 a double-leg donor–acceptor molecular elevator where the distance between two aromatic platforms could be controlled thanks to the translational motion of the macrocycles along a bis-threaded axle (Scheme 2).15
Scheme 2.
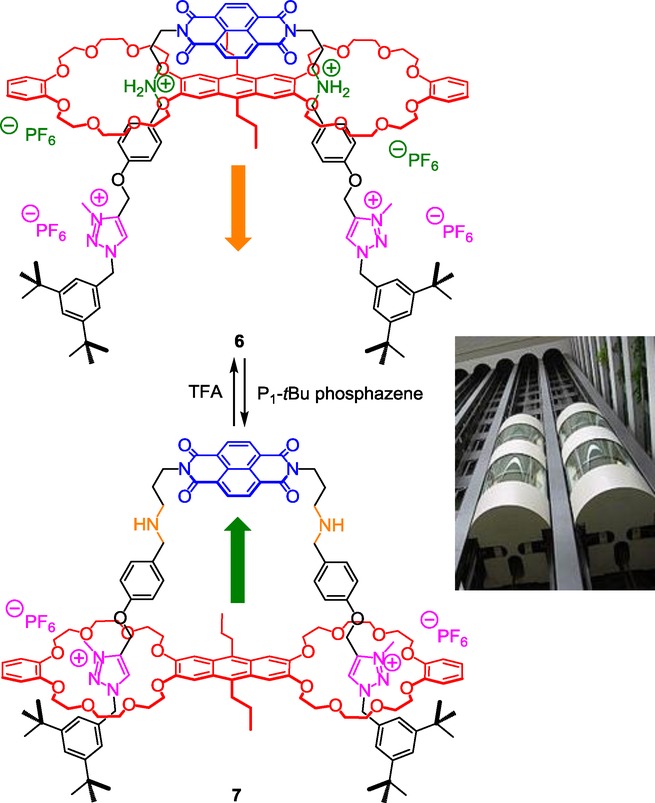
Actuation of a triazolium-containing double-leg molecular elevator.
At the protonated state 6, the bis-crown ether resides around the ammonium molecular stations where it interacts through hydrogen-bonding, ion-dipole, and additional charge-transfer interactions. Indeed, in this peculiar co-conformation, in addition to the already discussed weak interactions, and as already reported with a simpler example devoid of triazolium units,16 the electron-deficient naphtalenediimide interacts with the electron-rich anthracene moiety through a charge transfer. Noteworthy, the fluorescence of the interlocked molecule 6 is quenched by the proximity of these two moieties. Furthermore, in addition to hydrogen-bonding and ion–dipole interactions, this charge transfer improves the prior semi-rotaxane formation, so that the latter can be efficiently prepared using only a stoichiometric amount of each element to be assembled. The deprotonation of rotaxane 6 necessitated a strong base derived from phosphazene, because of the higher pKa of the ammonium group with respect to anilinium. The action of the base induces the shuttling of the bis-crown ether toward the triazolium stations, moving the anthracene and the naphtalenediimide away from each other, therefore restoring at the same time the fluorescence intensity of the anthracene. In summary, the molecular machine acts as a molecular “elevator”17 in which the distance between the anthracene and the naphtalenediimide moieties platform can be adjusted precisely. Authors suggest using the switchable cavity of this molecule in order to bind a guest molecule between the two rigs under the assistance of the two platforms.
Mimicking the flipping motion of butterfly wings
Another example of “chemical design” was reported by Chen et al. in 2014.18 Inspired by smart bionic machines, they proposed the synthesis, through CuAAC click reaction followed by methylation of the triazoles, of a [2](2)rotaxane containing a pentiptycene-derived bis(crown ether), two ammonium stations, and two N-methyltriazolium stations. The pH-sensitive molecular machinery that is inherent to this doubly-threaded molecule mimicks the flapping motion of the wings of a butterfly (Scheme 3).
Scheme 3.
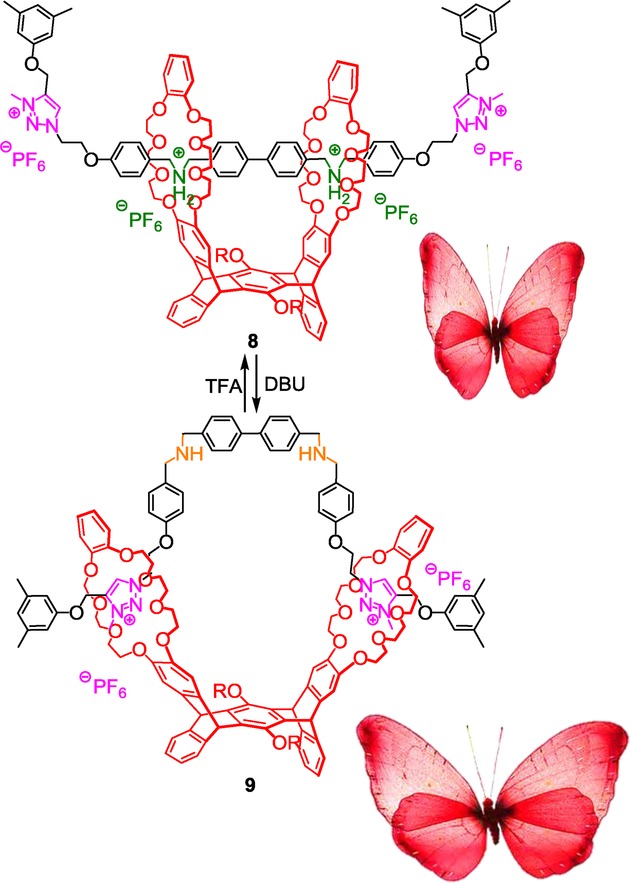
Mimicking the wing-flapping motion of a butterfly using a triazolium-containing pentiptycene bis(crown ether)-based [2](2)rotaxane.
Deprotonation of the ammonium-containing rotaxane 8 forces the DB24C8 rings to interact with the triazolium stations. Since the pure bending of the molecular axle seems impossible to reach without any torsion of the pentiptycene derivative, this being probably due to steric hindrance and constrains, the authors proposed that the twist of the host is necessary for the DB24C8 to reside around the triazolium units. The process can be reversed by simply adding trifluoroacetic acid (TFA). This sequential pH-dependent moving apart or approaching from each other of the two linked DB24C8 moieties can closely approximate the wing-flapping motion of a butterfly, where the DB24C8 are the wings.
A pH-sensitive molecular pulley
The knowledge acquired by Chen et al. on triptycene-derived crown ether hosts19 gave them the opportunity to mimic a molecular pulley (Scheme 4).[20] In their ingenious example, the triptycene moiety serves as a D3h symmetrical scaffold that bears three crown ethers. The three DB24C8 are all threaded by one single molecular axle that contains alternatively three ammonium and three N-methyltriazolium stations. In 10, the three DB24C8 derivatives encircle the ammonium stations. Deprotonation of the ammoniums triggers the forward shuttle of the cable-like thread until each DB24C8 interacts with the triazolium units. The reverse motion is possible through reprotonation of the amines using TFA. Due to the tridimensional geometry of the triptycene, the axle undergoes both translation and rotation motions, like in bi-stable [2]rotaxane and [2]catenane. The authors suggest the future incorporation of loads on either the molecular cable or on the wheel component, in analogy to the operation of a pulley.
Scheme 4.
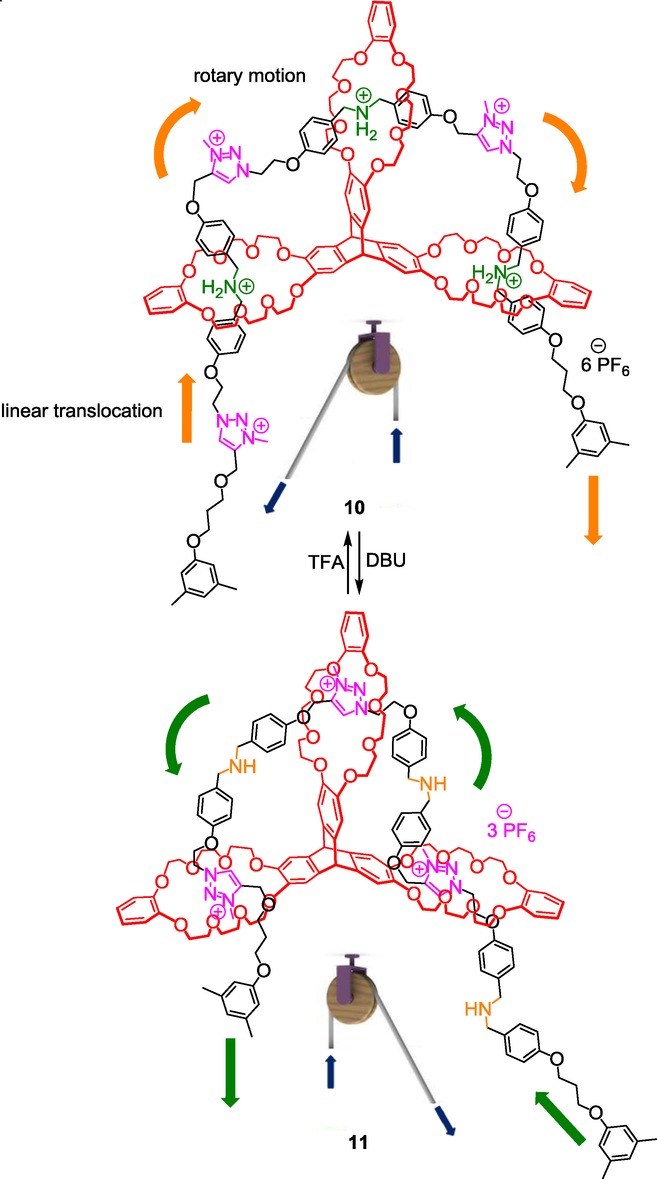
Mimicking a molecular pulley in a triply interlocked triazolium-containing rotaxane molecular machine.
Examples of triazolium-containing rotaxane molecular machines that have found utility as switchable organic catalysts or as switchable fluorophores are highlighted in the following section.
Utilization of Triazolium-Containing Molecular Machines
As switchable pH-sensitive [2]rotaxane catalysts
Inspired by how nature can control enzymatic syntheses through trigger-induced effects, Leigh et al. successfully used the crown ether- and triazolium-ammonium-based molecular machinery in order to prepare appealing switchable organic rotaxane catalysts.21 The aim was to turn “on” or “off” the activity of a [2]rotaxane catalyst through the concealing or the revealing of the catalytic motif, depending on the pH-sensitive localization of a macrocycle along a thread. The first switchable catalyst 13 they proposed consists of a [2]rotaxane molecular machine that contains one ammonium and two N-methyltriazolium stations for the DB24C8 (Scheme 5).22
Scheme 5.
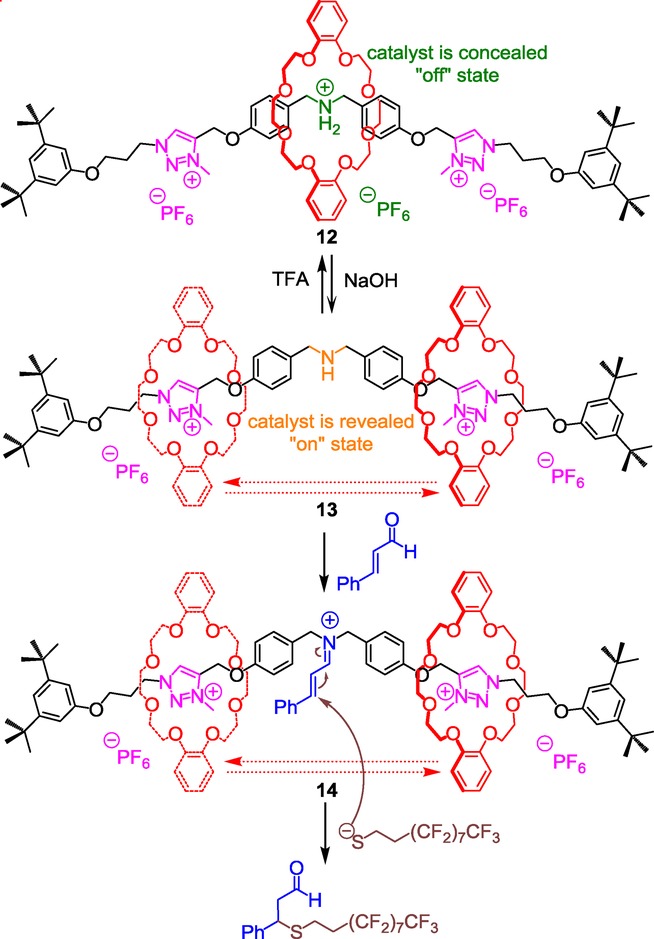
Switching “on” or “off” a triazolium-containing [2]rotaxane catalyst for Michael-type addition.
