

Abstract
Dr. David Barker first popularized the concept of fetal origins of adult disease (FOAD). Since its inception, FOAD has received considerable attention. The FOAD hypothesis holds that events during early development have a profound impact on one’s risk for development of future adult disease. Low birth weight, a surrogate marker of poor fetal growth and nutrition, is linked to coronary artery disease, hypertension, obesity, and insulin resistance. Clues originally arose from large 20th century, European birth registries. Today, large, diverse human cohorts and various animal models have extensively replicated these original observations. This review will focus on the pathogenesis related to FOAD and examines Dr. David Barker’s landmark studies, along with additional human and animal model data. Implications of the FOAD extend beyond the low birth weight population and include babies exposed to stress, both nutritional and non-nutritional, during different critical periods of development, which ultimately result in a disease state. By understanding FOAD, health care professionals and policy makers will make this issue a high healthcare priority and implement preventative measures and treatment for those at higher risk for chronic diseases.
Introduction
David Barker’s keen observations have been popularized as the “Barker hypothesis,” or “Fetal Origins of Adult Disease” (FOAD). It was his group that noted that low birth weight (LBW) serves as proxy not just for fetal, but also adult health. Today, LBW is associated with a host of chronic diseases ranging from coronary artery disease (CAD), Type II diabetes mellitus (T2DM), cancer, and osteoporosis to various psychiatric illnesses (See Table I.).1–7 FOAD is based on the premise of “developmental plasticity”—a single genotype, influenced by specific intrauterine events, has the capability to produce different phenotypes. This theory relies on the fact that there exist specific developmental periods whereby an organism is “plastic” or “sensitive” to its environment. Diversity is maximized to provide the best fit between phenotype and environment. For example, when faced with the adversity of malnutrition, a fetus will undergo remodeling thereby altering structure and function of various organs to preserve neurodevelopment and promote survival. These adaptations prepare the fetus for extrauterine life where additional stressors may be encountered. It must be recognized, however, that over time, this evolutionary advantage of “plasticity” is lost, and one’s response to environmental or pathological challenges becomes constrained. This phenomenon, known as “programming,” refers to the fact that stimuli, when applied during early development, generates permanent changes that persist throughout one’s lifespan. Programming is not just limited to the in-utero environment, but extends into childhood, where different organs and systems continue to adapt to various cues.
Table I.
Chronic diseases associated with the Fetal Origins of Adult Disease (FOAD) hypothesis
| Chronic diseases attributed to “Developmental Origins” |
| Diabetes Mellitus |
| Obesity |
| Dyslipidemia |
| Hypertension |
| Coronary Artery Disease |
| Stroke |
| Kidney Failure - glomerulosclerosis |
| Liver Failure – cholestasis, steatosis |
| Lung Abnormalities – BPD, reactive airway disease |
| Immune Dysfunction |
| Reduced Bone Mass |
| Alzheimer’s Disease |
| Depression, Anxiety, Bipolar Disorder, Schizophrenia |
| Cancer |
The FOAD theory was originally supported by large birth registries and human cohorts where gestating women and their offspring faced severe malnutrition in the form of famines.1,4,8,9 These large registries recorded the birth history of men and women, and these subjects were then identified later in life. This allowed investigators to correlate birth weight and childhood growth with adult onset diseases. These findings were found to be independent of confounders such as tobacco use, diet, exercise, socioeconomic status, and family history. Although numerous epidemiological studies across various cultures and ethnicities support the link between LBW and future adult disease, LBW is not necessarily a prerequisite.10–13 Those with “normal” birth weights, or appropriate for gestational age (AGA), may still be at risk depending on the type, timing, and duration of the original insult. Moreover, insults are not limited to malnutrition. Famines or severe calorie restriction merely provide a model for other afflictions. Alterations in diet composition, inflammation, infection, glucocorticoids, hypoxia, stress, and toxins also play a vital role in shaping the adult phenotype.14–20 Although the literature tends to focus on the LBW, or small for gestational age (SGA) baby, special consideration must be given to the stressed AGA, large for gestational age (LGA), and premature neonate. And lastly, the transgenerational and socioeconomic implications of FOAD have far-reaching repercussions that cannot be underestimated; further research is still needed to unravel its complexities.21–23
The aim of this review is to provide the general clinician with a foundation to better understand how nutritional and non-nutritional perturbations result in chronic disease. Since Barker’s original observations, numerous epidemiological and human studies, further reinforced by animal models, have provided a plethora of support.12,19, 24–31 Mice, rats, sheep, and monkeys have been subjected to adverse conditions during gestation to mimic the human condition. Models include global calorie or specific macronutrient or micronutrient restriction, hypoxic-ischemia induced by uterine artery ligation, and glucocorticoid exposure. The data, as a whole, force the clinician and researcher to question and discover how developmental stressors permanently alter structure, metabolism, and genetic expression resulting in cognitive, behavioral, and body composition changes ultimately leading to future disease. The literature points to opportunities for public health prevention, future pharmaceutical therapies, and innovative alternative nutritional/environmental strategies that could maximize population based well-being.
Background
The Helsinki and Hertfordshire cohorts from the1930s and 1940s, which comprised over 20,000 subjects collectively, linked poor fetal growth with CAD, hypertension, and insulin resistance in adult men and women.1,2,9 Differential mortality rates have been associated with variation in birth weights (Fig 1). Specifically, a LBW in either gender or a documented lower weight for males at 1 year of age was associated with an increased death rate from CAD. A similar trend was noted in both sexes for insulin resistance.2 Moreover, these effects have been noted to extend beyond the first generation of offspring (F1 generation), with both short- and long-term health implications in future generations.21–23
Figure 1.
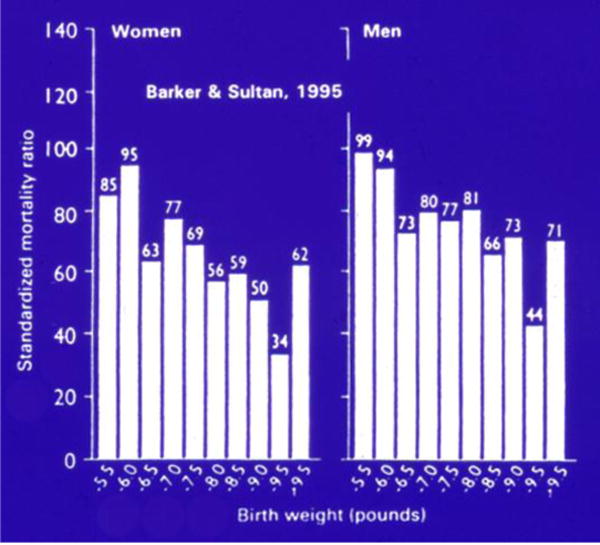
Standardized mortality ratio by gender based on birth weight in pounds. (Reprinted with permission from Barker DJ, Sultan HY. Fetal programming of human disease. In: Hanson M, et al, editors. Fetus and neonate physiology and clinical applications: growth. Vol. 3. Cambridge: Cambridge University Press; 1995.)
During the relatively short Dutch famine of 1944–1945, the daily nutritional intake of pregnant women was reduced to approximately 400–1000 calories. The timing during pregnancy of this famine (early, mid, or late gestation) was linked to differential birth weights and subsequent development of adult disease. Infants who were subjected to mid or late gestation calorie restriction were lighter, while those who endured the famine in early gestation had normal birth weights. In addition, adults whose mothers were exposed to the famine during mid or late gestation demonstrated reduced glucose tolerance, while those whose mothers were exposed to the famine during early gestation revealed a more atherogenic lipid profile and higher body mass index (BMI).4,9 Thus, although birth weight in some cases serves as a surrogate marker for the development of adult disease, it is the in utero environment that sets the trajectory for the subsequent acquisition of childhood or adult diseases.
Conversely, the Leningrad famine spanned more than 800 days and provided yet another opportunity to study how severe malnutrition during pregnancy and early infancy, both periods of critical development, affect adult disease. During this siege, fetuses subjected to severe calorie restriction also experienced malnutrition during infancy. However, these subjects, unlike their Dutch counterparts, did not demonstrate increased rates of insulin resistance, dyslipidemia, hypertension, or CAD.8 These two famines, although similar at first glance, differed in the duration and severity of the malnutrition the population endured. These two infamous natural disasters, with effects documented in large European databases, provided David Barker and others with clues to what subsequently was described by the term FOAD, or known as the “Barker hypothesis.” Even today, despite a plethora of human and animal data, scientists continue to debate the mechanistic link(s) between abnormal growth and adult disease. In addition, in humans, the genetic make-up and environment cannot be dismissed, and it has been a challenge to separate these influences from each other.
“The Mismatch Concept”
The contrast between two natural disasters, namely the Dutch famine and Leningrad siege, highlights the “mismatch concept.” Fetal survivors of the Leningrad siege developed a “thrifty phenotype” that served them well in their extrauterine life.32 In other words, the superimposed stress of malnutrition in their intrauterine environment was well-matched to the malnutrition experienced outside the womb, and may have offered a form of “protection” from future adult disease. The Dutch, by contrast, exhibited “catch-up” or compensatory growth during infancy. Their intrauterine environment which supported short-term survival was ill matched with their subsequent extrauterine environment. Thus, the Dutch fetuses paid the price for their intrauterine adaptation—“there is no short term gain without long term pain.” In other words, when there is a “match” between the predicted and the actual environment, survival is maximized. Conversely, when the two environments are “mismatched” a predisposition to disease may result; an individual’s full potential may not be realized because of maladaptation to the new disparate extrauterine environment predisposing to disease states (Fig 2).
Figure 2.
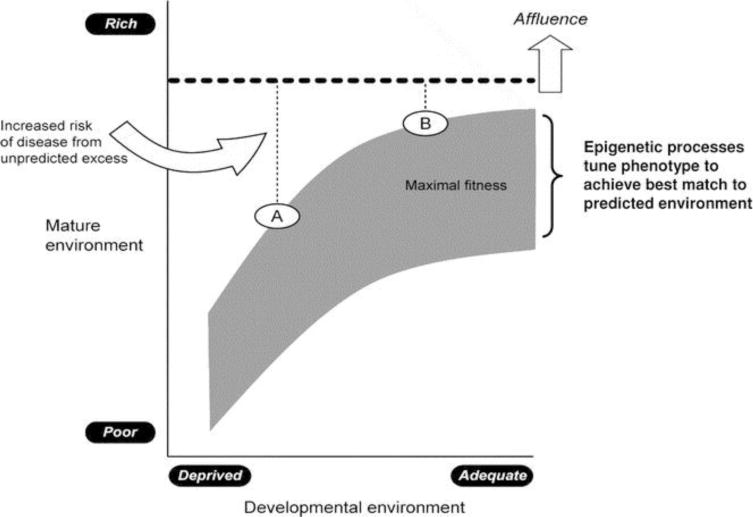
The “thrifty hypothesis.” Schematic representation of risk of disease based on the “mismatch concept,” which focuses on the degree of disparity between the intra- and extrauterine environment. During the period of developmental plasticity, epigenetic processes are thought to alter gene expression in the fetus based on maternal environmental cues to produce phenotypes in the offspring best suited for the environment. Greater mismatch between pre- and post birth environments results in greater risk of disease (e.g., development in a poor environment followed by transition to an affluent Western lifestyle with unpredicted excessive richness of high calorically dense food with sedentary lifestyle as seen in A versus B). (Reprinted with permission from Godfrey KM, et al. Epigenetic mechanisms and the mismatch concept of the developmental origins of the health and disease, Pediatr Res 61:5R, 2007).
This concept of “mismatch,” and its subsequent consequences, also applies to contemporary times. The World Health Organization estimates that over 30 million infants are born LBW, defined as a weight less than 2.5 kg.33 In addition, many poorly resourced countries are being industrialized or swept over by the “Mc Donald’s” fast food phenomenon. This “mismatch” is not confined to poor resourced nations; it also plagues well resourced societies with access to modern health care and high standards of living. High-calorie and high-fat diets coupled with limited active play and excessive media viewing have become the norm in Western nations. In the USA, obesity has more than doubled among children and adolescents. In comparing the two periods 1976–1980 and 2003–2006, the percentage of obese adolescents increased from 5% to 17.6%.34 Insulin resistance is intimately linked to T2DM, CAD, hypertension, and dyslipidemia. T2DM is now a global epidemic affecting over 170 million people; by 2030 this number is expected to double.35
Considering these statistics, one may ask—what developmental “hits” are considered detrimental? And during which time periods? When is too much truly too much? And practically speaking, how should the pediatrician, obstetrician, and neonatal-perinatal subspecialist promote growth during pregnancy and the various postnatal periods (neonatal, infancy, and childhood)? How should clinicians monitor the fetus, and later the infant, child, and adult when an adverse environment is experienced early in gestation? Can these negative outcomes be prevented or reversed?
