

Abstract
A dual-action ligand targeting both integrin αVβ3 and vascular endothelial growth factor receptors (VEGFRs), was synthesized via conjugation of a cyclic peptidomimetic αVβ3 Arg-Gly-Asp (RGD) ligand with a decapentapeptide. The latter was obtained from a known VEGFR antagonist by acetylation at the Lys13 side chain. Functionalization of the precursor ligands was carried out in solution and in the solid phase, affording two fragments: an alkyne VEGFR ligand and the azide integrin αVβ3 ligand, which were conjugated by click chemistry. Circular dichroism studies confirmed that both the RGD and VEGFR ligand portions of the dual-action compound substantially adopt the biologically active conformation. In vitro binding assays on isolated integrin αVβ3 and VEGFR-1 showed that the dual-action conjugate retains a good level of affinity for both its target receptors, although with one order of magnitude (10/20 times) decrease in potency. The dual-action ligand strongly inhibited the VEGF-induced morphogenesis in Human Umbilical Vein Endothelial Cells (HUVECs). Remarkably, its efficiency in preventing the formation of new blood vessels was similar to that of the original individual ligands, despite the worse affinity towards integrin αVβ3 and VEGFR-1.
Keywords: Angiogenesis, dual-action ligands, integrins, ligand conjugation, VEGFR
Introduction
Angiogenesis is an important physiological event, consisting of the formation of new blood vessels from the preexisting microvasculature network, and is also involved in pathological processes such as inflammation, tumor growth, and metastasis. Tumor angiogenesis plays a key role in cancer development, as neovascularization is necessary to supply oxygen and nutrients in order to support tumor cell proliferation.1 Substances capable of blocking tumor-related angiogenesis and, therefore, retarding cancer progression are termed antiangiogenic agents.
Angiogenesis is regulated by a number of receptors, whose expression is related to the conditions of the cell environment, such as pH and supply of oxygen and nutrients.1c Among the proteins involved in the angiogenic process, integrins play an important role by promoting endothelial cell attachment and migration onto the surrounding extracellular matrix, cell-to-cell interaction, and intracellular signal transduction.2 Integrins are heterodimeric glycoproteins composed of two noncovalently associated α and β transmembrane subunits, which recognize and bind their ligands through contiguous tripeptide sequences. The recognition motif Arg-Gly-Asp (RGD) allows endogenous ligands to interact with several integrins (e.g. αVβ3, αVβ5, and α5β1) that are very important for tumor progression, metastasis, and angiogenesis.3 A number of peptide and peptidomimetic integrin ligands containing the RGD sequence have been developed, some of which display a strong affinity towards these receptors.4 X-ray analysis of integrin αvβ3 cocrystallized with the cyclic RGD pentapeptide cyclo-[Arg-Gly-Asp-d-Phe-N(Me)-Val], Cilengitide, provided the structural basis to rationalize the observed binding properties.5 Activity and selectivity of these integrin ligands are linked to an extended conformation of the RGD sequence, with a distance of about 9 Å between the Cβ atom of aspartic acid and arginine residues, whose side chains form an “electrostatic clamp”5 crucial for ligand binding to the integrin receptor.
Our research group has recently reported a library of new cyclic RGD peptidomimetic integrin ligands, containing bifunctional diketopiperazine (DKP) scaffolds,6 that displayed low nanomolar IC50 values in inhibiting the binding of biotinylated vitronectin to integrin αVβ3. In vitro biological studies performed on compound 1 (Figure 1) showed that this ligand is able to inhibit angiogenesis in Human Umbilical Vein Endothelial Cells (HUVEC), both under basal conditions and in the presence of pro-angiogenic growth factors7 or of pro-inflammatory chemokine interleukin-8 (IL-8).8
Figure 1.
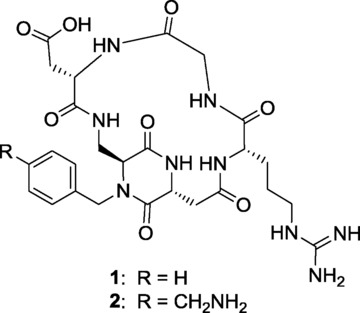
The potent and selective integrin αVβ3 ligand cyclo[DKP-RGD] (1), and its derivative cyclo[DKP-RGD]-CH2NH2 (2).
The observed antiangiogenic activity was likely due to the disruption of endothelial cell–extracellular matrix attachment, induced by integrin engagement by compound 1. This was further confirmed by the inhibition of the phosphorylation of Akt, a serine/threonine-specific protein kinase that plays a key role in the regulation of vascular homeostasis and angiogenesis. Unlike angiogenesis, other parameters such as cell viability, cell proliferation, and mRNA levels of αV, β3, or β5 integrin subunits were not affected by the administration of compound 1.8,9
Other cell surface receptors besides integrins are involved in tumor angiogenesis, and the pathways mediated by the different receptors are deeply intertwined. Such a “crosstalk” stimulates the angiogenic process through both direct and indirect association of the involved receptors. Interactions between integrins and vascular endothelial growth factor receptors (VEGFRs) have been investigated and suggested to be crucial for tumor growth and invasion.10 VEGFRs are members of the receptor tyrosine kinase (RTK) superfamily, and their dimerization and activation are induced by the binding of endogenous homodimeric vascular endothelial growth factors (VEGF) to the receptor extracellular domain. The biological activity of the VEGF–VEGFR system is closely related to tumor angiogenesis and progression: the hypoxic conditions present in tumors induce both VEGF gene expression and upregulation of VEGFRs.11 Small molecule inhibitors of VEGF-A receptor tyrosine kinase (receptor intracellular segments) have been shown to inhibit the biological function of this growth factor and are currently being used as drugs (Sorafenib and Sunitinib) for antiangiogenic therapy.12 Other inhibitors of the VEGF's biological activity interact with the VEGFRs’ extracellular segment, and most members of the latter family are peptides or peptidomimetics. Among them, it is worth mentioning the peptoid ligands developed by Kodadek and co-workers,13 the helical peptides derived from VEGF,14 and Vammin hotspots,15 a 17-amino acid cyclopeptide that was isolated from a phage display library.16
The reported colocalization of integrin αVβ3 and VEGFR on the surface of HUVEC upon VEGF stimulation demonstrates that a “crosstalk” between these receptors occurs.10f Following these findings, in a pioneering study, Cochran and co-workers reported that a dual specific fusion protein was able to bind simultaneously integrin αVβ3 and VEGFR-2, and to inhibit VEGF-mediated capillary tube formation in HUVEC and murine blood vessel formation within implanted Matrigel plugs.17a Inspired by this contribution, we speculated that a dual-action integrin–VEGFR small molecule ligand could also represent a novel antiangiogenic and antitumor strategy offering more effective multiple targeting to tumor cells and tumor vasculature.17b We thus designed a new dual-action ligand (compound 5 in Figure 2) targeting integrin αVβ3 and VEGFR. As integrin binding moiety, we exploited cyclo[DKP-RGD]-CH2NH2 (2) (Figure 1), a functionalized analog of ligand 1 that we had previously used for conjugation to paclitaxel18 and SMAC mimetic molecules.19 As VEGFR ligand moiety we selected the α-helical decapentapeptide 3 (Figure 2), recently reported by D′Andrea and co-workers,14 because of its simple preparation and efficacy in inhibiting angiogenesis in vivo. However, for conjugation to compound 2 we employed compound 4 (Figure 2), a derivative of 3 acetylated at the Lys13 side chain,20 in order to avoid any possible interference of the Lys13 free amino group with the “electrostatic clamp” used by the cyclo[DKP-RGD] moiety for binding to its αVβ3 integrin target. Previous studies clearly demonstrated that neither the N-terminal Lys1 side chain nor the Lys13 side chain are involved in receptor recognition.14b Accordingly, while both positions could in principle be exploited for conjugation, in this paper we used the N-terminal Lys1 side chain.
