

Abstract
Background
No efficient vaccine against plague is currently available. We previously showed that a genetically attenuated Yersinia pseudotuberculosis producing the Yersinia pestis F1 antigen was an efficient live oral vaccine against pneumonic plague. This candidate vaccine however failed to confer full protection against bubonic plague and did not produce F1 stably.
Methodology/Principal Findings
The caf operon encoding F1 was inserted into the chromosome of a genetically attenuated Y. pseudotuberculosis, yielding the VTnF1 strain, which stably produced the F1 capsule. Given orally to mice, VTnF1 persisted two weeks in the mouse gut and induced a high humoral response targeting both F1 and other Y. pestis antigens. The strong cellular response elicited was directed mostly against targets other than F1, but also against F1. It involved cells with a Th1—Th17 effector profile, producing IFNγ, IL-17, and IL-10. A single oral dose (108 CFU) of VTnF1 conferred 100% protection against pneumonic plague using a high-dose challenge (3,300 LD50) caused by the fully virulent Y. pestis CO92. Moreover, vaccination protected 100% of mice from bubonic plague caused by a challenge with 100 LD50 Y. pestis and 93% against a high-dose infection (10,000 LD50). Protection involved fast-acting mechanisms controlling Y. pestis spread out of the injection site, and the protection provided was long-lasting, with 93% and 50% of mice surviving bubonic and pneumonic plague respectively, six months after vaccination. Vaccinated mice also survived bubonic and pneumonic plague caused by a high-dose of non-encapsulated (F1-) Y. pestis.
Significance
VTnF1 is an easy-to-produce, genetically stable plague vaccine candidate, providing a highly efficient and long-lasting protection against both bubonic and pneumonic plague caused by wild type or un-encapsulated (F1-negative) Y. pestis. To our knowledge, VTnF1 is the only plague vaccine ever reported that could provide high and durable protection against the two forms of plague after a single oral administration.
Author Summary
Yersinia pestis, the agent of plague, is among the deadliest infectious agents affecting humans. Injected in the skin by infected fleas, Y. pestis causes bubonic plague, which occasionally evolves into the very lethal and contagious pneumonic plague. Y. pestis is also a dangerous potential bioweapon but no plague vaccine is available. The current study describes the development of a vaccine highly efficient against plague in both its bubonic and pneumonic forms. The strategy consists of a live, avirulent, genetically modified Yersinia pseudotuberculosis that produces the capsule antigen of Y. pestis, named F1. The goal was to propose a vaccine that would be both easy to produce rapidly in large amounts with high quality, and easy to administer to individuals via a single oral dose. The VTnF1 strain described fulfills these demands. The immune response generated is long-lasting, involving both antibodies and memory cells directed against F1 and other antigens. We conclude that VTnF1 is a very promising candidate vaccine against plague.
Introduction
Plague has been one of the deadliest bacterial infections in human history, causing millions of deaths during three major historical pandemics and leaving an indelible mark engraved in human's collective memory. In addition to ancient foci of the disease in Asia and Africa, the last pandemic (plague of modern times), which started one century ago, allowed plague to develop new foci in previously unaffected territories such as Madagascar, Southern Africa, and the Americas. Despite considerable progress in its prevention and cure during the 20th century, plague has recently made a new comeback, causing close to 50,000 human cases during the last twenty years [1], including cases in countries where plague was thought to be extinct [2]. Therefore, plague is categorized by WHO (World Health Organization) as a re-emerging disease [1, 3].
The etiologic agent of plague, Yersinia pestis, is a highly pathogenic Gram-negative bacillus, very recently derived from the much less virulent enteropathogen Yersinia pseudotuberculosis [4]. Transmission of the plague bacillus to humans generally starts with the bite of an infected flea, causing bubonic plague, the most frequent clinical form of the disease. Y. pestis occasionally reaches the airways, and the resulting secondary pneumonic plague is highly contagious due to the emission of infected aerosols, causing inter-human transmission of pneumonic plague. This pneumopathy is systematically lethal in usually less than three days if no treatment is administered.
The possible use of the plague bacillus as a bioterrorist weapon is also a serious threat due to its pathogenicity and human-to-human transmission. Y. pestis has been classified by the Centers for Disease Control (CDC) of the USA among Tier 1 select biological agents. Different strains of Y. pestis showing resistance to antibiotics currently used to treat patients have been identified in Madagascar [5]. Antibiotherapy can therefore no longer be considered as sufficient against the natural and intentional danger of plague. Facing such a public health risk, vaccines may be one of the only remaining alternatives to limit the death toll in humans. A plague vaccine should confer protection against bubonic plague, the most frequent form of the disease in nature [1], at the origin of pneumonic plague outbreaks. The vaccine should also protect against pneumonic plague, the most contagious and fatal form of the disease.
No plague vaccine is currently licensed. The live attenuated Y. pestis strain EV76 and its derivatives have previously been used in humans [6, 7], and were found to confer protection. However, the genetic instability of Y. pestis represents a major obstacle in its use as live vaccine [4, 8]. Several molecular vaccine candidates have been recently developed, among which two molecular vaccines (RypVaxtm and rF1Vtm) are the most advanced in clinical trials [9, 10]. These vaccines rely on a combination of two peptides: the F1 antigen composing the Y. pestis capsule and the LcrV component of the Type Three Secretion System (TTSS) [9, 10], which are efficient targets of protective immunity against plague [6, 11]. Molecular vaccines are generally adjuvanted with alum, and thus are good inducers of antibody production but poor inducers of cellular immune response [12, 13]. Cellular immunity is, however, important for plague protection [14], and a weak cellular response could explain why F1-V vaccinated African Green Monkeys were poorly protected despite adequate antibody titers [15].
We recently proposed a vaccine strategy against plague based on an oral vaccination with a live, attenuated strain of Y. pseudotuberculosis [16, 17]. Because this species is a recent ancestor of Y. pestis, the two species are genetically almost identical, whereas Y. pseudotuberculosis has much lower pathogenicity and much higher genomic stability [4]. Due to their immunogenicity and antigenic complexity, live vaccines generate both humoral and cell-mediated immune responses without addition of adjuvant, and the response is directed against multiple target antigens, thus inducing an immunological response that could not be circumvented by genetic engineering of Y. pestis. In addition, a live vaccine, once developed and validated, could easily and rapidly enter mass production, and would be well suited in response to an emergency need. As a proof of concept, we had reported that oral inoculation of two doses of a live, naturally attenuated Y. pseudotuberculosis could provide 88% protection against bubonic plague [16].
The initial Y. pseudotuberculosis strain that we tested was not genetically defined [16]. To develop a vaccine strain both avirulent and genetically defined, the virulent Y. pseudotuberculosis IP32953 strain, whose genome is known [4], was irreversibly attenuated by deletion of genes encoding three essential virulence factors (the High pathogenicity island, YopK, and the pH6 antigen (PsaA [17]). To increase vaccine efficiency, an F1-encapsulated derivative was constructed. This was obtained by cloning the Y. pestis F1-encoding caf operon into a plasmid, and introducing this plasmid into the attenuated V674 Y. pseudotuberculosis, thus producing the V674pF1 strain. Oral vaccination using 108 CFU protected 100% against pneumonic plague caused by a challenge with 33 LD50 Y. pestis and 80% against a high-dose challenge (3,300 LD50) [17]. However, we subsequently found that vaccination with strain V674pF1 protected only 81% of the mice against bubonic plague caused by subcutaneous injection of a moderate dose (100 LD50) of Y. pestis, a level of protection that was judged insufficient. We also observed that production of F1 was not stable and homogenous in the V674pF1 vaccine strain, possibly accounting for the incomplete protection conferred.