Catch-up Growth
Today, numerous human and animal studies have revealed that LBW followed by exponential childhood growth increases the risk for the metabolic syndrome, which is characterized by obesity, insulin resistance, dyslipidemia, and hypertension.2,32,36,37 This tendency to “catch-up” in growth reflects the body’s natural response to nutrient deprivation. Moreover, both the parents and clinician may not understand the dangers of rapid weight gain and may actually promote it. Catch-up growth, however, is associated with increased visceral adiposity.38,39 Recent findings in a group of racially diverse American children mimicked those of the 1930–1940s’ English cohort revealing an association between the tendency to store fat intra-abdominally and LBW. In this study specific markers of central adiposity, as measured by central-to-peripheral and subscapular-to-tricep skinfolds, were increased in LBW children as early as 5–11 years of age.38 Similarly in a cohort of Hispanic and non-Hispanic Caucasians, LBW was associated with elevated fasting insulin levels and truncal obesity. For each tertile decrease in birth weight, the risk for “insulin resistance syndrome” increased 1.72.10
Studies of children with T2DM have revealed that many of them were characterized as being small at birth, but during childhood and adolescence they began to cross growth percentiles. The odds ratio for developing T2DM was 1.38 for each 1 kg decrease in birth weight, and 1.39 for each increase in weight standard deviation between 7 and 15 years of age.37 Therefore, the thin toddler who begins to rapidly gain weight, or finally “catches-up” to his peers, is at the same risk, if not greater, for developing T2DM compared to his overweight age-matched peer.
Anthropometric data for over 1,400 men and women from Delhi, India, was collected during the first 3 decades of their life from 1970 to 2000. This Indian study conducted during modern times is reminiscent of the Dutch famine. The authors describe this period in India as a “transition” to a more Western lifestyle—a lifestyle marked by increased food availability and reduced physical activity. Results of this study revealed that those individuals with increased insulin resistance were more likely to be small at birth, grow slowly during the first couple of years in life, and then develop “an adiposity rebound”.12 These children however were not described as overweight or obese with this “rebound” growth. Instead, they were described as rapidly gaining body mass. This “catch-up fat” during childhood and early adulthood occurs at a disproportionately faster rate in comparison with lean body mass, and is one of the main promoters of early insulin resistance.
This “nutritional transition” is not solely limited to India but has occurred on all continents. The quantity and quality of the world’s food supply varies from country to country, and even neighborhood to neighborhood; while some individuals face starvation, others consume high-calorie, high-fat meals that lack significant nutritional value, which in itself is a different form of starvation.
The tendency for the LBW infant to store fat, particularly in the abdominal compartment, has received considerable attention.10,38,40,41 LBW infants are known to have adipocytes that demonstrate increased numbers of insulin receptors, glucose uptake, and basal and insulin-stimulated insulin-receptor substrate 1 (IRS-1)-associated phosphatidylinositiol 3-kinase (PI3K).42,43 Moreover, these adipocytes are resistant to insulin and demonstrate serine phosphorylation of IRS-1, resulting in a state of anti-lipolysis.43 In addition, increased central adiposity is associated with increased free fatty acids, which stimulate the production of cholesterol and glucose, which in turn decrease insulin sensitivity.24 As fat deposition progresses in the liver, permanent structural hepatic changes occur.44
This molecular “switch” in insulin signaling “protects” the LBW fetus and neonate by promoting storage of fat, a calorically dense nutrient. However, over time this altered metabolism sets the stage for the development of disease. Considering its impact on childhood growth, a clinician may then question whether replacing this quick “rebound” with slow steady measured growth could result in increased lean body mass rather than fat accumulation. Moreover, could any of these cellular aberrations resulting in chronic disease be avoided?
Biological Basis
Growth is largely determined by a mother’s ability to nourish her fetus. The provision of this nourishment depends on the maternal diet, the placenta’s ability to supply nutrients (amino acids, glucose, fat, oxygen, and growth-stimulating hormones), and the mother’s own in utero experience.21–23 In other words, the effect of FOAD extends beyond the first (F1) generation. Considering the medical and socio-economic implications of programming, it is critical that all health professionals promote healthy pregnancies to maximize each child’s potential and growth trajectory.
So why would growth or other insults in early life alter one’s disease potential? First, cell number and growth are exquisitely sensitive to their metabolic and hormonal milieu. Undernourished animals and babies have reduced renal volumes and nephrons, which can result in hypertension. Autopsies of hypertensive humans who died in motor vehicle accidents have demonstrated reduced numbers of, but hypertrophic, glomeruli.45–47 Similar to humans, growth-restricted animals have decreased numbers of glomeruli and develop chronic renal disease later on in life. Moreover, measurements of mammalian target of rapamycin, a nutrient sensing biomarker, are reduced in growth-restricted kidneys.48 Second, any stress can alter hormonal concentrations, hormone receptors, and hormonal responses. Receptors can be down- or up-regulated and mutations may result in structural transformations. Hormonal responses can be blunted, or exaggerated.
Consistent with this paradigm is the SGA child who later develops T2DM. These individuals have a reduced number and function of pancreatic β-cells resulting in decreased insulin production and consequently a high glucose/insulin ratio. This outcome leads to abnormal skeletal muscle, liver, and adipocyte insulin signaling and/or glucose production with each of these perturbations resulting in insulin resistance. The same is true with leptin, a critical hormone secreted by adipocytes. Leptin acts on the central nervous system to decrease food intake and increase energy expenditure. The SGA neonate has decreased leptin concentrations at birth, but with increased adiposity becomes hyperleptinemic during the period of “catch-up” exponential growth. This hyperleptinemic state is associated with decreased and/or resistant hypothalamic leptin receptors, which result in hyperphagia and decreased energy expenditure. Dysregulated insulin and leptin serve as two examples of how hormonal perturbations predispose the SGA infant to T2DM and other co-morbidities.
Small for Gestational Age Infant
It is important to acknowledge the spectrum of LBW, which includes SGA, intrauterine growth restriction (IUGR), and very low birth weight (VLBW) infants, some who are not only born prematurely but at times also with superimposed IUGR. While SGA is defined as a birth weight or birth length less than the 10th percentile with respect to gestational age, IUGR refers to an infant who does not reach his or her predetermined genetic potential because of some pathologic insult. This insult can be categorized as maternal, placental, or fetal, with the most common etiology thought to be placental insufficiency. Although IUGR infants may be SGA, all SGA infants are not necessarily IUGR.
Not surprisingly, prenatal and postnatal periods are exquisitely sensitive to any factor that can affect growth as these are periods of exponential cell replication, division, and growth. Infants born SGA are at increased risk for developing obesity, T2DM, CAD, hypertension, kidney disease, premature pubarche, and Polycystic Syndrome (PCOS), dyslipidemia, short stature, and osteoporosis.1–4,13,32,37,45,46,49–50 Many of these phenotypic changes are known to be secondary to or associated with insulin resistance.
Height
Although fetal growth is usually discussed in terms of birth weight, it is important not to dismiss the aspect of birth length. Infants with low birth length, LBW, or premature birth are more likely to be short adults.49–52 Most neonates who fall 2 standard deviations below the growth curve for length, however, are likely to “catch-up” to their peers during childhood. At 1 year and 18 years of age, respectively, only 13.4% and 7.9% of short babies remain stunted.53 Those that remain short often are referred after the age of 2 to the pediatric endocrinologist for growth hormone therapy or at an older age for evaluation of the metabolic syndrome because of their increased risk of obesity.
It is difficult to separate out the individual contributions of low birth length, LBW, prematurity, and other important factors such as parental height on the individual’s ultimate height and the associated complications, such as obesity and T2DM. Each characteristic may have an additive, synergistic, or independent effect. When only females were examined, being short for gestational age was associated with a fivefold increase in short adult stature, while being SGA was associated with only a twofold increase. Interestingly, females who are short at birth, not SGA, are at higher risk for obesity.49 At the other extreme, larger and longer babies are even more likely to become heavier adults in comparison with light and short babies.54 As is seen in infants born to mothers with gestational diabetes (GDM) secondary to maternal hyperglycemia, the GDM fetus becomes hyperinsulinemic, resulting in accelerated in utero growth. For every 1 kg increase in birth weight, full-term babies have a 50% increase in adolescent obesity.55 Short and large babies, both at risk for insulin resistance, are two distinct, separate populations, and are most likely the result of two different programming mechanisms.
Bone growth affects not only one’s ultimate height and risk for obesity, but also bone disease. Osteoporosis is linked to fractures, which are linked to a high risk for morbidity and mortality later in life. In a study of a Finnish cohort of patients admitted to the hospital for fractures, low birth length and slow growth were correlated with increased fracture risk during adult life.56 SGA status and slow growth during the first year of life predict decreased adult bone mass even after adjustment for other predictors of osteoporosis and osteopenia.6 Studies of twins have proven to be very useful in supporting the FOAD hypothesis. Twin studies are valuable as they help correct for well-known genetic and environmental confounders. The dizygotic or monozygotic co-twin with LBW is more likely to have a lower bone mineral density at specific sites.57 This data, along with results from other twin studies, suggest that even in genetically identical monozygotic twins, the co-twin with the lower birth weight is predisposed to lower adult bone mass and to developing the metabolic syndrome.58
Growth hormone, other insulin-like growth factors, and vitamin D are among many factors that influence a child’s growth and bone mass. The growth hormone/insulin-like growth factor-1 axis and receptors are most likely altered in individuals who fail to realize their growth potential. Adults with decreased bone mineral density are more likely to have decreased growth hormone and to have been smaller children.59 Other investigators have explored this area even further and have discovered a strong interaction between SGA, poor adult bone mass, and certain single nucleotide polymorphisms (SNPs) in the growth hormone, vitamin D, and calcium-sensing receptor.60–64 In a SNP analysis, the insulin-like growth factor-1 receptor was altered in 2 short children with low birth weights and postnatal growth restriction.64 Reduced insulin-like growth factor receptors impact not only a child’s ultimate height, but also the child’s ability to maintain glucose homeostasis and sustain good brain growth. In summary, like weight, length at birth and height serve as predictors for future disease.
Insulin Resistance/Diabetes
While LBW is undoubtedly associated with insulin resistance, the search for a mechanistic link continues. Many investigators suggest that the pancreas is “programmed” resulting in decreased β-cell mass and function and over time skeletal muscle becomes insulin resistant. Some authors, by contrast, have failed to replicate or generate data to support this theory and believe that insulin resistance, not production, is the primary culprit.65, 66 In their opinion, once insulin resistance sets in, the pancreas increasingly is stressed to produce more insulin and over time responds to its overwhelming exhaustion with progressive failure of its β-cells.