Figure 2.
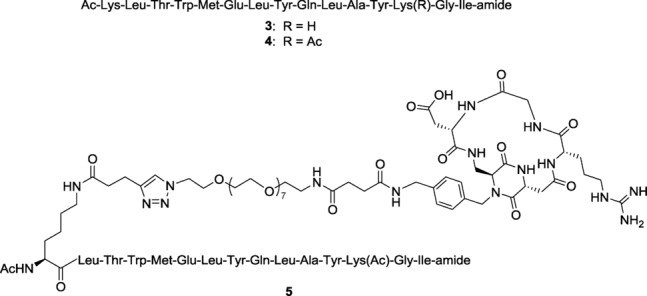
The α-helical peptide 3, its derivative 4, and the novel small-molecule dual-action ligand 5.
Results and Discussion
Synthesis of the dual-action compound 5
The dual-action ligand 5 (Figure 2) was obtained from the two fragments 6 and 7, that were joined by copper-catalyzed azide-alkyne cycloaddition (CuAAC) reaction (Scheme 1). Alkyne 6 was obtained from the resin-supported peptide 8, which was readily synthesized by microwave-assisted solid phase peptide synthesis (SPPS). Treating the on-bead decapentapeptide resin with a 94:5:1 dichloromethane/triisopropylsilane/trifluoroacetic acid (v/v/v) cleavage mixture resulted in the selective removal of 4-methyltrityl (Mtt) protecting group from Lys1 side chain. The free amine moiety in 9 was reacted on solid phase with 4-pentinoyc acid, after activation of the carboxylic acid moiety in the presence of the condensing agents, 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU) and 1-hydroxy-7-azabenzotriazole (HOAt), and N,N-diisopropylethylamine (DIPEA) as base, obtaining fragment 6. Fragment 7 was prepared starting from the commercially available bifunctional PEG8 compound 10 (Figure 3 A), which was selected because of its monodisperse structure profile.21 Amino azide 10 was first elongated with succinic anhydride, affording the azido acid 11. Cyclo[DKP-RGD]-CH2NH2 2 (Figure 1) was coupled with the spacer 11 in acetonitrile/phosphate buffer, controlling the pH of the medium in order to have the free benzylic amine of 2 acting as nucleophile. Finally, the synthesis of the dual-action ligand 5 was achieved by coupling fragments 6 and 7 on solid phase via CuAAC reaction in the presence of copper iodide and sodium ascorbate, and then by cleaving the adduct 12 from the resin.
Scheme 1.
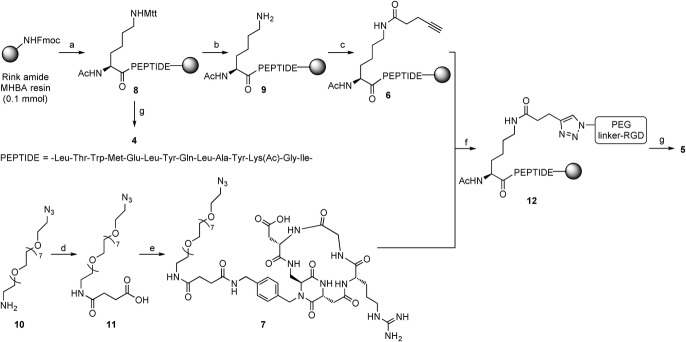
Reagents and conditions: a) SPPS: 1) 25 % piperidine in DMF, 2) Fmoc-AA-OH (4 eq), DIC, HOAt, DIPEA, DMF, 3) 25 % Ac2O in DMF; b) CH2Cl2/TIS/TFA 94:5:1 v/v/v, r.t., 12×2 min; c) 4-pentynoic acid, HATU, HOAt, DIPEA, DMF, r.t., o/n; d) succinic anhydride, DMAP, DIPEA, CH2Cl2, r.t., 18 h, 96 %; e) N-hydroxysuccinimide, DIC, DMF, r.t., 2 h, then 2, CH3CN, phosphate buffer, pH 7.3–7.6, 0 °C, 18 h, 65 % over 2 steps; f) 6+7, CuI, sodium ascorbate, DIPEA, DMF, 72 h, r.t.; g) TFA/EDT/H2O/TIS 94:2.5:2.5:1 v/v/v/v, 3 h, r.t., 5 % (4, over 16 steps) and 6 % (5, over 19 steps). Mtt=4-methyltrityl.
Figure 3.
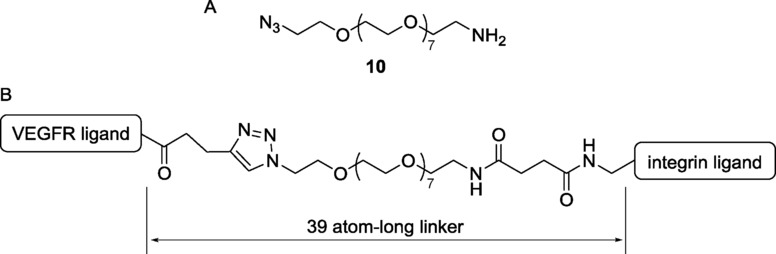
Bifunctional PEG8 amino azide 10 (A), and the distance between the two ligand moieties of conjugate 5 (B).
The 39-atom-long linker system of conjugate 5 (Figure 3 B) is of length commensurate with the linker used by Cochran and co-workers for their dual-specific proteins (14 amino acid residues, that is, 42 atoms, between the two ligand moieties).17a The decapentapeptide 4 (Figure 2), used as a reference compound, was synthesized by cleaving peptide 8 from the resin. The nonacetylated peptide 3 was also synthesized by SPPS, with a procedure similar to the one followed for compound 4 (see the Supporting Information for details).