The aim of this study was to generate a new vaccine strain that would allow a homogenous production of F1, and to evaluate the immune response elicited and its protective performances against both bubonic and pneumonic plague.
Methods
Ethics statement
Animals were housed in the Institut Pasteur animal facility accredited by the French Ministry of Agriculture to perform experiments on live mice (accreditation B 75 15–01, issued on May 22, 2008), in compliance with French and European regulations on the care and protection of laboratory animals (EC Directive 86/609, French Law 2001–486 issued on June 6, 2001). The research protocol was approved by the French Ministry of Research (N° CETEA 2013–0038) and was performed in compliance with the NIH Animal Welfare Insurance (#A5476-01 issued on 02/07/2007).
Bacterial strains and culture conditions
The Y. pseudotuberculosis and Y. pestis isolates used in this study and their derivatives are described in S1 Table [17]. Bacteria were grown at 28°C in Luria-Bertani (LB) broth or on LB agar plates supplemented with 0.002% (w/v) hemin (LBH). Bacterial concentrations were evaluated by spectrometry at 600 nm and plating on LBH or LB plates. Chloramphenicol (Cm, 25 μg/ml), ampicillin (Amp, 100 μg/ml), kanamycin (Km, 30 μg/ml), spectinomycin (Spec, 50 μg/ml), or irgasan (0.1 μg/ml) were added to the media when necessary. All experiments involving Y. pestis strains were performed in a BSL3 laboratory.
Cloning of the Y. pestis caf operon
To introduce the caf operon into the Y. pseudotuberculosis chromosome, the Tn7 transposition tool was used [18]. First, a mini-Tn7 containing a Cm resistance cassette was constructed by cloning the Cm-FRT KpnI-fragment from pFCM1 into pUCR6Kmini-Tn7 digested with KpnI. The resulting plasmid, named pUCR6K-miniTn7-Cm-FRT, was then digested by ApaI and EcoRI and ligated to the 5 kb-PCR fragment containing the entire caf operon from Y. pestis amplified with primers A (5’-ATAAGAATGAATTCGTGACTGATCAATATGTTGG-3’) and B (5’-CGTTAGGGCCCGTCAGTCTTGCTATCAATGC-3’), which added ApaI and EcoRI sites at the extremities of the fragment. To insert the caf locus into the Y. pseudotuberculosis V674 chromosome, plasmids pUC18R6KTn7-caf-CmR and pTNS2 (transposase provider) were introduced together into V674 by electroporation. Transposants were selected on LB agar plates containing chloramphenicol and verified for their sensitivity to ampicillin. Presence of the transposon Tn7-caf-CmR at the chromosomal att-Tn7 site was verified by PCR, using two pairs of primers: A (5’-CACAGCATAACTGGACTGATTTC-3’) and B (5’- GCTATACGTGTTTGCTGATCAAGATG-3’) for the left junction, and C (5’-ATTAGCTTACGACGCTACACCC-3’) and D (5’- ACGCCACCGGAAGAACCGATACCT-3’) for the right junction.
The recombinant Y. pseudotuberculosis strain containing the Tn7-caf-CmR region in its chromosome was initially named V674TnF1, and its use as a vaccine against plague is protected by patent application PCT/IB2012/001609, issued on August 7, 2012. For simplicity, we hereafter refer to this strain as "VTnF1".
To analyze F1 capsule production, bacteria were visualized by optical microscopy in contact with India ink, and an ELISA assay quantifying the F1 capsule on bacteria was performed as previously described [17].
Construction of the bioluminescent Y. pestis CO92 Tn7ail-lux
A mini-Tn7 transposon containing a Km resistance cassette was constructed by cloning the Km-FRT SacI-fragment from pFKM1 into pUCR6Kmini-Tn7 digested with SacI. The recombinant pUCR6KTn7-Km plasmid was then digested with ApaI and XmaI, and ligated to the 5878 bp ApaI/XmaI fragment from pGEN-lux (LuxCDABE provider;[19]). The resulting pUCR6KTn7-luxCDABE plasmid was then digested with SpeI and XmaI and ligated to the 109 bp promoter region of ail (YPO2905), obtained from the Y. pestis DNA template by PCR with primers E (5'- CGCACTAGTTGGAATACTGTACGAATATCC-3') and F (5'- ataCCCGGGccagattgttataacaatacc). The resulting plasmid was named pUCR6KTn7-Pail-lux. For transposition of Tn7-Pail-lux into the Y. pestis chromosome, plasmids pUCR6KTn7-Pail-lux and pTNS2 were introduced into CO92 bacterial cells by electroporation. Transposants were selected for with LBH agar plates containing Km. Verification of the CO92::Tn7-Pail-lux recombinant was performed by PCR using the primer pairs A/B and C/D for chromosomal integration, and by measurement of photon emission using a Xenius plate reader (SAFAS Monaco) for bioluminescence activity. The virulence of the recombinant CO92::Tn7-Pail-lux derivative upon s.c. injection was checked and was found to be similar to that of the wild-type strain, with a median lethal dose (LD50) of 10 CFU. In vivo imaging was performed with an In Vivo Imaging System (IVIS 100, Caliper Life Sciences).
Animal immunization and in vivo analyses
Mouse vaccinations were performed in a BSL3 animal facility as described previously [17]. Animals were seven-week-old OF1 female mice or (when specified) C57BL/6 mice from Charles River France. Bacterial suspensions (200 μl in saline) were given intragastrically (i.g.) to mice using a curved feeding needle. Animals were monitored for suffering (prostration, ruffled hair) every other day and were weighed to estimate the impact of vaccination. The virulence of the VTnF1 strain by the oral route was tested by infecting i.g. groups of mice (four per dose) with increasing doses of bacteria in order to determine the LD50. The LD50 value was calculated by the Spearman-Karber method [20]. In vivo dissemination of the VTnF1 strain was examined as described previously [17]. Feces (two fecal pellets) were collected from live mice and were homogenized in PBS using disposable homogenizers (Piston Pellet from Kimble Chase, Fisher Sci.). Peyer's patches, spleen, liver and mesenteric lymph nodes were collected aseptically from euthanized mice. They were homogenized in sterile PBS using three mm glass beads and an electric mill (TissueLyser, Qiagen). The bacterial load was determined by plating serial dilutions of the homogenates.
Plague challenge experiments
Mice were challenged either four weeks or six months after vaccination. Y. pestis strains were grown at 28°C on LBH plates, and suspensions in saline were prepared for infections. Mice were infected by s.c. injection (100 μl) in the ventral skin on the linea alba. The LD50 of the CO92Δcaf strain by this route was determined by infecting mice (six per dose) with serial dilutions of bacterial suspensions, and was found to be 100 CFU. To induce pneumonic plague, anesthetized mice were infected i.n. as previously described [17] by instillating 10 μl of bacterial suspensions in nostrils (5 μl each). Animal survival was followed for 21 days.