Pancreas
In support of the former theory, many studies have demonstrated decreased insulin secretion and normal insulin sensitivity in young SGA adults. In one study of adults born with LBW, insulin secretion was reduced by 30%, while insulin sensitivity was determined to be normal.27 In examining this phenomenon more closely, the IUGR population was noted to have pancreatic dysfunction even before birth; this was evidenced by decreased insulin, glucose, insulin/glucose ratios, and β-cell number and function.67–69 Interestingly, in some studies, this reduction in β-cell function was initially accompanied by increased insulin sensitivity.70 These changes persisted after birth and were accompanied by further alterations. When rats are subjected to calorie restriction in-utero, β-cell mass is initially reduced by approximately 35%. When caloric restriction extends into the neonatal period, impaired β-cell development persists with a 40–66% reduction. Later as adults, these animals are described as glucose intolerant.68,71 Moreover, pancreatic duodenal homeobox-1 (Pdx-1), a well-known transcription factor controlling pancreatic development, is reduced. At 28 days of life, insulin secretion and Pdx-1 protein expression is reduced when compared with controls.30
In another commonly used IUGR model, the uterine artery of pregnant rats is ligated, resulting in a severe, abrupt reduction of uteroplacental blood flow. These IUGR rats are growth-restricted at birth but later demonstrate accelerated weight gain, surpassing their age-matched controls. As they age, the rats become insulin resistant and glucose intolerant. Studies have demonstrated that early on, their β-cell mass is equivalent to that of controls. However by 26 weeks, the rats pancreatic mass is notably decreased to approximately 50% of their control counterparts. Using this same model, deterioration in the rats’ β-cell function is accompanied by mitochondrial dysfunction, as evidenced by decreased adenosine triphosphate (ATP) production, increased reactive oxygen species, and mitochondrial point mutations. In addition, all of these markers demonstrated a steady increase over the first 15 weeks of life.19 When faced with undernutrition, cell division is slowed (as evidenced by decreased Pdx-1 and β-cell-mass) and cell function is altered (as evidenced by decreased hormonal supply and cellular damage).
An alternative hypothesis to FOAD is the “fetal insulin hypothesis.” T2DM is known to have a strong genetic component. According to this theory, because insulin is key for fetal growth, any genetic variant that impairs insulin secretion may reduce birth weight and simultaneously result in T2DM. In other words, the genotype, not LBW, predisposes one to T2DM. Supporting this theory, Freathy et al, using SNPs, demonstrated that alleles for increased T2DM risk that reside at two separate loci, CDKAL1 and HHEX-IDE, also were associated with LBW.72
Although one cannot ignore the genetic component to T2DM, SGA humans and growth restricted animal studies clearly have served as models for FOAD and insulin resistance. Evidence is now accumulating about specific nutritional and non-nutritional alterations that may or may not alter growth and result in a similar pancreatic phenotype. This is the case with essential amino acids, glucose, and micronutrient deficiencies.14,73,74 Thus, clinicians must be aware that perturbations, small or large, in the fetal and postnatal environment can have long-term consequences.
Skeletal muscle, liver, and fat
Although the debate still continues whether the pancreas or skeletal muscle is the key instigator behind the T2DM FOAD theory, evidence from both camps is convincing. Taken together, human and animal studies highlight the importance of crosstalk between pancreas, skeletal muscle, liver, adipose tissue, and brain. Skeletal muscle, one of the principal sites of action for glucose and insulin, is prone to dysregulation in T2DM. LBW infants have decreased lean body mass and increased visceral adiposity.75–77 This “sarcopenic obesity” is considered one of the major culprits in the development of T2DM.39,75–77 Fat tends to accumulate viscerally and in ectopic sites, such as the liver. When children with nonalcoholic fatty liver disease were examined, close to 40% of the population was born SGA and this subset also was more likely to progress to non-alcoholic steatohepatitis.44 In addition to an increased risk for hepatic disease, it is hypothesized that LBW infants and later adults have increased hepatic glucose production. In fact, in the uterine artery ligation model, IUGR rats exhibited increased hepatic glucose production as evidenced by impaired oxidation of pyruvate and increased phosphoenolpyruvate carboxykinase before the onset of T2DM.78
Like many of the features in FOAD, there is subtle evidence of future pathology before clinical presentation. With respect to skeletal muscle, programming affects multiple areas—skeletal muscle development and/or fiber composition, glucose uptake, downstream insulin signaling molecules, glycolysis, and glycogen synthesis. Non-diabetic SGA young adults, when compared with age matched controls, demonstrate hyperinsulinemia at baseline and during oral glucose tolerance tests.24 When challenged with a hyperinsulinemic-euglycemic clamp, those born small have decreased peripheral glucose uptake and reduced activation of PI3K/Akt proteins.24,79,80 Looking more closely at muscle histology, LBW was associated with increased fast-twitch, glycolytic type IIx fibers, and larger type IIa and type I slow-twitch, oxidative fibers.81 Larger fiber size and a shift toward more type II fibers are associated with skeletal muscle insulin resistance. When examining the insulin signaling pathway, we and others have noted decreased protein expression of several key proteins, protein kinase C (PKC) ζ, p85, and insulin responsive glucose transporter (GLUT4), in both LBW adults and growth restricted rats.28,30,79,80 No difference was noted in insulin receptor expression, suggesting the defect lies downstream.80 To examine muscle glycolysis in LBW adult females, investigators analyzed ATP production with 31P-magnetic resonance spectroscopy and found that ATP production was reduced in LBW adults subjected to exercise.82
With regards to adipose tissue, adipocytes are smaller in the SGA/IUGR fetus and neonate and with aging increase in size and number.83 Moreover, as the SGA child accumulates more adipose tissue in extraneous sites, alterations in lipid metabolism occur and derangements in total cholesterol, low-density lipoprotein, and high-density lipoprotein become apparent.13,25,84–86 When the SGA population is examined as a whole, those who become obese adults are more likely to have abnormally high total cholesterol and low-density lipoprotein serum concentrations.86
How birth weight alters adipokines, adiponectin and leptin, and the implications of these changes remains an area of active research. Adiponectin, an insulin-sensitizing and anti-inflammatory protein, is inversely correlated with fat mass and is decreased in diabetic states. Although adiponectin’s role in adult disease is well-defined, its role in children and specifically in LBW infants is less clear. SGA fetuses have increased circulating adiponectin, and over time concentrations decrease. As expected, in the SGA population adiponectin is negatively correlated with weight, height, and BMI.87–89 In a series of studies, however, no difference in adiponectin in SGA and AGA children was noted.87–89 When a sub-group analysis was conducted, however, adiponectin was statistically higher in SGA subjects who did not exhibit “catch-up” growth or who were severely growth restricted at birth.87
In contrast to adiponectin, leptin concentrations correlate closely with fat mass and insulin resistance. Leptin, a satiety factor, acts on the hypothalamus and is responsible for regulating food intake and energy expenditure via a negative feedback loop. At birth IUGR neonates have decreased leptin, and by 1 year of age, leptin is increased compared with AGA controls.90–92 All neonates, undergo a “leptin surge,” and this surge occurs prematurely in growth-restricted rodents and sheep.26,93 In normal two-week-old sheep, leptin concentrations are inversely related to feeding activity. In other words, as leptin decreases, feeding activity increases. However, in growth-restricted sheep, this relationship is reversed, and plasma leptin correlates with increased suckling.26 As adults, LBW infants have abnormally high leptin concentrations even when BMI is taken into account.94 This hyperleptinemia, at various stages of the life cycle, may represent a form of “leptin resistance,” which acts to increase energy intake and decrease energy expenditure. When faced with the stress of a high fat diet, growth-restricted rats become obese and have higher leptin concentrations and decreased energy expenditure when compared to growth-restricted rats consuming standard chow or non-growth restricted rats on a high fat diet.26
Perinatal stress has been shown to alter neuronal circuitry. Studies have shown that intrauterine nutritional imbalances affect the rat/mouse hypothalamic orexigenic peptide, neuropeptide Y and anorexic peptide, cocaine- and amphetamine-regulated transcript.95 In addition, emerging evidence supports perturbations of circadian oscillator genes, or “clock genes.” Recently, a hyperphagic, hypoactive, preobese mouse was found to have altered clock genes in the brain and liver.29 Thus, undernourished fetuses are programmed for abnormal eating behaviors and energy metabolism and bear a predisposition towards obesity.
As emphasized above the FOAD hypothesis centers around reshaping the individual’s metabolism and body composition. To promote survival during periods of starvation, food intake increases, basal metabolic rate decreases, and energy is preferentially stored as fat. To increase glucose availability, pancreatic insulin production decreases, skeletal muscle and adipose tissue become insulin resistant, and hepatic glucose production is increased. This results in a state of hyperglycemia and increased de novo lipogenesis, which is further complicated by impaired action of growth and steroid hormones and leptin resistance. Overall, these events create a vicious cycle of adipose deposition (Fig 3). These adaptations are aimed at conserving energy and diverting scarce nutrients to vital organs. However, if there is “mismatch” in environments, or a transition from a nutritionally poor to nutritionally rich state, organ dysfunction and later disease become apparent.
Figure 3.
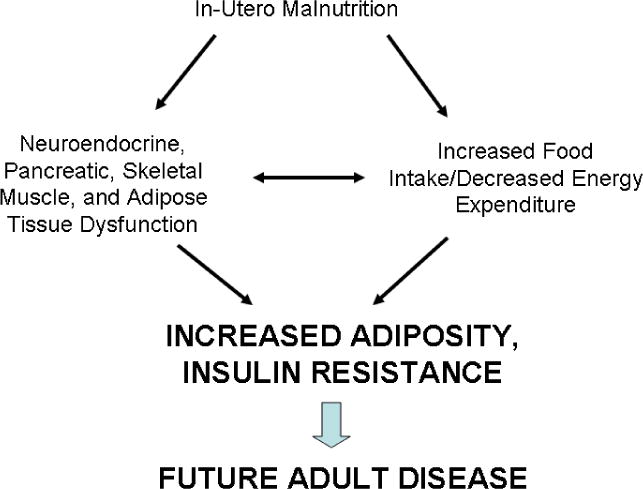
Schematic representing the pathogenesis of Fetal Origins of Adult Disease (FOAD.) In-utero malnutrition results in neuroendocrine, pancreatic, skeletal muscle, and adipose tissue dysfunction, and increased food intake and decreased energy expenditure. This leads to increased adiposity and insulin resistance, and ultimately future adult disease.
A LBW neonate, symbolic of a perturbed in utero state, is at increased risk for developing insulin resistance/T2DM. In addition, the individual’s growth during infancy, childhood, and adolescence, whether it is rapid, slow, or steady, at any stage, can further modify the eventual phenotype. The trajectory of one’s growth and future disease profile may be significantly impacted very early, even before conception. However, we have demonstrated that interventions with exercise and drug therapy suppress hepatic glucose production and increase skeletal muscle insulin responsiveness in the IUGR animal.96,97 Understanding how and why insulin resistance occurs in this population is key to developing strategies to reduce the burden of the metabolic syndrome and other associated diseases.
Puberty
Considering the implications of reduced fetal growth on the endocrine axis, it appears plausible that LBW children could have gonadal dysfunction and disturbed sexual maturation. In one study of SGA females, puberty and menarche occurred earlier than in the control population.98,99 Other studies, however, have demonstrated that SGA children enter puberty at the appropriate age, but progress more rapidly through puberty.100 In contrast to these observations, other studies have demonstrated that birth weight had no effect on the timing or progression through puberty.101,102 When height was taken into account, it was noted that SGA children enter puberty at “the normal age,” but this was not “normal” when considering their short stature.103 There has also been a recent trend, specifically in Western countries, for earlier menarche. As a result, it is difficult to dissect environmental influences, perhaps related to exposure to endocrine disruptors, from the programming effect. There are limited studies on how LBW affects fertility and menopause in females, and the onset of puberty in males.
Ibanez et al published a collection of articles describing the hormonal dysfunction of Northern Catalonian Spanish girls. In these studies, LBW status was linked with precocious pubarche and adrenarche.104,105 Adrenarche is characterized by the maturation of adrenal glands, and the production of sex steroids, such as dehydroepiandrosterone, which are responsible for pubarche. The hallmark of pubarche is the appearance of pubic hair, which usually occurs after the age of 8. These reports describe a “sequence of events” beginning with reduced growth, which progresses to pronounced adrenarche and precocious puberty, followed by ovarian hyperandrogenism, and finally insulin resistance. In these studies SGA girls have higher concentrations of dehydroepiandrosterone compared with AGA girls. Girls diagnosed with precocious puberty are more likely to have LBW status, and those with precocious puberty are more likely to have hyperandrogenism and hyperinsulinemia/insulin resistance.104,105
It is unclear if the link between precocious puberty and polycystic ovarian syndrome with SGA is secondary to early alterations of endocrine set points and/or a consequence of rapid weight gain during childhood.50 In an Australian cohort of children with precocious puberty, 65% were obese and 35% were SGA.98 As described above, exponential childhood growth is associated with central adiposity and insulin resistance, which is known to increase androgen and IGF-1 production.10,38,39
Overall, the link between LBW and premature precocious puberty and associated abnormalities appears plausible, but studies are limited by small, non-heterogeneous populations. Moreover, two other studies by different investigators have failed to replicate some of these results.101,102 Further work is needed before unequivocal conclusions can be reached.