Structural investigation of the synthesized compounds
The circular dichroism (CD) spectrum of peptide 4 measured in water (Figure 4) is consistent with that published by D′Andrea and co-workers for the parent peptide 3.14 The position and the intensities of the two negative maxima indicate the presence of a relevant population of α-helical structures. However, the negative maximum of the amide π→π* electronic transition, located at 203 nm, is significantly blue-shifted with respect to the canonical position (208 nm) of an α-helical conformation. This finding, together with the intensity decrease of the band at about 222 nm, can be safely assigned to a non-negligible participation of unordered conformations, also called “polyproline type-II”. We extended our analysis to two additional solvents, namely trifluoroethanol (TFE) and methanol, roughly mimicking the hydrophobic biological environment in which the peptide would probably display its action.22 These CD spectra (Figure 4) are characterized by two negative maxima typical of an α-helical conformation. This increase in the helical content, moving from water to the alcoholic solvents, clearly indicates that peptide 4 is not very rigid, as its 3 D-structure is highly dependent on the environment. The same is true for the parent peptide 3, whose CD spectra undergo a parallel modification in the cited solvents (see the Supporting Information).
Figure 4.
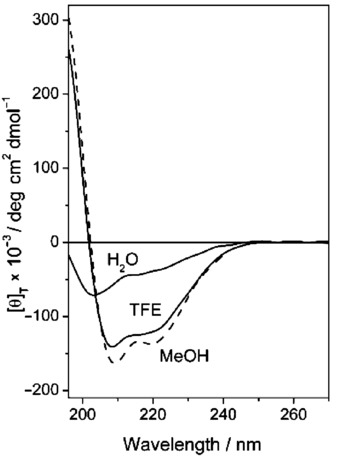
CD spectra of peptide 4 in water, 2,2,2-trifluoroethanol (TFE) and methanol (MeOH) (0.1 mm).
These conclusions, based on the analysis of the amide absorption region (190–250 nm), must be taken with caution as the aromatic chromophores of Tyr and Trp may contribute in an unpredictable way. Indeed, despite being achiral, they might display induced CD signals, whose intensities depend on the rigidity of the molecule. To evaluate this possible interference, we analyzed the region above 250 nm, where the amide absorption is negligible. In the UV absorption spectra of peptides 3 and 4, recorded at the same peptide concentration of the CD measurements, a band centered at 275 nm is clearly visible in all solvents investigated (data not shown). Conversely, in the corresponding CD spectra, no bands above 250 nm could be detected. On this basis, we can conclude that the contribution of the aromatic moieties to the CD curves is marginal.
The ratio R between the ellipticity values (θ) of the two negative maxima (R=[θ]222/[θ]208) can be used to discriminate the contribution to the CD curves of α- and 310-helical structures.22 In particular, an R value smaller than 0.5 is diagnostic of an important contribution from 310-helical conformations, while a value closer to 1 is indicative of an α-helical structure. The R value of peptide 4 in water is not significant because, as stated above, the wavelengths of the negative maxima in this solvent are too far from those of a canonical helix. In TFE and in methanol the R values are 0.84 and 0.86, respectively, thus highlighting the presence of a predominantly α-helical conformation.
The CD spectra in water of compounds 2 and 7 are shown in Figure 5. The two curves have trends usually attributed to type β folding, in particular β-turn type II, characterized by two maxima: a positive maximum at ≈203 nm and a negative one at ≈226 nm. Similar spectra were observed in cyclic tripeptides known to adopt β-turn structures. Interestingly, the relatively modest differences of the two curves suggest that the PEG8 moiety does not alter significantly the structure of compound 7 with respect to the precursor molecule 2.
Figure 5.
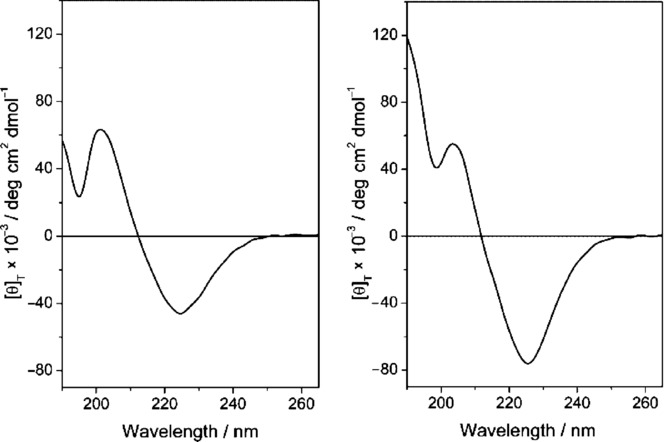
CD spectrum of 2 (left) and 7 (right) in H2O (0.1 mm).
In a previous work, this class of compounds was already demonstrated via NMR and computational studies to adopt a β-turn-like conformation (Figure 6).6
Figure 6.
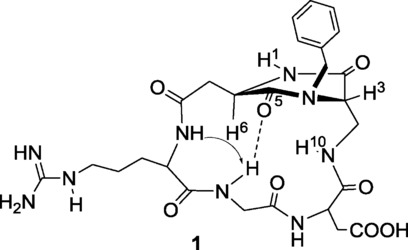
The preferred intramolecular hydrogen-bonded pattern proposed for compound 1 on the basis of NMR spectroscopic data. The arrow indicates a significant nuclear Overhauser effect (NOE) contact. Computational studies assessed that more than 90 % of the conformations sampled during restrained mixed-mode Metropolis Monte Carlo/Stochastic Dynamics simulations adopted an extended arrangement of the RGD sequence characterized by a pseudo-β-turn type II at DKP−Arg and the formation of the corresponding hydrogen bond between the NH−Gly and C(5)=O.
In Figure 7, the CD spectrum of 5 and the spectrum obtained by the algebraic sum of the spectra of 4 and 7 are reported.
Figure 7.
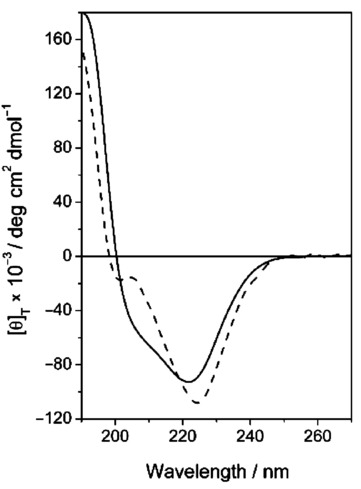
CD spectrum of 5 (solid line) in H2O (0.1 mm) superimposed to the sum CD spectrum of 4+7 (dashed line).
