

Abstract
Early treatment with heart failure drugs lisinopril and spironolactone improves skeletal muscle pathology in Duchenne muscular dystrophy (DMD) mouse models. The angiotensin converting enzyme inhibitor lisinopril and mineralocorticoid receptor (MR) antagonist spironolactone indirectly and directly target MR. The presence and function of MR in skeletal muscle have not been explored. MR mRNA and protein are present in all tested skeletal muscles from both wild-type mice and DMD mouse models. MR expression is cell autonomous in both undifferentiated myoblasts and differentiated myotubes from mouse and human skeletal muscle cultures. To test for MR function in skeletal muscle, global gene expression analysis was conducted on human myotubes treated with MR agonist (aldosterone; EC50 1.3 nM) or antagonist (spironolactone; IC50 1.6 nM), and 53 gene expression differences were identified. Five differences were conserved in quadriceps muscles from dystrophic mice treated with spironolactone plus lisinopril (IC50 0.1 nM) compared with untreated controls. Genes down-regulated more than 2-fold by MR antagonism included FOS, ANKRD1, and GADD45B, with known roles in skeletal muscle, in addition to NPR3 and SERPINA3, bona fide targets of MR in other tissues. MR is a novel drug target in skeletal muscle and use of clinically safe antagonists may be beneficial for muscle diseases.—Chadwick, J. A., Hauck, J. S., Lowe, J. , Shaw, J. J., Guttridge, D. C., Gomez-Sanchez, C. E., Gomez-Sanchez, E. P., Rafael-Fortney, J. A. Mineralocorticoid receptors are present in skeletal muscle and represent a potential therapeutic target.
Keywords: aldosterone, gene expression microarray, muscular dystrophy, spironolactone, steroid hormone receptors
Duchenne muscular dystrophy (DMD) is the most common and severe form of muscular dystrophy, affecting ∼1 in 5000 boys (1) and is characterized by progressive muscle degeneration and weakness. Most patients are wheelchair bound by 12 yr of age and have an average life span of only 25 yr (2). Currently, the only available standard-of-care treatment is glucocorticoid therapy [glucocorticoid receptor (GR) agonists], which has been able to defer severity of some symptoms and prolong mobility an average of 2 yr (3). However, these drugs are not specific for striated muscles, do not exhibit long-term efficacy, and have numerous deleterious side effects (4–6). We previously published that a combinatorial treatment with the mineralocorticoid receptor (MR) antagonist spironolactone and angiotensin-converting enzyme inhibitor lisinopril improved skeletal muscle function and pathology in a mouse model of DMD (7).
Preclinical studies using dystrophin-deficient, utrophin-haploinsufficient (het) (utrn+/−; mdx) dystrophic mice (8) showed lisinopril plus spironolactone treatment from 4 to 20 wk of age to be effective in preventing ongoing skeletal muscle damage. Mice treated with a combination of these drugs showed 80% of normal muscle force generation in both respiratory and limb muscles compared with only 40% of normal force observed in untreated het mice (7). In addition, limb muscle damage was significantly reduced and myocardial disease was almost entirely absent.
Lisinopril and spironolactone are U.S. Food and Drug Administration (FDA)-approved drugs with minimal adverse effects and a long history of safety and efficacy for treatment of heart diseases (9, 10). However, their benefit in skeletal muscle had never been studied. Lisinopril and spironolactone target the renin–angiotensin–aldosterone system at 2 points, formation of angiotensin II and antagonism of the MR, which binds aldosterone (11, 12). Angiotensin II has several functions, including increasing production of aldosterone, and therefore it has an indirect effect on MR downstream functions. Therefore, both spironolactone and lisinopril directly and indirectly target MR, respectively. However, we have recently shown that lisinopril alone improves histopathology, but not skeletal muscle function of dystrophic mice (13). Gaining insight into the mechanism by which spironolactone benefits dystrophic muscle will facilitate optimization of long-lasting therapies.
The MR is the largest member of a family of nuclear steroid receptors, which includes the GR, androgen receptor, and progesterone receptor (14–16). These ligand-dependent transcription factors remain dormant in the cytosol until activated by a ligand. Once ligand bound, the receptors form dimers and translocate to the nucleus, where they direct transcription of target genes (17). As a result of the high homology with GR, MR is activated by both mineralocorticoids (aldosterone, deoxycorticosterone) and glucocorticoids (cortisol in humans, corticosterone in mice) and forms functional heterodimers with GR (18, 19). MR specificity varies between cell types and is dependent on numerous factors. These factors include protection from glucocorticoids in aldosterone target tissues by type 2 11β-hydroxysteroid dehydrogenase enzymes that metabolize cortisol to inactive cortisone, and corticosterone to 11-dehydrocorticosterone; ligand-induced selectivity for hormone response elements on the DNA; and interaction with nuclear coregulators (17). MR is best known for its roles in maintaining electrolyte homeostasis in the kidneys and blood pressure in the heart, but it has also been shown to act in other organs including brain, pancreas, colon, ovaries, and adipose tissue (18, 20–23). However, the presence of MR in skeletal muscles has never been investigated.
One study has shown spironolactone is able to improve insulin-stimulated glucose transport in skeletal muscle by reducing reactive oxygen species in rats overexpressing renin, but a direct effect on skeletal muscle cells was not investigated (24). Despite long-standing clinical use of MR antagonists in cardiology, only Serpina-3 (serpin peptidase inhibitor clause A), a serine protease inhibitor, has been validated as a bona fide direct transcriptional target of MR in cardiomyocytes and patented as a potential biomarker of MR activation (25, 26). We investigated whether MR is present in skeletal muscle and is functional in downstream gene expression. These studies will help begin to elucidate the mechanism behind the efficacy of these drugs in dystrophic skeletal muscles.
MATERIALS AND METHODS
Animals
All protocols were approved by the Institutional Laboratory Animal Care and Use Committee. For this study, we used tissue from several DMD mouse models: dystrophin-deficient mdx mice (27, 28), het mice (8), and dystrophin/utrophin-deficient double knockout (dko) mice (29), in addition to 10(J)/10J (JAX 00665; The Jackson Laboratory, Bar Harbor, ME, USA) wild-type control mice. Skeletal muscles and heart were removed from 8- or 20-wk-old mice bred and genotyped as described previously (8, 29, 30). Samples for protein isolation were flash frozen; they were not directly processed because of the necessity of obtaining multiple age-matched mice for each genotype, even though this method is known to increase protein degradation of the MR (22).
Mammalian myogenic cell culture
Mouse C2C12 myoblasts (American Type Culture Collection, Manassas, VA, USA) were grown in high-glucose DMEM (Invitrogen, Grand Island, NY, USA), supplemented with 10% fetal bovine serum (Atlanta Biologicals, Norcross, GA, USA) and 100 U/ml penicillin–streptomycin (Invitrogen) and cultured at 37°C in 5% CO2. To generate myotubes, myoblasts were serum restricted in differentiation medium [DMEM supplemented with 2% horse serum (Invitrogen) and 100 U/ml penicillin–streptomycin] for 7 d. Cells were collected in 250 µl of Newcastle buffer: 75 mM Tris pH 6.8, 3.8% SDS, 4 M urea, 20% glycerol (Invitrogen), 1 mM PMSF, 1 mM benzamidine, 0.5 µg/ml leupeptin, and 0.2 U/ml aprotinin (all reagents were purchased from Sigma-Aldrich, St. Louis, MO, USA, unless specified otherwise).