Premature Infant
Equally challenging for the clinician is the preterm infant. The VLBW infant, and specifically the extremely low birth weight infant, is at high risk for postnatal growth failure during their intensive care stay, which is intimately linked to poor cognitive outcomes. VLBW is defined as a birth weight less than 1.5 kg; extremely low birth weight infant is defined as a birth weight less than 1 kg. In one investigation of infants studied at 36 weeks corrected gestational age, 100% of neonates with a birth weight < 1000 g were characterized as having postnatal growth restriction, and the majority remained restricted at 36 months.106 Despite aggressive, early parenteral nutrition and the introduction of enteral nutrition, the smallest infants remain small throughout childhood and neurodevelopmentally delayed.106–109
On the converse, preterm babies whose growth does not falter early demonstrate better cognitive outcomes. With the introduction of preterm formula, which is higher in calories and protein compared to term formula or breast milk, as an intervention in a preterm population, data revealed that males had higher verbal IQs and larger caudate volumes; however, both sexes demonstrated increased insulin resistance with higher split proinsulin levels and also higher diastolic blood pressures.110,111 In another study of VLBW infants as young adults, they were found to have a 6.7% increase in their 2 hour glucose concentration, a 16.7% increase in their fasting insulin concentration, a 40% increase in their 2-hour insulin concentration, and a 4.8 mm Hg increase in systolic blood pressure when compared to controls.112 However in another study, individuals with better weight gain post-hospitalization or after reaching term were noted to have better intelligence scores, and a small, possibly clinically irrelevant increase in blood pressure.113
So the question remains, when and how should postnatal growth be fostered? Breast milk is considered the gold standard nutrition for the full-term, AGA infant. However, if term formula does not meet the needs of the growing premature infant, will breast milk? Does fortified breast milk carry any unforeseen risks? In a recent study LBW babies were assigned to exclusive breast milk feedings or fortified breast milk. Those that received exclusive breast milk demonstrated less weight gain, but had lower fasting glucose and insulin concentrations at 3 months of age.114 Others have demonstrated that breast milk protects against rapid, exponential childhood growth.115 However, how that applies to the VLBW and/or premature population remains unclear, and longitudinal follow-up studies are still needed.
Before the 1950s, few preterm infants survived. Today, with use of mechanical ventilation, surfactant replacement, antibiotics, and state-of-the-art neonatal intensive care nurseries, these children are reaching adulthood. Opportunities now are available for investigators to study the programming effects of prematurity in children, adolescents, and adults. These effects are far-reaching, and extend beyond cardiovascular, metabolic, and cognitive perturbations, and affect the family and society as a whole. Follow-up studies of VLBW infants have revealed that they are less likely to graduate from high school or obtain a university degree; however, rates of employment do not differ between those who were VLBW and non-VLBW infants.116–118 Researchers have found that thoughts of depression and anxiety are more common in the follow-up of female VLBW infants, while males who were VLBW infants are more likely to exhibit attention deficit and hyperactivity disorder.116,119,120 In studies of the Helsinki Cohort, depressive symptoms increased in a linear fashion with decreased gestational length, and growth restriction was associated with increased anxiety traits.121 Additional evidence from a large Swedish cohort confirmed that infants born prematurely were more likely to suffer from psychiatric disorders and exhibit suicidal behavior.122,123 Thus, while the provision of suboptimal nutrition has deleterious effects perhaps in a gender-specific manner on the preterm’s cognitive development, prematurity also is associated with serious psychiatric implications. This is not necessarily surprising given that a good portion of CNS development occurs outside the womb in a foreign environment.
Other stressors, such as infections, hypoxia, and exogenous glucocorticoids, can permanently alter the neuroendocrine system.15,16,124 When faced with chronic or repeated perinatal and postnatal stress, the hypothalamic pituitary axis (HPA) is activated, resulting in elevated adrenocorticotrophic hormone (ACTH) and adrenal hypertrophy.125,126 An altered HPA carries significant long-term implications for atherosclerosis, dyslipidemia, T2DM, and immunosuppression. Prenatal glucocorticoids have been demonstrated to reduce hepatic production of insulin-like growth factor binding protein 1, which affects the child’s height, can alter hepatic gluconeogenesis and increase blood pressure.127 Providing yet another link between birth weight and adult insulin resistance, studies have demonstrated that LBW infants have elevated plasma cortisol concentrations as adults.128
Considering that the HPA is intimately tied to behavior, it is not surprising that HPA programming can lead to aberrant behavior and thoughts. If subjected to a hostile environment, a pregnant mother produces a “stress response,” which is transmitted to the fetus. When gestating rats were exposed to light and noise stress, their offspring had increased corticosterone levels and demonstrated impaired hippocampal function. This effect is most likely secondary to a reduction in pyramidal neurons and decreased synaptogenesis.17
Postnatal manipulations, like prenatal manipulations, can result in imprinting. When fetal rats are exposed to high levels of glucocorticoids in utero, their birth weights are reduced. As adults their HPA is chronically activated, and they exhibit anxiety-like and depressive behaviors.15 Increased in utero exposure to glucocorticoids, which can be seen with maternal protein restriction, is associated with increased blood pressure in offspring.129 There now is a growing body of evidence linking viral CNS infections with the development of schizophrenia, and glucose deprivation with autistic-like behaviors.130,131
Large for Gestational Age Infant
The other end of this spectrum finds the neonate born LGA, or with a birth weight greater than the 90th percentile secondary to gestational diabetes or idiopathic macrosomia. The obese or diabetic pregnant mother represents another form of programming. LGA, like SGA, is linked to future obesity and insulin resistance. LGA children are less likely to receive breast milk and exhibit rapid weight gain in the first 6 months of life.112,132 Although exclusive breast milk feeding appears to provide some protection from obesity, not all epidemiological studies consistently support this finding. LGA infants have been tracked to be heavier children with increased mid upper arm circumference at the level of the triceps and subscapular sites, and they are more likely to develop hypertension and hypertriglyceridemia and become obese adults.55,132–136
Mothers with GDM are at increased risk for delivering an LGA or SGA neonate. GDM is a spectrum; severe GDM is characterized by vasculopathy and nephropathy, both of which are known to produce IUGR fetuses. In most cases, however, GDM produces an increased delivery of glucose and other macronutrients to the fetus, resulting in increased fetal production of insulin, the dominant fetal growth hormone. Infants born to mothers with GDM have increased adiposity and elevated insulin and leptin levels.137,138 GDM also is hallmarked by maternal transmission; GDM mothers are more likely to have been born to mothers with T2DM, and GDM offspring are more likely to develop T2DM.31,139
It is important to remember that birth weight is merely a “symptom” of FOAD. In other words, it may or may not be a presenting feature or sign. It is imperative to recognize and acknowledge the AGA neonate born to a GDM mother. Although they appear “normal,” they too were subjected to in utero compromise and are at increased risk for future disease.31 Their in utero environment results in an increased secretion of hormones that promote the deposition of adipose tissue and increased insulin secretion early in life, thereby setting the fetus up for an abnormal body composition and future disease.
Cancer
In addition to being at increased risk for developing metabolic derangements, being LGA at birth is associated with an increased risk of cancer. As a result, fetal determinants are receiving new attention. Birth weight has been speculated to be associated with the development of breast, ovarian, prostate, testicular, and colon cancers. While fetal growth factors, such as insulin, are regulated by maternal substrates, it is also known that maternal concentrations of estrogen and testosterone can alter the offspring’s hypothalmic-pituitary-testicular/ovarian axis. By altering a specific tissue’s exposure to nutrition and steroid hormones, the risk for tissue dysplasia and subsequent cancer can be altered. Unlike insulin resistance, which demonstrates a U-shaped curve with respect to birth weight, colon cancer exhibits a J-shaped curve, with the heaviest babies carrying the largest risk.140,141 In a population-based cohort of over 10,000 men and women, macrosomia carried a hazard ratio of 2.57, while VLBW infants carried a modest, yet statistically significant, increase in incidence.141 Studies have demonstrated that when rats fed excessive calories were then exposed to carcinogens, they exhibited an increased number of colorectal tumors in comparison with similar controls who received a calorically appropriate diet.142
The strongest evidence for cancer and FOAD exists for breast cancer.5,143–150 A case-control study nested within the US Nurses’ Study revealed that those individuals at birth who weighed less than 2.5 kg were half as likely as those who weighed greater than 4 kg to subsequently develop breast cancer.5 Similarly, in a cohort of 2221 British women, a birth weight > 3.5 kg was associated with an increased risk for cancer, specifically pre-menopausal breast cancer, even after adjusting for confounders.145 Trichopoulos first proposed that in utero exposure to high levels of estrogens, either endogenous or exogenous, increased the number of stem cells and/or mitogenic activity of undifferentiated breast tissue. Consistent with this theory, mothers with advanced age, dizygotic gestations, or macrosomic or preterm babies demonstrate higher concentrations of estrogen. As a consequence, their fetuses are at an increased risk for breast cancer development.148,149 Conversely, mothers diagnosed with pregnancy-induced hypertension have decreased estrogen concentrations, and therefore, their offspring are relatively protected from breast cancer. In fact, in one study, pre-eclampsia/eclampsia was associated with a substantially lower breast cancer rate ratio of 0.24.150 Moreover, women who are taller in childhood and have higher growth rates seem to carry an even higher risk. For every 5-cm increase in height, there is an 11% increase in breast cancer, thus implicating growth factors, such as the insulin-like growth factor, potentially yet another carcinogenic culprit.146
Epigenetics
Cancer, like the metabolic syndrome, is a complex and multifactorial disease. Genetic mutations and environmental triggers are not sufficient to explain the pathogenesis and the rising incidence of cancer, obesity, and T2DM. Epigenetics, which means “on top of genes,” describes how the environment interacts with the genome to produce heritable changes resulting in phenotypic variation without altering the DNA of the genome. Epigenetic processes include DNA methylation and demethylation and post translational processes such as, acetylation phosphorylation and methylation of core histones, which result in an altered histone code. The DNA methylation of cytosine-guanine dinucleotides is commonly described in the literature. DNA methyltransferase enzyme isoforms (Dnmt 1, 3a, 3b) methylate DNA, causing the DNA to be tightly coiled or wound (i.e., heterochromatin). This state results in transcriptional repression and is maintained by heterochromatin protein, HP1. Methylation is maintained by the methyl donors, methionine, choline, and various cofactors, such as folic acid. By contrast, loosely wound DNA, known as euchromatin, allows active transcription (Fig 4).
Figure 4.
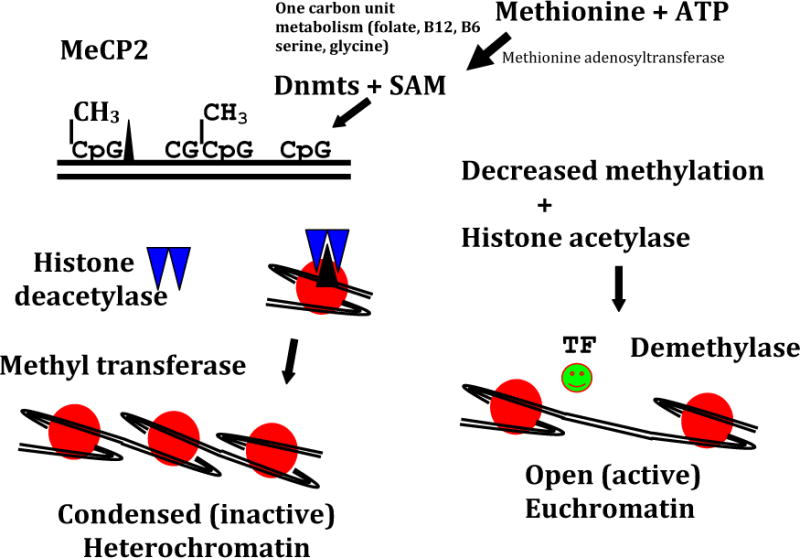
Simplistic scheme showing methylation (CH3) of CpG islands achieved by DNA methyltransferases (Dnmts) and S-adenosyl-L-methionione. Methylated DNA attracts methylated CpG binding protein (MeCP2), which recruits histone deacetylases and histone methylases, resulting in histone deacetylation and methylation. This process occuring in a gene promoter causes heterochromatin formation, resulting in failure of gene expression, which further is stabilized by the binding of the heterochromatin protein (HPI). In contrast, hypomethylation of DNA attracts histone actetylases and demethylases, which results in the oppositie effect with increased euchromatin formation, transcription factor binding, and the activation of gene transcription adenosine triphosphate (ATP). (Reprinted with permission from Devaskar SU, Thamotharan M. Metabolic programming in the pathogenesis of insulin resistance. Rev Endocr Metab Disord 2007; 8:105).