As the two spectra display rather similar features (band position, sign, and intensity), we tentatively conclude that no dramatic conformational changes occur. However, we cannot exclude the intervention of minor but significant structure modifications. The CD lineshape of the dual ligand 5 changes when water is replaced by either TFE or methanol (Figure 8), somehow mirroring the behavior of 4 (Figure 4). Therefore, this change in the CD pattern should be ascribed mainly to peptide 4. Indeed, in a comparative analysis we observed that the conformation of the cyclic, constrained, RGD ligand is not affected by the solvent (Supporting Information). In summary, the conformational behavior of the two isolated ligands is not substantially altered upon conjugation in the dual ligand 5. Yet, even minor structural changes can somehow impact on highly stereospecific activities such as receptor binding.
Figure 8.
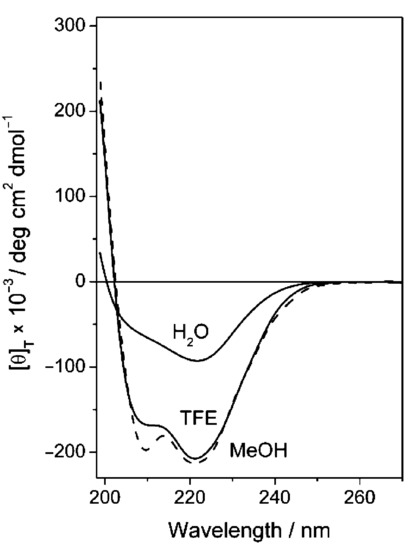
CD spectra of 5 in TFE, MeOH and H2O (0.1 mm).
Biological studies
In vitro binding assays on isolated integrins and VEGF receptor
The binding of the dual-action ligand 5 to the isolated integrin and VEGF receptors was measured. In the case of integrins, the ability of compound 5 to inhibit biotinylated vitronectin binding to the purified αVβ3 and αVβ5 extra-cellular integrin domains (IC50) was determined and compared with that of reference compounds 16b and cyclo[RGDfV] 13 (Figure 9).23
Figure 9.
The potent αVβ3 integrin ligand c[RGDfV] (13) (see Ref. 23).
Similar to its parent ligand 1, conjugate 5 showed a remarkable selectivity for integrin αVβ3 compared with αVβ5 (Table 1). In the case of 5, the affinity for integrin αVβ3 remained in the nanomolar range, although it was about 20 times worse than that of unconjugated ligand 1, possibly because of the increased steric hindrance or of interference with the decapentapeptide VEGFR ligand. These binding experiments demonstrate that the cyclo[DKP-RGD] moiety retains its ability to bind to the αVβ3 integrin receptor, even after conjugation.
Table 1.
In vitro binding assays on isolated αVβ3 and αVβ5 receptors.
IC50 values were calculated as the concentration of compound required for 50 % inhibition of biotinylated vitronectin binding as estimated by GraphPad Prism software; all values are the arithmetic mean±SD of triplicate determinations.
Ref. 6b.
The conjugate ligand 5, together with the VEGFR ligands 3 and 4, were also evaluated for their ability to compete with biotinylated VEGF165 for the binding to the extracellular domain (D1–D7) of recombinant VEGFR-1. The test was accomplished according to the procedure reported by Vidal, Inguimbert, and co-workers.15,24 Unlabelled VEGF165, used as reference compound, showed an IC50 value of 146 pm, comparable with the value reported by the same authors.24
Unfortunately, for compounds 3 and 4, it was not possible to obtain a dose-response curve because, at high concentrations (above 0.1 mm), the wells showed an anomalously high chemiluminescence readout (even higher than that of the positive control). In this way, we could only observe the initial inhibition of the binding of biotinylated VEGF165 by compounds 3 and 4 at 10 μm concentration. The reasons for this behavior are currently unclear, and modifications in the sample preparation protocol (e.g. change of the amount of dimethylsulfoxide, DMSO, used for solubilizing the peptide) did not solve the problem. It should be noted that the IC50 binding value of compound 3 to VEGFR-1 reported in the literature was not determined with this kind of competitive binding protocol. Rather, the affinity value was measured by NMR titration on VEGFR-1D2 (Kd=46 μm).14b
The dual-action compound 5 did not show this anomalous behavior in competitive binding assay, and its affinity towards VEGFR-1 was determined as 57 %±10 % inhibition at 500 μm concentration. Therefore, conjugate 5 is about ten times less potent than peptide 3 (or its acetylated derivative 4) in inhibiting VEGF binding to VEGFR-1. In summary, dual-action compound 5 showed affinity towards both its receptor targets (integrin αVβ3 and VEGFR-1), although with one order of magnitude (10/20 times) loss of potency.
In vitro morphogenesis assays on HUVEC
HUVECs represent a valid in vitro model providing seminal insights into the cellular and molecular events leading to neovascularization in response to inflammation and hypoxia in cancer, ischemic events, and in embryogenesis.25 Integrins αVβ3 and αVβ5 are expressed on HUVEC26 and these human endothelial-derived cells represent a suitable model to investigate the effects of integrin ligands on angiogenesis.
To assess the activity of the compounds targeting only one cell surface receptor, namely integrin ligand 1 and VEGFR ligands 3 and 4, as well as the dual-action ligand 5, HUVEC were incubated with these compounds under basal (absence of stimuli) or stimulated (VEGF165) conditions. Under basal conditions, HUVEC did not show any network formation, but the addition of VEGF165 induced significant morphogenesis (Figure 10 A). The addition of integrin ligand 1, VEGFR ligands 3 and 4 and dual-action ligand 5 markedly decreased the new capillary network formation (see for instance Figure 10 B) whereas the addition of negative control 14 (Ac-Lys-Gln-Met-Tyr-Leu-Glu-Leu-Gly-Tyr-Ala-Thr-Ile-Lys-Trp-Leu-amide), a peptide containing the same amino acids as 3 and 4 but in a scrambled sequence, did not affect morphogenesis to a significant extent (Figure 10 C).
Figure 10.

Representative phase contrast photomicrographs of HUVEC plated on Matrigel in the presence of: A) VEGF165 (10 ng mL−1); B) VEGF165 (10 ng mL−1)+5 (1 μm); C) VEGF165 (10 ng mL−1)+14 (1 μm). Images were elaborated by phase-contrast microscopy using a fluorescence microscope. Frames are approximately 10 μm wide × 10 μm tall.