Human skeletal muscle myoblasts isolated from normal males (HSMM; Lonza, Walkersville, MD, USA) were grown in skeletal muscle cell growth medium (SkGM-2 bullet kit; Lonza), containing 1% bovine serum albumin, 1% fetuin, 1% insulin, 0.1% human epidermal growth factor, 0.1% dexamethasone, and 0.1% gentamicin–amphotericin B and cultured at 37°C in 5% CO2. Lots 0000418971 and 0000424745 were combined to help minimize false-positive gene expression changes specific to a single individual. Cells were serum restricted in differentiation medium (above) for 5 d, followed by 48 h or 5 d treatments with aldosterone (10 µM; EC50 1.3 nM), eplerenone (2 µM; IC50 81 nM; Pfizer Compound Transfer Program), spironolactone (10 µM; IC50 1.6 nM) (drugs were purchased from Sigma-Aldrich and dissolved in 100% ethanol unless specified otherwise) or ethanol only to serve as untreated controls. Drugs were added directly to existing differentiation medium and refreshed every 2.5 d (for cells treated 5 d). Cells were collected in 250 µl of cellular extract buffer: 10 mM HEPES pH 7.6 (Fisher Scientific, Robinson Township, PA, USA), 60 mM potassium chloride, 1 mM EDTA, 0.25% Tergitol-type NP-40, 2.5 µg/ml leupeptin, 2.5 µg/ml aprotinin, 2.5 µg/ml pepstatin A, 1 µM DTT, and 1 mM PMSF (all reagents were purchased from Sigma-Aldrich unless specified otherwise).
Protein extraction
Snap-frozen mouse tissues were pulverized in liquid nitrogen using a mortar and pestle and vortexed in cellular extract buffer, 1 ml buffer per 100 mg tissue. Protein concentration was determined by Dc Protein Assay (Bio-Rad, Hercules, CA, USA), and samples were stored at −80°C.
Western blot analysis
A total of 35 µg per lane of total protein from cell extracts or tissue homogenates was probed with a combination of MR-specific monoclonal antibodies, MRN 2B7 and rMR 1-18 1D5 (mouse tissue and cells) or MRN 2B7 and rMR 1-18 6G1 (human cells) (31), or with antibodies against Ankrd1 (ankyrin repeat domain 1; Proteintech, Chicago, IL, USA) and actin (α-sarcomeric; Sigma-Aldrich). Membranes were incubated with anti-mouse or -rabbit horseradish peroxidase (Jackson Immunoresearch, Grove, PA, USA) secondary antibody, and signals were detected with ECL 2 Western blot analysis substrate (Pierce, Grand Island, NY, USA) followed by film (Blue Ultra; GeneMate, Kaysville, UT, USA) exposure.
RNA preparation
RNA was isolated using TRIzol reagent (Life Technologies, Grand Island, NY, USA), according to the manufacturer’s instructions, followed by incubation with DNAse I (RQ1; Promega, Madison, WI, USA). DNAse-treated RNA concentration was determined spectrophotometrically, and samples were stored at −80°C. Samples for microarray were further purified using the RNeasy mini kit (Qiagen, Valencia, CA, USA) cleanup protocol, and final RNA concentrations were determined.
Microarray
RNA integrity was interrogated using the Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA). A 100 ng aliquot of total mRNA was linearly amplified. Then 5.5 µg of cDNA was labeled and fragmented using the GeneChip WT Plus reagent kit (Affymetrix, Santa Clara, CA, USA) following the manufacturer's instructions. Labeled cDNA targets were hybridized to Affymetrix GeneChip Human Transcriptome Array 2.0 or Mouse Genome 430 Array for human and mouse gene expression, respectively, for 16 h at 45°C rotating at 60 rpm. The arrays were washed and stained using the Fluidics Station 450 and scanned using the GeneChip Scanner 3000. Arrays were normalized using RMA algorithm in Expression Console, and comparisons were made in Transcriptome Analysis Console (Affymetrix). The microarray data have been deposited in the NCBI Gene Expression Omnibus (human: GSE70822; mouse: GSE70984). Gene groups were determined using the functional annotation clustering tool from the Database for Annotation, Visualization, and Integrated Discovery (DAVID).
DNA sequencing
cDNA was generated using the Reverse Transcriptase High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Grand Island, NY, USA) according to the manufacturer’s instructions and were validated using positive-control CASK 5′-ATTGGTGGCAGGGTAAACTGG-3′ (forward), 5′-TTTGGAGGTCTGGTTGTATGTGG-3′ (reverse) and dystrophin 5′-GCACTCCGACTACATCCGGAGAAGA-3′ (forward), 5′-CTCCAGCTGTTTATTGTGGTCTTCC-3′ (reverse) primers. PCR was performed using an Applied Biosystems GeneAmp 9700 Thermal cycler, with MR-specific primers designed using MacVector: human MR 5′- TCATGGAAATCACACGGCGA-3′ (forward) and 5′- CGATCTCCAGCTCAAGGCAA-3′ (reverse) and mouse MR 5′-TCTGTGTGCTGGAAGAAATGACTG-3′ (forward) and 5′-CCAACTCAAGGCAAACGATGATAG-3′ (reverse). PCR products were isolated on a gel and further purified using the Wizard SV Gel and PCR Clean-up System (Promega) according to the manufacturer’s instructions. Products were sequenced by The Ohio State Nucleic Acid/Microarray Shared Resource.
Real-time PCR
A total of 1 µg of DNased RNA from each sample was used to generate cDNA as described above. Relative quantitation real-time PCR was performed using Power SYBR Green PCR Master Mix (Applied Biosystems) inoculated with 1 µl of the cDNA reaction plus 40 nM of each primer. Reactions were run on an Applied Biosystems 7300 Real-Time PCR System, version 1.4. Primers used to amplify mouse Ankrd1 were 5′-ACAGCGCGTGTCCTTTTGAT-3′ and 5′-GCCAAGGAAAACTGAGTTCGA-3′, and 18S were 5′-CGCCGCTAGAGGTGAAATTC-3′ and 5′-TCTTGGCAAATGCTTTCGC-3′. Expression levels of Ankrd1 were normalized to 18S rRNA, and the C57 with the highest level of Ankrd1 expression was normalized to 1× and used to determine fold changes in the other samples. Non–reverse-transcribed RNA was used as a negative control for each sample.
Immunofluorescence
For immunofluorescence staining of cells in culture, cells were grown and differentiated in glass chamber slides (2-chamber slides; Thermo Scientific, Marietta, OH, USA) coated with Mouse Laminin Protein (Life Technologies). Primary antibodies against Ankrd1 (Proteintech) or a combination of MR-specific antibodies (MRN 2B7, rMR 1-18 6G1, and MR MID 6D1) were used with secondary antibodies Alexa 555 anti-rabbit or Alexa 488 anti-mouse (Life Technologies). Samples were mounted using Vectashield mounting medium (Vector Laboratories, Burlingame, CA, USA) with 0.2 µl/ml DAPI and viewed using an Olympus FV1000 Spectral Confocal microscope (Olympus, Tokyo, Japan); z-stack images are shown.
RESULTS
MR is expressed in skeletal muscle
To determine whether MR is present in skeletal muscles and whether MR antagonism could exert a direct effect on this cell type, we investigated protein and gene expression in mouse skeletal muscles and human and mouse skeletal muscle cultures. We first carried out Western blot analysis on tissue homogenates from 10(J)/10 wild-type mice using a combination of MR-specific monoclonal antibodies (31). MR protein was present at abundant levels in all mouse skeletal muscles tested, including diaphragm, quadriceps, soleus, gastrocnemius, tibialis anterior, and extensor digitorum longus (Fig. 1A).
Figure 1.
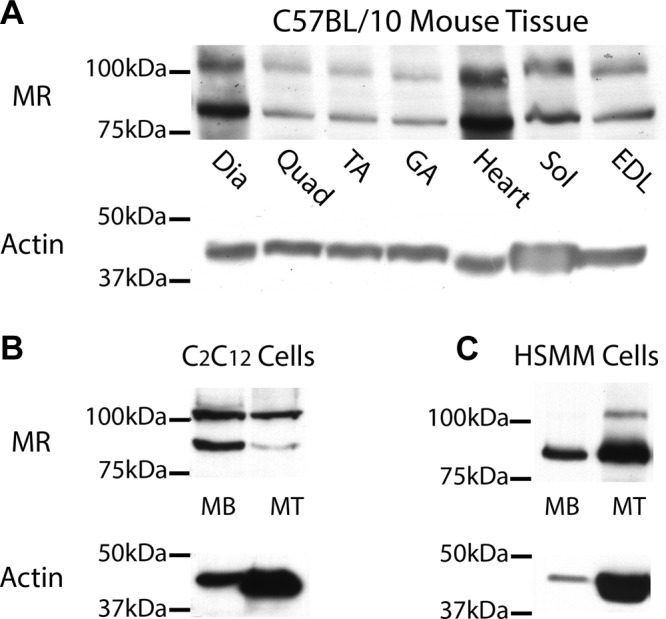
MR is expressed in mouse skeletal muscle tissues and both mouse and human cultured myoblasts (MB) and myotubes (MT). Western blots using combination of MR-specific monoclonal antibodies MR1-18 1D5 and MRN 2B7 (31) show protein at predicted molecular weight for full-length MR of ∼107 kDa in all normal mouse (C57BL/10) skeletal muscles analyzed, and in heart (A). Second lower molecular weight isoform is also present in all tissues. Two MR protein bands are also present in both mouse (B) and human (C) myoblasts, but full-length MR becomes predominant in differentiated mouse myotubes. MR protein expression was detected using equivalent amounts (35 µg) of C57BL/10 protein homogenates, and C2C12 and human primary muscle (HSMM) cell lysates. Myotubes were differentiated for either 5 (HSMM) or 7 d (C2C12). Standardization of loading was verified using α-sarcomeric actin antibody; predicted molecular weight, ∼42 kDa. Differentiated myotubes contain higher levels of actin than undifferentiated myoblasts per microgram of total protein. Dia, diaphragm; Quad, quadriceps; TA, tibialis anterior; GA, gastrocnemius; Sol, soleus; EDL, extensor digitorum longus.
To determine whether MR is expressed in a cell-autonomous manner in skeletal muscle cells or whether it is only being detected from the other cell types present in whole skeletal muscle known to express MR, such as endothelial cells and fibroblasts, we investigated MR protein in myogenic cultures. MR was present in both undifferentiated myoblasts and differentiated myotubes of the mouse C2C12 myogenic cell line (Fig. 1B). Because our ultimate goal is to translate MR antagonism into a treatment for patients, we also showed that MR was expressed in human primary undifferentiated and differentiated myogenic cultures (Fig. 1C). A band for full-length MR at the predicted molecular weight, 107 kDa (22), was detected in all mouse skeletal muscle tissues and in both mouse and human myogenic cultures. A second protein band, at a lower molecular weight compared with the full-length MR, was also detected and may be due to alternative splicing or a known proteolytic product resulting from snap freezing of samples before processing (22).
Due to the instability of MR and the high homology it shares with GR (22, 32), sequencing analysis of reverse-transcribed PCR (RT-PCR) RNA from mouse skeletal muscle and human myotubes was used to confirm full-length MR expression and identify a transcript that may account for the second protein band (Fig. 2A). Sequence data confirmed expression of both full-length mouse (NM_001083906) and human (M16801) MR and identified a known human MR splice variant (transcript variant 2; NM_001166104) with a deletion of exon 5 in human myotubes (Fig. 2B). However, PCR with primers spanning exon 5 (Fig. 2A) in mouse showed no alternative splicing in that region (Fig. 2C).
Figure 2.

Sequencing analysis verified presence of MR in skeletal muscle. A) Linearized structure of full-length MR gene (mouse and human MR genes are comparable) and human splice variant NR3C2 with exon 5 deletion. Translation is initiated in exon 2; exons 3, 4, and small portion of exon 5 encode DNA-binding domain; and exons 5–9 encode ligand-binding domain. Region spanned by human and mouse MR primers used for PCR and subsequent sequencing analysis are depicted by arrows. B) RT-PCR amplifying human MR from human myotubes yielded 2 bands corresponding to full-length MR (upper band) and NR3C2 transcript variant 2 (lower band) with deletion of exon 5 and truncation of DNA and ligand binding domains. C) Only 1 band corresponding to full-length MR was detected from RT-PCR of mouse MR using mouse quadriceps muscle; primers spanning exon 5 did not detect any splice variants with exon 5 deletion.
To test whether MR levels were maintained in dystrophic muscle and with treatment, we ran additional Western blot analyses comparing protein levels between wild-type and dystrophic models (Fig. 3A), as well as agonist- and antagonist-treated cells (Fig. 3B). MR protein was present in all of the same skeletal muscles in dystrophic models as in 10(J)/10 mice (Fig. 1A). Quadriceps muscles were shown as representative data because they are important targets for therapeutic intervention. Immunofluorescence of human myotubes treated with MR agonist or antagonist also showed no significant changes in MR staining pattern (Fig. 3C). MR levels did not vary significantly between wild-type and dystrophic muscle or with treatment, supporting MR as a potential therapeutic target for skeletal muscle for the treatment of dystrophy.
Figure 3.

MR protein levels are maintained in dystrophic muscle and agonist vs. antagonist treated human cells. A) Representative Western blots of quadriceps muscles from 3 biologic replicates are shown comparing MR protein levels from equivalent amounts of protein homogenates from C57BL/10 wild-type (C57) mice, dystrophin-deficient mdx (MDX) mice, het (HET) mice, and dystrophin/utrophin-deficient double knockout (DKO) mice. B) MR was also detected by Western blots of human primary muscle (HSMM) myotubes differentiated for 5 d (MT) and then treated for 5 d with 10 µM of MR antagonist spironolactone (S-10), MR agonist aldosterone (A-10), or ethanol vehicle untreated control (UC). For (A) and (B), 35 µg of total protein was used for each sample. Western blots used combination of MR-specific monoclonal antibodies MR1-18 1D5 or MR1-18 6G1 and MRN 2B7 (31) (full-length MR predicted molecular weight, ∼107 kDa) or α-sarcomeric actin antibody (loading control; predicted molecular weight, ∼42 kDa). C) MR localization is similar after treatment with MR antagonist and agonist. MR was detected by immunofluorescence staining (green) on human primary (HSMM) myotubes differentiated for 5 d then treated for 48 h with 10 µM of MR antagonist spironolactone or MR agonist aldosterone. Staining of subset of nuclei was present in all 3 groups. MR staining is similar with longer 5 d treatments (data not shown). Scale bars, 50 µm.
MR is functional in skeletal muscle
We next sought to determine whether MR in skeletal muscle is functional as a steroid hormone receptor with ligand-dependent transcriptional activity. We treated normal, differentiated human myogenic cultures with an MR agonist (aldosterone) or antagonist (spironolactone) for 48 h and then compared global gene expression. Microarray analysis showed 53 gene expression differences, greater than 2-fold, between myotubes treated with the MR antagonist spironolactone vs. the MR agonist aldosterone. The enriched functional related gene groups listed in Table 1 suggested that MR is an important transcription factor in normal differentiated myotubes. Microarray analysis comparing spironolactone-treated vs. untreated normal human myotubes revealed almost no gene expression changes (data not shown). These data support the notion that gene expression changes observed between antagonist (spironolactone)- and agonist (aldosterone)-treated myotubes were aldosterone dependent and not caused by cortisol in the media.
TABLE 1.
Fold changes of genes from microarray comparing human primary skeletal muscle myotubes treated with the MR antagonist spironolactone vs. the agonist aldosterone
| Function | Gene symbol | Full gene name | Fold change (antagonist/ agonist) |
|---|---|---|---|
| Response to oxidative stress | GPX3 | Glutathione peroxidase 3 (plasma) | −6.3 |
| PTGS2 | Prostaglandin–endoperoxide synthase 2 (prostaglandin G/H synthase and cyclooxygenase) | −2.0 | |
| TLR4 | Toll-like receptor 4 | −2.3 | |
| NRCAM | Neuronal cell adhesion molecule | −5.1 | |
| FOS | FBJ murine osteosarcoma viral oncogene homolog | −2.6 | |
| Serine protease inhibitor | SERPINA3 | Serpin peptidase inhibitor, clade A (α-1 antiproteinase, antitrypsin), member 3 | −2.2 |
| SERPING1 | Serpin peptidase inhibitor, clade G (C1 inhibitor), member 1 | −2.1 | |
| TFPI2 | Tissue factor pathway inhibitor 2 | 2.0 | |
| EGF-like type 3 | FBN2 | Fibrillin 2 | −3.5 |
| NID1 | Nidogen 1 | −2.6 | |
| ADAM12 | ADAM metallopeptidase domain 12 | 2.3 | |
| Defense response | SAMHD1 | SAM domain and HD domain 1 | −2.6 |
| ABCA8 | ATP-binding cassette, subfamily A (ABC1), member 8 | −2.2 | |
| DPT | Dermatopontin | −2.1 | |
| TNFAIP6 | Tumor necrosis factor, α-induced protein 6 | 2.4 | |
| Extracellular matrix | COL8A1 | Collagen, type VIII, α 1 | −2.6 |
| COL11A1 | Collagen, type XI, α 1 | −2.8 | |
| VIT | Vitrin | −2.3 | |
| Angiogenesis | ANGPT1 | Angiopoietin 1 | −2.0 |
| ID1 | Inhibitor of DNA binding 1, dominant negative helix-loop-helix protein | −2.6 | |
| RHOB | ras homolog family member B | −2.1 | |
| Apoptosis | GADD45B | Growth arrest and DNA-damage-inducible, β | −2.2 |
| ZBTB16 | Zinc finger and BTB domain containing 16 | −2.1 | |
| DRAM1 | DNA-damage regulated autophagy modulator 1 | 2.5 | |
| GREM1 | Gremlin 1 | 2.4 | |
| Ion binding/homeostasis | STEAP4 | STEAP family member 4 | −2.2 |
| CPM | Carboxypeptidase M | −3.9 | |
| MGP | Matrix Gla protein | −3.4 | |
| MT1X | Metallothionein 1X | −2.5 | |
| MT2A | Metallothionein 2A | −2.2 | |
| NPR3 | Natriuretic peptide receptor C/guanylate cyclase C | −2.5 | |
| SCN7A | Sodium channel, voltage-gated, type VII, α subunit | −2.3 | |
| Regulation of transcription | ANKRD1 | Ankyrin repeat domain 1 (cardiac muscle) | −2.2 |
| EGR1 | Early growth response 1 | −4.6 | |
| NFYB | Nuclear transcription factor Y β | −2.7 | |
| POLR2A | Polymerase (RNA) II (DNA directed) polypeptide A | 2.0 | |
| FKBP5 | FK506 binding protein 5 | −5.2 | |
| Drug metabolism | ADH1C | Alcohol dehydrogenase 1C (class I), γ polypeptide | −2.7 |
| ADH1B | Alcohol dehydrogenase 1B (class I), β polypeptide | −16.2 | |
| FMO2 | Flavin containing monooxygenase 2 (non-functional) | −3.5 | |
| MAOA | Monoamine oxidase A | −2.8 |
Myotubes were differentiated 5 d and then treated 48 h with 10 µM aldosterone or spironolactone. Gene functions were assigned and clustered using the functional annotation clustering tool from DAVID. Positive fold changes indicate gene expression was increased with antagonist compared with agonist treatment and vice versa.
Identification of potential MR downstream targets
Global gene expression analysis was used to uncover whether gene changes identified in antagonist- vs. agonist-treated myotubes contributed to the therapeutic effects previously observed from treatment of dystrophic het mice with lisinopril plus spironolactone (7). Gene expression microarray was used to compare quadriceps muscles from lisinopril plus spironolactone-treated het mice with untreated het controls (Table 2). Cluster analysis of gene expression differences between treated and untreated mice identified several of the same functional related gene groups seen in microarray data from treated primary myogenic cultures, including ion binding, defense response, apoptosis, and regulation of transcription.
TABLE 2.
Fold changes of gene expression differences from microarray comparing quadriceps muscle from lisinopril plus spironolactone-treated het mice vs. untreated het controls
| Function | Gene symbol | Full gene name | Fold change (treated/ untreated) |
|---|---|---|---|
| Hypertrophic/dilated cardiomyopathy | Itga5 | Integrin α 5 (fibronectin receptor α) | −2.1 |
| Myh6 | Myosin, heavy polypeptide 6, cardiac muscle, α | −4.4 | |
| Myl2 | Myosin, light polypeptide 2, regulatory, cardiac, slow | −15.1 | |
| Myl3 | Myosin, light polypeptide 3 | −2.5 | |
| Tpm3 | Tropomyosin 3, γ | −6.1 | |
| Tnnc1 | Troponin C, cardiac/slow skeletal | −2.4 | |
| Mybph | Myosin binding protein H | 2.1 | |
| Hormone responsive | Mid1ip1 | Mid1 interacting protein 1 [gastrulation specific G12-like (zebrafish)] | −2.5 |
| Inhbb | Inhibin β-B | −2.2 | |
| Serpina3m | Serine (or cysteine) peptidase inhibitor, clade A, member 3M | −2.3 | |
| Thrsp | Thyroid hormone responsive SPOT14 homolog (Rattus) | 2.1 | |
| Retn | Resistin | 2.2 | |
| Glucose metabolic process | Gpd1 | Glycerol-3-phosphate dehydrogenase 1 (soluble) | 2.2 |
| Pck1 | Phosphoenolpyruvate carboxykinase 1, cytosolic | 2.1 | |
| Ppp1r3c | Protein phosphatase 1, regulatory (inhibitor) subunit 3C | 2.1 | |
| Ion binding | Lrg1 | Leucine-rich α-2-glycoprotein 1 | −2.0 |
| Adamts1 | A disintegrin-like and metallopeptidase (reprolysin type) with thrombospondin type 1 motif, 1 | −2.0 | |
| Sik1 | Salt inducible kinase 1 | −2.3 | |
| Cdo1 | Cysteine dioxygenase 1, cytosolic | 2.2 | |
| Cyp2e1 | Cytochrome P450, family 2, subfamily e, polypeptide 1 | 4.0 | |
| Slc25a25 | Solute carrier family 25 (mitochondrial carrier, phosphate carrier), member 25 | 3.0 | |
| Itgb1bp3 | Integrin β 1 binding protein 3 | 2.1 | |
| Kcng4 | Potassium voltage-gated channel, subfamily G, member 4 | 2.4 | |
| Regulation of transcription | Ankrd1 | Ankyrin repeat domain 1 (cardiac muscle) | −2.2 |
| Tgif1 | TGFB-induced factor homeobox 1 | −2.0 | |
| Hivep3 | Human immunodeficiency virus type I enhancer binding protein 3 | −2.4 | |
| Fos | FBJ osteosarcoma oncogene | −2.3 | |
| Cited4 | Cbp/p300-interacting transactivator, with Glu/Asp-rich carboxy-terminal domain, 4 | 2.3 | |
| Mbd2 | Methyl-CpG binding domain protein 2 | 3.8 | |
| Apoptosis | Ahr | Aryl-hydrocarbon receptor | −2.1 |
| Cidec | Cell death–inducing DFFA-like effector c | 2.4 | |
| Gadd45g | Growth arrest and DNA-damage-inducible 45 γ | 2.1 | |
| Defense response | Cxcl1 | Chemokine (C-X-C motif) ligand 1 | −3.7 |
| Prg4 | Proteoglycan 4 (megakaryocyte stimulating factor, articular superficial zone protein) | −3.2 | |
| Npr3 | Natriuretic peptide receptor 3 | −2.4 | |
| Cytoskeleton and tight junction | Tjp2 | Tight junction protein 2 | −2.0 |
| Lmod2 | Leiomodin 2 (cardiac) | −2.4 | |
| Myo5a | Myosin VA | −2.1 |
Gene functions were assigned and clustered using the functional annotation clustering tool from DAVID. Positive fold changes indicate gene expression was increased with treatment, and vice versa.
Microarray analysis of lisinopril plus spironolactone-treated vs. untreated het mice revealed that many of the genes with altered expression in patients with DMD and mouse models (33, 34) were normalized by treatment. Genes associated with hypertrophy and dilated cardiomyopathy, which are increased in dystrophic skeletal muscles, were down-regulated with treatment. Treatment also normalized several metabolic genes, immunoresponsive genes, and genes associated with cell signaling and transcriptional regulation.
Several genes identified by the microarray analysis of antagonist- vs. agonist-treated human myotubes were also changed in lisinopril plus spironolactone-treated versus untreated dystrophic mice (Table 3). Genes conserved between both data sets, including GADD45B (growth arrest and DNA-damage-inducible, β), ANKRD1, NPR3 (natriuretic peptide receptor 3), and FOS (FBJ (Finkel–Biskis–Jinkins) osteosarcoma oncogene), represent potential direct MR downstream targets (Table 3). Two bona fide MR targets from cardiomyocytes and renal cells, SERPINA3 (25) and FKBP5 (35), respectively, were also differentially expressed with MR antagonism in skeletal muscle, further validating the microarray results.
TABLE 3.
Subset of genes conserved between both microarray data sets
| Full gene name | Gene symbol | Functional relationship to treatment | Fold change (antagonist/agonist) |
|---|---|---|---|
| Serpin peptidase inhibitor, clade A, member 3 | SERPINA3 | Known downstream target of MR in cardiac cells (25) | −2.2 |
| FBJ murine osteosarcoma viral oncogene homolog | FOS | Up-regulated by aldosterone and GR-overexpression (25, 50) | −2.6 |
| Growth arrest and DNA-damage-inducible, β | GADD45B | Up-regulated in stressful growth conditions (52) | −2.2 |
| Ankyrin repeat domain 1 | ANKRD1 | Transcript levels altered in numerous muscle diseases (36) | −2.2 |
| Natriuretic peptide receptor C/guanylate cyclase C | NPR3 | Involved in MR-regulated pathways (46) | −2.5 |
These gene changes are likely caused directly by MR modulation; gene function or regulation related to treatment is listed.
ANKRD1 was selected for additional analysis because it represents a promising MR gene target candidate as a result of its role in transcriptional regulation in response to skeletal muscle myofibrillar stress (36). Real-time RT-PCR was used to validate microarray results and determine whether Ankrd1 transcript levels varied between normal and dystrophic muscle. Ankrd1 transcript levels were higher in dystrophic mouse muscle relative to wild-type (Fig. 4), which is consistent with previous findings in the literature (36). Despite elevated Ankrd1 mRNA levels in dystrophic muscle, relative to wild-type, there are no gross changes in relative Ankrd1 protein levels (Fig. 5A) comparable with previous studies of dystrophic models (36). To determine whether ANKRD1 protein levels are reduced after treatment with MR antagonist vs. agonist, and to confirm that these reductions are occurring via MR, we treated human myotubes for 5 d with the MR agonist aldosterone and with either spironolactone, which antagonizes both the MR and androgen receptor, or the more specific MR antagonist eplerenone, which does not block androgen receptor. Eplerenone was not used for initial studies because of its much lower solubility. Aldosterone treatment led to increased ANKRD1 protein levels relative to treatment with MR antagonist eplerenone (Fig. 5B). Immunofluorescence of human myotubes treated with MR agonist or antagonist also showed a reduction of ANKRD1 staining in eplerenone-treated cells (Fig. 5C). Cells used for immunofluorescence and collected for Western blot analysis were treated for 5 d to ensure adequate time for protein levels to accumulate.
Figure 4.

Real-time RT-PCR revealed Ankrd1 mRNA levels are up-regulated in dystrophic muscle. Quadriceps muscles from MDX (dystrophin deficient) and HET (utrn+/−; mdx) mice relative to C57 (wild-type). Each bar represents mean of 3 technical triplicates ± se. Three biologic replicates are shown for each genotype; all samples are normalized to mean of C57-3. 18S was used as normalization control for all samples.
Figure 5.

ANKRD1 protein levels are conserved between normal and dystrophic mouse muscles but altered by treatment with MR agonists and antagonists in human cultured myotubes. A) Representative samples (quadriceps muscle) are shown comparing level of Ankrd1 protein (∼43 kDa) expression on equivalent amounts (35 µg) of protein homogenates from: C57BL/10 wild-type (C57) mice, dystrophin-deficient mdx (MDX) mice, het (HET) mice, and dystrophin/utrophin-deficient double knockout (DKO) mice (n = 3). These Western blots were run in conjunction with MR blots from Fig. 3 using same actin loading control. B) Human primary muscle (HSMM) myotubes (MT) were differentiated for 5 d and then treated for 5 d with 10 µM of MR agonist aldosterone (A-10), MR antagonist spironolactone (S-10), 2 µM of MR antagonist eplerenone (E-2), or ethanol vehicle untreated control (UC); 35 µg of total protein from cell lysates were used for each sample. Standardization of loading was verified using α-sarcomeric actin antibody; predicted molecular weight was ∼42 kDa. C) ANKRD1 staining is altered by treatment with MR antagonist. Ankrd1 was detected by immunofluorescence staining (red) on human primary (HSMM) myotubes differentiated for 5 d and then treated for ∼5 d with 10 µM of MR agonist aldosterone, MR antagonist spironolactone, or 2 µM of more selective MR antagonist eplerenone. Scale bars, 50 µm.
DISCUSSION
We showed that MR was expressed and functional in skeletal muscle fibers. These data suggest that FDA-approved MR antagonists, with their long history of safe and effective use in cardiology, may also be efficacious for skeletal muscle disorders. In addition to full length MR, human skeletal muscle myotubes express an alternatively spliced form of MR with a deletion of exon 5 (NR3C2, transcript variant 2). This variant has been shown to be present in numerous human tissues including lung, heart, kidney, lymphocytes, and hippocampus (37). It is unclear whether this deletion affects ligand binding because exon 5 includes part of the ligand-binding domain, according to the crystal structure (38). However, only amino acids 804 to 874 of the ligand binding domain, which is downstream from the deleted region of amino acids 672 to 788, were found to be necessary for high-affinity binding of the antagonist spironolactone and agonist aldosterone in previous studies (39). NR3C2, transcript variant 2, may function like hMRΔ5, 6, another known human MR splice variant, with an exon 5 and 6 deletion that functions ligand independently and appears to modulate transcriptional activation of wild-type full-length human MR (37). Sequencing analysis did not detect an exon 5 deletion in mouse skeletal muscle. The additional band detected by Western blot analysis may represent a truncated form of the protein or a degradation product of this protein known to be highly sensitive to proteolysis (22). Future experiments, including the role of MR alternative splicing, and the levels of 11β-hydroxysteroid dehydrogenase and other MR coregulators will be needed to fully define the function of MR in skeletal muscle in vivo.
Microarray comparing antagonist- vs. agonist-treated human myotubes showed decreased expression of several genes associated with oxidative stress, including ADH1B, ADH1C, and GPX. This result is consistent with previous studies, which found spironolactone prevents aldosterone-induced inflammation and oxidative stress in cardiac cells; the authors speculated that it may be able to prevent oxidative stress that contributes to cardiac muscle wasting (40, 41). A previous study found that cells under oxidative stress up-regulate the alcohol dehydrogenases ADH1B and ADH1C in order to convert retinol to retinoic acid, which has been shown to protect muscle cells (42). Retinoic acid in turn up-regulates glutathione peroxidase GPX3, which is a regulator of its antioxidant effects. Cells under oxidative stress have also been reported to have reduced cell adhesion (42). This stress could account for the up-regulation of the neuronal cell adhesion molecule gene NRCAM and the secreted matrix genes MGP, COL11A1, COL8A1, and NID1 in agonist-treated cells. However, given the disparity between the effectiveness of spironolactone and published antioxidant therapies, it is very unlikely that spironolactone only protects against oxidative stress; it likely is targeting other pathways.
We have identified potential direct MR downstream targets, which may help elucidate the mechanism behind the efficacy of these drugs in dystrophic skeletal muscles and serve as biomarkers of treatment (Table 3). Genes conserved between both microarray data sets are likely to be caused by treatment and are not just a secondary result of efficacy. We found that SERPINA3, a known direct target of MR in cardiac cells (25), was also down-regulated by spironolactone in skeletal muscle. Although its physiologic function is not well understood, it is known to inhibit neutrophil cathepsin G and mast cell chymase (43), both of which are involved in the renin–angiotensin–aldosterone pathway. Although mast cells are traditionally thought of as proinflammatory, there is a growing body of evidence suggesting that they can also suppress inflammation (44). SERPINA3 is also involved in tissue protection. Glucocorticoid treatment of lung cells led to increased SERPINA3 expression, which was postulated to cause some of the adverse effects seen with long-term glucocorticoid therapy (45). NPR3 helps maintain blood pressure and extracellular fluid volume (46), which are known MR-regulated pathways (47). Mutations in NPR2 are known to effect muscle growth (48), but NPR3 has not previously been investigated in skeletal muscle. FOS, a regulator of cell proliferation and differentiation in skeletal muscle and other cell types (49), has previously been shown to be up-regulated by aldosterone and GR overexpression (25, 50). GADD45B, a regulator of cell death (51), has been linked to oxidative stress and is known to be up-regulated in stressful growth conditions (52). GADD45B is down-regulated in MR antagonist-treated human myotubes but up-regulated in dystrophic mice treated with lisinopril and spironolactone. In vitro models of muscle wasting found that TNF suppresses GADD45B expression, which leads to increased cell death (53). Dystrophic mice may have suppressed GADD45B levels, which are normalized with treatment (53).
ANKRD1 is a transcriptional cofactor and sarcomeric component altered in a number of muscle diseases, including muscular dystrophies, congenital myopathies, muscle wasting, and motor neuron diseases (36, 54). Previous studies have recognized its potential as a biomarker for muscular dystrophies because Ankrd1 is consistently and significantly up-regulated in animal models for multiple forms of muscular dystrophy (54), and levels have been shown to correlate with creatine kinase activity in peroxisome proliferator-activated receptor–induced skeletal myopathies (55). Changes in Ankrd1 gene expression do not correlate with dramatic changes in steady-state protein levels. Ankrd1 transcript levels were up-regulated in the dystrophin-deficient mdx4Cv mouse model, but protein levels were comparable to wild-type (54). ANKRD1 protein is only present at high levels in newly regenerating fibers of patients with DMD (36, 56). Ankrd1 protein levels were elevated with transient muscle denervation but returned to basal levels when reinnervation occurred, suggesting that Ankrd1 transcript levels may be elevated so protein can be rapidly translated. Ankrd1 functions as a link between stress sensing and response (36). Therefore, protein levels could be temporarily elevated in response to myofibrillar stress and then degraded or recycled once downstream targets are activated. ANKRD1 protein levels were up-regulated in aldosterone (agonist)-treated myotubes compared with those treated with eplerenone (antagonist). This result suggests that Ankrd1 regulation is due to the MR and not the androgen receptor that is also antagonized by spironolactone, but not eplerenone. Additional studies will be required to confirm whether ANKRD1 and the other genes with altered expression between agonist and antagonist treatment are direct MR targets. In addition, future studies will be needed to confirm whether Ankrd1 or one or possibly several of these other gene expression changes, contributes to the efficacy observed with MR antagonist treatment of dystrophic skeletal muscle (7, 30).
MRs represent a novel therapeutic target for treating both the skeletal and cardiac muscle pathologies of DMD. Current treatments are limited, and although emerging therapies such as exon skipping, mutation suppression, and gene supplementation through viral vectors are promising, they may not be available for current patients with DMD, and some are targeted only toward skeletal muscle. Both lisinopril and spironolactone are FDA-approved drugs with minimal side effects and a long history of safety and efficacy for treatment of heart failure. Early use of angiotensin-converting enzyme inhibition has already been shown in clinical trials to slow cardiomyopathy progression in DMD (57). A recent clinical trial has demonstrated further improvement in cardiac function in patients with DMD by the addition of an MR antagonist (58). Here, we demonstrate for the first time the presence of MR in skeletal muscles and that MR antagonism can lead to beneficial gene expression changes. Use of these clinic-ready drugs can likely be rapidly developed into an effective regimen that, we hope, will slow disease progression and improve the quality of life of patients with DMD.
Acknowledgments
This work was supported by the U.S. National Institutes of Health [R01 NS082868 and R01 HL116533 (to J.R.F.), and T32 NS077984 (to J.A.C.)], Department of Defense [MD120063 (to J.R.F.)], and the Center for Muscle Health and Neuromuscular Disorders at The Ohio State University (OSU) and Nationwide Children’s Hospital. The authors thank The Ohio State Microarray Core, and in particular S. Warner, for microarray processing and analysis; Pfizer for supplying eplerenone under their Compound Transfer Program; K. J. Ladner (OSU) for RNA preparation for mouse microarray; A. Wodarcyk (OSU) for technical assistance in mouse maintenance, dissections, and genotyping; and S. Swager (OSU) for assistance with confocal imaging.
Glossary
- Ankrd1/ANKRD1
ankyrin repeat domain 1
- DAVID
Database for Annotation, Visualization, and Integrated Discovery
- DMD
Duchenne muscular dystrophy
- FDA
U.S. Food and Drug Administration
- Fos/FOS
FBJ (Finkel–Biskis–Jinkins) osteosarcoma oncogene
- Gadd45b/GADD45B
growth arrest and DNA-damage-inducible, β
- GR
glucocorticoid receptor
- het
dystrophin-deficient utrophin-haploinsufficient
- HSMM
human skeletal muscle myoblast
- MR
mineralocorticoid receptor
- NPR3
natriuretic peptide receptor 3
- RT-PCR
reverse-transcribed PCR
- SERPINA3
serpin peptidase inhibitor clade A
REFERENCES
- 1.Mendell J. R., Shilling C., Leslie N. D., Flanigan K. M., al-Dahhak R., Gastier-Foster J., Kneile K., Dunn D. M., Duval B., Aoyagi A., Hamil C., Mahmoud M., Roush K., Bird L., Rankin C., Lilly H., Street N., Chandrasekar R., Weiss R. B. (2012) Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann. Neurol. 71, 304–313 [DOI] [PubMed] [Google Scholar]
- 2.Emery A. E. (2002) The muscular dystrophies. Lancet 359, 687–695 [DOI] [PubMed] [Google Scholar]
- 3.DeSilva S., Drachman D. B., Mellits D., Kuncl R. W. (1987) Prednisone treatment in Duchenne muscular dystrophy. Long-term benefit. Arch. Neurol. 44, 818–822 [DOI] [PubMed] [Google Scholar]
- 4.Bauer R., Straub V., Blain A., Bushby K., MacGowan G. A. (2009) Contrasting effects of steroids and angiotensin-converting-enzyme inhibitors in a mouse model of dystrophin-deficient cardiomyopathy. Eur. J. Heart Fail. 11, 463–471 [DOI] [PubMed] [Google Scholar]
- 5.Lo Cascio V., Kanis J. A., Beneton M. N., Bertoldo F., Adami S., Poggi G., Zanolin M. E. (1995) Acute effects of deflazacort and prednisone on rates of mineralization and bone formation. Calcif. Tissue Int. 56, 109–112 [DOI] [PubMed] [Google Scholar]
- 6.Angelini C., Peterle E. (2012) Old and new therapeutic developments in steroid treatment in Duchenne muscular dystrophy. Acta Myol. 31, 9–15 [PMC free article] [PubMed] [Google Scholar]
- 7.Rafael-Fortney J. A., Chimanji N. S., Schill K. E., Martin C. D., Murray J. D., Ganguly R., Stangland J. E., Tran T., Xu Y., Canan B. D., Mays T. A., Delfín D. A., Janssen P. M., Raman S. V. (2011) Early treatment with lisinopril and spironolactone preserves cardiac and skeletal muscle in Duchenne muscular dystrophy mice. Circulation 124, 582–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou L., Rafael-Fortney J. A., Huang P., Zhao X. S., Cheng G., Zhou X., Kaminski H. J., Liu L., Ransohoff R. M. (2008) Haploinsufficiency of utrophin gene worsens skeletal muscle inflammation and fibrosis in mdx mice. J. Neurol. Sci. 264, 106–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Messaoudi S., Azibani F., Delcayre C., Jaisser F. (2012) Aldosterone, mineralocorticoid receptor, and heart failure. Mol. Cell. Endocrinol. 350, 266–272 [DOI] [PubMed] [Google Scholar]
- 10.Chitnis A. S., Aparasu R. R., Chen H., Johnson M. L. (2012) Comparative effectiveness of different angiotensin-converting enzyme inhibitors on the risk of hospitalization in patients with heart failure. J. Comp. Eff. Res. 1, 195–206 [DOI] [PubMed] [Google Scholar]
- 11.Tamargo J., Solini A., Ruilope L. M. (2014) Comparison of agents that affect aldosterone action. Semin. Nephrol. 34, 285–306 [DOI] [PubMed] [Google Scholar]
- 12.Delyani J. A., Rocha R., Cook C. S., Tobert D. S., Levin S., Roniker B., Workman D. L., Sing Y. L., Whelihan B. (2001) Eplerenone: a selective aldosterone receptor antagonist (SARA). Cardiovasc. Drug Rev. 19, 185–200 [DOI] [PubMed] [Google Scholar]
- 13.Lowe J., Wodarcyk A. J., Floyd K. T., Rastogi N., Schultz E. J., Swager S. A., Chadwick J. A., Tran T., Raman S. V., Janssen P. M. L., Rafael-Fortney J. A. (2015) The angiotensin converting enzyme inhibitor lisinopril improves muscle histopathology but not contractile function in a mouse model of Duchenne muscular dystrophy. J. Neuromusc. Dis. In press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weinberger C., Giguère V., Hollenberg S. M., Thompson C., Arriza J., Evans R. M. (1987) Human steroid receptors and erb-A gene products form a superfamily of enhancer-binding proteins. Clin. Physiol. Biochem. 5, 179–189 [PubMed] [Google Scholar]
- 15.Arriza J. L., Weinberger C., Cerelli G., Glaser T. M., Handelin B. L., Housman D. E., Evans R. M. (1987) Cloning of human mineralocorticoid receptor complementary DNA: structural and functional kinship with the glucocorticoid receptor. Science 237, 268–275 [DOI] [PubMed] [Google Scholar]
- 16.Evans R. M. (1988) The steroid and thyroid hormone receptor superfamily. Science 240, 889–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang J., Young M. J. (2009) The mineralocorticoid receptor and its coregulators. J. Mol. Endocrinol. 43, 53–64 [DOI] [PubMed] [Google Scholar]
- 18.Hawkins U. A., Gomez-Sanchez E. P., Gomez-Sanchez C. M., Gomez-Sanchez C. E. (2012) The ubiquitous mineralocorticoid receptor: clinical implications. Curr. Hypertens. Rep. 14, 573–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Odermatt A., Kratschmar D. V. (2012) Tissue-specific modulation of mineralocorticoid receptor function by 11β-hydroxysteroid dehydrogenases: an overview. Mol. Cell. Endocrinol. 350, 168–186 [DOI] [PubMed] [Google Scholar]
- 20.Gomez-Sanchez E. P., Gomez-Sanchez M. T., de Rodriguez A. F., Romero D. G., Warden M. P., Plonczynski M. W., Gomez-Sanchez C. E. (2009) Immunohistochemical demonstration of the mineralocorticoid receptor, 11beta-hydroxysteroid dehydrogenase-1 and -2, and hexose-6-phosphate dehydrogenase in rat ovary. J. Histochem. Cytochem. 57, 633–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gomez-Sanchez E. P. (2004) Brain mineralocorticoid receptors: orchestrators of hypertension and end-organ disease. Curr. Opin. Nephrol. Hypertens. 13, 191–196 [DOI] [PubMed] [Google Scholar]
- 22.Gomez-Sanchez C. E., Warden M., Gomez-Sanchez M. T., Hou X., Gomez-Sanchez E. P. (2011) Diverse immunostaining patterns of mineralocorticoid receptor monoclonal antibodies. Steroids 76, 1541–1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Viengchareun S., Le Menuet D., Martinerie L., Munier M., Pascual-Le Tallec L., Lombès M. (2007) The mineralocorticoid receptor: insights into its molecular and (patho)physiological biology. Nucl. Recept. Signal. 5, e012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lastra G., Whaley-Connell A., Manrique C., Habibi J., Gutweiler A. A., Appesh L., Hayden M. R., Wei Y., Ferrario C., Sowers J. R. (2008) Low-dose spironolactone reduces reactive oxygen species generation and improves insulin-stimulated glucose transport in skeletal muscle in the TG(mRen2)27 rat. Am. J. Physiol. Endocrinol. Metab. 295, E110–E116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Latouche C., Sainte-Marie Y., Steenman M., Castro Chaves P., Naray-Fejes-Toth A., Fejes-Toth G., Farman N., Jaisser F. (2010) Molecular signature of mineralocorticoid receptor signaling in cardiomyocytes: from cultured cells to mouse heart. Endocrinology 151, 4467–4476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jaisser, F., Farman, N., Sainte-Marie, Y., Latouche, C., Steenman, M. (2010) Biomarkers of mineralocorticoid receptor activation. U.S. Patent Application No. 20110257140
- 27.Bulfield G., Siller W. G., Wight P. A., Moore K. J. (1984) X chromosome–linked muscular dystrophy (mdx) in the mouse. Proc. Natl. Acad. Sci. USA 81, 1189–1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sicinski P., Geng Y., Ryder-Cook A. S., Barnard E. A., Darlison M. G., Barnard P. J. (1989) The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science 244, 1578–1580 [DOI] [PubMed] [Google Scholar]
- 29.Deconinck A. E., Rafael J. A., Skinner J. A., Brown S. C., Potter A. C., Metzinger L., Watt D. J., Dickson J. G., Tinsley J. M., Davies K. E. (1997) Utrophin–dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell 90, 717–727 [DOI] [PubMed] [Google Scholar]
- 30.Janssen P. M., Murray J. D., Schill K. E., Rastogi N., Schultz E. J., Tran T., Raman S. V., Rafael-Fortney J. A. (2014) Prednisolone attenuates improvement of cardiac and skeletal contractile function and histopathology by lisinopril and spironolactone in the mdx mouse model of Duchenne muscular dystrophy. PLoS One 9, e88360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gomez-Sanchez C. E., de Rodriguez A. F., Romero D. G., Estess J., Warden M. P., Gomez-Sanchez M. T., Gomez-Sanchez E. P. (2006) Development of a panel of monoclonal antibodies against the mineralocorticoid receptor. Endocrinology 147, 1343–1348 [DOI] [PubMed] [Google Scholar]
- 32.Clyne C. D., Chang C. Y., Safi R., Fuller P. J., McDonnell D. P., Young M. J. (2009) Purification and characterization of recombinant human mineralocorticoid receptor. Mol. Cell. Endocrinol. 302, 81–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baker P. E., Kearney J. A., Gong B., Merriam A. P., Kuhn D. E., Porter J. D., Rafael-Fortney J. A. (2006) Analysis of gene expression differences between utrophin/dystrophin-deficient vs. mdx skeletal muscles reveals a specific upregulation of slow muscle genes in limb muscles. Neurogenetics 7, 81–91 [DOI] [PubMed] [Google Scholar]
- 34.Bakay M., Zhao P., Chen J., Hoffman E. P. (2002) A Web-accessible complete transcriptome of normal human and DMD muscle. Neuromuscul. Disord. 12(Suppl 1), S125–S141 [DOI] [PubMed] [Google Scholar]
- 35.Ueda K., Fujiki K., Shirahige K., Gomez-Sanchez C. E., Fujita T., Nangaku M., Nagase M. (2014) Genome-wide analysis of murine renal distal convoluted tubular cells for the target genes of mineralocorticoid receptor. Biochem. Biophys. Res. Commun. 445, 132–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kojic S., Radojkovic D., Faulkner G. (2011) Muscle ankyrin repeat proteins: their role in striated muscle function in health and disease. Crit. Rev. Clin. Lab. Sci. 48, 269–294 [DOI] [PubMed] [Google Scholar]
- 37.Zennaro M. C., Souque A., Viengchareun S., Poisson E., Lombès M. (2001) A new human MR splice variant is a ligand-independent transactivator modulating corticosteroid action. Mol. Endocrinol. 15, 1586–1598 [DOI] [PubMed] [Google Scholar]
- 38.Bledsoe R. K., Madauss K. P., Holt J. A., Apolito C. J., Lambert M. H., Pearce K. H., Stanley T. B., Stewart E. L., Trump R. P., Willson T. M., Williams S. P. (2005) A ligand-mediated hydrogen bond network required for the activation of the mineralocorticoid receptor. J. Biol. Chem. 280, 31283–31293 [DOI] [PubMed] [Google Scholar]
- 39.Rogerson F. M., Yao Y., Smith B. J., Fuller P. J. (2004) Differences in the determinants of eplerenone, spironolactone and aldosterone binding to the mineralocorticoid receptor. Clin. Exp. Pharmacol. Physiol. 31, 704–709 [DOI] [PubMed] [Google Scholar]
- 40.Burniston J. G., Saini A., Tan L. B., Goldspink D. F. (2005) Aldosterone induces myocyte apoptosis in the heart and skeletal muscles of rats in vivo. J. Mol. Cell. Cardiol. 39, 395–399 [DOI] [PubMed] [Google Scholar]
- 41.Young M. J., Rickard A. J. (2012) Mechanisms of mineralocorticoid salt-induced hypertension and cardiac fibrosis. Mol. Cell. Endocrinol. 350, 248–255 [DOI] [PubMed] [Google Scholar]
- 42.El Haddad M., Jean E., Turki A., Hugon G., Vernus B., Bonnieu A., Passerieux E., Hamade A., Mercier J., Laoudj-Chenivesse D., Carnac G. (2012) Glutathione peroxidase 3, a new retinoid target gene, is crucial for human skeletal muscle precursor cell survival. J. Cell Sci. 125, 6147–6156 [DOI] [PubMed] [Google Scholar]
- 43.Rubin H., Wang Z. M., Nickbarg E. B., McLarney S., Naidoo N., Schoenberger O. L., Johnson J. L., Cooperman B. S. (1990) Cloning, expression, purification, and biological activity of recombinant native and variant human alpha 1-antichymotrypsins. J. Biol. Chem. 265, 1199–1207 [PubMed] [Google Scholar]
- 44.Caughey G. H. (2011) Mast cell proteases as protective and inflammatory mediators. Adv. Exp. Med. Biol. 716, 212–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lannan E. A., Galliher-Beckley A. J., Scoltock A. B., Cidlowski J. A. (2012) Proinflammatory actions of glucocorticoids: glucocorticoids and TNFα coregulate gene expression in vitro and in vivo. Endocrinology 153, 3701–3712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Anand-Srivastava M. B. (2005) Natriuretic peptide receptor-C signaling and regulation. Peptides 26, 1044–1059 [DOI] [PubMed] [Google Scholar]
- 47.Lother A., Moser M., Bode C., Feldman R. D., Hein L. (2015) Mineralocorticoids in the heart and vasculature: new insights for old hormones. Annu. Rev. Pharmacol. Toxicol. 55, 289–312 [DOI] [PubMed] [Google Scholar]
- 48.Vasques G. A., Arnhold I. J., Jorge A. A. (2014) Role of the natriuretic peptide system in normal growth and growth disorders. Horm. Res. Paediatr. 82, 222–229 [DOI] [PubMed] [Google Scholar]
- 49.Okazaki K., Sagata N. (1995) The Mos/MAP kinase pathway stabilizes c-Fos by phosphorylation and augments its transforming activity in NIH 3T3 cells. EMBO J. 14, 5048–5059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rincon Garriz J. M., Suarez C., Capponi A. M. (2009) c-Fos mediates angiotensin II–induced aldosterone production and protein synthesis in bovine adrenal glomerulosa cells. Endocrinology 150, 1294–1302 [DOI] [PubMed] [Google Scholar]
- 51.Takekawa M., Saito H. (1998) A family of stress-inducible GADD45-like proteins mediate activation of the stress-responsive MTK1/MEKK4 MAPKKK. Cell 95, 521–530 [DOI] [PubMed] [Google Scholar]
- 52.Kim J. H., Qu A., Reddy J. K., Gao B., Gonzalez F. J. (2014) Hepatic oxidative stress activates the Gadd45b gene by way of degradation of the transcriptional repressor STAT3. Hepatology 59, 695–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stewart C. E., Newcomb P. V., Holly J. M. (2004) Multifaceted roles of TNF-alpha in myoblast destruction: a multitude of signal transduction pathways. J. Cell. Physiol. 198, 237–247 [DOI] [PubMed] [Google Scholar]
- 54.Laure L., Suel L., Roudaut C., Bourg N., Ouali A., Bartoli M., Richard I., Danièle N. (2009) Cardiac ankyrin repeat protein is a marker of skeletal muscle pathological remodelling. FEBS J. 276, 669–684 [DOI] [PubMed] [Google Scholar]
- 55.Casey W. M., Brodie T., Yoon L., Ni H., Jordan H. L., Cariello N. F. (2008) Correlation analysis of gene expression and clinical chemistry to identify biomarkers of skeletal myopathy in mice treated with PPAR agonist GW610742X. Biomarkers 13, 364–376 [DOI] [PubMed] [Google Scholar]
- 56.Nakada C., Tsukamoto Y., Oka A., Nonaka I., Takeda S., Sato K., Mori S., Ito H., Moriyama M. (2003) Cardiac-restricted ankyrin-repeated protein is differentially induced in Duchenne and congenital muscular dystrophy. Lab. Invest. 83, 711–719 [DOI] [PubMed] [Google Scholar]
- 57.Duboc D., Meune C., Lerebours G., Devaux J. Y., Vaksmann G., Bécane H. M. (2005) Effect of perindopril on the onset and progression of left ventricular dysfunction in Duchenne muscular dystrophy. J. Am. Coll. Cardiol. 45, 855–857 [DOI] [PubMed] [Google Scholar]
- 58.Raman S. V., Hor K. N., Mazur W., Halnon N. J., Kissel J. T., He X., Tran T., Smart S., McCarthy B., Taylor M. D., Jefferies J. L., Rafael-Fortney J. A., Lowe J., Roble S. L., Cripe L. H. (2015) Eplerenone for early cardiomyopathy in Duchenne muscular dystrophy: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 14, 153–161 [DOI] [PMC free article] [PubMed] [Google Scholar]