Evaluation of the immune response
Mouse blood was collected three weeks after vaccination from live animals by puncture of the maxillary artery with a Goldenrod lancet (Medipoint, USA). Microtiter plates (NUNC) were coated either with F1 antigen, or a sonicate of the Y. pestis CO92Δcaf strain (10 μg/ml). The F1 antigen was obtained from Y. pestis as described previously [21]. The sonicate of the Y. pestis CO92Δcaf strain was obtained by sonication of bacteria grown at 37°C on LB agar as previously described [17]. The Yops antigens were purified as previously described [22], and assays were performed as described before [17]. Briefly, plates were blocked with 5% defatted dry milk and 0.1% Tween 20 in PBS. Sera serially diluted in PBS containing 0.1% BSA were incubated in wells and bound antibodies were detected using horseradish peroxydase (HRPO)–coupled rat antibodies specific for mouse IgG (Bio-Rad). HRPO activity was revealed using TMB substrate (OptiEIA, BD Pharmingen). Antibody (Ab) titers were calculated as the reciprocal of the lowest sample dilution giving a signal equal to two times the background. To analyze immunoglobulin isotypes, horseradish peroxidase (HRP)-coupled probes directed against mouse IgG1, IgG2a, IgG2b and IgM (Caltag) were used. IgG3 were detected using an uncoupled goat antibody (Abcam), and revealed with an HRP-coupled rabbit antibody against goat IgG (BioRad). For Western blotting, Y. pestis CO92, wild-type and Δcaf, S1 Table) were boiled in Laemmli sample buffer (Thermo) and were loaded on 12% acrylamide gel for SDS-PAGE separation using a MiniProtean device (BioRad). Migrated material was transferred from the gel onto a PVDF membrane (Amersham). All subsequent steps were performed following the western immunoblotting protocol recommended by Cell Signaling Technology (USA). Membrane strips were incubated overnight in pooled 1/100 diluted sera from naïve mice, or mice vaccinated one month earlier. Bound IgG were revealed using a secondary goat anti-mouse IgG coupled to horseradish peroxidase (BioRad). ECL Plus kit (Pierce) was used for peroxidase revelation, and membranes were photographed using a ChemiDoc apparatus (BioRad).
To evaluate the cellular memory in vaccinated animals, splenocytes were cultured as described previously [17]. Briefly, spleens from euthanized animals were dissociated and erythrocytes were lyzed using Gey’s hemolytic solution [23]. Cells extensively washed with cold PBS were resuspended in RPMI 1640 + Glutamax (Invitrogen) supplemented with 5% fetal bovine serum, penicillin + streptomycin, and 10 mM ß-mercaptoethanol. Cells (5x106/condition) were stimulated with either a sterile Y. pestis CO92Δcaf sonicate (5 μg/ml), sterile F1 antigen (5 μg/ml), or Concanavalin A (1 μg/ml; Sigma) as a positive control. After three days, the supernatant was collected and the cytokine content was determined using IFNγ, IL-1β, IL-10, and IL-17 assays (Duosets, R&D Systems).
Statistical analyses
The Log-rank (Mantel-Cox) test was used to compare survival curves. Unpaired Mann-Whitney or Student's t tests were used to compare bacteria numbers, animal weights, antibody titers, and cytokine production. The paired Mann-Whitney test was used for bioluminescence results. Analyses were performed with Prism 6.0 software (GraphPad Software).
Results
Construction of a Y. pseudotuberculosis strain stably producing the F1 capsule
Strain VTnF1 was constructed by inserting the caf operon encoding F1 [17] into the chromosome of the attenuated Y. pseudotuberculosis V674, using mini-Tn7 transposon technology [18]. F1 capsule production by recombinant VTnF1 grown at 37°C in LB broth was tested using an F1-specific rapid dipstick test [21], which was clearly F1 positive (S1 Fig). Microscopic visualization of VTnF1 in India ink revealed that all VTnF1 bacterial cells produced the F1 capsule, as visualized by the repulsion of ink particles with comparable thickness (Fig 1A). This contrasted with V674pF1 cultures, which displayed encapsulated and non-encapsulated bacteria. To quantify F1 capsule production, isolated colonies obtained after growth in LB broth at 37°C were tested using an F1-specific ELISA. All VTnF1 colonies were F1 positive and their F1 levels were homogenous (Fig 1B), whereas V674pF1 colonies exhibited various levels of F1 at their surface, indicating both heterogeneity and instability of F1 production. VTnF1 produced as much F1 as Y. pestis, and this production was temperature-dependent (Fig 1C).
Fig 1. F1 capsule production.

(A) V674, V674pF1, and VTnF1 bacteria grown at 37°C were re-suspended in India ink and observed by microscopy. (B) To evaluate the stability of F1 production, isolated colonies of Y. pestis CO92, or Y. pseudotuberculosis V674pF1, and VTnF1 were obtained after three subcultures in vitro. Isolated VTnF1 colonies were also obtained by culturing an homogenate of Peyer’s patches taken from mice previously inoculated with VTnF1 (108 CFU i.g.; noted "in vivo"). Serial dilutions of bacteria were tested using an F1-specific ELISA and the F1 index shown was calculated as 1/CFU yielding DO450nm = 1 in the ELISA. The un-encapsulated V674 was used as a negative control. (C) F1 production by VTnF1 was compared to that of Y. pestis CO92 after growth at 28°C or 37°C, using an F1-specific ELISA, and CO92Δcaf was used as negative control.
Stability of F1 production was additionally tested after VTnF1 growth in vivo in mice. To this aim, VTnF1 was injected i.g. to animals, and five days later, bacteria were recovered from Peyer’s patches. All VTnF1 colonies were positive for F1 by ELISA (Fig 1B). F1 production was homogenous and comparable to F1 levels of colonies obtained after in vitro culture. This demonstrates that VTnF1 stably and homogenously produced F1 after growing in vivo in mice.
VTnF1 is rapidly cleared from orally inoculated mice
The V674 strain used to construct VTnF1 was strongly attenuated after intragastric inoculation into mice (LD50 >1010 CFU;[17]). VTnF1 exhibited an attenuation of virulence similar to V674 and the previous vaccine V674pF1, with an LD50 >1010 CFU. Mice vaccinated with VTnF1 at a dose of 108 CFU presented transient signs of infection (ruffled hair), and a slight delay in weight gain from day three to seven post-vaccination (average 1.7 g), but they recovered a normal weight from day 10 (S2 Fig). The F1 capsule is dispensable for Y. pestis virulence in many mouse strains [24–27], but not in others such as C57BL/6 [28]. When a high dose of VTnF1 (4x109 CFU) was inoculated orally to C57BL/6 mice (N = 7), no lethality was observed during the three weeks of follow-up. This confirmed that VTnF1 was strongly attenuated, even for C57BL/6 mice that are more susceptible to F1 action.
After oral inoculation of VTnF1 (108 CFU), the bacteria were detected on day five-six in the feces and Peyer's patches of all mice (Fig 2A and 2B), and in the spleen, liver and mesenteric lymph nodes (MLN) of most animals (Fig 2C and 2E). However, bacteria were almost completely cleared from the spleen, Peyer's patches and MLN after 15 days, and from the liver after 26 days. Feces tested monthly during the following 5 months remained negative, indicating a vaccine clearance from visceral organs and the gut lumen.
Fig 2. In vivo dissemination of VTnF1 after oral vaccination.
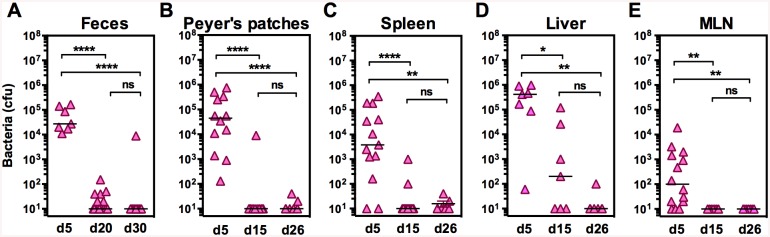
Groups of mice were inoculated orally with the VTnF1 vaccine (108 CFU) and were sacrificed at the indicated times to evaluate the VTnF1 loads in: (A) feces (two pellets/mouse), (B) Peyer’s patches (two patches/mouse), (C) the spleen (whole organ), (D) the liver (whole organ), and (E) mesenteric lymph nodes (all). Samples were minced and dilutions were plated on selective agar plates containing kanamycin to count colonies, with a detection limit of 10 CFU/sample. Shown are individual values from 7–14 mice per condition. The horizontal line indicates the median. The Mann Whitney test was used for statistical analysis: *: p ≤0.05, **: p <0.01, ***: p<0.001.