A dose-response study (Figure 1) revealed that all the tested ligands display a similar profile of activity, reducing the morphogenesis already at nanomolar concentration. This effect is in agreement with previous reports using ligand 1 on HUVEC, albeit with different stimuli,8 and ligand 3.14 Interestingly, also peptide 4, bearing an acetyl group at the nitrogen atom of Lys13 side chain, proved capable of inhibiting angiogenesis with an efficacy similar to that of its parent compound 3 (Figure 1 C). Therefore, Lys13 side chain acetylation does not affect significantly the biological properties of peptide 3. The dual-action compound 5 (Figure 1 D) proved comparably active, inhibiting morphogenesis in HUVECs with a similar concentration profile and effect: all the ligands tested decreased the length of branches in newly formed vessels to a 40 % with respect to the stimulation with VEGF (Figure 2). In addition, the co-administration of ligands 1 and 4 was investigated (Figure 1 E). Also in this case no difference in concentration and effect profile could be noticed. Finally, the scrambled peptide 14 (negative control), tested at the 1 μm concentration, did not significantly affect VEGF-induced morphogenesis (82 %±21 % of VEGF alone, calculated probability value p>0.05) and confirmed that the effects observed for all the other compounds are due to their specific interaction with the targeted receptors. The overall picture designed by these data refers to a very active set of ligands which, once combined in a dual-action system fully retain the antiangiogenic activity. Remarkably, the efficiency of the dual-action ligand 5 in preventing the formation of new blood vessels was comparable to that of ligands 1 and 4, singly or jointly administered, despite its worse affinity towards integrin αVβ3 and VEGFR-1 (one order of magnitude loss of potency). This finding might be interpreted as the result of a synergy between the two covalently linked binding motifs, in analogy with the results on fusion proteins in which the mutated protein containing both the VEGFR and integrin binding sequences displayed a more pronounced effect of morphogenesis inhibition in HUVEC.17a
Figure 11.
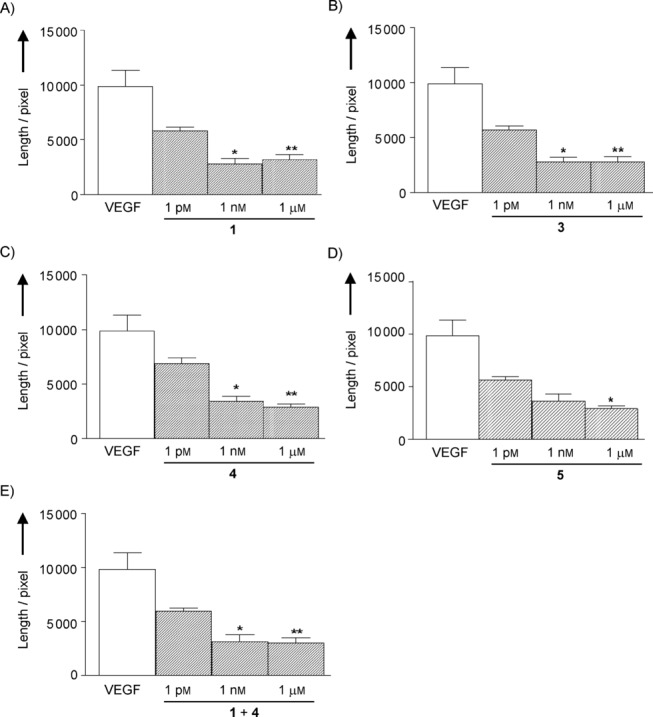
Effect of incubation of HUVEC for 5 h with the ligands 1 (A), 3 (B), 4 (C), 5 (D), and 1+4 (E) on VEGF-induced morphogenesis. Tube formation was evaluated as length of branches. Data are presented as mean±S.D. of 4–10 separate experiments (*=p<0.05 and **=p<0.01 vs. VEGF alone).
Figure 12.
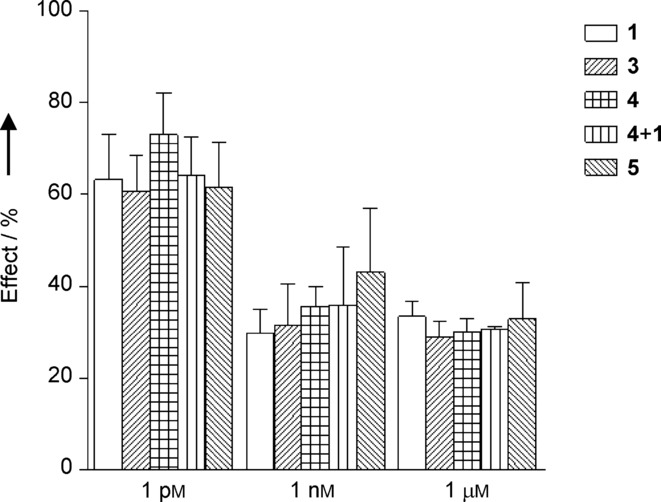
Inhibitory effects exerted by incubation with the different ligands on VEGF-induced morphogenesis. Results are presented as % of the effect normalized to VEGF alone and data are expressed as mean±S.D. for 4–10 separate experiments.
Conclusions
Tumor angiogenesis is a crucial phenomenon for cancer development and metastasis, and is regulated by a number of cell surface receptors, such as integrin αVβ3 and VEGFRs, which are known to specifically interact (receptor–receptor “crosstalk”). A dual-action ligand 5 was synthesized via conjugation of the potent peptidomimetic αVβ3 RGD ligand 1 with the decapentapeptide 4, a derivative acetylated at the Lys13 side chain of known peptide 3, a VEGF receptor antagonist with antiangiogenic activity. Ligand 5, targeting both integrin αVβ3 and VEGFRs, was designed with the aim of inhibiting both receptors and possibly blocking their “crosstalk”. The secondary structure of dual-action ligand 5 was studied by circular dichroism spectroscopy, and both the RGD portion and the decapentapeptide in the conjugate were found to substantially retain their respective conformations. However, even minor structural changes can somehow impact on highly stereospecific activities such as receptor binding.
Dual-action ligand 5 was tested in vitro for its ability to bind to isolated integrin αVβ3 and VEGFR-1 receptors. Conjugate 5 showed affinity towards both its receptor targets (integrin αVβ3 and VEGFR-1), although with one order of magnitude (10/20 times) loss of potency compared to ligands 1 and 4, targeting only one receptor. Conjugate 5 showed a potent antiangiogenic activity in VEGF-stimulated morphogenesis assays on HUVEC. Remarkably, its efficiency in preventing the formation of new blood vessels was similar to that of ligands 1 and 4, singly or jointly administered, despite its worse affinity towards integrin αVβ3 and VEGFR-1. This finding might be interpreted as the result of a synergy between the two covalently linked binding motifs.17a However, in this particular case (no real advantage in the morphogenesis assays over ligands 1 and 4), the extra effort for the synthesis of conjugate 5 is not justified, and further investigations are necessary to: 1) design and develop more efficient dual-action compounds targeting integrins and VEGFR and 2) confirm the beneficial effect of a dual ligand with in vivo experiments.
Experimental Section
The detailed procedure for the synthesis of compounds 1 and 2, and for conjugation of 2 to other molecular entities was previously described.6b,18
Solid phase peptide synthesis (SPPS): The SPPS was accomplished using the semiautomatic synthesizer Biotage Initiator (Uppsala, Sweden) assisted by microwaves; fluorenylmethyloxycarbonyl (Fmoc) strategy and Rink amide MHBA resin (100–200 mesh; loading: 0.5 mmol g−1) were used. Each coupling step consisted of 1) activation of the Fmoc-protected amino acid, 2) addition of the activated amino acid to the resin in the synthesizer, in order to perform the coupling reaction, and 3) capping, deprotection, and washing steps.