Epigenetic changes, previously well-described in the field of cancer, are now implicated in the pathogenesis of obesity and insulin resistance. Epigenetic modifications have been implicated in the SGA neonate, affecting promoters of glucose transporter 4, Pdx-1, glucocorticoid receptor, and peroxisomal proliferator activated receptor-α genes.151,152 Whether methyl donor supplementation before or during pregnancy can reverse the SGA phenotype remains to be elucidated. While epigenetic modifications have been described in various animal studies, replications in human samples have proven to be challenging. This is perhaps related to the fact that human samples have been limited to blood cells while most of the epigenetic changes have been observed to be tissue-specific.153
Summary
To curb the epidemic of rising chronic diseases, some attention must be given to FOAD. David Barker’s observations continue to stand the test of time across multicultural populations subjected to various constraints. The FOAD hypothesis has far-reaching implications as indicated by the following statement by the World Health Organization, “The global burden of death, disability and loss of human capital as a result of impaired fetal development is huge and affects both developed and developing countries.”154 The purpose of this review was to summarize some of the epidemiological, human, and animal data and provide pediatricians with a more complete understanding of the pathogenesis of chronic disease that are making their appearance earlier in childhood rather than waiting until adulthood.
Footnotes
The authors have no disclosures.
References
- 1.Barker DJP, Osmond C, Kajantie E, Eriksson J. Growth and chronic disease: findings in the Helsinki Birth Cohort. Ann Hum Biol. 2009;36:445–58. doi: 10.1080/03014460902980295. [DOI] [PubMed] [Google Scholar]
- 2.Barker DJ. The developmental origins of adult disease. J Am Coll Nutr. 2004;23:588S95S. doi: 10.1080/07315724.2004.10719428. [DOI] [PubMed] [Google Scholar]
- 3.Barker DJ. The developmental origins of insulin resistance. Horm Res. 2005;64(Suppl 3):2–7. doi: 10.1159/000089311. [DOI] [PubMed] [Google Scholar]
- 4.Roseboom TJ, van der Meulen JH, Ravelli AC, Osmond C, Barker DJ, Bleker OP. Effects of prenatal exposure to the Dutch famine on adult disease in later life: an overview. Mol and Cell Endocrinol. 2001;185:93–8. doi: 10.1016/s0303-7207(01)00721-3. [DOI] [PubMed] [Google Scholar]
- 5.Michels KB, Trichopoulos D, Robins JM, Rosner BA, Manson JE, Hunter DJ, et al. Birthweight as a risk factor for breast cancer. Lancet. 1996;348:1542–6. doi: 10.1016/S0140-6736(96)03102-9. [DOI] [PubMed] [Google Scholar]
- 6.Cooper C, Fall C, Egger P, Hobbs R, Eastell R, Barker D. Growth in infancy and bone mass in later life. Ann Rheum Dis. 1997;56:17–21. doi: 10.1136/ard.56.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lahti J, Raikkonen K, Pesonen AK, Heinonen K, Kajantie E, Forsén T, et al. Prenatal growth, postnatal growth and trait anxiety late adulthood – the Helsinki Birth Cohort. Acta Psychiatr Scand. 2010;121:227–35. doi: 10.1111/j.1600-0447.2009.01432.x. [DOI] [PubMed] [Google Scholar]
- 8.Stanner SA, Bulmer K, Andrès C, Lantseva OE, Borodina V, Poteen VV, et al. Does malnutrition in utero determine diabetes and coronary heart disease in adulthood? Results from the Leningrad siege study, a cross sectional study. BMJ. 1997;315:1342–9. doi: 10.1136/bmj.315.7119.1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ravelli GP, Stein ZA, Susser MW. Obesity in young men after famine exposure in utero and early infancy. N Engl J Med. 1976;293:349–53. doi: 10.1056/NEJM197608122950701. [DOI] [PubMed] [Google Scholar]
- 10.Valdez R, Athens MA, Thompson GH, Bradshaw BS, Stern MP. Birthweight and adult health outcomes in a biethnic population in the USA. Diabetologia. 1994;37:624–31. doi: 10.1007/BF00403383. [DOI] [PubMed] [Google Scholar]
- 11.Kulkarni ML, Mythri HP, Kulkarni AM. ‘Thinfat’ phenotype in newborns. Indian J Pediatr. 2009;76:369–373. doi: 10.1007/s12098-009-0010-8. [DOI] [PubMed] [Google Scholar]
- 12.Bhargava SK, Sachdev HS, Fall CH, Osmond C, Lakshmy R, Barker DJ, et al. Relation of serial changes in childhood body-mass index to impaired glucose tolerance in young adulthood. N Engl J of Med. 2004;350:865–75. doi: 10.1056/NEJMoa035698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tenhola S, Martikainen A, Rahiala E, Herrgârd E, Halonen P, Voutilainen R. Serum lipid concentrations and growth characteristics in 12-year old children born small for gestational age. Pediatr Res. 2000;48:623–8. doi: 10.1203/00006450-200011000-00012. [DOI] [PubMed] [Google Scholar]
- 14.Yajnik CS, Deshpande SS, Panchanadikar AV, Naik SS, Deshpande JA, Coyaji KJ, et al. Maternal and total homocysteine concentration and neonatal size in India. Asia Pac J Clin Nutr. 2005;14:179–81. [PubMed] [Google Scholar]
- 15.Welberg LA, Seckl JR, Holmes MC. Inhibition of 11β-hydroxysteriod dehydrogenase, the foeto-placental barrier to maternal glucocorticoids, permanently programs amygdala GF mRNA expression and anxiety-like behavior in the offspring. Eur J of Neurosci. 2000;12:1047–54. doi: 10.1046/j.1460-9568.2000.00958.x. [DOI] [PubMed] [Google Scholar]
- 16.Dalman C, Allebeck P, Gunnel D, Harrison G, Kristensson K, Lewis G, et al. Infections in CNS during childhood and the risk of subsequent psychotic illness: a cohort study of more than one million Swedish subjects. Am J Psychiatry. 2008;165:59–83. doi: 10.1176/appi.ajp.2007.07050740. [DOI] [PubMed] [Google Scholar]
- 17.Fride E, Dan Y, Feldon J, Halevy G, Weinstock M. Effects of prenatal stress on vulnerability to stress in prepubertal and adult rats. Physiol Behav. 1986;37:681–7. doi: 10.1016/0031-9384(86)90172-1. [DOI] [PubMed] [Google Scholar]
- 18.Nyirenda MJ, Lindsay RS, Kenyon CJ, Burchell A, Seckl JR. Glucocorticoid exposure in late gestation permanently programs rat hepatic phosphoenolpyruvate carboxykinase and glucocorticoid receptor expression and causes glucose intolerance in the adult offspring. J Clin Invest. 1988;101:2174–81. doi: 10.1172/JCI1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simmons RA, Suponitsky-Kroyter I, Selak MA. Progressive accumulation of mitochrondrial DNA mutations and decline in mitochondrial function lead to β-cell failure. J Biol Chem. 2005;280:28785–91. doi: 10.1074/jbc.M505695200. [DOI] [PubMed] [Google Scholar]
- 20.Ho SM, Tang WY, Belmonte de Frausto J, Prins GS. Developmental exposure to estradiol and bisphenol A increases susceptibility to prostate carcinogenesis and epigentically regulates type 4 variant 4. Cancer Res. 2006;66:5264–32. doi: 10.1158/0008-5472.CAN-06-0516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Painter RC, Osmond C, Gluckman P, Hanson M, Phillips DI, Roseboom TJ. Transgenerational effects of prenatal exposure to the Dutch famine on neonatal adiposity and health later in life. BJOG. 2008;115:1243–9. doi: 10.1111/j.1471-0528.2008.01822.x. [DOI] [PubMed] [Google Scholar]
- 22.Jimenez-Chillaron JC, Isganaitis E, Charalambous M, Gesta S, Pentiant-Pelegrin T, Faucette RR, et al. Intergenerational transmission of glucose intolerance and obesity by in utero undernutrition in mice. Diabetes. 2009;8:460–8. doi: 10.2337/db08-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thamotharan M, Garg M, Oak S, Rogers LM, Pan G, Sangiorgi F, et al. Transgenerational inheritance of the insulin-resistant phenotype in embyo-transferred intrauterine growth-restricted adult female rat offspring. Am J Physiol Endocrinol Metab. 2007;292:E1270–9. doi: 10.1152/ajpendo.00462.2006. [DOI] [PubMed] [Google Scholar]
- 24.Jaquet D, Gaboriau A, Czernichow P, Levy-Marchal C. Insulin resistance early adulthood in subjects born with intrauterine growth retardation. J of Clin Endocrinol Metab. 2000;85:1401–6. doi: 10.1210/jcem.85.4.6544. [DOI] [PubMed] [Google Scholar]
- 25.Reinehr T, Kleber M, Toschke AM. Small for gestational age status is associated with metabolic syndrome in overweight children. Eur J of Endocrinol. 2009;160:579–84. doi: 10.1530/EJE-08-0914. [DOI] [PubMed] [Google Scholar]
- 26.De Blasio MJ, Blache D, Gatford KL, Robinson JS, Owens JA. Placental restriction increases adipose leptin gene expression and plasma leptin and alters their relationship to feeding activity in the young lab. Pediatr Res. 2010;67:603–8. doi: 10.1203/PDR.0b013e3181dbc471. [DOI] [PubMed] [Google Scholar]
- 27.Jenson CB, Storgaard H, Dela F, Holst JJ, Madsbad S, Vaag AA. Early differential defects of insulin secretion and actions in 19-year-old Caucasian men who had low birth weight. Diabetes. 2002;51:1271–80. doi: 10.2337/diabetes.51.4.1271. [DOI] [PubMed] [Google Scholar]
- 28.Oak SA, Tran C, Pan G, Thamotharan M, Devaskar SU. Perturbed skeletal muscle insulin signaling in the adult female intrauterine growth-restricted rat. Am J Physiol Endocrinol Metab. 2006;290:E1321–30. doi: 10.1152/ajpendo.00437.2005. [DOI] [PubMed] [Google Scholar]
- 29.Sutton GM, Centanni AV, Butler AA. Protein malnutrition during pregnancy in C57BL/6J mice results in offspring with altered circadian physiology before obesity. Endocrinology. 2010;151:1570–80. doi: 10.1210/en.2009-1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arantes VC, Teixeira VP, Reis MA, Latorraca MQ, Leite AR, Carneiro EM, et al. Expression of PDX-1 is reduced in pancreatic islets from pups of rat dams fed low protein diet during gestation and lactation. J Nutr. 2002;132:3030–5. doi: 10.1093/jn/131.10.3030. [DOI] [PubMed] [Google Scholar]
- 31.Thamotharan M, McKnight RA, Thamotharan S, Kao DJ, Devaskar SU. Aberrant insulin-induced GLUT 4 translocation predicts glucose intolerance in the offspring of a diabetic mother. Am J Physiol Endocrinol Metab. 2003;284:E901–14. doi: 10.1152/ajpendo.00516.2002. [DOI] [PubMed] [Google Scholar]
- 32.Hales CN, Barker DJ. The thrifty phenotype hypothesis. Br Med Bull. 2001;60:5–20. doi: 10.1093/bmb/60.1.5. [DOI] [PubMed] [Google Scholar]
- 33.http://www.who.int.nutrition/topics/lbw_strategy_background.pdf