The humoral immune response elicited by VTnF1 targets multiple antigens, including F1, and is long-lasting
The humoral immune response elicited by vaccination with VTnF1 (108 CFU orally) was first evaluated by quantifying serum antibodies against purified F1 at different times post-vaccination over a period of six months. Anti-F1 IgG were detectable as early as four days after vaccination, and reached plateau values after 7 days (Fig 3A). They then maintained at high levels without significant evolution, as shown by the fact that titers observed after six months (d180) were not significantly different from those at d30 (p = 0.08, 14 mice per group). Analysis of the immunoglobulin isotypes revealed that both IgG1, IgG2a, IgG2b and IgG3 contributed to this humoral response, whereas IgM peaked rapidly after vaccination and then fell to low levels (Fig 3B).
Fig 3. Humoral immune response induced by vaccination.

(A) The antibody production induced by VTnF1 vaccination (108 CFU) was quantified at various days post vaccination using an ELISA measuring IgG directed against purified F1 antigen. Shown are the means ± sem of titers observed in two experiments performed during the first month post-vaccination (Exp.1, 7 mice) and during the six-months post-vaccination period (Exp.2, 14 mice). Mean IgG titers at each time point were compared statistically to that on day zero using the Mann-Whitney test, and all were significantly higher (p<0.001), except those on day 1. (B) The different Ig mouse isotypes present in sera were analyzed at sequential times. (C) The IgG response against antigens other than F1 was analyzed by ELISA against a Y. pestis CO92Δcaf sonicate, and (D) by western blotting. The Y. pestis CO92 wild type or CO92Δcaf (noted F1-) were used as source of blotted antigens and sera pooled either from naive mice or mice vaccinated 30 days earlier (noted VTnF1). The Caf1 band is indicated by an asterisk and the Caf1M band by a triangle. (E) The response against purified Yops was also analyzed by ELISA. Shown are results from 14 individual animals (dots), and group medians (horizontal line). The Mann-Whitney test was used for statistical analysis: ***: p<0.001.
IgG recognizing Y. pestis antigens other than F1 were quantified by ELISA using a sonicate of Y. pestis CO92Δcaf as target. All vaccinated mice had high amounts of IgG against Y. pestis CO92Δcaf antigens (Fig 3C). Western blotting analysis (Fig 3D) confirmed that in addition to F1, at least 12 target antigens were recognized by immune sera. Because conformational epitopes were lost due to the denaturing conditions used for electrophoresis, actual targets were probably more numerous. Among antigens strongly recognized are two components of the Caf operon (absent from the CO92Δcaf strain, Fig 3D): the Caf1 and Caf1M antigens (MW 15.6 and 28.7 kDa respectively). Finally, IgG against purified Yops were also measured because these molecules are essential Y. pestis virulence factors. Such IgG were observed in almost all mice, but with varying levels (Fig 3E). Altogether, our results indicate that the humoral immune response developed rapidly after a single-dose vaccination and involved all main IgG isotypes. The antibodies recognized several antigens other than F1 and were maintained at high levels for extended periods of time without recall vaccination.
VTnF1-induced memory cells directed toward F1 and other antigens display a pro-inflammatory functional profile
To evaluate the antigen-specific T cell memory elicited by VTnF1, splenocytes taken from vaccinated animals were re-stimulated in vitro with either purified F1 antigen, or with a sonicate of the un-encapsulated Y. pestis CO92Δcaf. Cells from mice vaccinated one month earlier with VTnF1 produced IFNγ, IL-17, and IL-10 in response to F1 (Fig 4). Although these levels were low, they were significantly higher than those of control mice (Fig 4), indicating that vaccination mobilized F1-specific memory T cells producing both pro- (IFNγ, IL-17) and anti-inflammatory (IL-10) cytokines. IL-1β production was also measured, but levels were very low (<0.02 ng/ml) in all conditions.
Fig 4. Cellular immune response of vaccinated mice.
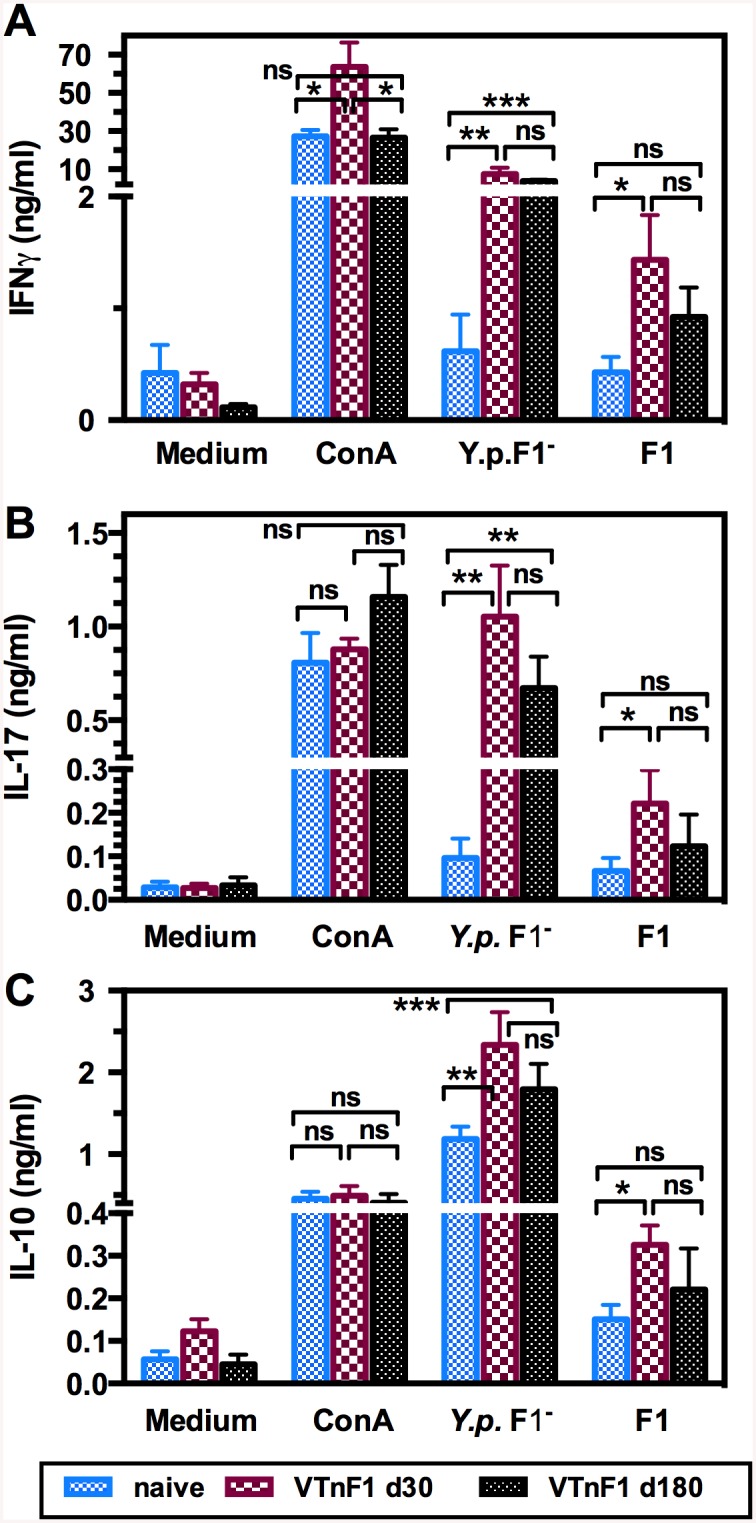
Splenocytes isolated from mice vaccinated orally with VTnF1 (108 CFU) 30 days (dark red boxes), or 180 days (black boxes) earlier, or from unvaccinated (naive) mice (blue boxes), were stimulated in vitro with 5 μg/ml of either an antigenic preparation of Y. pestis CO92Δcaf (noted Y.p.F1-) or purified F1 antigen, or the mitogen Concanavalin A (ConA, 1 μg/ml) as positive control. Supernatants taken three days after stimulation were tested for the presence of IFNγ (A), IL-17 (B), and IL-10 (C) by ELISA. Shown are the mean ± s.e.m. of 14 mice per condition (two pooled experiments). The unpaired Mann Whitney test was used for statistical analysis: *: p<0.05, **: p<0.01, ***: p<0.001, ns: not significant. Comparable results were obtained in two other experiments using naive and 30-days vaccinated mice (16 per group).
In contrast, cells from VTnF1 vaccinated mice produced high amounts of IFNγ and IL-17 in response to non-F1 Y. pestis antigens, reaching levels at least 10 times higher than those observed for cells from naive mice (Fig 4A and 4B). Strikingly, levels of IL-17 were comparable to those induced by the mitogenic lectin ConA, used as a positive control, indicating that a large proportion of responding splenocytes of vaccinated animals recognized Yersinia antigens and displayed potent pro-inflammatory functions. This cellular response was also much higher than that stimulated by the F1 antigen alone, reflecting the mobilization of T cells directed against multiple Y. pestis antigenic targets.
Y. pestis antigens induced production of IL-10 by splenocytes from both naive and vaccinated mice, probably due to the response of innate immunity cells such as macrophages. However, splenocytes from vaccinated mice produced significantly higher levels of IL-10 than cells from naive mice stimulated with both F1 and other antigens, revealing the recall response of Y. pestis-specific memory cells with anti-inflammatory activity (Fig 4C).
To evaluate the durability of the cell-mediated response, splenocytes from mice vaccinated six months earlier were also tested. Levels of IFNγ, IL-17 and IL-10 in response to F1 and non-F1 antigens were lower on day 180 than on day 30 post-vaccination, but this difference was not statistically significant. Thus, although slightly reduced, the cell-mediated memory persisted after six months.
VTnF1 confers high protection against both bubonic and pneumonic forms of plague
To determine the protective efficacy of VTnF1 against pneumonic plague, vaccinated mice were challenged intranasally with the fully virulent Y. pestis CO92 strain four weeks after a single oral vaccination (108 CFU of VTnF1). 100% of mice challenged with 105 CFU (33 LD50) of Y. pestis CO92 survived (Fig 5A). Vaccination also protected 100% of the animals exposed to an extremely severe challenge with 107 CFU CO92 (i.e. 3,300 LD50; Fig 5A), whereas the previous V674pF1 vaccine strain protected only 80% of the mice infected with this dose [17]. VTnF1 thus appears more protective.
Fig 5. Protection against bubonic and pneumonic plague of mice vaccinated with VTnF1.

Mice having received a single oral dose of VTnF1 (108 CFU) were challenged via i.n. or s.c. routes, with various doses of bacteria as indicated. (A-D) Animals were challenged four weeks after vaccination by injection of (A, B) Y. pestis CO92, or (C, D) the un-encapsulated CO92Δcaf. (E) Vaccinated mice were challenged six months after vaccination. Mouse survival was recorded daily for 21 days. The number of mice surviving / number of animals tested is indicated above the corresponding bar for each condition. The Fisher Exact test was used for statistical analysis: *: p≤0.05; ***: p<0.001.
To evaluate the protective efficacy of VTnF1 against bubonic plague, vaccinated mice were infected s.c. with Y. pestis CO92, four weeks after vaccination. A single oral dose (108 CFU) of the VTnF1 vaccine protected 100% of the mice against 103 CFU (100 LD50) of CO92 (Fig 5B). Compared to V674pF1 which only protected 81% (13/16) of animals against bubonic plague in these conditions, VTnF1 was again more protective. When animals vaccinated with VTnF1 received a very severe challenge by s.c. injection of 105 CFU CO92 (10000 LD50), 93% of mice (13/14) were still protected.
Because the immune memory is known to decline with time, the protection conferred by VTnF1 was evaluated six months (one third of an OF1 mouse's lifespan [29] after a single-dose oral vaccination with VTnF1. VTnF1 was completely undetectable in mice's feces at day 30 post vaccination and the following months. Upon s.c. challenge with 100 LD50 of Y. pestis CO92, 93% of the animals were still protected against bubonic plague. Furthermore, 50% of the mice survived an i.n. challenge with 33 LD50 of Y. pestis CO92 six months after vaccination (Fig 5D). This indicates that the immune memory elicited by the vaccine persisted and provided a long-lasting protection.
The capacity of VTnF1 vaccination to confer protection against bubonic plague caused by a non-encapsulated Y. pestis was also evaluated by challenging vaccinated mice with Y. pestis CO92Δcaf. A single oral dose of VTnF1 conferred 100% protection against a severe intranasal challenge with 107 CFU of Y. pestis (107 CFU, i.e. 3,300 LD50, Fig 5C). The same vaccination protected 93% of vaccinated mice against a severe s.c. challenge with 105 CFU (104 LD50; Fig 5D). Thus, VTnF1 conferred a strong protection against Y. pestis in the absence of the F1 pseudocapsule, indicating that, even if plague was caused by a natural or genetically modified F1-negative Y. pestis, vaccination with VTnF1 would provide a high level of protection.
Vaccinated mice rapidly control Y. pestis systemic spread
To determine the stage of the infectious process at which immunity provided by VTnF1 controls Y. pestis proliferation, infection by a bioluminescent Y. pestis strain (CO92 Tn7ail-lux) was followed in vivo in live mice. The strain carries in its chromosome the lux operon under the control of the ail promoter, known to be very active during bubonic plague [30]. In unvaccinated animals, Y. pestis multiplied at the site of injection and spread to other organs, causing the death of two mice out of five at 92h post-injection. (Fig 6A). In VTnF1-vaccinated mice, the bioluminescence signal was much lower after 20 hours at the site of injection (Fig 6B) and after 44 hours it was no longer visible. Dissemination outside of the injection site was not observed in vaccinated mice. Therefore, vaccination induced fast-acting immune mechanisms that prevented the dissemination of Y. pestis from the site of injection.
Fig 6. Vaccination with VTnF1 prevents Y. pestis dissemination from the site of infection.

(A) Mice vaccinated with VTnF1 (108 CFU i.g.) were infected four weeks later by s.c. injection of 103 CFU of the bioluminescent Y. pestis CO92::Tn7-Pail-lux in the ventral skin. Whole body bioluminescence of the animals was recorded at regular time intervals using an IVIS camera. Shown are white light photographs merged with the bioluminescence signal, scaled in the right margin. (B) The bioluminescence intensity emitted at the site of injection was measured for each animal and shown are means ± s.e.m. for each group. Animals missing at time point 92h died from infection before this bioluminescence recording. The paired Mann-Whitney test was used for statistical analysis: **: p<0.01.
Discussion
To meet the demand for a vaccine able to confer protective immunity against both bubonic and pneumonic plague, we previously constructed the genetically modified Y. pseudotuberculosis V674pF1 candidate plague vaccine, which produces the Y. pestis F1 antigen [17]. Although this vaccine protected against inhalational exposure to Y. pestis, protection against bubonic plague was not complete. This lack of full protection was potentially explained by the observation that production of the F1 antigen was unstable. The objective of the present work was therefore to improve the vaccine by generating a new strain with improved robustness and efficiency.