Resin preparation and storage: Before starting the SPPS, the resin was swollen in dimethylformamide (DMF) (3.0 mL) at r.t. for 45 min, then the solvent was drained. Two deprotection steps were carried out adding 25 % piperidine in DMF (3.0 mL for each step) to the beads: the reaction was performed at r.t. under inert atmosphere for 5 min and 15 min for the first and the second deprotection step, respectively. The resin was then washed with DMF. At the end of each step, the liquid phase was drained. Whenever it was necessary to stop the SPPS, the beads were stored in DMF (2–3 mL) at −20 °C, with the terminal amino group of the peptide left Fmoc-protected.
General procedure for Fmoc-AA-OH activation and coupling cycle: To a solution of the desired Fmoc-AA-OH (4.0 eq with respect to the resin) in DMF (3.5 mL) DIC (4.0 eq), HOAt (4.0 eq) and DIPEA (8.0 eq) were added successively at 0 °C, under stirring and inert atmosphere. After 25 min, the reaction mixture was added to the resin in the reaction vessel of the synthesizer, and a cycle of coupling–capping–deprotection–washing was effected. At the end of the cycle, it is possible to add another residue or to effect a capping–washing cycle if the peptide sequence is complete.
General procedure for Kaiser test: A few resin beads were taken and put in a glass test tube. The beads were washed with EtOH, then three drops of each of the following solutions were added: 1) ≈80 % phenol in EtOH, 2) KCN in pyridine [prepared by diluting a 0.1 mm aqueous solution of KCN with pyridine (2 % v/v)], and 3) 6 % ninhydrin in EtOH. The mixture was shaken and heated in a water bath. The resin beads and the solution turned dark blue when a free primary amine was present (positive result). Resin beads and solution maintained their yellow color when free primary amino groups were absent (negative result).
General procedure for final cleavage and deprotection: The protected-on-beads peptide was swollen first with DMF (3.5 mL), then with CH2Cl2 (3.5 mL). The beads were treated three times with the cleavage cocktail 94:2.5:2.5:1 TFA/H2O/EDT/TIS (v/v/v/v). After 1 h, the liquid was filtered off under nitrogen flow and collected in a round-bottom flask. The combined filtered fractions were concentrated and poured in cold diethyl ether, provoking precipitation of the product. Diethyl ether was removed with a syringe, affording the crude product, which was purified with reverse-phase HPLC.
Circular dichroism spectroscopy: The CD measurements were registered on a Jasco J-715 instrument (Easton, USA) with Hellma 0.1 cm quartz cell in milliQ H2O as solvent. The spectra were elaborated with Origin and the Jasco instrument associated software. The values are reported as total molar ellipticity [θ]T (deg×cm2×dmol−1).
Solid-phase receptor-binding assays on integrin receptors: Purified αVβ3 and αVβ5 receptors (Chemicon International, Inc., Temecula, USA) were diluted to 0.5 μg mL−1 in coating buffer containing 20 mmol L−1 Tris-HCl (pH 7.4), 150 mmol L−1 NaCl, 1 mmol L−1 MnCl2, 2 mmol L−1 CaCl2, and 1 mmol L−1 MgCl2. An aliquot of diluted receptors (100 μL/well) was added to 96-well microtiter plates (NUNC MW 96F Maxisorp Straight) and incubated overnight at 4 °C. The plates were then incubated with blocking solution (coating buffer plus 1 % bovine serum albumin) for additional 2 h at r.t. to block nonspecific binding followed by 3 h-incubation at r.t. with various concentrations (10−12–10−5 m) of test compounds in the presence of 1 μg mL−1 vitronectin biotinylated using EZ-Link Sulfo-NHS-Biotynilation kit (Pierce, Rockford, USA). After washing, the plates were incubated for 1 h at r.t. with streptavidin-biotinylated peroxidase complex (Amersham Biosciences, Uppsala, Sweden) followed by 30 min incubation with Substrate Reagent Solution (100 μL, R&D Systems, Minneapolis, USA) before stopping the reaction by addition of 2 n H2SO4 (50 μL) Absorbance at 415 nm was read in a Synergy HT Multi-Detection Microplate Reader (BioTek Instruments, Inc., Winooski, USA). Each data point is the result of the average of triplicate wells and was analyzed by nonlinear regression analysis with Prism software version 5 (GraphPad Software, Inc., La Jolla, USA).
Solid-phase receptor-binding assays on VEGFR-1: The surface of white high-binding 96-well microplates (Corning Life Sciences, Netherlands) was coated with of phosphate-buffered saline solution (100 μL, PBS, pH 7.4) containing 200 ng mL−1 of VEGFR-1 ECD/Fc chimera (R&D Systems, Minneapolis, USA) and incubated overnight at 4 °C. After three washes with PBS 0.1 %, (150 μL, v/v) Tween 20 (buffer A), the plate was blocked by PBS (160 μL) with 3 % (w/v) of BSA and incubated at r.t. for 2 h. The plate was washed three times with buffer A. Then, 100 μL of a solution of btVEGF165 at 131 pm (5 ng mL−1) and the tested compounds at various concentrations diluted in PBS containing 5 % DMSO were added to each well. After 3 h at 37 °C, the plate was washed three times with buffer A and streptavidin-horseradish peroxidase (100 μL) diluted at 1:1000 in PBS containing 0.1 % (v/v) Tween 20 and 0.3 % (w/v) BSA were added per well. After 1 h of incubation at r.t., the plate was washed five times with buffer A (150 μL), and SuperSignal West Pico Chemiluminescent Substrate (100 μL, Pierce, Rockford, USA) was added. The remaining bt-VEGF165 was detected by chemiluminescence, which was quantified with a Synergy HT Multi-Detection Microplate Reader (BioTek Instruments, Inc., Winooski, USA). The percentages of btVEGF165 displacement were calculated by the following formula: percentage of displacement=100×[1−(S−NS)/(MS−NS)], where S is the signal measured, NS is the nonspecific binding signal, and MS is the maximum binding signal observed with btVEGF165 without tested compounds.
Cell culture: HUVEC were cultured in a medium supplemented with fetal bovine serum (FBS, 2 %), l-glutamine (10 mm), heparin sulfate (0.75 U mL−1), VEGF (15 ng mL−1), EGF (5 ng mL−1), FGF2 (5 ng mL−1), IGF-I (15 ng mL−1), and ascorbic acid (50 μg mL−1) at 37 °C, in a moist atmosphere of 5 % CO2. HUVEC were used for the experiment between passages 2 to 10.