- 34.Prevalence of Obesity Among US Children and Adolescents (Aged 2 – 19 Years) – National Health and Nutrition Examination Surveys, NHANES, 1976–1980 and 2003–2006) Centers for Disease Control and Prevention; Atlanta GA: Nov, 2009. [Google Scholar]
- 35.http://www.who.int/mediacentre/news/releases/2004/pr31/en
- 36.Law CM, Shiell AW, Newsome CA, Syddall HE, Shinebourne EA, Fayers PM, et al. Fetal, infant and childhood growth and adult blood pressure: a longitudinal study from birth to 2 years of age. Circulation. 2002;105:1088–92. doi: 10.1161/hc0902.104677. [DOI] [PubMed] [Google Scholar]
- 37.Forsén T, Eriksson J, Tuomilehto J, Reunanen A, Osmond C, Barker D. The fetal and childhood growth of persons who develop type 2 diabetes. Ann Intern Med. 2000;133:176–82. doi: 10.7326/0003-4819-133-3-200008010-00008. [DOI] [PubMed] [Google Scholar]
- 38.Okosun IS, Liao Y, Rotimi CN, Dever GE, Cooper RS. Impact of birth weight on ethnic variations in subcutaneous and central adiposity in American children aged 5–11 years. A study from the National Health and Nutrition Examination Survey. Int J of Obes Relat Metab Disord. 2000;24:479–84. doi: 10.1038/sj.ijo.0801182. [DOI] [PubMed] [Google Scholar]
- 39.Law CM, Barker DJ, Osmond C, Fall CH, Simmonds SJ. Early growth and abdominal fatness in adult life. J of Epidemiol and Community Health. 1992;46:184–6. doi: 10.1136/jech.46.3.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barker M, Robinson S, Osmond C, Barker DJ. Birth weight and body fat distribution in adolescent girls. Arch Dis Child. 1997;77:381–3. doi: 10.1136/adc.77.5.381. [DOI] [PubMed] [Google Scholar]
- 41.Ozanne SE, Dorling MW, Wang CL, Petry CJ. Depot-specific effects of early growth retardation on adipocyte insulin action. Horm Metab Res. 2000;32:71–5. doi: 10.1055/s-2007-978592. [DOI] [PubMed] [Google Scholar]
- 42.Ozanne SE, Nave BT, Wang C, Shephard PR, Prins J, Smith GD. Poor fetal nutrition causes long-term expression of insulin signaling components in adipocytes. Am J Physiol. 1997;273:E46–51. doi: 10.1152/ajpendo.1997.273.1.E46. [DOI] [PubMed] [Google Scholar]
- 43.Ozanne SE, Dorling MW, Wang CL, Nave BT. Impaired PI3-kinase activation in adipocytes from early growth restricted male rats. Am J Physiol. 2001;280:E534–9. doi: 10.1152/ajpendo.2001.280.3.E534. [DOI] [PubMed] [Google Scholar]
- 44.Nobili V, Marcellini M, Marchesini G, Vanni E, Manco M, Villani A, et al. Intrauterine growth retardation, insulin resistance, and nonalcoholic fatty liver disease in children. Diabetes Care. 2007;30:2638–40. doi: 10.2337/dc07-0281. [DOI] [PubMed] [Google Scholar]
- 45.Keller G, Zimmer G, Mall G, Ritz E, Amann K. Nephron number in patients with primary hypertension. N Engl J of Med. 2003;348:101–8. doi: 10.1056/NEJMoa020549. [DOI] [PubMed] [Google Scholar]
- 46.Hallan S, Euser AM, Irgens LM, Finken MJ, Holmen J, Dekker FW. Effect of intrauterine growth restriction on kidney function at young adult age: the Nord Trondelag Health (HUNT 2) study. Am J Kidney Dis. 2008;51:10–20. doi: 10.1053/j.ajkd.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 47.Wlodek ME, Westcokk K, Siebel AL, Owens JA, Moritz KM. Growth restriction before or after birth reduces nephron number and increases blood pressure in male rats. Kidney Int. 2008;74:187–95. doi: 10.1038/ki.2008.153. [DOI] [PubMed] [Google Scholar]
- 48.Nijland MJ, Schlabritz-Loutsevitch NE, Hubbard GB, Nathanielsz PW, Cox LA. Non-human primate fetal kidney transciptome analysis indicates mammalian target of rapamycin (mTOR) is a central nutrient-responsive pathway. J Physiol. 2007;579:643–56. doi: 10.1113/jphysiol.2006.122101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lundgren EM, Cnattinguis S, Jonsson B, Tuvemo T. Prediction of adult height and risk of overweight females born small-for-gestational age. Paediatr and Perinat Epidemiol. 2003;17:156–63. doi: 10.1046/j.1365-3016.2003.00489.x. [DOI] [PubMed] [Google Scholar]
- 50.Ibanez L, Jaramilli A, Enriquez G, Miro E, Lopez-Bermejo A, Dunger D, et al. Polycystic ovaries after precocious pubarche: relation to prenatal growth. Human Repro. 2007;22:395–400. doi: 10.1093/humrep/del395. [DOI] [PubMed] [Google Scholar]
- 51.Eide MG, Oyen N, Skjaerven R, Nilsen ST, Bjerkedal T, Tell GS. Size at birth and gestational age as predictors of adult height and weight. Epidemiology. 2005;16:175–81. doi: 10.1097/01.ede.0000152524.89074.bf. [DOI] [PubMed] [Google Scholar]
- 52.Tuvemo T, Cnattingius S, Jonsson B. Prediction of male adult stature using anthropometric data at birth: a nationwide population-based study. Pediatr Res. 1990;46:491–5. doi: 10.1203/00006450-199911000-00001. [DOI] [PubMed] [Google Scholar]
- 53.Karlberg J, Albertsson-Wikland K. Growth in full-term small-for-gestational age infants: from birth to final height. Pediatr Res. 1995;38:733–9. doi: 10.1203/00006450-199511000-00017. [DOI] [PubMed] [Google Scholar]
- 54.Leger J, Levy-Marchael C, Bloch J, Pinet A, Chevenne D, Porquet D, et al. Reduced final height and indications for insulin resistance in 20 year olds born small for gestational age: regional cohort study. BMJ. 1997;315:341–7. doi: 10.1136/bmj.315.7104.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gilman MW, Rifas-Siman SL, Berkley CS, Field AE, Colditz GA. Maternal gestational diabetes, birth weight, and adolescent obesity. Pediatrics. 2003;111:e221–6. doi: 10.1542/peds.111.3.e221. [DOI] [PubMed] [Google Scholar]
- 56.Javid MK, Eriksson JG, Kajantie E, Forsen E, Osmond C, Barker DJP, et al. Growth in childhood predicts hip fracture in later life. Osteoporos Int. 2011;22:69–73. doi: 10.1007/s00198-010-1224-3. [DOI] [PubMed] [Google Scholar]
- 57.Antoniades L, MacGregor AJ, Andrew T, Spector TD. Association of birth weight with osteoporosis and osteoarthritis in adult twins. Rheumatol. 2003;42:791–6. doi: 10.1093/rheumatology/keg227. [DOI] [PubMed] [Google Scholar]
- 58.Poulsen P, Vaag A, Beck-Nielsen H. Does zygosity influence the metabolic profile of twins? A population based cross sectional study. BMJ. 1999;310:151–4. doi: 10.1136/bmj.319.7203.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fall C, Hindmarsh P, Dennison E, Kellingray S, Barker D, Cooper C. Programming of growth hormone secretion and bone mineral density in elderly men: a hypothesis. J Clin Endocrinol Metab. 1998;83:135–9. doi: 10.1210/jcem.83.1.4487. [DOI] [PubMed] [Google Scholar]
- 60.Lips MA, Syddall HE, Gaunt TR, Rodriguez S, Day IN, Cooper C, et al. Interaction between birthweight and polymorphism in the calcium-sensing receptor gene in determination of adult bone mass: the Hertfordshire cohort study. J Rheumatol. 2007;34:769–75. [PMC free article] [PubMed] [Google Scholar]
- 61.Dennison EM, Syddall HE, Rodriguez S, Voropanov A, Day NM, Cooper C. Polymorphism in the growth hormone gene, weight in infancy, and adult bone mass. J Clin Endocrinol Met. 2004;89:4898–903. doi: 10.1210/jc.2004-0151. [DOI] [PubMed] [Google Scholar]
- 62.Dennison EM, Dyddall HE, Jameson KA, Sayer AA, Gaunt TR, Rodriguez S, et al. A study of relationships between single nucleotide polymorphisms from the growth hormone-insulin like growth factor axis and bone mass: the Hertfordshire cohort study. J Rheumatol. 2009;36:1520–6. doi: 10.3899/jrheum.081061. [DOI] [PubMed] [Google Scholar]
- 63.Dennison EM, Arden NK, Keen RW, Sydall H, Day IN, Spector TD, et al. Birthweight, vitamin D receptor genotype and the programming of osteoporosis. Paediatr Peri Epidemiol. 2001;15:211–9. doi: 10.1046/j.1365-3016.2001.00350.x. [DOI] [PubMed] [Google Scholar]
- 64.Abuzzahab MJ, Schneider A, Goddard A, Grigorescu F, Lautier C, Keller E, et al. IGF-I receptor mutations resulting in intrauterine and postnatal growth restriction. N Engl J Med. 2003;349:2211–22. doi: 10.1056/NEJMoa010107. [DOI] [PubMed] [Google Scholar]
- 65.Jaquet D, Dhevenne D, Czernichow P, Levy-Marchal C. No evidence for major beta-cell dysfunction in young adults born with intra-uterine growth retardation. Pediatr Diabetes. 2000;1:181–5. doi: 10.1046/j.1399543x.2000.010402.x. [DOI] [PubMed] [Google Scholar]
- 66.Flanagan DE, Moore VM, Godsland IF, Cockington RA, Robinson JS, Phillips DI. Fetal growth and the physiological control of glucose tolerance in adults: a minimal model analysis. Am J Physiol Endocrinol Metab. 2000;278:E700–6. doi: 10.1152/ajpendo.2000.278.4.E700. [DOI] [PubMed] [Google Scholar]
- 67.Econimides DL, Prodler A, Nicolaides KH. Plasma insulin in appropriate and small for gestational age fetuses. Am J Obstet Gynecol. 1989;160:1091–4. doi: 10.1016/0002-9378(89)90167-1. [DOI] [PubMed] [Google Scholar]
- 68.Garofano A, Czernichow P, Bréant B. In utero undernutrition impairs beta-cell development. Diabetolgia. 1997;40:1231–4. doi: 10.1007/s001250050812. [DOI] [PubMed] [Google Scholar]
- 69.Limesand S, Rozance P, Zerbe G, Hutton J, Hay WW., Jr Attenuated insulin release and storage in fetal sheep pancreatic islets with intrauterine growth restriction. Endocrinology. 2006;147:1488–97. doi: 10.1210/en.2005-0900. [DOI] [PubMed] [Google Scholar]
- 70.Seita S, Sridhar MG, Bhat V, Chaturvedula L, Vinayagamoorti R, John M. Insulin sensitivity and insulin secretion at birth in intrauterine growth retarded infants. Pathology. 2006;38:236–8. doi: 10.1080/00313020600696256. [DOI] [PubMed] [Google Scholar]
- 71.Schwitzgebel V, Somm E, Klee P. Modeling intrauterine growth retardation in rodents: impact on pancreas development and glucose homestasis. Mol and Cell Endo. 2009;304:78–83. doi: 10.1016/j.mce.2009.02.019. [DOI] [PubMed] [Google Scholar]
- 72.Freathy R, Bennett AJ, Ring SM, Shields B, Groves CJ, Timpson NJ, et al. Type 2 diabetes risk alleles are associated with reduced size at birth. Diabetes. 2009;58:1428–33. doi: 10.2337/db08-1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ganguly A, Devaskar SU. Glucose transporter isoform-3 null heterozygous mutation causes sexually dimorphic adiposity with insulin resistance. Am J Physiol Endocrinol Metab. 2008;294:E1144–51. doi: 10.1152/ajpendo.90251.2008. [DOI] [PubMed] [Google Scholar]