Loss of F1 production by V674pF1 mainly resulted from plasmid instability. Others who used Salmonella as receiver of the caf operon had to apply a sustained antibiotic pressure in vitro to ensure plasmid persistence, but this pressure could not be maintained in vivo [31]. Here, the caf operon was transposed into the chromosome [18], and we show that production of F1 capsule by VTnF1 was comparable to that of Y. pestis, whereas F1 production by V674pF1 was much more heterogeneous and unstable. The stability of VTnF1 characteristics will allow the large-scale production of the live vaccine according to good manufacturing procedures.
VTnF1 provides high protection against both bubonic and pneumonic plague, and is more efficient than V674pF1 used at the same dose (108 CFU) and tested in the same conditions [17]. We had previously reported that only 80% of the mice vaccinated with V674pF1 survived a high-dose pneumonic plague challenge (107 CFU CO92), or a moderate bubonic plague challenge (103 CFU). In contrast, VTnF1 provided complete protection against commonly used bacterial challenges, and almost complete protection against very high challenges. Because V674pF1 and VTnF1 are both derivatives of the V674 attenuated strain, the only possible explanation for this difference of protection is the more homogenous and sustained production of the F1 pseudocapsule by VTnF1. The F1 antigen can activate macrophages [32], an adjuvant effect favorable to the adaptive immune response. Therefore, the efficiency of VTnF1 may result from a stronger stimulation of macrophages, and possibly also of dendritic cells which belong to the same lineage, thus fostering immunity more efficiently.
The high-level protection observed is especially remarkable as it was obtained with a single oral dose of vaccine. Such a very simple procedure is a key advantage as compared to the repeated injections required by most vaccines to confer a protection extended in time. Difficult to perform in the field, repeated injections are considered by public health authorities as a limitation for mass vaccination.
Mice vaccinated with VTnF1 very rapidly control Y. pestis at the skin entry site. This suggests that easily mobilizable effectors such as antibodies and phagocytes reach the infected tissue. Antibodies might play an essential role in protection conferred by VTnF1, as suggested by the high antibody titers of vaccinated mice. VTnF1 displays much more antigenic diversity than plague molecular vaccines currently under development, which are composed of only F1 and LcrV antigens. The immune response induced by VTnF1 targets various Y. pestis proteins in addition to Caf1 and Caf1M antigens. This target diversity is valuable to protect against bacteria, which can lose target antigens via gene deletions, as observed for the Caf1/F1 antigen [33]. The anti-F1 IgG are known to provide protection by opsonizing Y. pestis, facilitated by F1 abundance and surface localization [25, 26, 34]. Whereas the abundant anti-F1 IgG induced by VTnF1 probably play a central protective role against wild type (F1-encapsulated) Y. pestis [25, 27, 34, 35], the resistance of vaccinated mice to plague caused by an F1-negative Y. pestis strain demonstrates that the antibodies directed against other antigens also contribute to protection.
Y. pestis F1-specific IgG induced by VTnF1 are detectable as early as four days after vaccination, a fast kinetic comparable to that observed after vaccination with soluble, recombinant F1 [36]. This prompt onset of IgG production indicates that VTnF1 rapidly interacts with lymphoid cells, probably in Peyer's patches, mesenteric lymph nodes and spleen where VTnF1 was observed. The fast switch of the humoral response from IgM toward antibodies of isotypes IgG1, two and three, generally of high affinity, indicates a strong T-cell dependent response. The presence of abundant IgG3 also indicates that carbohydrate targets are recognized [37]. This diversity is favorable for opsonization via all Fcγ receptors and suggests the involvement of the various B-cells subtypes to the vaccine-induced response [38]. Because IgG levels escalate during the first week to reach the plateau levels observed during the following six months, their contribution to protection against plague at day 30 could already be available at day seven.
Cellular immunity plays an important role against plague [39, 40] and performs critical protective functions during humoral defense against pneumonic plague [41]. Molecular vaccines adjuvanted with alum are poor inducers of this part of the immune response [12, 13]. In contrast, VTnF1 triggered a strong cell-mediated response without adjuvant. Part of the recall cell response was directed against F1, but the most important part was directed toward non-F1 antigens. Its intensity, with IL-17 comparable to a mitogenic stimulation by Concanavalin A, indicates the engagement of a high percentage of splenocytes that are likely recognizing multiple antigenic targets. Memory cells produced IFNγ, IL-17, and IL-10, composing a mixed Th1-Th17 profile. An IFNγ-dependent type 1 immune response is essential for vaccine-induced protection against plague [39]. IFNγ derived from memory T cells instruct potent innate cell activation, resulting in a fast protective immunity against invading microorganisms [42]. IL-17 producing T lymphocytes (Th17 cells) are essential to cure pneumonic plague [43] due to the essential role played by IL-17 in the induction of antimicrobial peptides and attraction of polymorphonuclear leukocytes [44]. The memory response induced by VTnF1 also involves production of the anti-inflammatory cytokine IL-10, which balances potentially harmful effects of IL-17 [45]. In addition to these direct functions, T cells play an important role in antibody-dependent immunity [40] and thus potentiate the humoral response. Altogether, the immune response induced by VTnF1, by combining humoral and cellular mechanisms, has the characteristics required to efficiently clear Y. pestis.
The sustained humoral and cellular immunity six months after vaccination is unlikely to result from a prolonged stimulation of immunity by live bacteria because VTnF1 was undetectable in feces and organs of most mice one month after vaccination. IgG could be produced by long-lived plasma cells, which differ from so-called memory B cells, and produce abundant IgG without re-stimulation [46]. VTnF1 could have persisted in the gut after one month, for example by durably colonizing the cecum as recently reported [47], however this is unlikely because only virulent strains cause cecum foci, and they yield high levels of cultivable bacteria in feces.
The protective immunity provided by VTnF1 after a six-month period (93% against bubonic plague and 50% against pneumonic plague) is remarkably long-lasting since six months correspond to one third of the mouse life [29], which is comparable to #30 years in human lifespan. Considering that both antibody titers and cellular responsiveness remained high after six months, the almost full protection against bubonic plague may result from either compartment or more likely a synergy of the two [14]. The lower protection against pneumonic plague indicates that lung immunity is the most demanding and requires one or more components of the immune response, that are mandatory for protection but are the first to decline with time. The nature of this component of acquired immunity is yet to determine, but does not seem related to Ig isotype switching or the decline of antibody or IFNγ/IL-17 production. One possible explanation is that aging causes a modification of alveolar macrophages functions, with spontaneous activation and reduced responsiveness to external stimuli, thus contributing to lung susceptibility to infection [48, 49].
Live attenuated Y. pestis plague vaccines (EV76 and subclones) has been previously used with success in humans, but could generate strong side effects [6, 7], and, as all Y. pestis strains, are subject to easy genetic rearrangements due to high numbers of insertion sequences in the genome [4, 8]. In addition to genetic stability, VTnF1 combines the known advantages of replicating vaccines: elicitation of humoral and cell-mediated immune responses, robustness against mutant microorganisms, easiness of mass production and use, limited cost, etc., whilst providing guarantees in terms of attenuation, stability, and efficacy against both bubonic and pneumonic plague. Preparedness plans against bioterrorist attacks imply stockpiling millions of vaccine doses. However, stockpiles have a finite lifespan and thus demand regular production of new doses, a rapidly expensive strategy [50]. Live vaccines can be rapidly produced in mass amounts, and are now viewed as a valuable alternative.
In conclusion, we propose here a vaccine providing high-level protection against both bubonic and pneumonic plague after a single-dose immunization. VTnF1 is an easy-to-produce, genetically stable and irreversibly attenuated vaccine, providing a long-lasting and highly efficient protection against both wild type and un-encapsulated (F1-negative) Y. pestis. To our knowledge, VTnF1 is the only plague vaccine ever reported that could provide high and long lasting protection against both bubonic and pneumonic plague after a single oral administration.
Supporting Information
To determine the F1 capsule production, a dipstick test devised to detect F1 [21] was used on a cell suspension of the recombinant VTnF1 vaccine strain grown at 37°C in LB broth. The parental V674 strain was used as negative control because it does not possess a caf locus, and the Y. pestis CO92 strain was used as positive control. A positive band at the same position as the Y. pestis control was observed whereas no signal was detected with the V674 Y. pseudotuberculosis strain, thus indicating that VTnF1 synthesizes the F1 antigen.
(DOCX)
Mice were weighed at regular intervals after oral vaccination with VTnF1 (108 CFU) or left unvaccinated (Naïve). Shown are the means of 16 naïve mice and 32 vaccinated mice. Groups were compared at each time point using the unpaired Student’s t test. *: p <0.05; **: p <0.01. ns: not significant.
(DOCX)
(DOCX)
Acknowledgments
The authors thank Sofia Filali and Rym Bagga for technical advice, and Pierre Goossens and Xavier Montagutelli for helpful discussions.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
The authors received no specific funding for this work.
References
- 1.WHO. Plague. World Health Organization. 2005;(267).
- 2. Bertherat E, Bekhoucha S, Chougrani S, Razik F, Duchemin JB, Houti L, et al. Plague reappearance in Algeria after 50 years, 2003. Emerging Infectious Diseases. 2007;13(10):1459–62. 10.3201/eid1310.070284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schrag SJ, Wiener P. Emerging infectious diseases: what are the relative roles of ecology and evolution? Trends Evolution and Ecology. 1995;10:319–24. [DOI] [PubMed] [Google Scholar]
- 4. Chain PS, Carniel E, Larimer FW, Lamerdin J, Stoutland PO, Regala WM, et al. Insights into the evolution of Yersinia pestis through whole-genome comparison with Yersinia pseudotuberculosis . Proc Natl Acad Sci USA. 2004;101(38):13826–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Guiyoule A, Gerbaud G, Buchrieser C, Galimand M, Rahalison L, Chanteau S, et al. Transferable plasmid-mediated resistance to streptomycin in a clinical isolate of Yersinia pestis . Emerging Infectious Diseases. 2001;7(1):43–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Girard G. Immunity in Plague. Acquisitions Supplied by 30 Years of Work on the "Pasteurella Pestis Ev" (Girard and Robic) Strain. Biol Med (Paris). 1963;52:631–731. [PubMed] [Google Scholar]
- 7. Feodorova VA, Motin VL. Plague Vaccines In: Feodorova VA, Motin VL, editors. Vaccines against Bacterial Biothreat Pathogens. Kerala, India: Research Signpost; 2011. p. 175–233. [Google Scholar]
- 8. Cui Y, Yang X, Xiao X, Anisimov AP, Li D, Yan Y, et al. Genetic variations of live attenuated plague vaccine strains (Yersinia pestis EV76 lineage) during laboratory passages in different countries. Infect Genet Evol. 2014;26:172–9. 10.1016/j.meegid.2014.05.023 [DOI] [PubMed] [Google Scholar]
- 9. Heath DG, Anderson GW Jr., Mauro JM, Welkos SL, Andrews GP, Adamovicz J, et al. Protection against experimental bubonic and pneumonic plague by a recombinant capsular F1-V antigen fusion protein vaccine. Vaccine. 1998;16(11–12):1131–7. [DOI] [PubMed] [Google Scholar]
- 10. Williamson ED, Eley SM, Griffin KF, Green M, Russell P, Leary SE, et al. A new improved sub-unit vaccine for plague: the basis of protection. FEMS Immunol Med Microbiol. 1995;12(3–4):223–30. [DOI] [PubMed] [Google Scholar]
- 11. Burrows TW. An Antigen determining Virulence in Pasteurella pestis . Nature. 1956;177:426–7. [DOI] [PubMed] [Google Scholar]
- 12. Mannhalter JW, Neychev HO, Zlabinger GJ, Ahmad R, Eibl MM. Modulation of the human immune response by the non-toxic and non-pyrogenic adjuvant aluminium hydroxide: effect on antigen uptake and antigen presentation. Clinical and experimental immunology. 1985;61(1):143–51. [PMC free article] [PubMed] [Google Scholar]
- 13. Dinc G, Pennington JM, Yolcu ES, Lawrenz MB, Shirwan H. Improving the Th1 cellular efficacy of the lead Yersinia pestis rF1-V subunit vaccine using SA-4-1BBL as a novel adjuvant. Vaccine. 2014;32(39):5035–40. 10.1016/j.vaccine.2014.07.015 [DOI] [PubMed] [Google Scholar]
- 14. Smiley ST. Current challenges in the development of vaccines for pneumonic plague. Expert Review of Vaccines. 2008;7(2):209–21. 10.1586/14760584.7.2.209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pitt L. Nonhuman Primates as a Model for Pneumonic Plague. Gaithersburg, Maryland, USA. 2004 [cited 2014]. http://www.fda.gov/cber/minutes/workshop-min.htm.
- 16. Blisnick T, Ave P, Huerre M, Carniel E, Demeure CE. Oral vaccination against bubonic plague using a live avirulent Yersinia pseudotuberculosis strain. Infect Immun. 2008;76(8):3808–16. 10.1128/IAI.00034-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Derbise A, Cerdà Marín A, Ave P, Blisnick T, Huerre M, Carniel E, et al. An encapsulated Yersinia pseudotuberculosis is a highly efficient vaccine against pneumonic plague. PLoS NTD. 2012;6(2):e1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Choi KH, Gaynor JB, White KG, Lopez C, Bosio CM, Karkhoff-Schweizer RR, et al. A Tn7-based broad-range bacterial cloning and expression system. Nat Methods. 2005;2(6):443–8. Epub 2005/05/24. [DOI] [PubMed] [Google Scholar]
- 19. Lane MC, Alteri CJ, Smith SN, Mobley HL. Expression of flagella is coincident with uropathogenic Escherichia coli ascension to the upper urinary tract. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(42):16669–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hamilton MA, Russo RC, Thurston RV. Trimmed Spearman-Karber method for estimating median lethal concentrations in toxicity assays. Environmental Science and Technology. 1977;11(7):714–9. [Google Scholar]
- 21. Chanteau S, Rahalison L, Ralafiarisoa L, Foulon J, Ratsitorahina M, Ratsifasoamanana L, et al. Development and testing of a rapid diagnostic test for bubonic and pneumonic plague. Lancet. 2003;361(9353):211–6. [DOI] [PubMed] [Google Scholar]
- 22. Benoit C, Guiyoule A, Carniel E. Sérodiagnostic des infections humaines à Yersinia pathogènes. Presse Médicale. 1996;25(34):1627–30. [PubMed] [Google Scholar]
- 23. Mishell B, Shiigi S. Selected Methods in Cellular Immunology. San Francisco: W.H. Freeman and Co; 1980. 23–4 p. [Google Scholar]
- 24. Davis KJ, Fritz DL, Pitt ML, Welkos SL, Worsham PL, Friedlander AM. Pathology of experimental pneumonic plague produced by fraction 1-positive and fraction 1-negative Yersinia pestis in African green monkeys (Cercopithecus aethiops). Arch Pathol Lab Med. 1996;120(2):156–63. [PubMed] [Google Scholar]
- 25. Friedlander AM, Welkos SL, Worsham PL, Andrews GP, Heath DG, Anderson GW, et al. Relationship between virulence and immunity as revealed in recent studies of the F1 capsule of Yersinia pestis . Clin Infect Dis. 1995;21(Suppl. 2):S178–S81. [DOI] [PubMed] [Google Scholar]
- 26. Quenee LE, Cornelius CA, Ciletti NA, Elli D, Schneewind O. Yersinia pestis caf1 variants and the limits of plague vaccine protection. Infection and Immunity. 2008;76(5):2025–36. 10.1128/IAI.00105-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Welkos SL, Davis KM, Pitt LM, Worsham PL, Friedlander AM. Studies on the contribution of the F1 capsule-associated plasmid pFra to the virulence of Yersinia pestis In: Ravagnan G, Chiesa C, editors. Yersiniosis: Present and Future. Contributions to Microbiology and Immunology. 13. Postfach, CH-4009 Basel, Switzerland: Karger; 1995. p. 299–305. [PubMed] [Google Scholar]
- 28. Weening EH, Cathelyn JS, Kaufman G, Lawrenz MB, Price P, Goldman WE, et al. The Dependence of the Yersinia pestis Capsule on Pathogenesis Is Influenced by the Mouse Background. Infection and Immunity. 2011;79(2):644–52. 10.1128/IAI.00981-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Flurkey K, Currer J, Harrison DE. Mouse Models in Aging Research In: Fox JG, Davisson M, Quimby FW, Barthold SW, Newcomer CE, Smith AL, editors. Mouse in Biomedical Research (second edition). Burlington: Academic Press; 2007. p. 637–72. [Google Scholar]
- 30. Sebbane F, Lemaitre N, Sturdevant DE, Rebeil R, Virtaneva K, Porcella SF, et al. Adaptive response of Yersinia pestis to extracellular effectors of innate immunity during bubonic plague. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(31):11766–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Maskell DJ, Sweeney KJ, O'Callaghan D, Hormaeche CE, Liew FY, Dougan G. Salmonella typhimurium aroA mutants as carriers of the Escherichia coli heat-labile enterotoxin B subunit to the murine secretory and systemic immune systems. Microb Pathog. 1987;2(3):211–21. [DOI] [PubMed] [Google Scholar]
- 32. Sodhi A, Sharma RK, Batra HV, Tuteja U. Recombinant fraction 1 protein of Yersinia pestis activates murine peritoneal macrophages in vitro. Cellular Immunology. 2004;229(1):52–61. [DOI] [PubMed] [Google Scholar]
- 33. Winter CC, Cherry WB, Moody MD. An Unusual Strain of Pasteurella pestis Isolated from a Fatal Human Case of Plague. Bulletin de l'Organisation Mondiale de la Santé. 1960;23:408–9. [PMC free article] [PubMed] [Google Scholar]
- 34. Anderson GW, Worsham PL, Bolt CR, Andrews GP, Welkos SL, Friedlander AM, et al. Protection of mice from fatal bubonic and pneumonic plague by passive immunization with monoclonal antibodies against the F1 protein of Yersinia pestis. Am J Trop Med Hyg. 1997;56(4):471–3. [DOI] [PubMed] [Google Scholar]
- 35. Carr KW, Heath DG, Friedlander AM. Antiphagocytic effect of F1 capsular protein of Yersinia pestis . Abstracts of the General Meeting of the American Society for Microbiology. 1997;97:96. [Google Scholar]
- 36. Levy YY, Flashner Y, Tidhar A, Zauberman A, Aftalion M, Lazar S, et al. , editors. Rapid onset of protective immunity against plague is induced by the F1 but not by the LcrV antigen Yersinia 11: the 11th International Symposium on Yersinia; 2013; Suzhou, China: Cold Spring Harbor Laboratory. [Google Scholar]
- 37. Perlmutter RM, Hansburg D, Briles DE, Nicolotti RA, Davie JM. Subclass restriction of murine anti-carbohydrate antibodies. J Immunol. 1978;121(2):566–72. [PubMed] [Google Scholar]
- 38. Swanson CL, Pelanda R, Torres RM. Division of labor during primary humoral immunity. Immunol Res. 2013;55(1–3):277–86. 10.1007/s12026-012-8372-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Elvin SJ, Williamson ED. Stat 4 but not Stat 6 mediated immune mechanisms are essential in protection against plague. Microbial Pathogenesis. 2004;37(4):177–84. [DOI] [PubMed] [Google Scholar]
- 40. Levy Y, Flashner Y, Tidhar A, Zauberman A, Aftalion M, Lazar S, et al. T cells play an essential role in anti-F1 mediated rapid protection against bubonic plague. Vaccine. 2011;29(40):6866–73. 10.1016/j.vaccine.2011.07.059 [DOI] [PubMed] [Google Scholar]
- 41. Parent MA, Wilhelm LB, Kummer LW, Szaba FM, Mullarky IK, Smiley ST. Gamma interferon, tumor necrosis factor alpha, and nitric oxide synthase 2, key elements of cellular immunity, perform critical protective functions during humoral defense against lethal pulmonary Yersinia pestis infection. Infection and Immunity. 2006;74(6):3381–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Soudja SM, Ruiz AL, Marie JC, Lauvau G. Inflammatory monocytes activate memory CD8(+) T and innate NK lymphocytes independent of cognate antigen during microbial pathogen invasion. Immunity. 2012;37(3):549–62. Epub 2012/09/04. 10.1016/j.immuni.2012.05.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lin JS, Kummer LW, Szaba FM, Smiley ST. IL-17 contributes to cell-mediated defense against pulmonary Yersinia pestis infection. J Immunol. 2011;186(3):1675–84. Epub 2010/12/22. 10.4049/jimmunol.1003303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Littman DR, Rudensky AY. Th17 and regulatory T cells in mediating and restraining inflammation. Cell. 2010;140(6):845–58. Epub 2010/03/23. 10.1016/j.cell.2010.02.021 [DOI] [PubMed] [Google Scholar]
- 45. McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, et al. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol. 2007;8(12):1390–7. Epub 2007/11/13. [DOI] [PubMed] [Google Scholar]
- 46. Manz RA, Lohning M, Cassese G, Thiel A, Radbruch A. Survival of long-lived plasma cells is independent of antigen. International immunology. 1998;10(11):1703–11. [DOI] [PubMed] [Google Scholar]
- 47. Fahlgren A, Avican K, Westermark L, Nordfelth R, Fallman M. Colonization of cecum is important for development of persistent infection by Yersinia pseudotuberculosis . Infect Immun. 2014;82(8):3471–82. 10.1128/IAI.01793-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li Z, Li J, Bu X, Liu X, Tankersley CG, Wang C, et al. Age-induced augmentation of p38 MAPK phosphorylation in mouse lung. Exp Gerontol. 2011;46(8):694–702. 10.1016/j.exger.2011.04.005 [DOI] [PubMed] [Google Scholar]
- 49. Boyd AR, Orihuela CJ. Dysregulated inflammation as a risk factor for pneumonia in the elderly. Aging Dis. 2011;2(6):487–500. [PMC free article] [PubMed] [Google Scholar]
- 50. Metzer MI. The economics of Planning and Preparing for Bioterrorism In: Fong IW, Alibek K, editors. Bioterrorism and Infectious agents A new dilemma for the 21st century. New-York: Springer Sciences+Business Media, Inc; 2005. p. 237–57. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
To determine the F1 capsule production, a dipstick test devised to detect F1 [21] was used on a cell suspension of the recombinant VTnF1 vaccine strain grown at 37°C in LB broth. The parental V674 strain was used as negative control because it does not possess a caf locus, and the Y. pestis CO92 strain was used as positive control. A positive band at the same position as the Y. pestis control was observed whereas no signal was detected with the V674 Y. pseudotuberculosis strain, thus indicating that VTnF1 synthesizes the F1 antigen.
(DOCX)
Mice were weighed at regular intervals after oral vaccination with VTnF1 (108 CFU) or left unvaccinated (Naïve). Shown are the means of 16 naïve mice and 32 vaccinated mice. Groups were compared at each time point using the unpaired Student’s t test. *: p <0.05; **: p <0.01. ns: not significant.
(DOCX)
(DOCX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.