In vitro morphogenesis assays on HUVEC: To assess antiangiogenic activity, HUVEC (2.5×104 cells) were seeded in a 24-well plate coated with 100 μL/well of Matrigel previously polymerized for 1 h at 37 °C. Cells were then incubated for 5 h at 37 °C in a moist atmosphere of 5 % CO2 without or with tested compounds under either resting (cell cultured in EndoGRO medium alone, without FBS and all the growth factors) or stimulated conditions (addition of VEGF, 10 ng mL−1). Network formation was evaluated by phase-contrast microscopy using a fluorescence microscope (Axiovert 40CFL, Carl Zeiss S.p.A., Milan, Italy). Network formation was finally quantified in terms of total length of the branches. For the purpose of the analysis, open ramifications were considered as branches. The total branch length (pixels) was quantified using the ImageJ image analysis software (http://rsbweb.nih.gov/ij/).
Statistical analysis: Data are shown as means±standard deviation (S.D.) unless indicated otherwise. Statistical significance of the differences was assessed by two-tailed Student's t test for paired data. Calculations were performed using a commercial software (GraphPad Prism version 5.00 for Windows, GraphPad Software, La Jolla, USA, www.graphpad.com).
Acknowledgments
S. Z. and A. D. C. thank the University of Milan for their PhD fellowship. The authors also gratefully acknowledge “Ministero dell′Università e della Ricerca” (PRIN project 2010NRREPL: Synthesis and biomedical applications of tumor-targeting peptidomimetics) and Fondazione CARIPLO (RE-D DRUG TRAIN) for financial support. The valuable collaboration of Monica Pinoli (PhD Course in Clinical Medicine and Medical Humanities, Center for Research in Medical Pharmacology, University of Insubria) is gratefully acknowledged.
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
References
- 1a.Mizejewski GJ. Proc. Soc. Exp. Biol. Med. 1999;222:124–138. doi: 10.1177/153537029922200203. [DOI] [PubMed] [Google Scholar]
- 1b.Danhier F, Le Breton A, Préat V. Mol. Pharm. 2012;9:2961–2973. doi: 10.1021/mp3002733. [DOI] [PubMed] [Google Scholar]
- 1c.Johannessen TC, Wagner M, Straume O, Bjerkvig R, Eikesdal HP. Expert Opin. Ther. Targets. 2013;17:7–20. doi: 10.1517/14728222.2013.730522. [DOI] [PubMed] [Google Scholar]
- 2a.Barczyk M, Carracedo S, Gullberg D. Cell Tissue Res. 2010;339:269–280. doi: 10.1007/s00441-009-0834-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b.Hynes RO. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 2c.Shimaoka M, Springer TA. Nat. Rev. Drug Discovery. 2003;2:703–716. doi: 10.1038/nrd1174. [DOI] [PubMed] [Google Scholar]
- 2d.Rathinam R, Alahari SK. Cancer Metastasis Rev. 2010;29:223–237. doi: 10.1007/s10555-010-9211-x. [DOI] [PubMed] [Google Scholar]
- Plow EF, Haas TA, Zhang L, Loftus J, Smith JW. J. Biol. Chem. 2000;275:21785–21788. doi: 10.1074/jbc.R000003200. [DOI] [PubMed] [Google Scholar]
- 4a.Avraamides CJ, Garmy-Susini B, Varner JA. Nat. Rev. Cancer. 2008;8:604–617. doi: 10.1038/nrc2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b.Paolillo M, Russo MA, Serra M, Colombo L, Schinelli S. Mini Rev. Med. Chem. 2009;9:1439–1446. doi: 10.2174/138955709789957404. [DOI] [PubMed] [Google Scholar]
- 4c.Auzzas L, Zanardi F, Battistini L, Burreddu P, Carta P, Rassu G, Curti C, Casiraghi G. Curr. Med. Chem. 2010;17:1255–1299. doi: 10.2174/092986710790936301. [DOI] [PubMed] [Google Scholar]
- 5a.Gottschalk KE, Kessler H. Angew. Chem. Int. Ed. 41:3767–3774. doi: 10.1002/1521-3773(20021018)41:20<3767::AID-ANIE3767>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002;114 [Google Scholar]
- 5b.Xiong J-P, Stehle T, Zhang R, Joachimiak A, Frech M, Goodman SL, Arnaout MA. Science. 2002;296:151–155. doi: 10.1126/science.1069040. [DOI] [PubMed] [Google Scholar]
- 6a.da Ressurreição ASM, Vidu A, Civera M, Belvisi L, Potenza D, Manzoni L, Ongeri S, Gennari C, Piarulli U. Chem. Eur. J. 2009;15:12184–12188. doi: 10.1002/chem.200902398. [DOI] [PubMed] [Google Scholar]
- 6b.Marchini M, Mingozzi M, Colombo R, Guzzetti I, Belvisi L, Vasile F, Potenza D, Piarulli U, Arosio D, Gennari C. Chem. Eur. J. 2012;18:6195–6207. doi: 10.1002/chem.201200457. [DOI] [PubMed] [Google Scholar]
- Vascular endothelial growth factor (VEGF), epidermal growth factor (EGF), fibroblast growth factor (FGF) and insulin-like growth factor-I (IGF-I) were used as pro-angiogenic factors (see Ref. [8])
- Fanelli R, Schembri L, Piarulli U, Pinoli M, Rasini E, Paolillo M, Galiazzo MC, Cosentino M, Marino F. Vascular Cell. 2014;6:11. doi: 10.1186/2045-824X-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panzeri S, Zanella S, Arosio D, Vahdati L, Dal Corso A, Pignataro L, Paolillo M, Schinelli S, Belvisi L, Gennari C, Piarulli U. Chem. Eur. J. 2015;21:6265–6271. doi: 10.1002/chem.201406567. Compound 1 displayed inhibitory effects on the FAK/Akt integrin activated transduction pathway and on integrin mediated cell infiltration processes, and induced apoptosis after 72 h, in human U373 glioblastoma cells, see: [DOI] [PubMed] [Google Scholar]
- 10a.Eliceiri BP. Circ. Res. 2001;89:1104–1110. doi: 10.1161/hh2401.101084. [DOI] [PubMed] [Google Scholar]