- 74.Bhasin KK, van Nas A, Martin LJ, Davis RC, Devaskar SU, Lusis AJ. Maternal low-protein diet or hypercholesterolemia reduces circulating essential amino acids and leads to intrauterine growth restriction. Diabetes. 2009;58:559–63. doi: 10.2337/db07-1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ylihãrsilã H, Kajantie E, Osmond C, Forsén T, Barker DJ, Eriksson JG. Birth size, adult body composition and muscle strength in later life. Int J of Obes. 2007;31:1392–99. doi: 10.1038/sj.ijo.0803612. [DOI] [PubMed] [Google Scholar]
- 76.Eriksson J, Forsén T, Tuomilehto J, Osmond C, Barker D. Size at birth, fat-free mass and resting metabolic rate in adult life. Horm Metab Res. 2002;34:72–6. doi: 10.1055/s-2002-20518. [DOI] [PubMed] [Google Scholar]
- 77.Sayer AA, Syddall HE, Gilbody HJ, Dennison EM, Cooper C. Birth weight, weight at l year of age, and body composition in older men: findings from the Hertfordshire Cohort Study. Am J Clin Nutr. 2004;80:166–203. doi: 10.1093/ajcn/80.1.199. [DOI] [PubMed] [Google Scholar]
- 78.Peterside I, Selak M, Simmons RA. Impaired oxidative phosphorylation in hepatic mitochondria. Am J Physiol Endocrinol Metab. 2003;285:E1258–66. doi: 10.1152/ajpendo.00437.2002. [DOI] [PubMed] [Google Scholar]
- 79.Jenson CB, Malgorzata S, Martin-Gronert M, Storgaard H, Madsbad S, Vaag A, et al. Altered PI3-kinase/Akt signaling in skeletal muscle of young men with low birth weight. PLos ONE. 2008;3:e3738. doi: 10.1371/journal.pone.0003738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ozanne SE, Olsen GS, Hansen LL, Tingey KJ, Nave BT, Wang CL, et al. Early growth restriction leads to downregulation of protein kinase C zeta and insulin resistance in skeletal muscle. J Endocrinol. 2003;177:235–41. doi: 10.1677/joe.0.1770235. [DOI] [PubMed] [Google Scholar]
- 81.Jenson CB, Storgaard H, Madsbad S, Richter E, Vaag A. Altered skeletal muscle fiber composition and size precedes whole-body insulin resistance in young men with low birth weight. J Clin Endocrinol Metab. 2007;92:1530–4. doi: 10.1210/jc.2006-2360. [DOI] [PubMed] [Google Scholar]
- 82.Taylor DJ, Thompson CH, Kemp GJ, Barnes PR, Sanderson AL, Radda GK, et al. A relationship between impaired fetal growth and reduced muscle glycolysis revealed b 31P magnetic resonance spectroscopy. Diabetologia. 1995;38:1205–12. doi: 10.1007/BF00422370. [DOI] [PubMed] [Google Scholar]
- 83.Enzi G. Intrauterine growth and adipose tissue and development. Am J Clin Nutr. 1981;34:1785–90. doi: 10.1093/ajcn/34.9.1785. [DOI] [PubMed] [Google Scholar]
- 84.Tenhola S, Rahiala E, Martikainen A, Halonen P, Voutilainen R. Blood pressure, serum lipids, fasting insulin, and adrenal hormones in 12-year-old children born with maternal preeclampsia. J Clin Endocrinol Metab. 2003;88:1217–22. doi: 10.1210/jc.2002-020903. [DOI] [PubMed] [Google Scholar]
- 85.Skidmore P, Hardy R, Kuh D, Langenberg C, Wadsworth M. Birth weight and lipids in a national cohort study. Arterioscler Thromb Vasc Biol. 2004;34:588–94. doi: 10.1161/01.ATV.0000116692.85043.ef. [DOI] [PubMed] [Google Scholar]
- 86.Amigo H, Bustos P, Alvarado M, Barbieri M, Bettiol H, da Silva A, et al. Size at birth and lipoprotein concentrations in adulthood: two prospective studies in Latin American cities. J Epidmiol Community Health. 2010;64:855–9. doi: 10.1136/jech.2008.078345. [DOI] [PubMed] [Google Scholar]
- 87.Evagelidou EN, Giapros VI, Challa AS, Kiortsis DN, Tsatsoulis AA, Andronikou SK. Serum adiponectin levels, insulin resistance, and lipid profiles in children born small for gestational age. Eur J Endocrinol. 2007;156:271–7. doi: 10.1530/eje.1.02337. [DOI] [PubMed] [Google Scholar]
- 88.Sancakli O, Darendeliler F, Bas F, Gokcay G, Disci R, Semih A, et al. Insulin, adiponectin, IGFBP-1 levels and body composition in small for gestational age born non-obese children during pre-pubertal ages. Clinical Endocrinology. 2008;69:88–92. doi: 10.1111/j.1365-2265.2007.03138.x. [DOI] [PubMed] [Google Scholar]
- 89.Challa A, Evagelidou E, Cholveas V, Kiortsis D, Giapros V, Drougia A, et al. Growth factors and adipocytokines in prepubertal children born small for gestational age. Diabetes Care. 2009;32:714–19. doi: 10.2337/dc08-1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Matsuda J, Yokota I, Iida M, Murakami T, Naito E, Ito M, et al. Serum leptin concentration in cord blood: relationship to birthweight and gender. J Clin Endocrinol Metab. 1997;82:1642–44. doi: 10.1210/jcem.82.5.4063. [DOI] [PubMed] [Google Scholar]
- 91.Schrubing C, Prohaska F, Prohaska A, Englaro P, Blum W, Kratzch J, et al. Levels of leptin in maternal serum, amniotic fluid, and arterial and venous cord blood: relation to neonatal and placental weight. J Clin Endocrinol Metab. 1997;82:1480–3. doi: 10.1210/jcem.82.5.3935. [DOI] [PubMed] [Google Scholar]
- 92.Jaquet D, Leger J, Tabone P, Czernichow P, Levy-Marchal C. High serum leptin concentrations during catch-up growth of children born with intrauterine growth retardation. J Clin Endocrinol Metab. 1999;84:1949–55. doi: 10.1210/jcem.84.6.5744. [DOI] [PubMed] [Google Scholar]
- 93.Yura S, Itoh H, Sagawa N, Yamatot H, Masuzaki H, Nakao K, et al. Role of premature leptin surge in obesity resulting from intrauterine undernutrition. Cell Metab. 2005;1:371–7. doi: 10.1016/j.cmet.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 94.Phillips DI, Fall CH, Cooper C, Norman FJ, Robinson JS, Owens PC. Size at birth and plasma leptin concentrations in adult life. Int J Obes Relat Metab Disord. 1990;23:1025–9. doi: 10.1038/sj.ijo.0801050. [DOI] [PubMed] [Google Scholar]
- 95.Kristensen P, Judge ME, Thim L, Ribel U, Christjansen KN, Wulff BS, Clausen JT, et al. Hypothalmic CART is a new anorectic peptide regulated by leptin. Nature. 1998;7:72–6. doi: 10.1038/29993. [DOI] [PubMed] [Google Scholar]
- 96.Garg M, Thamotharan M, Oak SA, Pan G, Maclaren DC, Lee PW, Devaskar SU. Early exercise regimen improves insulin sensitivity in the intrauterine growth restricted adult female rat offspring. Am J Physiol Endocrinol Metab. 2009;296:E272–81. doi: 10.1152/ajpendo.90473.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Garg M, Thamotharan M, Pan G, Lee PW, Devaskar SU. Early exposure of the pregestational intrauterine and postnatal growth restricted offspring to a peroxisome proliferator-activated receptor-{gama} agonist. Am J Physiol Endocrinol Metab. 2010;298:E498–98. doi: 10.1152/ajpendo.00361.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Neville KA, Walker JL. Precious pubarche is associated with SGA, prematurity, weight gain, and obesity. Arch Dis Child. 2005;90:258–61. doi: 10.1136/adc.2004.053959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Persson I, Ahlsson F, Ewald U, Tuvemo T, Quingyan M, von Rosen D, et al. Influences of perinatal factors on the onset of puberty in boys and girls: implications for interpretation of link with risk of long term diseases. Am J Epidemiol. 1999;150:747–55. doi: 10.1093/oxfordjournals.aje.a010077. [DOI] [PubMed] [Google Scholar]
- 100.Ibáñez L, Ferrer A, Marcos MV, Hierro FR, de Zegher F. Early puberty: rapid progression and reduced final height in girls with low birth weight. Pediatrics. 2000;106:E72. doi: 10.1542/peds.106.5.e72. [DOI] [PubMed] [Google Scholar]
- 101.Jaquet D, Leger J, Chevenne D, Czernichow P, Levy-Marchal C. Intrauterine growth retardation predisposes to insulin resistance but not to hyperandrogenism in young women. J Clin Endocrinol Metab. 1999;84:3945–9. doi: 10.1210/jcem.84.11.6106. [DOI] [PubMed] [Google Scholar]
- 102.Boonstra VH, Mulder PG, de Jong FH, Hokken-Koelega AC. Serum dehydroepiandrosterone sulfate levels and pubarche in short children born small for gestational age before and during growth hormone treatment. J Clin Endocrinol Metab. 2004;89:712–7. doi: 10.1210/jc.2003-031160. [DOI] [PubMed] [Google Scholar]
- 103.Albetson-Wikland K, Karlberg J. Natural growth in children born small for gestational age with and without catch-up growth. Acta Paediatrica Scandinavica. 1994;399:64–70. doi: 10.1111/j.1651-2227.1994.tb13292.x. [DOI] [PubMed] [Google Scholar]
- 104.Ibáñez L, Potau N, Francois I, de Zegher F. Precocious pubarche, hyperinsulinsm, and ovarian hyperadrogenism in girls: relation to reduced fetal growth. J Clin Endocrinol Metab. 1998;83:3588–62. doi: 10.1210/jcem.83.10.5205. [DOI] [PubMed] [Google Scholar]
- 105.Ibáñez L, Potau N, Marcos MV, de Zegher F. Exaggerated adrenarche and hyperinsulinism in adolescent girls born small for gestional age. J Clin Endocrinol Metabol. 1999;84:4739–41. doi: 10.1210/jcem.84.12.6341. [DOI] [PubMed] [Google Scholar]
- 106.Dusick A, Poindexter BB, Ehrenkranz RA, Lemons JA. Growth failure in the preterm infant: can we catch up? Semin Perinatol. 2003;27:302–10. doi: 10.1016/s0146-0005(03)00044-2. [DOI] [PubMed] [Google Scholar]
- 107.Neubauer AP, Kattner Voss W. Outcomes of extremely low birth weight survivors at school age: the influence of perinatal parameters on neurodevelopment. Eur J Pediatr. 2008;167:87–9. doi: 10.1007/s00431-007-0435-x. [DOI] [PubMed] [Google Scholar]
- 108.Anderson P, Doyle LW. Cognitive and educational deficits in children born extremely preterm. Semin Perinatol. 2008;32:51–8. doi: 10.1053/j.semperi.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 109.Ehrenkranz RA, Younes N, Lemons JA, Fanaroff AA, Donovan EF, Wright LL, et al. Longitudinal growth of hospitalized very low birth weight infants. Pediatrics. 1999;104:280–9. doi: 10.1542/peds.104.2.280. [DOI] [PubMed] [Google Scholar]
- 110.Singhal A, Fewtrell M, Cole T, Lucas A. Low nutrient intake and early growth for later insulin resistance in adolescents born preterm. The Lancet. 2003;361:1089–97. doi: 10.1016/S0140-6736(03)12895-4. [DOI] [PubMed] [Google Scholar]
- 111.Isaacs E, Gadian D, Sabatini S, Chong W, Quinn B, Fishi B, Lucas A. The effect of early human diet on caudate volume and IQ. Pediatr Res. 2008;63:308–14. doi: 10.1203/PDR.0b013e318163a271. [DOI] [PubMed] [Google Scholar]
- 112.Hovi P, Andersson S, Erikksson J, Jarvenpaa A, Strang-Karlsson S, Makitie O, et al. Glucose regulation in young adults with very low birth weight. N Engl J Med. 2007;356:2053–63. doi: 10.1056/NEJMoa067187. [DOI] [PubMed] [Google Scholar]
- 113.Belfort M, Martin C, Smith V, Gillman M, McCormick M. Infant weight gain and school-age blood pressure and cognition in former preterm infants. Pediatrics. 2010;125:e1419–26. doi: 10.1542/peds.2009-2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gupta M, Zaheer Jora R, Kaul V, Gupta R. Breast feeding and insulin levels in low birthweight neonates: a randomized study. Indian J Pediatr. 2010;77:509–13. doi: 10.1007/s12098-010-0065-6. [DOI] [PubMed] [Google Scholar]
- 115.Panagiotakos DB, Papadimitriou A, Anthracopoulos MB, Konstantinidou M, Antonogeorgos G, Fretzayas A, et al. Birthweight, breast-feeding, parental weight and prevelance of obesity in schoolchildren aged 10–12 years, in Greece; the Physical Activity, Nutrition and Allergies in Children Examined in Athens (PANACEA) study. Pediatr Int. 2008;50:563–8. doi: 10.1111/j.1442-200X.2008.02612.x. [DOI] [PubMed] [Google Scholar]
- 116.Hack M. Young adult outcomes of very-low-birth weight children. Seminars in Fetal and neonatal Medicine. 2006;11:127–37. doi: 10.1016/j.siny.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 117.Moster D, Lie R, Markestad T. Long-term medical and social consequences of preterm birth. N Engl J of Med. 2008;359:262–72. doi: 10.1056/NEJMoa0706475. [DOI] [PubMed] [Google Scholar]
- 118.Hack M, Daniel C, Flannery J, Schluchter M, Cartar L, Borawki E, Klein N. Outcomes in young adulthood for very-low-birth weight infants. N Engl J of Med. 2002;246:149–57. doi: 10.1056/NEJMoa010856. [DOI] [PubMed] [Google Scholar]
- 119.Dahl L, Kaaresen P, Tunby J, Handlegard B, Kvernmo S, Ronning J. Emotional, behavioral, social and academic outcomes in adolescents born with very low birth weight. Pediatrics. 2006;118:e449–59. doi: 10.1542/peds.2005-3024. [DOI] [PubMed] [Google Scholar]
- 120.Hack M, Youngstrom E, Cartar L, Schluchter M, Taylor G, Flannery D, et al. Behavioral outcomes and evidence of psychopathology amoung very low birth weight infants at age 20 years. Pediatrics. 2004;114:932–40. doi: 10.1542/peds.2003-1017-L. [DOI] [PubMed] [Google Scholar]
- 121.Raikkonen K, Pesonen A, Kajantie E, Heinonen K, Forsén T, Phillips D, et al. Length of gestation and depressive symptoms at age 60. Br J of Psychiatry. 2007;190:469–74. doi: 10.1192/bjp.bp.106.022145. [DOI] [PubMed] [Google Scholar]
- 122.Selling K, Cartensen J, Finnstrom O, Josefssonk A. Hospitalizations in adolescence and early adulthood among Swedish men and women born preterm and small for gestational age. Epidemiology. 2008;19:63–70. doi: 10.1097/EDE.0b013e318159074b. [DOI] [PubMed] [Google Scholar]
- 123.Lindstrom K, Lindblad F, Hjern A. Psychiatric morbidity in adolescents and young adults born preterm: a Swedish national cohort. Pediatrics. 2009;123:e47–53. doi: 10.1542/peds.2008-1654. [DOI] [PubMed] [Google Scholar]
- 124.Nilsson C, Jennische E, Ho HP, Eriksson E, Bjorntorp P, Holmang A. Postnatal endotoxin exposure results in increased insulin sensitivity and altered activity of neuroendocrine axes in the adult female rats. Eur J Endocrinol. 2002;146:251–60. doi: 10.1530/eje.0.1460251. [DOI] [PubMed] [Google Scholar]
- 125.Ward HE, Johnson EA, Salm AK, Birkle DL. Effects of postnatal stress on defensive withdrawal behavior and corticotropin releasing factor in rat brains. Physiol Behav. 2000;70:359–66. doi: 10.1016/s0031-9384(00)00270-5. [DOI] [PubMed] [Google Scholar]
- 126.Weinstock M, Polyrev T, Schorer-Apelbaum D, Men D, McCarty R. Effect of prenatal stress on plasma corticosterone and catecholamine and catecholamines in response to footschock in rats. Physiol Behav. 1998;64:439–44. doi: 10.1016/s0031-9384(98)00056-0. [DOI] [PubMed] [Google Scholar]
- 127.Nyirenda M, Lindsay R, Kenyon C, Burchell A, Seckl J. Glucocorticoid exposure in late gestation permanently programs rat hepatic phosphoenolpyruvate carboxykinase and glucorticoid receptor expression and causes glucose intolerance in adult offspring. J Clin Invest. 1998;101:2174–81. doi: 10.1172/JCI1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Phillips DI, Walker BR, Reynolds RM, Flanagan DE, Wood PJ, Osmond C, et al. Low birth weight predicts elevated plasma cortisol concentrations in adults from three populations. Hypertension. 2000;35:1301–6. doi: 10.1161/01.hyp.35.6.1301. [DOI] [PubMed] [Google Scholar]
- 129.Langley SC, Jackson AA. Increased systolic blood pressure in adult rats induced by fetal exposure to maternal low protein diets. Clin Sci. 1994;86:217–22. doi: 10.1042/cs0860217. [DOI] [PubMed] [Google Scholar]
- 130.Zhao Y, Fung C, Shin D, Shin BC, Thamotharan S, Sankar R, Ehnigher D, Silva A, Devaskar SU. Neuronal glucose transporter isoform 3 deficient mice demonstrate features of autism disorders. Molecular Psychiatry. 2010;15:286–99. doi: 10.1038/mp.2009.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ross MC, Desai M, Khorran O, McKnight RA, Lane RH, Torday J. Gestational programming of offspring obesity: a potential contributor to Alzheimer’s disease. Curr Alzheimer Res. 2007;4:213–7. doi: 10.2174/156720507780362056. [DOI] [PubMed] [Google Scholar]
- 132.Taveras E, Rifas-Shiman S, Belfort M, Kleinman K, Oken E, Gillamn M. Weight status in the first 6 months of life and obesity at 3 years of age. Pedatrics. 2009;123:1177–83. doi: 10.1542/peds.2008-1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wang Y, Gao E, Wu J, Zhou J, Yang Q, Walker MC. Fetal macrosomia and adolescent obesity: results from a longitudinal cohort study. Int J Obes. 2009;33:923–8. doi: 10.1038/ijo.2009.131. [DOI] [PubMed] [Google Scholar]
- 134.Eriksson J, Forsén T, Tuomilehto J, Osmond C, Barker D. Size at birth, childhood growth, and obesity in adult life. Int J Obesity. 2001;25:735–40. doi: 10.1038/sj.ijo.0801602. [DOI] [PubMed] [Google Scholar]
- 135.Hediger ML, Overpeck MD, McGlynn A, Kuczmarski RJ, Maurer KR, Davis WW. Growth and fatness at three to six years of age of children born small- or large-for-gestational age. Pediatrics. 1999;104:e33. doi: 10.1542/peds.104.3.e33. [DOI] [PubMed] [Google Scholar]
- 136.Boney CM, Verma A, Tucker R, Vohr RR. Metabolic syndrome in childhood: association with birth weight, maternal obesity, and gestational diabetes mellitus. Pediatrics. 2005;115:e290–96. doi: 10.1542/peds.2004-1808. [DOI] [PubMed] [Google Scholar]
- 137.Catalono PM, Thomas A, Huston-Presley L, Amini SB. Increased fetal adiposity: a very sensitive marker of abnormal in utero development. Am J Obstet Gynecol. 2003;189:1698–1704. doi: 10.1016/s0002-9378(03)00828-7. [DOI] [PubMed] [Google Scholar]
- 138.Vela-Huera MM, San Vicente-Santoscoy EU, Guizar-Mendoza JM, Amador-Licona N, Aldana-Valenzuela C, Hernandez J. Leptin, insulin and glucose serum levels in large-for-gestational age infants of diabetic and non-diabetic mothers. J Pediatr Endocrinol Metab. 2008;21:17–22. doi: 10.1515/jpem.2008.21.1.17. [DOI] [PubMed] [Google Scholar]
- 139.McLean M, Chipps D, Cheung NW. Mother to child transmission of diabetes mellitus: does gestational diabetes program Type 2 diabetes in the next generation? Diabet Met. 2006;23:1213–5. doi: 10.1111/j.1464-5491.2006.01979.x. [DOI] [PubMed] [Google Scholar]
- 140.Nilsen TIL, Romundstad PR, Troisi R, Potischman N, Vatten LJ. Birth size and colorectal cancer risk: a prospective population based study. Gut. 2005;54:1728–32. doi: 10.1136/gut.2004.060475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sandhu MS, Luben R, Day NE, Khaw K. Self-reported birth weight and subsequent risk of colorectal cancer. Cancer Epidemiol Biomarkers Prevent. 2002;11:935–8. [PubMed] [Google Scholar]
- 142.Newberne PM, Bueche D, Suphiphat V, Schrager TF, Sahaphong S. The influence of pre- and postnatal caloric intake on colon carcinogenesis. Nutr Cancer. 1990;13:165–73. doi: 10.1080/01635589009514057. [DOI] [PubMed] [Google Scholar]
- 143.Kaijser M, Akre O, Cnattingius S, Ekbom A. Preterm birth, birth weight and subsequent risk of female cancer. Br J of Cancer. 2003;89:1664–6. doi: 10.1038/sj.bjc.6601357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Vatten LJ, Maehle BO, Lungd Nilsen TI, Tretli S, Hsieh CC, Trichopoulos D, et al. Birth weight as a predictor of breast cancer: a case-control study in Norway. Br J Cancer. 2002;86:89–91. doi: 10.1038/sj.bjc.6600011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.De Stavola BL, Hardy R, Kuh D, Silva I, Wadswork M, Swerdlow AJ. Birthweight, childhood growth and risk of breast cancer in a British cohort. Br J of Cancer. 2000;83:964–8. doi: 10.1054/bjoc.2000.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ahlgren M, Melbye M, Wohlfahrt J, Sorensen T. Growth patterns and the risk of breast cancer in women. N Engl J Med. 2004;35:1619–26. doi: 10.1056/NEJMoa040576. [DOI] [PubMed] [Google Scholar]
- 147.Kaijser M, Lichtenstein P, Granath F, Erlandsson G, Cnattingius S, Ekbom A. In utero exposures and breast cancer: a study of opposite sex twins. J Natl Cancer Inst. 2001;93:60–2. doi: 10.1093/jnci/93.1.60. [DOI] [PubMed] [Google Scholar]
- 148.Trichopoulos D. Hypothesis: does breast cancer originate in utero? Lancet. 1990;335:939–40. doi: 10.1016/0140-6736(90)91000-z. [DOI] [PubMed] [Google Scholar]
- 149.Trichopoulos D. Intrauterine environment, mammary gland mass and breast cancer risk. Breast Cancer Res. 2003;5:42–4. doi: 10.1186/bcr555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ekbom A, Hsieh C, Lipworth L, Wolk A, Pontein J, Adami H, et al. Evidence of prenatal influences on breast cancer. Lancet. 1992;340:1015–18. doi: 10.1016/0140-6736(92)93019-j. [DOI] [PubMed] [Google Scholar]
- 151.Raychaudhuri N, Raychaudhuri S, Thamotharan T, Devaskar SU. Histone code modifications repress glucose transporter 4 expression in the intrauterine growth-restricted offspring. J Biol Chem. 2008;283:13611–26. doi: 10.1074/jbc.M800128200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Simmons RA. Developmental origins of {beta}-cell failure in type 2 diabetes: the role of epigenetics. Pediatr Res. 2007;61:64R–7R. doi: 10.1203/pdr.0b013e3180457623. [DOI] [PubMed] [Google Scholar]
- 153.McDaniell R, Lee B, Song L, Liu Z, Boyle A, Erdos M, et al. Heritable individual-specific and allele-specific chromatin signatures in humans. Science. 2010;328:235–9. doi: 10.1126/science.1184655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.http://www.who.int/int/nutrition/topics/fetal_dev_report_EN.pdf