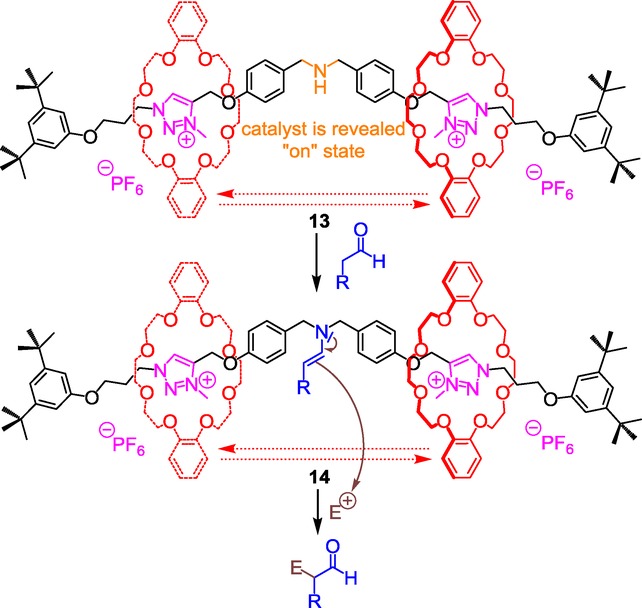
The ammonium moiety was chosen because of its ability, in a non-interlocked molecule, to catalyze Michael-type reactions either at the protonated or the deprotonated state. After having prepared the [2]rotaxane molecular machine 12 according to the two-step sequential chemical route highlighted in section 2.1, the authors investigated the possibility to reveal or conceal the secondary amino function via the shuttling of the DB24C8 along the threaded molecular axle. At the protonated state, the ammonium is encircled by the crown ether, which results in the concealing of the ammonium catalyst, hence its deactivation. At this stage, mixing 5 % of the [2]rotaxane 12 with trans-cinnamaldehyde and an aliphatic thiol did not yield to any Michael-type adduct, even after five days of reaction. However, washing the previous mixture with a 1 m aqueous solution of sodium hydroxide induces the motion of the DB24C8 toward the triazolium stations (rotaxane 13). In this new co-conformation, the amino moiety is revealed, bringing about the “on” state of the catalyst, hence the iminium catalysis.23 In this basic condition, the Michael addition could be carried out in a 66 % yield after only one hour!
By continuing their efforts, the same authors successfully extended the catalyzed Michael-type reaction of the same thiol compound to different α,β-unsaturated carbonyl compounds.24 They also reported the Michael addition using 1,3-dicarbonyl compounds as the nucleophilic agent. Enamine catalysis25 was also investigated using the same switchable rotaxane catalyst for nucleophilic substitution and nucleophilic addition reactions in the α position of an aldehyde (Scheme 6).
Scheme 6.
Utilization of a switchable triazolium-containing rotaxane catalyst for the introduction of an electrophile at the α position of an aldehyde.
In this case, both the α-chlorination of saturated aldehydes using N-chlorosuccinimide and the Michael addition to vinyl bis-sulfone were achieved using 13 as the catalyst (20 mol %) with respective ranges of conversion of 40–61 % and 23–70 % depending on the substitution of the aldehyde. No such reactions were observed in the absence of 13 or in the presence of the protonated rotaxane 12. One drawback still remains in the impossibility to extend these two catalyzed reactions to cyclic or acyclic ketones. The extension to tandem iminium–enamine catalyzed reaction was achieved in a 50 % overall conversion using 20 mol % of the deprotonated rotaxane catalyst 13. In that peculiar case, the trans-cinnamaldehyde was first subjected to iminium catalysis allowing the introduction of a thiol in the β position of the aldehyde. This led to an enamine which subsequently reacted with vinyl bis-sulfone in the α position of the aldehyde. Eventually, the efficacy of the switchable rotaxane catalyst 13 was successfully tested in a Diels–Alder reaction between a dienaldehyde and a dienophile.
The same authors recently extended their work by synthesizing the chiral pH-sensitive rotaxane molecular switch 15/16, which proved to catalyze conjugated additions, only at the deprotonated state, through the iminium ion activation and notably with high stereoselectivity (Scheme 7).26
Scheme 7.
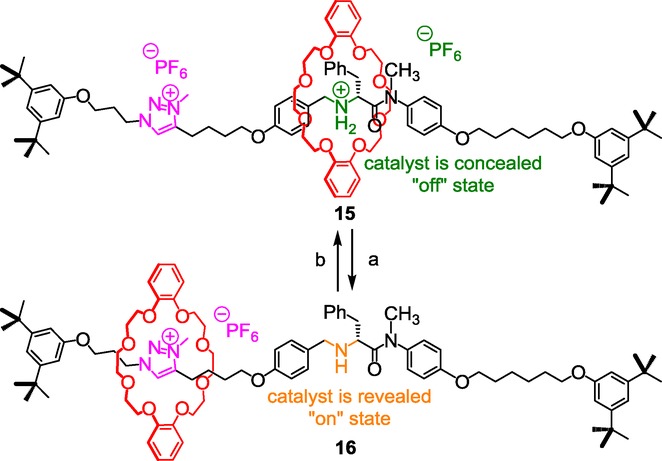
A chiral switchable triazolium-containing rotaxane catalyst for stereoselective conjugated addition. Reagents and conditions: a) NaOH; b) 1) HCl, 2) KPF6.
The principle is the same as already discussed, that is to say based on the revealing of the amine moiety through the shuttling of the DB24C8 in a basic medium. Although the literature mentioned that conjugated addition could be generally carried out efficiently with high stereoselectivity using cyclic amine as catalyst, here, it is interesting to note that it could be realized with an acyclic secondary amine derived from a (R)-phenylalanine residue. Enantiomeric excess as high as 88 % was obtained. This very attractive contribution now opens new avenues toward the selective sequential reactions of a mixture of a wide range of compounds using successive activation of switchable catalysts.
As switchable pH-sensitive fluorescent rotaxanes
In triazolium-containing [2]rotaxanes
The possibility for a macrocycle to be localized at a different place along a thread gives the opportunity to tune the fluorescence of an interlocked molecule depending on a stimulus. Qu et al. synthesized a switchable spiropyran-containing [2]rotaxane in which the threaded axle is end-capped by a fluorescent 4-morpholin-naphthalimide.27 The targeted pH-sensitive molecular machine 17 contains a DB24C8 macrocycle derivative and two ammonium and triazolium molecular stations. The DB24C8 macrocycle is substituted by two nitroaromatic spiropyran moieties that can tune the fluorescence of the 4-morpholin-naphthalimide stopper depending on their distance (Scheme 8).
Scheme 8.
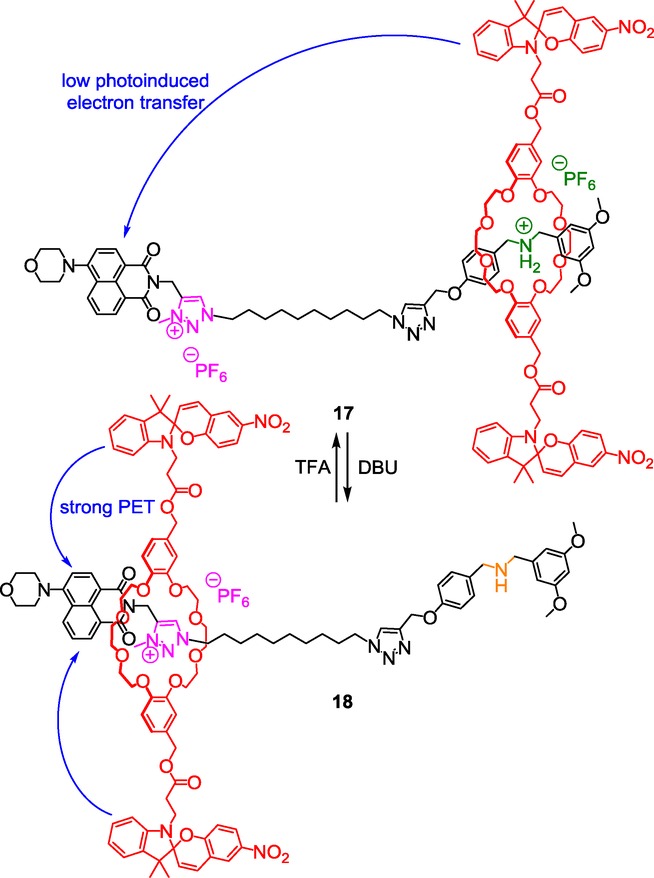
A triazolium-containing [2]rotaxane with fluorescent output.
Shuttling the macrocycle toward the triazolium recognition site was achieved through the addition of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) on rotaxane 17. Concomitantly, the fluorescent emission intensity decreased by 45 % in 18, with respect to 17. This can be ascribed to the higher proximity between the fluorophore and the two photochrome units in the deprotonated rotaxane 18. In this state, the quenching of the 4-morpholin-naphthalimide fluorophore, via the photo-induced transfer of one electron of the highest occupied molecular orbital (HOMO) of the spiropyran moieties to the vacancy of the HOMO of the excited 4-morpholin-naphthalimide, becomes more effective.
A somewhat similar example of controlled fluorescence in a [2]rotaxane molecular machine was published by Li et al. in 2011.28 They synthesized a tight rotaxane based on the ammonium/triazolium/DB24C8 system which operates in a basic environment. Here, the threaded axle holds at each extremity two chromophores as bulky stoppers (Scheme 9).
Scheme 9.
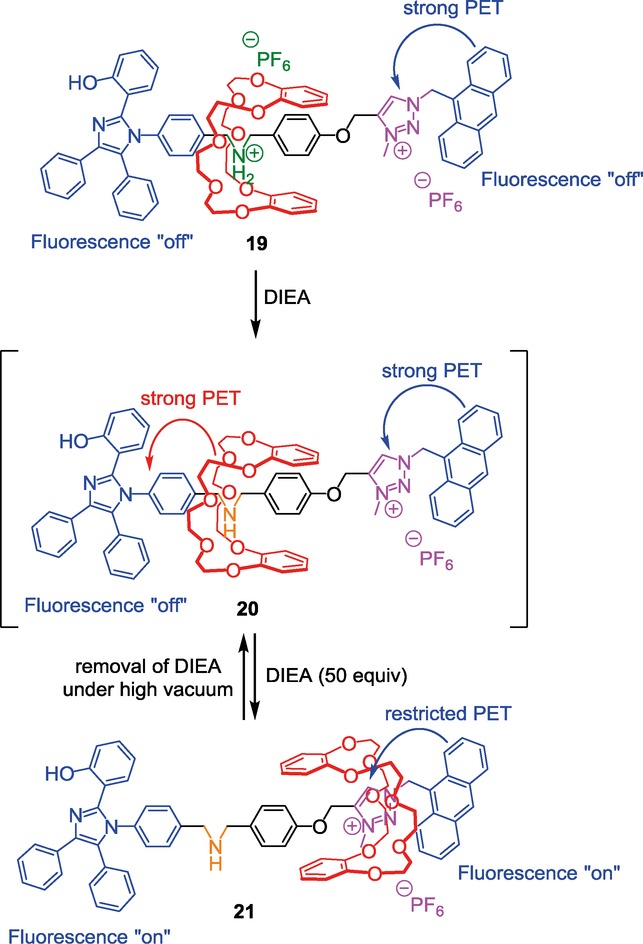
A triazolium-containing [2]rotaxane with two fluorescent outputs.
Surprisingly, the molecular machine 19 operated slightly differently with respect to the other reported examples. On one hand, deprotonation using the weak base N,N-diisopropylethylamine (DIEA) did not lead to the immediate displacement of the crown ether around the triazolium station in 20, unless a large amount of 50 equivalents of DIEA was used. On the other hand, removal of DIEA under high vacuum leads to the reversible motion of the DB24C8 around the NH site. The authors explained this result by the fact that anthracene is a bulky moiety which sterically disturbs the solvation of the N-methyltriazolium cation by acetonitrile. This results in a tighter ion-pair between the triazolium cation and the hexafluorophosphate anion that prevents the DB24C8 to interact optimally with the triazolium station. Adding the stronger Lewis base DIEA causes a better solvation of the triazolium cation, inducing the dissociation of the ion-pair, hence promoting the interaction between the DB24C8 and the triazolium recognition site (compound 21). Another reasonable explanation is that a small amount of DIEA is just not sufficient to deprotonate the ammonium quantitatively. Removal of the DIEA under vacuum increases the concentration of diisopropylethylammonium, thus leading to the reprotonation of the rotaxane. In any cases, what is interesting here is that the fluorescence of the two extremities can be switched on or off using a pH stimulus. In the protonated rotaxane 19, the fluorescence of the anthracenyl moiety is quenched because of the strong photoinduced electron transfer of the anthracene to the triazolium moiety. At the same time, the close distance between the DB24C8 and the hydroxyl-substituted tetraphenylimidazole (HPI) results in the weak fluorescence emission of the HPI moiety. At the deprotonated state, the DB24C8 moves toward the triazolium station. The concealing of the triazolium by the crown ether now prevents from any photoinduced electron transfer (PET) between the anthracenyl group and the triazolium. Therefore, fluorescence of the anthracene is restored. The same increase in fluorescence emission is observed for the HPI moiety because of the higher distance from the DB24C8 which now sits over the triazolium station.
In triazolium-containing [3]rotaxanes
Another example of tunable fluorescence in a more sophisticated [3]rotaxane molecular machine was reported by Liu et al.29 The internal motion of the DB24C8 along the thread is once again based on its different affinities for the ammonium and triazolium stations as anteriorly explained (Scheme 10).
Scheme 10.
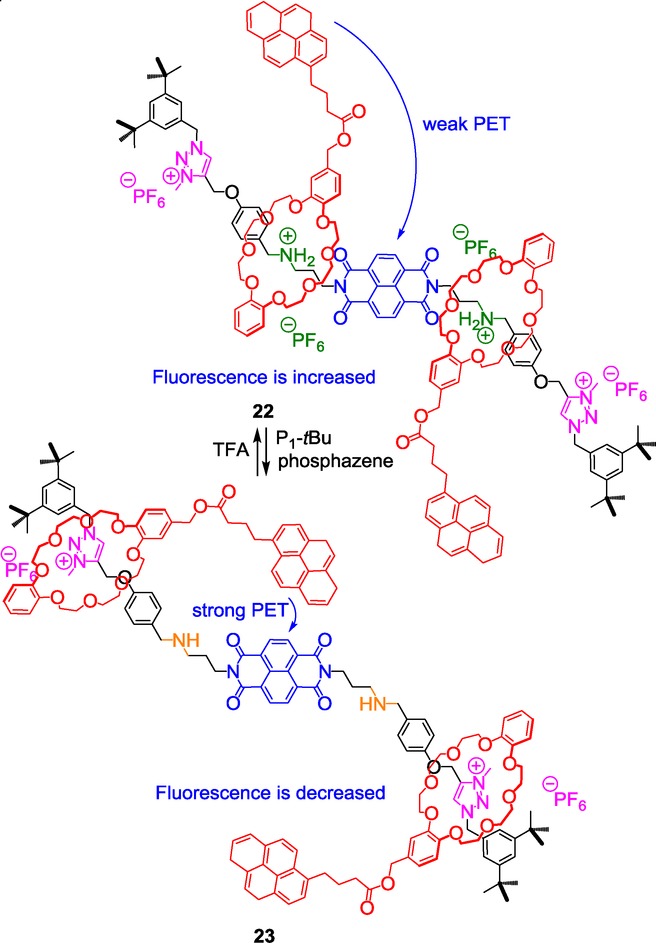
A triazolium-containing [3]rotaxane with fluorescent output.
An electron-deficient naphthalenediimide (NDI) is used as a linker between two branched [2]rotaxanes that both contain an ammonium and a triazolium station. The ammoniums are located close to the NDI core, whereas triazolium units lie at each extremity of the [3]rotaxane. Two DB24C8 initially surround the ammoniums of the thread at the protonated state 22. They both bear an electron-rich pyrene moiety that can be involved in a charge-transfer complex with the NDI moiety. The obtained results indicate that the fluorescence intensity of 22 decreases upon addition of the phosphazene strong base, whereas addition of TFA on 23 increases the fluorescence. This can be explained by an internal charge-transfer (ICT) process which depends on the spatial distance between NDI and pyrene moieties. Further experiments showed that the measured luminescence is only due in this case to the free pyrene. The authors used molecular energy minimization to evaluate the distance between the electron-rich and the electron-deficient moieties. In the protonated system 22, the distance between the pyrenes and the NDI appears larger than in 23, although the DB24C8 are located much closer to the NDI. This corroborates the π–π stacking interactions observed in 23 by 1H NMR and by UV/Vis absorption spectroscopies between the NDI and one pyrene unit.
Another fluorescent [3]rotaxane was published by Qu et al. in 2015. The symmetrical bi-stable bis-branched [3]rotaxane molecular machine contains a perylene bisimide (PBI) chromophore at the middle of the threaded axle and ferrocenyl units on the DB24C8.30 The molecular machinery is once again based on DB24C8 derivative and ammonium/triazolium molecular stations, so that two distinct co-conformational states can be easily reached upon variation of pH (Scheme 11).
Scheme 11.
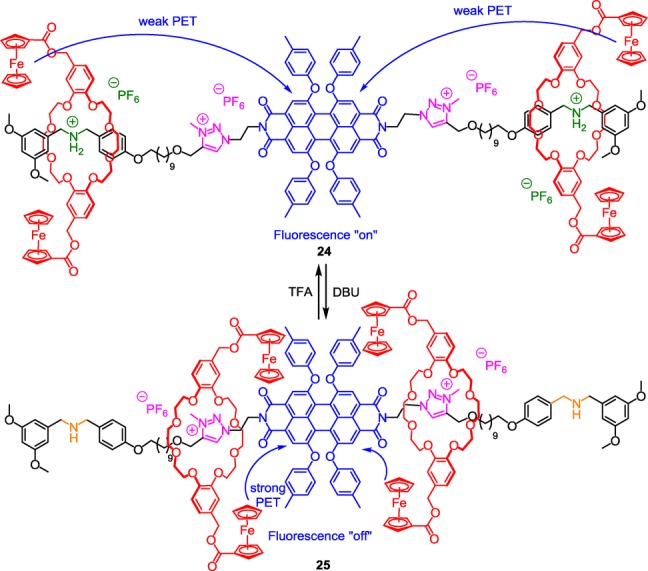
A triazolium-containing [3]rotaxane with fluorescent output.
However, contrary to the previous example, the ammonium station is located at the two extremities of the thread, while triazoliums are next to the PBI core. The remarkable pH-dependent tuning of the visual fluorescence of the PBI was achieved through its distance-dependent quenching by the ferrocenyl units. The change of this photophysical property was possible thanks to both the molecular machinery and the photoinduced electron transfer from the electron-rich ferrocene donors to the electron-deficient PBI fluorophore acceptor. At the protonated state (compound 24), the two macrocycles reside around the ammonium stations: in this co-conformation, the ferrocenyl moieties are located too far away from the PBI to allow any efficient PET. Hence, the fluorescence of the PBI is “on”. However, deprotonation of the ammoniums using 3 equiv of DBU causes the shuttling of the two macrocycles along the thread until the triazolium sites, which are close to the fluorescent PBI moiety. It results from these motions that ferrocenyl groups are now near the PBI core of the [3]rotaxane molecular machine, hence inducing a strong PET which is responsible for the decrease (80 %) of the emission intensity at 620 nm with respect to the original spectrum. Interestingly, this fluorescence quenching could be visualized with the naked eye. The authors also demonstrated that protonation of 25 using 6 equiv of TFA restored the fluorescence intensity. This cycle (protonation/deprotonation) could be repeated several times without noticing any degradation.
In triazolium-containing [1]rotaxanes
In 2013, Qu et al. published a fluorescence switch in the [1]rotaxane series.31 The principle is quite similar to the [3]rotaxane described above, although the motion of the molecular machine relies on the tightening or the loosening of a lasso (Scheme 12).
Scheme 12.
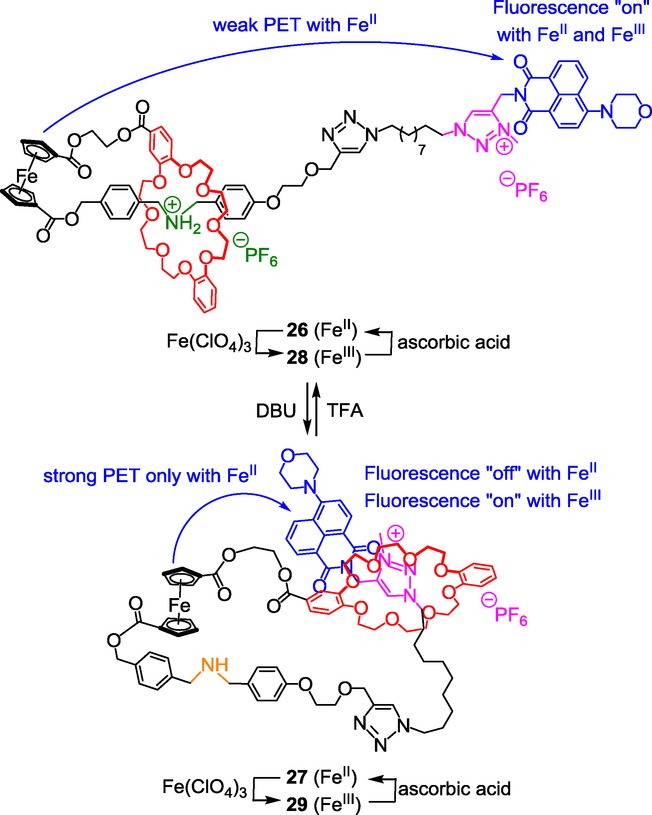
A triazolium-containing [1]rotaxane with fluorescent output.
The ferrocenyl unit, which is part of the lasso's loop, can be localized more or less close to the fluorescent stoppered molecular tail depending on the molecular machinery, using the difference of affinity of the DB24C8 for the ammonium and the triazolium station. In the protonated state, the DB24C8 has more affinity for the ammonium moiety, therefore forcing the lasso to adopt a tightened co-conformation. At this point, the ferrocenyl unit remains too far away from the morpholin-naphtalimide to quench its fluorescence, the latter being monitored by the naked eye. Deprotonation of the ammonium results in the shuttling of the DB24C8 around the triazolium, hence loosening the lasso. In this specific co-conformation, the ferrocenyl unit is much closer to the tail, now authorizing a strong PET from the electron-rich ferrocene to the electron-deficient naphthalimide. This PET affects the fluorescence emission intensity at 521 nm in 27 which decreases by 80 % with respect to the protonated tightened lasso 26. Using theoretical calculations, the authors managed to get data relative to the distance separating the iron atom and the nitrogen atom of the naphthalimide. The results obtained are in total agreement with the fluorescence observation. Indeed, in 26, the calculated distance between the two atoms is 35.66 Å, whereas in 27 it decreases to only 12.87 Å, therefore allowing a more efficient PET between the ferrocene and the naphthalimide, thus the quenching of the fluorescence.
The authors then explored the possible dual pH- and redox-dependent output signal after oxidizing/reducing the ferrocene unit, using respectively Fe(ClO4)3 or ascorbic acid. In rotaxane 29, after that the ferrocene unit of rotaxane 27 is oxidized, the fluorescence is dramatically restored to its original level. Furthermore, the oxidation of the protonated fluorescent rotaxane 26 leads to compound 28 without any significant change of emission intensity. These two last results can be ascribed to the fact that oxidation of the ferrocene units obviously affects its electron-donating ability, thus not allowing efficient PET in this case, whatever the distance between the considered moieties. Reducing the ferrocene unit with ascorbic acid gave rise to the controllable pH-switchable fluorescence output signal (rotaxanes 26/27). In summary, this molecular machine proved to operate upon a pH variation through the motion of the macrocycle along the threaded axle. The fluorescent output signal was found to be both pH- and redox-dependent. Quenching the fluorescence could be achieved using a pH stimulus (deprotonation) through actuation of the machinery, whereas restoring the original level of fluorescence was possible either by oxidation or by protonation. The authors present their system as an INHIBIT logic gate combining a “silent” and an “active” signal output which could be of real interest for the conception of logic circuits with memories or sequential functions. The same group extended this work to the synthesis and the study of two pH-sensitive star-shaped [1](n)rotaxane molecular machines with tri and tetrabranched cores that are linked to triazolium stations.32 Using the features of the two previously reported examples, 24 and 26, Qu et al. recently published an appealing bis-branched [1]rotaxane that can combine dual-mode molecular motions and tunable nanostructural morphologies (Scheme 13).33
Scheme 13. Scheme.
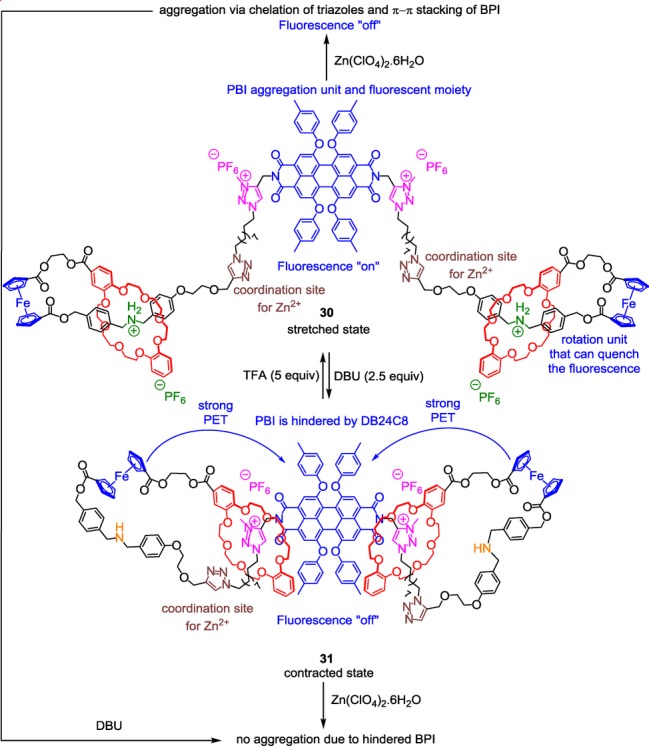
A triazolium-containing bis-branched [1]rotaxane with reversible fluorescence, stretching/contraction motion, and reversible aggregation.
The molecular machine 30 is based on the difference in affinity of ammonium and triazolium stations for the DB24C8 macrocycles. Moreover, it combines two ferrocenyl units that are used to connect the thread and the macrocycle for the bis[1]rotaxane architecture, on one hand, and for its electron-rich donor ability on the other hand. An electron-deficient PBI fluorophore lies in the core of the thread and possesses aggregation potential that can be tuned. 1,2,3-triazoles are located between ammonium and triazolium molecular stations and serve as coordinating sites for Zn2+ in order to tune the aggregation degree of the PBI moiety. As for the previously described examples, the DB24C8 can move along the thread upon a pH stimulus, here triggering the contraction and the stretching of the bis[1]rotaxane 30/31 because of its single molecular [1]rotaxane architecture. Interestingly, it was shown that the translational movement of the DB24C8 was accompanied by a simultaneous rotational motion of the ferrocene units, which makes this machine acting as a dual-mode movement type. The fluorescence of the PBI could be tuned either upon change in solvent polarity or variation in pH. Indeed, increasing the proportion of the apolar methylcyclohexane in a solution of 30 in acetone results in the dramatic decrease of the fluorescence, due to π–π stacking aggregation of the PBI. On the contrary, in 31, very little π–π stacking aggregation of the PBI was noticed. On the other hand, the fluorescence can be also controlled by the molecular machinery, via the distance-dependent photoinduced electron transfer of the ferrocene moieties to the PBI. Upon deprotonation, the fluorescence of PBI is quenched because the ferrocene units are located closer to the PBI. At the protonated state, it is also interesting to notice that addition of Zn2+ could induce the aggregation of the molecule through the cross-linking chelation of the cationic transition metal with the neutral triazole moieties. This chelation phenomenon helps the PBI moieties to self-interact through π–π stacking. Addition of the strong base DBU decreased the aggregation because of the shuttling of the DB24C8 around the triazolium, thus hampering π–π stacking due to steric hindrance. Using theoretical calculations, the authors estimated a percentage of molecular length change of 28.3 % between the two conformational states. This value is very close to the value observed in human muscles. Although the contraction and the stretching of the molecule can be compared with a muscle, the first molecular muscle reported by Sauvage et al., which was based on metal–ligand coordination, holds a slightly different doubly interlocked molecular architecture.34 Examples of such a topology, based on the system of molecular machinery that is highlighted in this review, are given in the following section.
Triazolium-Containing Molecular Muscles and Their Use as Building blocks in Polymers
Synthesis and study of a pH-sensitive molecular muscle
“Molecular muscle”35 is a generic term used for a molecule designed to adopt a contracted or a stretched co-conformation in response to a stimulus, in a similar manner to human muscles. Inspired by the pioneering work of Jean-Pierre Sauvage36 on doubly interlocked molecular architectures and by those of Fraser Stoddart on DB24C8-based [c2]daisy chain,37 we reported in 2008 the first pH-sensitive molecular muscle. A few other examples based on other systems have been reported since then.38 We actually extended the ammonium–triazolium system of recognition sites for DB24C8 that we previously published13 in the [2]rotaxane series to the preparation of a dimannosyl pH-sensitive molecular muscle (Scheme 14).39
Scheme 14.
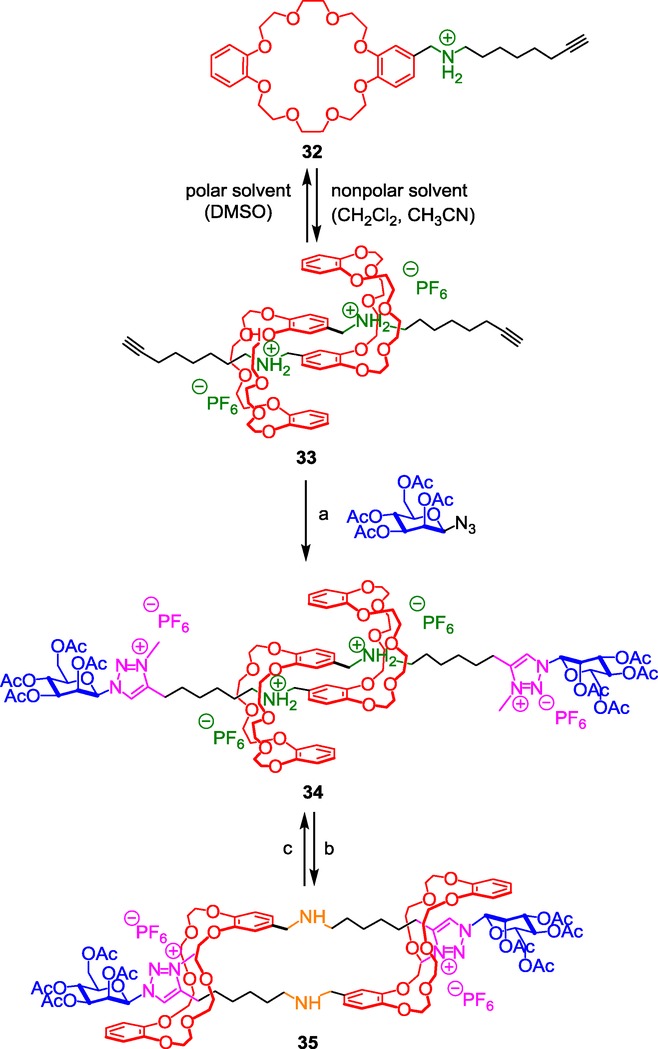
An ammonium/triazolium-containing pH-sensitive molecular muscle. Reagents and conditions: a) 1) mannosyl azide (pictured), Cu(CH3CN)4PF6, lutidine, 2) CH3I then NH4PF6; b) NaOH (aq)/CH2Cl2; c) 1) HCl/Et2O, 2) NH4PF6.
The so-called hermaphrodite compound 32 was synthesized and dissolved in several different solvents which differ in polarity. Whereas 32 remains unassembled in the more polar solvent DMSO, it self-assembles as a pseudo-rotaxane dimer in the less polar hydrogen-bond-promoting solvents like dichloromethane or acetonitrile. We pointed out the meso stereochemistry of the self-assembled [c2]daisy chain supramolecular arrangement 33, which is probably due to both steric hindrance and dipole-moment repulsion between the ionic chains linked to the threaded crown ether. The subsequent two-step sequence, 1) CuAAC click reaction and 2) methylation of the triazole, locked the interwoven supramolecular structure by capping the threads with mannosyl stoppering groups and also revealed the triazolium stations for the DB24C8. The isolated rotaxane dimer 34 was then studied in order to evidence the actuation of the molecular muscle. As usually observed with simpler interlocked architecture, at the protonated state 34, the molecule remains in an extended co-conformation because the two DB24C8 prefer to interact with the best ammonium molecular stations. However, deprotonation of the ammonium moieties using sodium hydroxide quantitatively yields to the contracted molecular muscle 35 through the gliding motion of the two DB24C8 along the treads until they reach the triazolium sites. The very efficient molecular machinery from the contracted to the stretched co-conformations matches with a maximum contraction ratio of 48 %. The distance between the two anomeric carbons of the stoppering mannose was evaluated up to 23 and 34 Å, respectively. This initial work paved the way to applications in the domain of molecular-muscle-containing supramolecular polymers.
Use of molecular muscles as building blocks for the synthesis of supramolecular polymers
The same specific system of molecular muscle, but with different stoppers, was employed later by Buhler, Giuseppone, et al. with the aim to construct a supramolecular polymer.40 To this end, they used terpyridine moieties both to lock the interwoven structure and to further have the possibility to assemble the molecular muscle building blocks through metal coordination (Scheme 15).
Scheme 15.
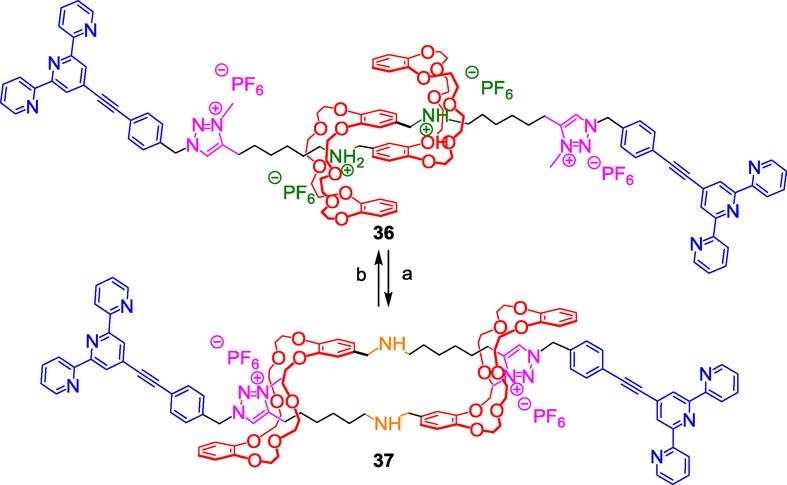
An ammonium/triazolium-containing pH-sensitive muscle-like monomer for the synthesis of a metallosupramolecular polymer. Reagents and conditions: a) NaOH (aq)/CH2Cl2; b) 1) HCl/Et2O, 2) NH4PF6.
The terpyridine moiety was already known to coordinate strongly FeII, ZnII,41 or other metals such as RuII or OsII.42 Moreover, Che et al., as well as Schubert et al., previously used terpyridines to prepare supramolecular polymers.43 In the present case, the molecular muscle building block 36 was prepared according to Coutrot's strategy and acted upon pH variation like the previously published compound 34. Adding either FeII or ZnII to 36 triggered the polymerization thanks to the formation of stable octahedral 2:1 complexes. A longer supramolecular polymer was obtained with FeII because of its better coordination for terpyridine than ZnII. Of particular interest is the length of the polymer that was obtained this way. No less than 3000 units have been assembled through coordination, which is a real achievement with respect to already published molecular-muscle-containing polymers that usually do not contain more than 22 units.44 The change of size of the metallosupramolecular polymer upon variation in pH was evaluated by small-angle neutron scattering. The contour length of the protonated polymer is long (15.9 μm) with respect to the deprotonated polymer (9.4 μm), while the decrease of the linear density is observed simultaneously. An amplification of almost four orders of magnitude in the mechanical output is observed in the polymer in comparison with the monomer. In other words, the length variation of the polymer between the two co-conformations is about 6.5 μm, while that of the monomer is only around 1 nm. With this work, a real step toward the macroscopic world has been done, even though these polymers should be bundled together in order to better mimic myofibrils of sarcomers. Very recent improvements in this way have been achieved by the same group. They reported the possibility to induce the fabrication of micrometer-long supramolecular fibers from the molecular muscle building block 38 (Scheme 16).45
Scheme 16.
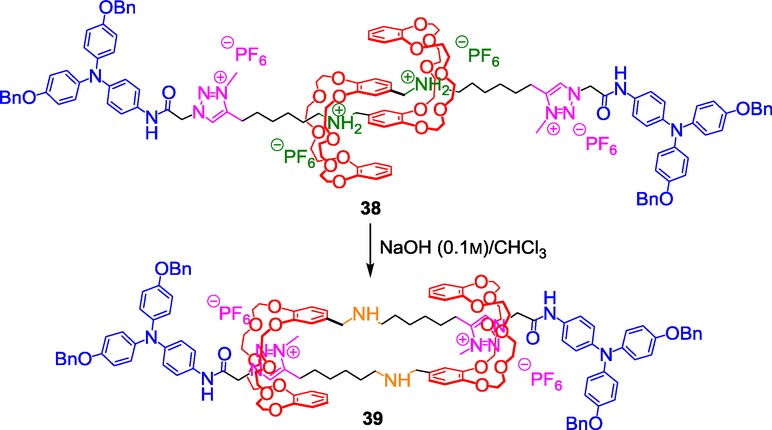
Triazolium-based building block for microfiber-induced formation.
The system works as an INHIBIT logic gate where both light and acidic medium trigger the formation of the supramolecular fiber. The Coutrot [c2]daisy chain 33 was functionalized this time with a triarylamine moiety that holds an amide functional group. The usual two-step chemical strategy was applied to lock the structure and to incorporate a triazolium station in each encircled thread. Importantly, light exposure in presence of chlorinated solvent was found to induce the self-assembly of the sole building block 38 through hydrogen bond, π–π stacking, and van der Waals interactions. The formation of bundled fibrils of several microns length was evidenced by NMR, dynamic light scattering, UV/Vis–near infrared (NIR) absorption, and transmission electron microscopy (TEM). In this case, the self-assembly is attributed to the formation and the stabilization of triarylamine radical cations, as well as to the necessary hydrogen bonding between the amide moieties. This latter point is crucial, since no aggregation of the triarylamine is possible in the absence of amide moiety. Interestingly, the same result is observed if the amide is hindered by the DB24C8 through molecular machinery at basic pH. For this reason, no such supramolecular self-assembly of the deprotonated molecular muscle building block 39 could be observed.
In order to enhance the differentiation, hence the physical property, of the two co-conformational states of supramolecular polymers, the increase of both the rigidity and the length of the spacer which links the two molecular stations might be envisaged in the next future.
Other methodological studies focused on the relative affinity of the triazolium station for the DB24C8 in three-station-based rotaxanes.
Relative Affinity of the DB24C8 for the Triazolium Station With Respect to Pyridinium Amide Station in Three-Station-Based Rotaxane Molecular Machines
In three-station-based [2]rotaxane molecular machines
The synthesis of three-station-based rotaxanes containing an anilinium, a pyridinium amide, and N-methyltriazolium stations provided new interesting behavior of the system and gave insights into the relative affinity of the DB24C8 for the two different weak interaction sites.46 Each efficiently synthesized rotaxane 40 or 41 holds three different molecular sites of interaction for the DB24C8 (Scheme 17).
Scheme 17.
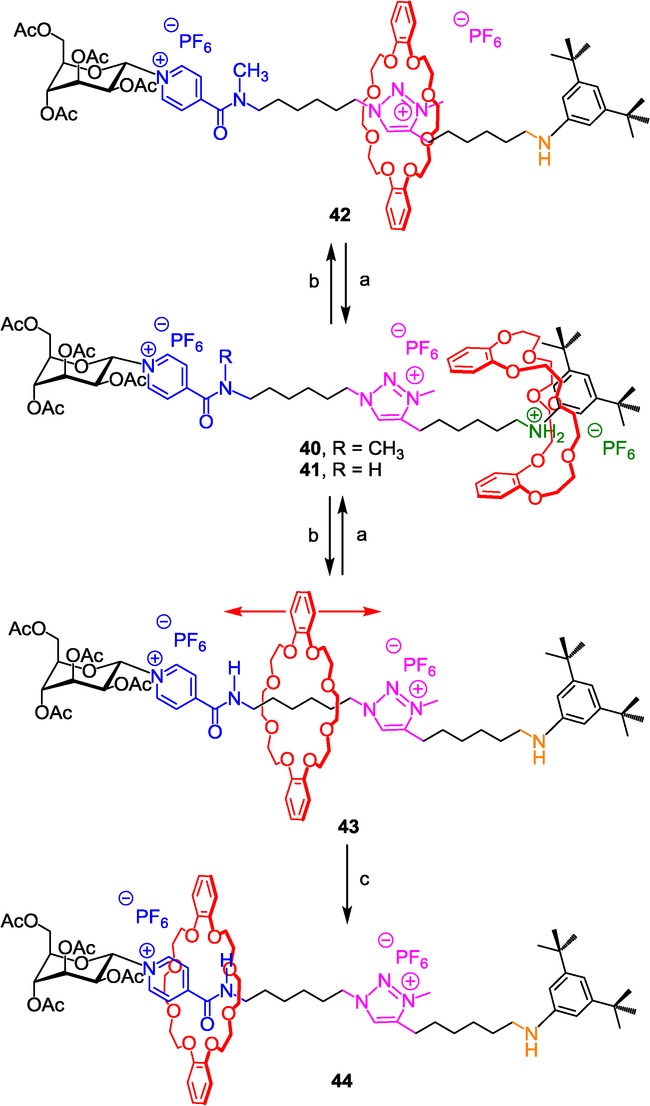
N-methyltriazolium as a molecular station for the DB24C8 in a three-station-based [2]rotaxane. Reagents and conditions: a) 1) HCl/Et2O, 2) NH4PF6, CH2Cl2/H2O, 30 min; b) DIEA; c) CD2Cl2, 223 K.
The only difference between these two components relies on the substitution of the amide group. At the protonated state, the behavior of the two rotaxanes 40 and 41 is identical because the substitution of the amide does not affect the localization of the DB24C8, which resides around the best anilinium station. However, the pH-sensitive molecular machines react very differently after the addition of diisopropylethylamine. Deprotonation of the anilinium induces distinct co-conformations in 42 and 43, because of the quite different affinities of the mono- and the disubstituted pyridinium amide moieties for the macrocycle. In 42, NMR studies proved that the DB24C8 is localized around the sole triazolium molecular station. It follows that the affinity of the DB24C8 is better for the N-methyltriazolium than for the disubstituted pyridinium amide. Deprotonation of rotaxane 41 resulted in a very different comportment of the molecular machine. In this case, the DB24C8 interacts with both the triazolium and the pyridinium amide molecular stations, and the two translational isomers are in fast exchange with respect to the NMR time scale. Extensive NMR studies were achieved to better characterize the translational Brownian motion along the thread between the two stations. At room temperature, in acetonitrile, the DB24C8 spends 62 % of its time around the monosubstituted pyridinium amide station for only 38 % around the triazolium. This clearly indicates that the monosubstituted pyridinium amide has a slightly better affinity for the DB24C8 than the triazolium. Decreasing the temperature triggers the gradual decrease of the proportion of the translational isomer in which the DB24C8 is localized around the triazolium site. At 223 K, the DB24C8 does not spend any time around the triazolium (44). In summary, in the disubstituted pyridinium amide series, a bi-stable molecular machine is observed in which the DB24C8 can only shuttle between the anilinium and the triazolium. On contrary, in the monosubstituted pyridinium amide series, depending on pH, the machine adopts either a stable protonated state or undergoes a controlled Brownian movement of the DB24C8 between the two weakest stations. In this latter case, the molecular machinery appears both pH- and temperature-dependent, with the two stimuli (i.e. acidification and temperature decrease) leading to two opposite localizations of the macrocycle along the thread.
In a molecular muscle architecture
The previous work was also extended to the more sophisticated molecular muscle architecture and furnished complementary results on the behavior of the triazolium moiety (Scheme 18).47
Scheme 18.
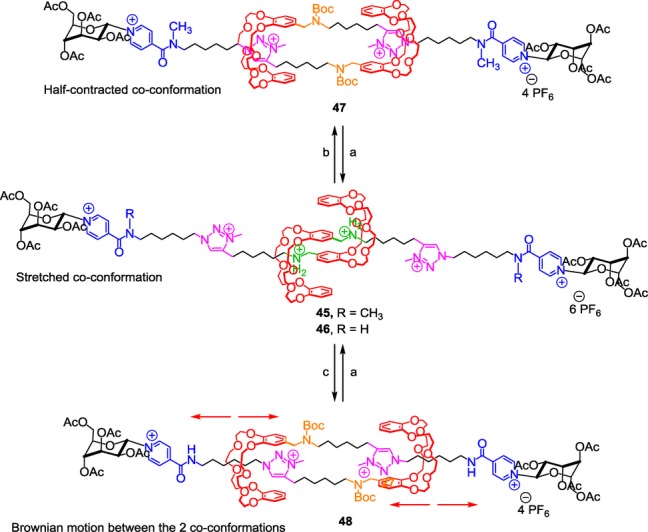
N-methyltriazolium as a molecular station for the DB24C8 in a three-station based molecular muscle. Reagents and conditions: a) 1) HCl/Et2O, 2) NH4PF6, H2O/CH2Cl2, RT, 30 min; b) DIEA, Boc2O, CH3CN, RT, 5 d; c) DIEA, Boc2O, CH3CN.
In the disubstituted pyridinium amide series (compounds 45 and 47), similarity was observed with respect to 40/42—in other words, the ammonium is the best molecular station, and among the weakest stations, the triazolium is a better station than the disubstituted pyridinium amide. Therefore, at the protonated state 45, the DB24C8 sits over the ammonium stations forcing the molecular muscle to adopt a stretched co-conformation. Unsurprisingly, in the carbamoylated48 molecular muscle 47, the DB24C8 reside around the triazolium stations, triggering a half-contracted co-conformation. In contrast, carbamoylation of the monosubstituted pyridinium amide-containing rotaxane dimer 46 results in a Brownian motion between the two triazolium and pyridinium amide sites as already observed in the simpler [2]rotaxane 43. The two translational isomers are in fast equilibrium between the contracted and the half-contracted co-conformations. However, in comparison with 43, it is noteworthy that important changes are noticed. Indeed, the ratio of the two translational isomers are inverted in 48, as if the triazolium seems to be now a better site of recognition for the DB24C8 than the monosubstituted pyridinium amide. In fact, this variation is directly linked to the doubly threaded architecture and is not correlated to a change of affinity of the two stations for the DB24C8. In the peculiar molecular muscle architecture, intramolecular repulsions are possible between the two triazolium cationic charges. The interactions between the DB24C8 and the monosubstituted pyridinium amide (which have been proved to be stronger than with the triazolium) should logically force the structure to adopt a contracted state. However, in this state, the two triazolium moieties would face each other, which is thermodynamically unfavorable. Then, the molecular muscle 48 rather prefers to adopt a favored half-contracted co-conformation where no such repulsion remains. Unsurprisingly, decreasing the temperature led to the displacement of the equilibrium between the two translational isomers 48 as observed in 43. Interestingly, the doubly interlocked architecture also makes possible the displacement of the translational equilibrium by tuning the repulsion through changing the polarity of the solvent. The repulsion between triazoliums proved to be the highest in a more polar solvent such as DMSO, hence favoring the half-contracted co-conformation at 97.5 %, whereas the lowest repulsion was noticed in the less polar solvent CDCl3, favoring the contracted co-conformation in a 88.5 % ratio. In summary, this study gave new complementary information concerning the relative affinity of a triazolium moiety for the DB24C8. Moreover, we have demonstrated that it is possible to trigger a wide range of co-conformational states upon pH-stimulus. Among them, a co-conformational state in which a Brownian motion between two stations can be generated, controlled by a temperature stimulus or a change in solvent polarity, while it can be switched off by acidification.
The repulsion between triazoliums in the doubly interlocked structure is a very interesting feature that has dramatic consequences on some motions observed in double-lasso architectures (see Section 8). The conversion of the triazole to the triazolium, in order to get a molecular station for crown ether, also gave us the opportunity to introduce a group of interest at the side-chain of the thread.
Dual Role of the Triazolium as Both a Molecular Station and a Kinetic Barrier for a Crown Ether
Triazolium has not only been utilized as a molecular station in a rotaxane molecular machine. It has also served as a kinetic molecular barrier that is able to compartmentalize1a,49 molecular machines by trapping the macrocycle around one of the two sides of a threaded axle. For that, we investigated the selective N-benzylation of triazole in order to incorporate such a molecular barrier and station at the middle of a threaded axle that contains two other molecular stations.50 The aim was either to trap the macrocycle around stations of weaker affinity than that of anilinium, or to know if and how the DB24C8 could interact only over one of the two sides of the bulky triazolium moiety (Section 7.1). A further reported application of this methodological study consisted in locking a mono-lasso molecular machine possessing a bigger benzometaphenylene-25-crown-8 (BMP25C8) macrocycle (Section 7.2).51
In a [2]rotaxane molecular architecture
First, the two-station-containing [2]rotaxane molecular machine 49 was efficiently prepared following the two-step sequence synthetic strategy we described in 2008 (Scheme 19).
Scheme 19.
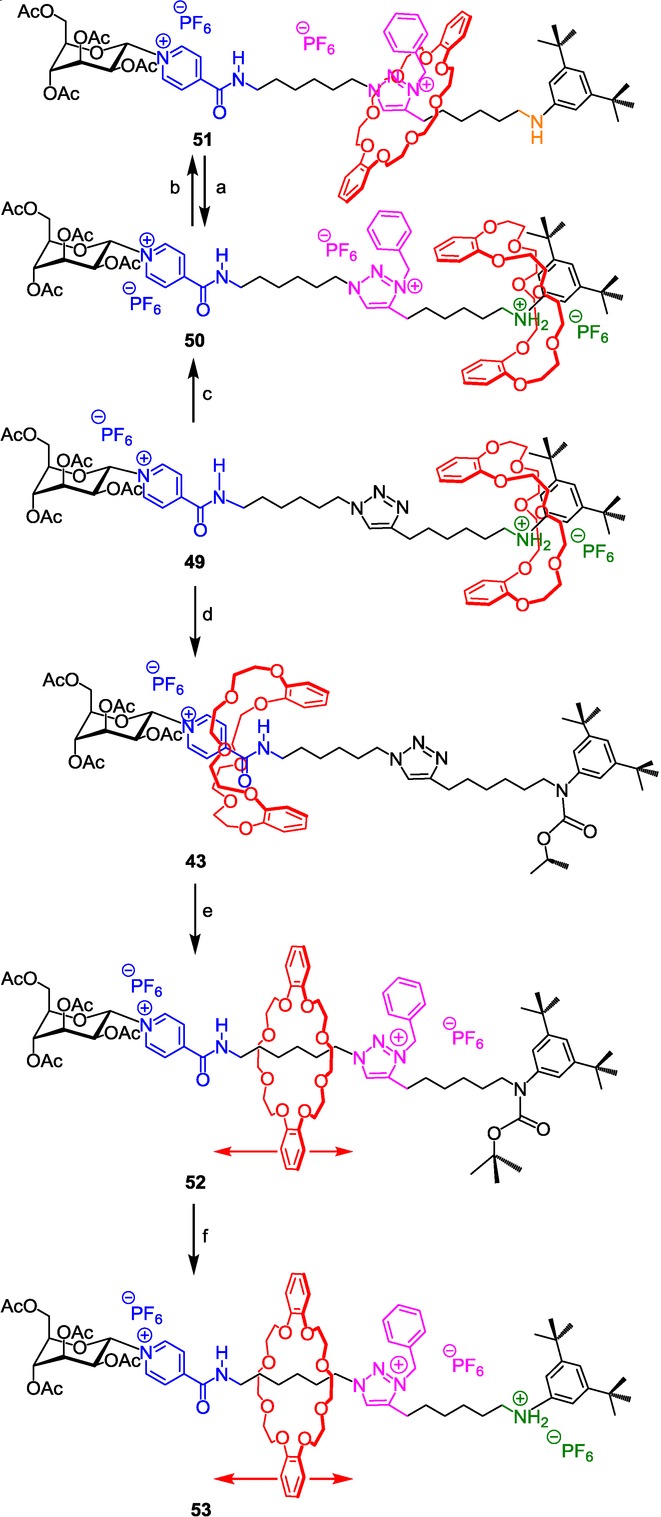
N-benzyltriazolium as both a molecular station and a barrier for the DB24C8 in a [2]rotaxane molecular machine. Reagents and conditions: a) 1) HCl, 2) NH4PF6; b) DIEA; c) 1) BnBr, CH2Cl2, 4 d, RT, 2) NH4PF6, CH2Cl2/H2O, 30 min, 79 %; d) Boc2O, DIEA, 15 h, RT, 93 %; e) 1) BnBr, CH2Cl2, 5 d, RT, 2) NH4PF6, CH2Cl2/H2O, 30 min, 65 %; f) 1) HCl, 1 h, RT, 2) NH4PF6, CH2Cl2/H2O, 30 min, 60 %.
Given the difference of the relative affinities of the DB24C8 for the two present sites of interactions,52 the DB24C8 resides around the best anilinium molecular station. The N-benzylation of the triazole afforded the three-station [2]rotaxane molecular machine 50, in which the N-benzyltriazolium proved to act as a molecular station. Indeed, deprotonation of 50 triggers the shuttling of the macrocycle as a result of the disappearance of the anilinium moiety. Whereas a kind of oscillating Brownian motion between the two stations of similar affinity for the DB24C8 (i.e. monosubstituted pyridinium amide and triazolium) was observed in the analogous N-methyltriazolium compound 43, the behavior of the macrocycle appears quite different in the present case. In 51, the DB24C8 only interacts with the N-benzyltriazolium station because the side bulky N-benzyl group acts as a molecular barrier that prevents the macrocycle from gliding across it. A so-called bi-stable pH-sensitive compartmentalized molecular machine is thus obtained, in which the DB24C8 shuttles from the ammonium to the triazolium site upon a pH-stimulus, without being able to reach the pyridinium amide site.
A diverted strategy from 43 was also applied in order to obtain 53, that is to say the translational isomer of 50, in which the DB24C8 is trapped on the left side of the threaded axle with respect to the benzyltriazolium barrier. A first carbamoylation of the aniline moiety triggered the large-amplitude displacement of the DB24C8 around the pyridinium amide molecular station. The subsequent N-benzylation of the triazole afforded, after decarbamoylation, the new compartmentalized molecular machine 53. This time, and contrary to 51, the DB24C8 undergoes a continuous motion between the pyridinium amide and the triazolium station, as already observed in the N-methyltriazolium [2]rotaxane 43. Nevertheless, the molecular machine 53 is here independent of any variation in pH, even if the anilinium is still the best site of interactions for the DB24C8. Indeed, a persistent Brownian co-conformational state is observed in 53 whatever the presence of the best anilinium station, due to the presence of the bulky molecular barrier.
Therefore, to summarize, the triazolium moiety can be used as both a molecular station and a barrier in the same molecular machine, bringing new insights for the conception of new molecular machines. A particular interest of this newly described feature of the triazolium moiety is the possibility to trap lasso molecules.
In lasso structures
Taking advantage of the possibility to use N-benzyltriazolium as both a barrier and a station at the same time in an interlocked molecular machine, we reported, in 2013, the preparation of lasso molecular architectures53,54 using a self-entanglement strategy55 of a “hermaphrodite” compound (Schemes 20 and 1).
Scheme 20.
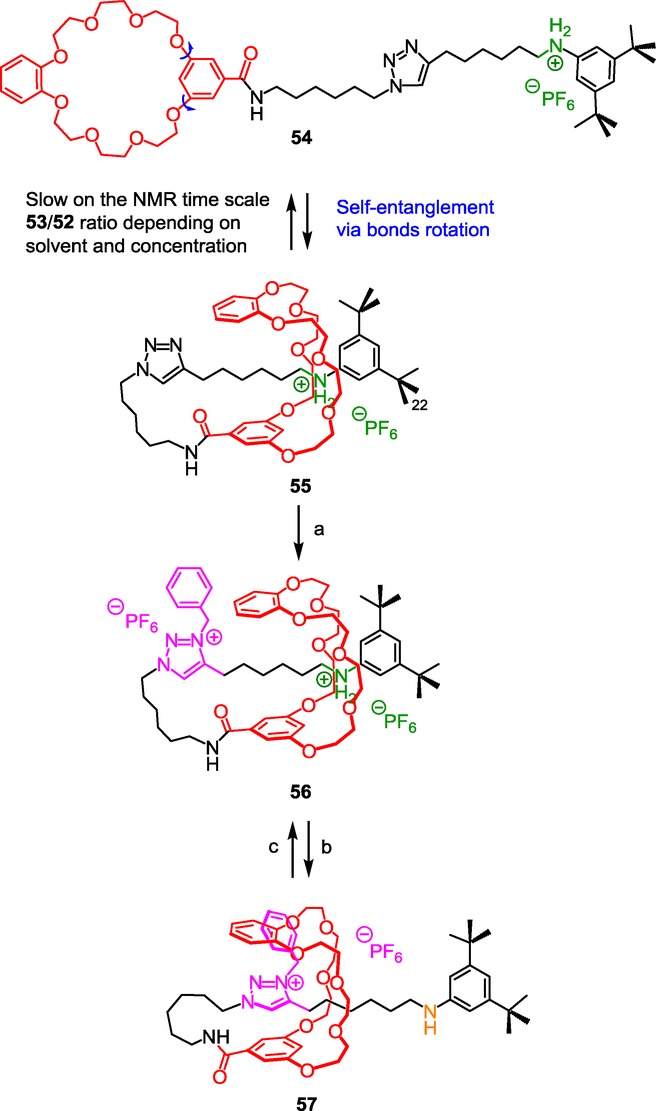
N-benzyltriazolium as both a molecular station and a barrier in a lasso molecular architecture. Reagents and conditions: a) 1) BnBr, CH2Cl2, <5×10−4m, 3 d, RT, 2) NH4PF6, H2O/CH2Cl2, RT, 30 min, 27 %; b) NaOH/H2O (1 m); c) 1) HCl/Et2O, 2) NH4PF6, CH2Cl2/H2O.
In the synthesized compound 54, the thread is linked to a BMP25C8 crown ether macrocycle and contains an anilinium station as a necessary template to envisage the efficient formation of the rotaxane. Contrary to the strategies previously described, the tail of the thread cannot go through the macrocycle because it already holds a stoppering ditert-butylphenyl end. Here, the only way to self-interlock is to use a large crown ether such as the BMP25C8 which allows the internal rotation of the metaphenylene aromatic ring. In this case, the rotation of the meta-substituted aromatic ring of the BMP25C8 around two σ-bonds allows the intramolecular threading of the anilinium-containing tail through self-entanglement. Obviously, the interactions between the template and the crown ether are weaker than with the shorter DB24C8 which fits much better. Nevertheless, they are sufficient to get, at high dilution, the pseudo[1]rotaxane 55 in a satisfactory yield of 45 %. The subsequent incorporation of the benzyl group on the triazole trapped the crown ether, hence locking the [1]rotaxane architecture 56 in a loosened conformation, due to the localization of the BMP25C8 around the best anilinium station. Treatment of the loosened lasso with a base causes the dramatic tightening of the lasso due to the shuttling of the BMP25C8 around the triazolium molecular station.
Using the same strategy, we then extended our studies to a peptide-containing lasso in which a GlyGlyGly tripeptide sequence is part of the loop of the lasso (Scheme 21).
Scheme 21.
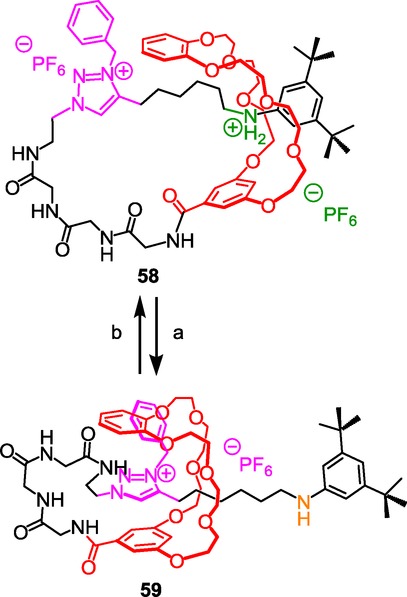
N-benzyltriazolium as both a molecular station and a barrier in a peptide-containing lasso molecular architecture. Reagents and conditions: a) DIEA; b) 1) HCl/Et2O, 2) NH4PF6, CH2Cl2/H2O.
Rotating-frame Overhauser enhancement (ROE) NMR experiments and drift-tube ion-mobility mass spectrometry proved the possibility to constrain more or less the peptide backbone of the lasso depending on molecular machinery. This first example of peptide-containing lasso now paves the way to many further possible applications in the selective targeting using peptide-containing lassos.
Triazolium-Containing Molecular Double Lassos: A Unique Spherical Molecular Muscle
In Sections 5 and 6, we described several examples of linear molecular muscles and their use as building blocks for polymers. In this section, we highlight a unique double-lasso56 molecular architecture that can act as a spherical muscle.57 Two distinct motions have been observed: a loosening–tightening of the double-lasso and a controllable molecular “jump rope” movement of a molecular chain around a [c2]daisy58 arrangement.
Tightening–loosening the double lasso
The molecular spherical muscle relies on a double-lasso molecular architecture that can be obtained by the chemical connection of the two ends of a linear molecular muscle (Scheme 22).
Scheme 22.
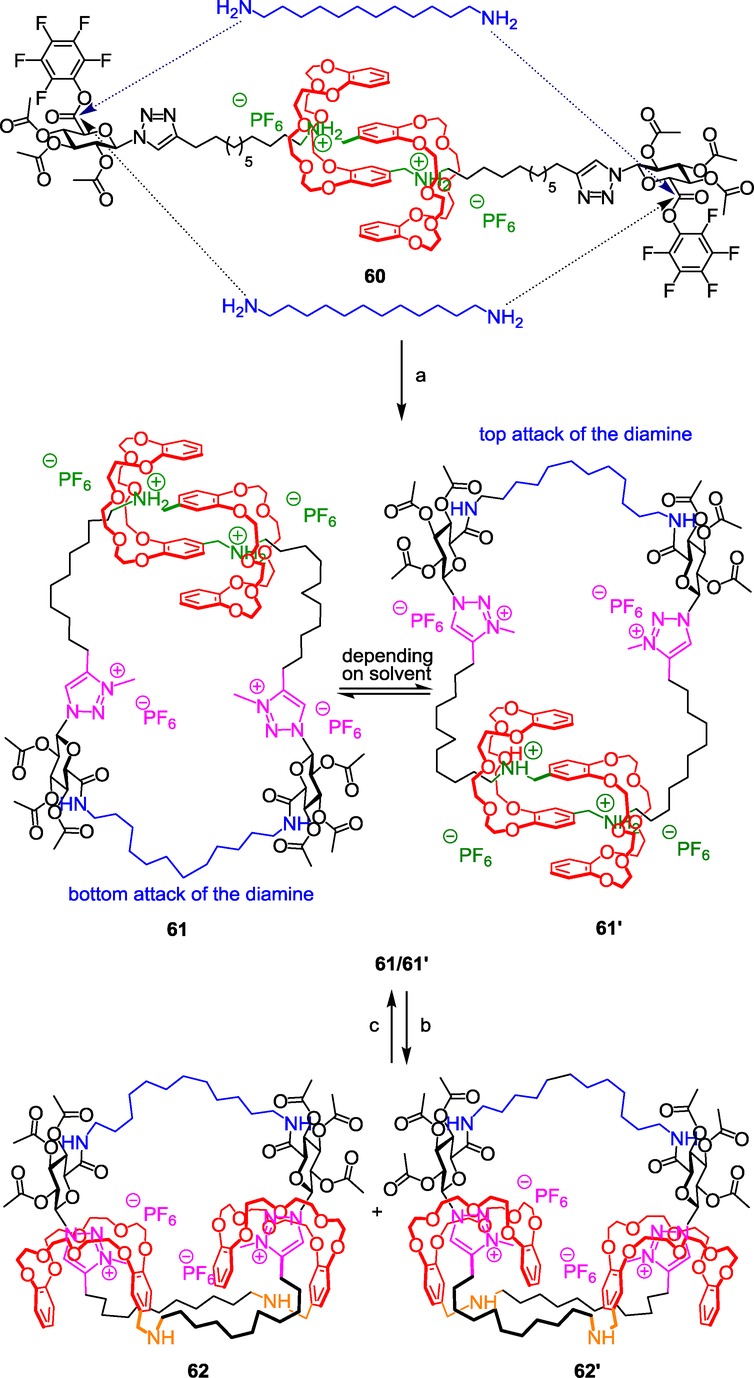
Triazolium-based double-lasso molecular architecture and loosening–tightening motion. Reagents and conditions: a) 1) H2N(CH2)12NH2 (1 equiv), CH2Cl2 (0.5 mm), 25 °C, 3 d, 2) CH3I, 4 d, 3) NH4PF6, H2O/CH2Cl2, 30 min; b) NaOH/H2O/CH2Cl2; c) 1) HCl/Et2O, 2) NH4PF6/H2O/CH2Cl2, quanti 61/61’.
The synthesized [c2]daisy-chain-incorporating compound 60 was designed in order to give enough flexibility to the two extremities in order to permit the cyclization. Thus, the length of the alkyl chain between the ammonium station and the triazole moiety was increased with respect to the previously published molecular muscle 34. Furthermore, two pentafluorophenyl active ester functions on the glucuronic acid derivative units serve as bulky stopper to lock the interwoven structure. They are also present as highly sensitive moieties toward amines, so that the dodecanediamine can close the double-lasso structure at high dilution, through the linking of the two extremities of the [c2]daisy chains 60. Subsequent alkylation of the triazoles reveals, in 61, the second molecular stations for the DB24C8 macrocycles. At this stage, it is interesting to notice that the attack of the diamine by the top or by the bottom of the molecule provides two atropoisomers, whose exchange can be controlled. This exchange is explained in detail in the following section. What interested us first was the deprotonation of the loosened double-lasso 61. As expected, it triggers the tightening of the double lasso through the shuttling of the macrocycle around the triazolium sites. In 62, two distinct double-lasso diastereomers were obtained. The size of the double-lasso molecules at the protonated and deprotonated states was evaluated thanks to diffusion-ordered spectroscopy (DOSY) experiments and molecular mechanics calculations. The results corroborate the impressive tightening-loosening internal motion of the lasso.
Designing new double-lasso molecular machines which are able to host a guest molecule with preferential interactions in one of the two co-conformational states is still an issue to tackle. One may imagine different kinds of applications of this new cyclic architecture in the future, like making molecular carriers that respond to a stimulus in order to release a guest molecule, or making switchable nanoreactors for asymmetric synthesis by taking advantage of the chiral helix-type double lasso. One may also think about using this sort of compounds for closing or opening a channel or even capturing molecules to decontaminate a medium.
Molecular jump-rope motion around the [c2]daisy moiety
Another interesting motion that can be controlled in the double lasso 61 concerns the rotation of the triazolium-containing chain around the [c2]daisy arrangement. We compared this movement to a molecular “jump rope” or to a molecular “turnstile”59 depending on if we consider the rotation of the cyclic chain with respect to the [c2]daisy arrangement or the inverse (Scheme 23).
Scheme 23.
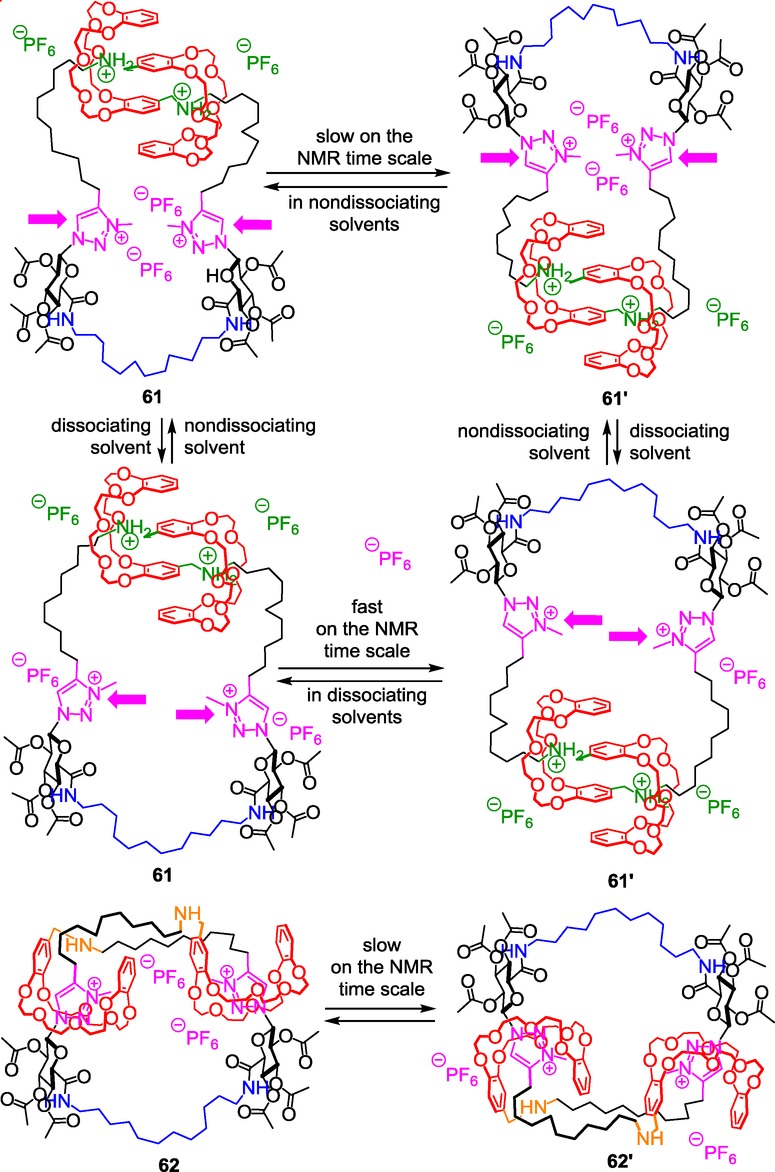
Molecular jump-rope motion in a triazolium-based double-lasso spherical molecular muscle.
As mentioned before, the possibility of the triazolium-containing chain to clear the [c2]daisy arrangement allows the exchange between isomers 61 and 61′. As long as the rotation is possible and fast, 61 and 61’ remain conformers, and only one set of 1H NMR resonances is observed. However, tightening the cavity of the double lasso results in the deceleration or the stopping of this internal rotation. In that case, one set of distinguishable 1H NMR signals is observed for each isomer.
Two different ways to tighten the double lasso were found in this unique structure. The first is a consequence of a pH stimulus, while the second results from the variation of the polarity of the solvent. Obviously, on one hand, adding a base induces the tightening of the spherical molecular muscle and therefore the impossibility for the cyclic chain to turn around the [c2]daisy chain. On the other hand, tuning the repulsion between the triazoliums that are facing each other in this singular structure enables us to disturb or favor the jump-rope movement in the loosened co-conformation 61. This was achieved through the variation of the polarity of the solvent, as already mentioned in the three-station-containing molecular muscle 48. The shape of the cavity of the double lasso appears larger in polar solvents like acetonitrile because the repulsion between triazoliums is higher. This causes a fast rotation of the molecular jump rope on the NMR time scale because no much steric hindrance disturbs the rotation around the [c2]daisy chain. It is exactly the contrary in dichloromethane where the two triazoliums sit much closer to each other, thus tightening the cavity of the double lasso in a new way (i.e. different from the tightening motion of the lasso ascribed to the gliding of the DB24C8) and to another shape than a basic reagent enforces. This singular tightening of the double lasso induces a slower rotation of the “jump rope” around the [c2]daisy arrangement due to steric hindrance. Interestingly, increasing progressively the proportion of one of the two solvents allows the progressive acceleration or deceleration of the motion.
Summary and Outlook
Since we first used the triazolium moiety in 2008 as a molecular station for the DB24C8 in a [2]rotaxane molecular machine, several examples of original molecular machines have emerged. From pure molecular design to applications in stereoselective catalysis or fluorescence output, from motion at the molecular scale to enhanced supramolecular polymer extension or contraction, and from simpler [2]rotaxane architectures to molecular muscles or lasso compounds, triazolium proved to be a molecular station and/or a kinetic molecular barrier of choice for the construction of a wide range of more or less sophisticated interlocked molecular machines. Its very easy access through a two-step sequence strategy, combined with its easy 1H NMR characterization in both its naked state or in interaction with crown ethers makes the triazolium a multipurpose and very friendly moiety to handle. In addition, the molecular machinery appears very simple to monitor. Although triazolium has a very poor intermolecular affinity for crown ethers, it can interact pretty well in a locked molecular architecture in the absence of a better site of affinity, which makes triazoliums an ideal second molecular station. Moreover, the possibility to reach triazolium-based interlocked components devoid of any other efficient template using a diverted strategy based on a “transporter” of macrocycles has been recently published, opening the way to a wide variety of new triazolium-containing interlocked molecules. Finally, the possibility to form triazolium through the linking of various moieties to the nitrogen atom of a triazole group opens a wide range of further possibilities. Investigating new properties of the triazolium moiety, like its behavior as a redox molecular station in an interlocked machine, should certainly be of high interest for the construction of multiresponsive systems.
Biography
Frédéric Coutrot received his PhD in 1999 from the University Henri Poincaré (Nancy, France). He then joined the group of Dr. Michel Marraud (Ecole Nationale Supérieure des Industries Chimiques, Nancy) as a temporary lecturer. In 2000, he moved to David Leigh's laboratory with a Marie-Curie Fellowship, first at Warwick University (England), then at King's Building in Edinburgh (Scotland). Since 2002, he has been an Assistant Professor at the University of Montpellier (France) and is currently the team leader of the Supramolecular Machines and Architectures Team at the Institut des Biomolécules Max Mousseron (IBMM). His research focuses on the synthesis and study of new interlocked molecular machines.
References
- 1a.Kay E, Leigh DA, Zerbetto F. Angew. Chem. Int. Ed. 46:72–191. doi: 10.1002/anie.200504313. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007;119 [Google Scholar]
- 1b.Balzani V, credi A, Raymo FM, Stoddart JF. Angew. Chem. Int. Ed. 39:3348–3391. doi: 10.1002/1521-3773(20001002)39:19<3348::aid-anie3348>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2000;112 [Google Scholar]
- 2a.Bissell RA, Cordova E, Kaifer AE, Stoddart JF. Nature. 1994;369:133–137. [Google Scholar]
- 2b.Martínez-Díaz MV, Spencer N, Fraser Stoddart J. Angew. Chem. Int. Ed. Engl. 36:1904–1907. [Google Scholar]; Angew. Chem. 1997;109 [Google Scholar]
- 2c.Ashton PR, Ballardini R, Balzani V, Baxter I, Credi A, Fyfe MCT, Gandolfi MT, Gomez-Lopez M, Martinez-Diaz MV, Piersanti A, Spencer N, Stoddart JF, Venturi M, White AJP, Williams DJ. J. Am. Chem. Soc. 1998;120:11932–11942. [Google Scholar]
- 2d.Garaudée S, Silvi S, Venturi M, Credi A, Flood AH, Stoddart JF. ChemPhysChem. 2005;6:2145–2152. doi: 10.1002/cphc.200500295. [DOI] [PubMed] [Google Scholar]
- Bottari G, Dehez F, Leigh DA, Nash PJ, Pérez EM, Wong JKY, Zerbetto F. Angew. Chem. Int. Ed. 42:5886–5889. doi: 10.1002/anie.200352176. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003;115 [Google Scholar]
- 4a.Zhang Z, Han C, Yu G, Huang F. Chem. Sci. 2012;3:3026–3031. [Google Scholar]
- 4b.Da Ros T, Guldi DM, Farran Morales A, Leigh DA, Prato M, Turco R. Org. Lett. 2003;5:689–691. doi: 10.1021/ol0274110. [DOI] [PubMed] [Google Scholar]
- 5a.Altieri A, Bottari G, Dehez F, Leigh DA, Wong JKY, Zerbetto F. Angew. Chem. Int. Ed. 42:2296–2300. doi: 10.1002/anie.200250745. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003;115 [Google Scholar]
- 5b.Leigh DA, Wong JKY, Dehez F, Zerbetto F. Nature. 2003;424:174–179. doi: 10.1038/nature01758. [DOI] [PubMed] [Google Scholar]
- 5c.Ceroni P, Credi A, Venturi M. Chem. Soc. Rev. 2014;43:4068–4083. doi: 10.1039/c3cs60400d. [DOI] [PubMed] [Google Scholar]
- 6a.Raehm L, Kern J-M, Sauvage J-P. Chem. Eur. J. 1999;5:3310–3317. [Google Scholar]
- 6b.Brouwer AM, Frochot C, Gatti FG, Leigh DA, Mottier L, Paolucci F, Roffia S, Wurpel GWH. Science. 2001;291:2124–2128. doi: 10.1126/science.1057886. [DOI] [PubMed] [Google Scholar]
- 6c.Altieri A, Gatti FG, Kay ER, Leigh DA, Martel D, Paolucci F, Slawin AMZ, Wong JKY. J. Am. Chem. Soc. 2003;125:8644–8654. doi: 10.1021/ja0352552. [DOI] [PubMed] [Google Scholar]
- 6d.Gaviña P, Sauvage J-P. Tetrahedron Lett. 1997;38:3521–3524. [Google Scholar]
- 6e.Armaroli N, Balzani V, Collin J-P, Gavina P, Sauvage J-P, Ventura B. J. Am. Chem. Soc. 1999;121:4397–4408. [Google Scholar]
- 6f.Collin J-P, Gavina P, Sauvage J-P. J. Chem. Soc., Chem. Commun. 1996:2005–2006. [Google Scholar]
- 7a.Tachibana Y, Kawasaki H, Kihara N, Takata T. J. Org. Chem. 2006;71:5093–5104. doi: 10.1021/jo0601563. [DOI] [PubMed] [Google Scholar]
- 7b.Suzuki S, Nakazono K, Takata T. Org. Lett. 2010;12:712–715. doi: 10.1021/ol902719m. [DOI] [PubMed] [Google Scholar]
- 7c.Koyama Y, Matsumura T, Yui T, Ishitani O, Takata T. Org. Lett. 2013;15:4686–4689. doi: 10.1021/ol401984j. [DOI] [PubMed] [Google Scholar]
- Aizpurua JM, Fratila RM, Monasterio Z, Pérez-Esnaola N, Andreieff E, Irastorza A, Sagartzazu-Aizpuruaa M. New J. Chem. 2014;38:474–480. [Google Scholar]
- 9a.Kolchinski AG, Busch DH, Alcock NW. J. Chem. Soc. Chem. Commun. 1995:1289–1291. [Google Scholar]
- 9b.Ashton PR, Campbell PJ, Glink PT, Philp D, Spencer N, Stoddart JF, Chrystal EJT, Menzer S, Williams DJ, Tasker PA. Angew. Chem. Int. Ed. Engl. 34:1865–1869. [Google Scholar]; Angew. Chem. 1995;107 [Google Scholar]
- 10a.Huisgen R. Pure. Appl. Chem. 1989;61:613–628. [Google Scholar]
- 10b.Huisgen R, Szeimies G, Möbius L. Chem. Ber. 1967;100:2494–2507. [Google Scholar]
- 10c.Huisgen R. Angew. Chem. Int. Ed. Engl. 2:565–598. [Google Scholar]; Angew. Chem. 1963;75 [Google Scholar]
- 10d.Huisgen R. Angew. Chem. Int. Ed. Engl. 2:633–645. [Google Scholar]; Angew. Chem. 1963;75 [Google Scholar]
- 11a.Kolb HC, Finn MG, Sharpless KB. Angew. Chem. Int. Ed. 40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2001;113 [Google Scholar]
- 11b.Tornøe CW, Christensen C, Meldal M. J. Org. Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- Chao S, Romuald C, Fournel-Marotte K, Clavel C, Coutrot F. Angew. Chem. Int. Ed. 53:6914–6919. doi: 10.1002/anie.201403765. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014;126 [Google Scholar]
- Coutrot F, Busseron E. Chem. Eur. J. 2008;14:4784–4787. doi: 10.1002/chem.200800480. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Guo J-B, Chen C-F. Org. Lett. 2010;12:4248–4251. doi: 10.1021/ol101920b. [DOI] [PubMed] [Google Scholar]
- Zhang Z-J, Han M, Zhang H-Y, Liu Y. Org. Lett. 2013;15:1698–1701. doi: 10.1021/ol400481r. [DOI] [PubMed] [Google Scholar]
- Jiang W, Han M, Zhang H-Y, Zhang Z-J, Liu Y. Chem. Eur. J. 2009;15:9938–9945. doi: 10.1002/chem.200901206. [DOI] [PubMed] [Google Scholar]
- 17a.Badjic JD, Balzani V, Credi A, Silvi S, Stoddart JF. Science. 2004;303:1845–1849. doi: 10.1126/science.1094791. [DOI] [PubMed] [Google Scholar]
- 17b.Badjic JD, Ronconi CM, Stoddart JF, Balzani V, Silvi S, Credi A. J. Am. Chem. Soc. 2006;128:1489–1499. doi: 10.1021/ja0543954. [DOI] [PubMed] [Google Scholar]
- Ma Y-X, Meng Z, Chen C-F. Org. Lett. 2014;16:1860–1863. doi: 10.1021/ol500149k. [DOI] [PubMed] [Google Scholar]
- Chen CF. Chem. Commun. 2011;47:1674–1688. doi: 10.1039/c0cc04852f. [DOI] [PubMed] [Google Scholar]
- Meng Z, Chen CF. Chem. Commun. 2015;51:8241–8244. doi: 10.1039/c5cc01301a. [DOI] [PubMed] [Google Scholar]
- Leigh DA, Marcos V, Wilson MR. ACS Catal. 2014;4:4490–4497. [Google Scholar]
- Blanco V, Carlone A, Hänni KD, Leigh DA, Lewandowski B. Angew. Chem. Int. Ed. 51:5166–5169. doi: 10.1002/anie.201201364. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012;124 [Google Scholar]
- Erkkilä A, Majander I, Pihko PM. Chem. Rev. 2007;107:5416–5470. doi: 10.1021/cr068388p. [DOI] [PubMed] [Google Scholar]
- Blanco V, Leigh DA, Lewandowska U, Lewandowski B, Marcos V. J. Am. Chem. Soc. 2014;136:15775–15780. doi: 10.1021/ja509236u. [DOI] [PubMed] [Google Scholar]
- Pihko PM, Majander I, Erkkilä A. In: Asymmetric Organocatalysis, Vol. 291. B. List., editor. Berlin: Springer; 2009. pp. 29–75. [Google Scholar]
- Blanco V, Leigh DA, Marcos V, Morales-Serna JA, Nussbaumer AL. J. Am. Chem. Soc. 2014;136:4905–4908. doi: 10.1021/ja501561c. [DOI] [PubMed] [Google Scholar]
- Zhou W, Zhang H, Li H, Zhang Y, Wang Q-C, Qu D-H. Tetrahedron. 2013;69:5319–5325. [Google Scholar]
- Yang W, Li Y, Zhang J, Yu Y, Liu T, Liu H, Li Y. Org. Biomol. Chem. 2011;9:6022–6026. doi: 10.1039/c1ob05726j. [DOI] [PubMed] [Google Scholar]
- Jiang Q, Zhang H-Y, Han M, Ding Z-J, Liu Y. Org. Lett. 2010;12:1728–1731. doi: 10.1021/ol100321k. [DOI] [PubMed] [Google Scholar]
- Cao Z-Q, Li H, Yao J, Zou L, Qu D-H, Tian H. Asian J. Org. Chem. 2015;4:212–216. [Google Scholar]
- Li H, Zhang JN, Zhou W, Zhang H, Zhang Q, Qu D-H, Tian H. Org. Lett. 2013;15:3070–3073. doi: 10.1021/ol401251u. [DOI] [PubMed] [Google Scholar]
- Li H, Li X, Agren H, Qu D-H. Org. Lett. 2014;16:4940–4943. doi: 10.1021/ol502466x. [DOI] [PubMed] [Google Scholar]
- Li H, Li X, Cao Z-Q, Qu D-H, Agren H, Tian H. ACS Appl. Mater. Interfaces. 2014;6:18921–18929. doi: 10.1021/am506283g. [DOI] [PubMed] [Google Scholar]
- 34a.Jiménez MC, Dietrich-Buchecker C, Sauvage J-P. Angew. Chem. Int. Ed. 39:3284–3287. doi: 10.1002/1521-3773(20000915)39:18<3284::aid-anie3284>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2000;112 [Google Scholar]
- 34b.Collin JP, Dietrich-Buchecker C, Gavina P, Jimenez-Molero MC, Sauvage JP. Acc. Chem. Res. 2001;34:477–487. doi: 10.1021/ar0001766. [DOI] [PubMed] [Google Scholar]
- 34c.Jimenez-Molero MC, Dietrich-Buchecker C, Sauvage JP. Chem. Eur. J. 2002;8:1456–1466. doi: 10.1002/1521-3765(20020315)8:6<1456::aid-chem1456>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 34d.Colasson BX, Dietrich-Buchecker C, Jimenez-Molero MC, Sauvage JP. J. Phys. Org. Chem. 2002;15:476–483. [Google Scholar]
- 34e.Jimenez-Molero MC, Dietrich-Buchecker C, Sauvage JP. Chem. Commun. 2003:1613–1616. doi: 10.1039/b302326p. [DOI] [PubMed] [Google Scholar]
- 34f.Collin JP, Heitz V, Bonnet S, Sauvage JP. Inorg. Chem. Commun. 2005;8:1063–1074. [Google Scholar]
- 34g.Bonnet S, Collin JP, Koizumi M, Mobian P, Sauvage JP. Adv. Mater. 2006;18:1239–1250. [Google Scholar]
- 35a.Niess F, Duplan V, Sauvage J-P. Chem. Lett. 2014;43:964–974. [Google Scholar]
- 35b.Bruns CJ, Stoddart JF. Acc. Chem. Res. 2014;47:2186–2199. doi: 10.1021/ar500138u. [DOI] [PubMed] [Google Scholar]
- Fraser Stoddart J. Chem. Soc. Rev. 2009;38:1521–1529. doi: 10.1039/b819336n. [DOI] [PubMed] [Google Scholar]
- Cantrill SJ, Youn GY, Stoddart JF, Williams DJ. J. Org. Chem. 2001;66:6857–6872. doi: 10.1021/jo010405h. [DOI] [PubMed] [Google Scholar]
- 38a.Leung JWu„KCF, Benitez D, Han JY, Cantrill SJ, Fang L, Stoddart JF. Angew. Chem. Int. Ed. 47:7470–7474. doi: 10.1002/anie.200803036. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008;120 [Google Scholar]
- 38b.Dawson RE, Lincoln SF, Easton CJ. Chem. Commun. 2008:3980–3982. doi: 10.1039/b809014a. [DOI] [PubMed] [Google Scholar]
- 38c.Bruns CJ, Li J, Frasconi M, Schneebeli ST, Iehl J, Jacquot de Rouville H-P, Strupp SI, Voth GA, Stoddart JF. Angew. Chem. Int. Ed. 53:1953–1958. doi: 10.1002/anie.201308498. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014;126 [Google Scholar]
- 38d.Bruns CJ, frasconi M, Iehl J, Hartlieb KJ, Schneebeli ST, Cheng C, Stupp SI, Stoddart JF. J. Am. Chem. Soc. 2014;136:4714–4723. doi: 10.1021/ja500675y. [DOI] [PubMed] [Google Scholar]
- Coutrot F, Romuald C, Busseron E. Org. Lett. 2008;10:3741–3744. doi: 10.1021/ol801390h. [DOI] [PubMed] [Google Scholar]
- 40a.Du G, Moulin E, Jouault N, Buhler E, Giuseppone N. Angew. Chem. Int. Ed. 51:12504–12508. doi: 10.1002/anie.201206571. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012;124 [Google Scholar]
- 40b.Bruns CJ, Fraser Stoddart J. Nat. Nanotechnol. 2013;8:9–10. doi: 10.1038/nnano.2012.239. [DOI] [PubMed] [Google Scholar]
- Hofmeier H, Schubert US. Chem. Soc. Rev. 2004;33:373–399. doi: 10.1039/b400653b. [DOI] [PubMed] [Google Scholar]
- Sauvage JP, Collin J-P, Chambron J-C, Guillerez S, Coudret C, Balzani V, Barigelletti F, De Cola L, Flamigni L. Chem. Rev. 1994;94:993–1019. [Google Scholar]
- 43a.Yu S-C, Kwok C-C, Chan W-K, Che C-M. Adv. Mater. 2003;15:1643–1647. [Google Scholar]
- 43b.Hofmeier H, Hoogenboom R, Wouters MEL, Schubert US. J. Am. Chem. Soc. 2005;127:2913–2921. doi: 10.1021/ja042919e. [DOI] [PubMed] [Google Scholar]
- 44a.Fang L, Hmadeh M, Wu J, Olson MA, Spruell JM, Trabolsi A, Yang Y-W, Elhabiri M, Albrecht-Gary A-M, Stoddart JF. J. Am. Chem. Soc. 2009;131:7126–7134. doi: 10.1021/ja900859d. [DOI] [PubMed] [Google Scholar]
- 44b.Clark PG, Day MW, Grubbs RH. J. Am. Chem. Soc. 2009;131:13631–13633. doi: 10.1021/ja905924u. [DOI] [PubMed] [Google Scholar]
- 44c.Hmadeh M, Fang L, Trabolsi A, Elhabiri M, Albrecht-Gary A-M, Stoddart JF. J. Mater. Chem. 2010;20:3422–3430. [Google Scholar]
- Wolf A, Moulin E, Cid J-J, Goujon A, Du G, Busseron E, Fuks G, Giuseppone N. Chem. Commun. 2015;51:4212–4215. doi: 10.1039/c4cc10331a. [DOI] [PubMed] [Google Scholar]
- Busseron E, Romuald C, Coutrot F. Chem. Eur. J. 2010;16:10062–10073. doi: 10.1002/chem.201000777. [DOI] [PubMed] [Google Scholar]
- Romuald C, Busseron E, Coutrot F. J. Org. Chem. 2010;75:6516–6531. doi: 10.1021/jo101234u. [DOI] [PubMed] [Google Scholar]
- Due to degradation of the compound in strong basic medium, carbamoylation was preferred to deprotonation.
- 49a.Chatterjee MN, Kay ER, Leigh DA. J. Am. Chem. Soc. 2006;128:4058–4073. doi: 10.1021/ja057664z. [DOI] [PubMed] [Google Scholar]
- 49b.Alvarez-Pérez M, Goldup SM, Leigh DA, Slawin MZ. J. Am. Chem. Soc. 2008;130:1836–1838. doi: 10.1021/ja7102394. [DOI] [PubMed] [Google Scholar]
- 49c.Carlone A, Goldup SM, Lebrasseur N, Leigh DA, Wilson AJ. J. Am. Chem. Soc. 2012;134:8321–8323. doi: 10.1021/ja302711z. [DOI] [PubMed] [Google Scholar]
- 49d.Kay ER, Leigh DA. Pure Appl. Chem. 2008;80:17–29. [Google Scholar]
- Busseron E, Coutrot F. J. Org. Chem. 2013;78:4099–4106. doi: 10.1021/jo400414f. [DOI] [PubMed] [Google Scholar]
- Coutrot F. In: Single Molecular Machines and Motors. C. Joachim., editor; G. Rapenne., editor. Berlin: Springer; 2015. pp. 35–64. [Google Scholar]
- Coutrot F, Busseron E. Chem. Eur. J. 2009;15:5186–5190. doi: 10.1002/chem.200900076. [DOI] [PubMed] [Google Scholar]
- Clavel C, Romuald C, Brabet E, Coutrot F. Chem. Eur. J. 2013;19:2982–2989. doi: 10.1002/chem.201203597. [DOI] [PubMed] [Google Scholar]
- Clavel C, Fournel-Marotte K, Coutrot F. Molecules. 2013;18:11553–11575. doi: 10.3390/molecules180911553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Z, Mayer MF. J. Am. Chem. Soc. 2010;132:3274–3276. doi: 10.1021/ja9077655. [DOI] [PubMed] [Google Scholar]
- Romuald C, Cazals G, Enjalbal C, Coutrot F. Org. Lett. 2013;15:184–187. doi: 10.1021/ol303186j. [DOI] [PubMed] [Google Scholar]
- Rotzler J, Mayor M. Chem. Soc. Rev. 2013;42:44–62. doi: 10.1039/c2cs35217f. [DOI] [PubMed] [Google Scholar]
- 59a.Lang T, Graf E, Kyritsakas N, Hosseini MW. Chem. Eur. J. 2012;18:10419–10426. doi: 10.1002/chem.201200562. [DOI] [PubMed] [Google Scholar]
- 59b.Guenet A, Graf E, Kyritsakas N, Allouche L, Hosseini MW. Chem. Commun. 2007:2935–2937. doi: 10.1039/b706527b. [DOI] [PubMed] [Google Scholar]
- 59c.Guenet A, Graf E, Kyritsakas N, Hosseini MW. Chem. Eur. J. 2011;17:6443–6452. doi: 10.1002/chem.201100057. [DOI] [PubMed] [Google Scholar]
- 59d.Lang T, Graf E, Kyritsakas N, Hosseini MW. Dalton Trans. 2011;40:3517–3523. doi: 10.1039/c1dt00004g. [DOI] [PubMed] [Google Scholar]
- 59e.Bedard TC, Moore JS. J. Am. Chem. Soc. 1995;117:10662–10671. [Google Scholar]
- 59f.Zigon N, Guenet A, Graf E, Kyritsakas N, Hosseini MW. Dalton Trans. 2013;42:9740–9745. doi: 10.1039/c3dt50809a. [DOI] [PubMed] [Google Scholar]