- 10b.Masson-Gadais B, Houle F, Laferrière J, Huot J. Cell Stress Chaperones. 2003;8:37–52. doi: 10.1379/1466-1268(2003)8<37:ivrfva>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10c.De S, Chen J, Narizhneva NV, Heston W, Brainard J, Sage EH, Byzova TV. J. Biol. Chem. 2003;278:39044–39050. doi: 10.1074/jbc.M304494200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10d.Mahabeleshwar GH, Feng W, Phillips DR, Byzova TV. J. Exp. Med. 2006;203:2495–2507. doi: 10.1084/jem.20060807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10e.Mahabeleshwar GH, Feng W, Reddy K, Plow EF, Byzova TV. Circ. Res. 2007;101:570–580. doi: 10.1161/CIRCRESAHA.107.155655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10f.Mahabeleshwar GH, Chen J, Feng W, Somanath PR, Razorenova OV, Byzova TV. Cell Cycle. 2008;7:335–347. doi: 10.4161/cc.7.3.5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10g.Desgrosellier JS, Cheresh DA. Nat. Rev. Cancer. 2010;10:9–22. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10h.West XZ, Meller N, Malinin NL, Deshmukh L, Meller J, Mahabeleshwar GH, Weber ME, Kerr BA, Vinogradova O, Byzova TV. PLOS One. 2012;7:31071. doi: 10.1371/journal.pone.0031071. e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11a.Ferrara N, Gerber HP, LeCouter J. Nat. Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 11b.Hoeben A, Landuyt B, Highley MS, Wildiers H, van Oosterom A, De Bruijin E. Pharmacol. Rev. 2004;56:549–580. doi: 10.1124/pr.56.4.3. [DOI] [PubMed] [Google Scholar]
- 11c.Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. Nat. Rev. Mol. Cell Biol. 2006;7:359–371. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- 11d.Carmeliet P, Jain RK. Nat. Rev. Drug Discovery. 2011;10:417–427. doi: 10.1038/nrd3455. [DOI] [PubMed] [Google Scholar]
- 11e.Shibuya M. J. Biochem. 2013;153:13–19. doi: 10.1093/jb/mvs136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musumeci F, Radi M, Brullo C, Schenone S. J. Med. Chem. 2012;55:10797–10822. doi: 10.1021/jm301085w. [DOI] [PubMed] [Google Scholar]
- Udugamasooriya DG, Dineen SP, Brekken RA, Kodadek T. J. Am. Chem. Soc. 2008;130:5744–5752. doi: 10.1021/ja711193x. [DOI] [PubMed] [Google Scholar]
- 14a.Basile A, Del Gatto A, Diana D, Di Stasi R, Falco A, Festa M, Rosati A, Barbieri A, Franco R, Arra C, Pedone C, Fattorusso R, Turco MC, D′Andrea LD. J. Med. Chem. 2011;54:1391–1400. doi: 10.1021/jm101435r. [DOI] [PubMed] [Google Scholar]
- 14b.Diana D, Di Stasi R, De Rosa L, Isernia C, D′Andrea L, Fattorusso R. J. Pept. Sci. 2013;19:214–219. doi: 10.1002/psc.2480. [DOI] [PubMed] [Google Scholar]
- García-Aranda MI, González-López S, Santiveri CM, Gagey-Eilstein N, Reille-Seroussi M, Martín-Martínez M, Inguimbert N, Vidal M, García-López MT, Jiménez MA, González-Muñiza R, Pérez de Vega M. Org. Biomol. Chem. 2013;11:1896–1905. doi: 10.1039/c3ob27312a. [DOI] [PubMed] [Google Scholar]
- Zilberberg L, Shinkaruk S, Lequin O, Rousseau B, Hagedorn M, Costa F, Caronzolo D, Balke M, Canron X, Convert O, Laïn G, Gionnet K, Gonçalvès M, Bayle M, Bello L, Chassaing G, Deleris G, Bikfalvi A. J. Biol. Chem. 2003;278:35564–35573. doi: 10.1074/jbc.M304435200. [DOI] [PubMed] [Google Scholar]
- 17a.Papo N, Silverman AP, Lahti JL, Cochran JR. Proc. Natl. Acad. Sci. USA. 2011;108:14067–14072. doi: 10.1073/pnas.1016635108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17b.Ma Y, Liang S, Guo J, Guo R, Wang H. J. Labelled Compd. Radiopharm. 2014;57:627–631. doi: 10.1002/jlcr.3222. Wang and co-workers recently developed a small molecule 18V3 F labeled RGD-peptide(ATWLPPR) dual ligand targeting integrin α β and neuropilin-1 for tumor imaging, see: and references therein. [DOI] [PubMed] [Google Scholar]
- 18a.Colombo R, Mingozzi M, Belvisi L, Arosio D, Piarulli U, Carenini N, Perego P, Zaffaroni N, De Cesare M, Castiglioni V, Scanziani E, Gennari C. J. Med. Chem. 2012;55:10460–10474. doi: 10.1021/jm301058f. [DOI] [PubMed] [Google Scholar]
- 18b.Dal Corso A, Caruso M, Belvisi L, Arosio D, Piarulli U, Albanese C, Gasparri F, Marsiglio A, Sola F, Troiani S, Valsasina B, Pignataro L, Donati D, Gennari C. Chem. Eur. J. 2015;21:6921–6929. doi: 10.1002/chem.201500158. [DOI] [PubMed] [Google Scholar]
- Mingozzi M, Manzoni L, Arosio D, Dal Corso A, Manzotti M, Innamorati F, Pignataro L, Lecis D, Delia D, Seneci P, Gennari C. Org. Biomol. Chem. 2014;12:3288–3302. doi: 10.1039/c4ob00207e. [DOI] [PubMed] [Google Scholar]
- Acetylation of Lys13 side chain did not substantially affect the conformational and biological properties of compound 43 compared with the parent peptide (see the CD analysis and the morphogenesis assay sections)
- Most commercially available medium- to long-chain PEG linkers are not monodisperse. In order to obtain a structurally well-defined molecular object rather than a statistical mixture of products differing in linker length, we selected one of the longest PEG linkers characterized by a monodisperse profile.
- Toniolo C, Formaggio F, Woody RW. Polavarapu PL. Nakanishi K. Woody RW. In: Comprehensive Chiroptical Spectroscopy: Applications in Stereochemical Analysis of Synthetic Compounds, Natural Products, and Biomolecules, Vol. 2. Berova N, editor; Hoboken: John Wiley & Sons, Inc; 2012. pp. 499–544. (Eds.:, pp. . [Google Scholar]
- Haubner R, Schmitt W, Höllzemann G, Goodman SL, Jonczyk A, Kessler H. J. Am. Chem. Soc. 1996;118:7881–7891. [Google Scholar]
- Goncalves V, Gautier B, Garbay C, Vidal M, Inguimbert N. Anal. Biochem. 2007;366:108–110. doi: 10.1016/j.ab.2007.03.027. [DOI] [PubMed] [Google Scholar]
- Hynes RO. J. Thromb. Haemostasis. 2007;5:32–40. doi: 10.1111/j.1538-7836.2007.02569.x. [DOI] [PubMed] [Google Scholar]
- Baranska P, Jerczynska H, Pawlowska Z, Koziolkiewicz W, Cierniewski CS. Cancer Genomics Proteomics. 2005;2:265–270. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary