

Abstract
Motivation: Today, the base code of DNA is mostly determined through sequencing by synthesis as provided by the Illumina sequencers. Although highly accurate, resulting reads are short, making their analyses challenging. Recently, a new technology, single molecule real-time (SMRT) sequencing, was developed that could address these challenges, as it generates reads of several thousand bases. But, their broad application has been hampered by a high error rate. Therefore, hybrid approaches that use high-quality short reads to correct erroneous SMRT long reads have been developed. Still, current implementations have great demands on hardware, work only in well-defined computing infrastructures and reject a substantial amount of reads. This limits their usability considerably, especially in the case of large sequencing projects.
Results: Here we present proovread, a hybrid correction pipeline for SMRT reads, which can be flexibly adapted on existing hardware and infrastructure from a laptop to a high-performance computing cluster. On genomic and transcriptomic test cases covering Escherichia coli, Arabidopsis thaliana and human, proovread achieved accuracies up to 99.9% and outperformed the existing hybrid correction programs. Furthermore, proovread-corrected sequences were longer and the throughput was higher. Thus, proovread combines the most accurate correction results with an excellent adaptability to the available hardware. It will therefore increase the applicability and value of SMRT sequencing.
Availability and implementation: proovread is available at the following URL: http://proovread.bioapps.biozentrum.uni-wuerzburg.de
Contact: frank.foerster@biozentrum.uni-wuerzburg.de
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Looking back just a decade, sequencing a genome was a time-consuming and expensive endeavor. The emergence of second-generation sequencers and their sequencing by synthesis have changed this drastically, thereby revolutionizing molecular biology. Today, a single run of a HiSeq2500 can generate as much as 600 Gb high-quality output data, which covers a human genome 200×. Unfortunately, this new technology came with a drawback. Compared with the traditional Sanger sequencing, resulting reads are short (150 bp). This became a major challenge for the assembly, especially in the case of large repetitive genomes. Accordingly, a plenitude of short read assemblers has been developed, e.g. Allpath-LG (Gnerre et al., 2011), the Celera Assembler (Miller et al., 2008; Myers et al., 2000) and SOAPdenovo (Li et al., 2010). Still, repeats longer than the short reads (SRs) can not be resolved, and therefore, the genome can not be reconstructed in these regions (Gnerre et al., 2011). For the assembly of repetitive genomes, a combination of short and long insert libraries and additional fosmid sequencing are therefore recommended (Gnerre et al., 2011).
In 2009, however, a new long read (LR) sequencing technology emerged: single-molecule real-time (SMRT) sequencing. Here, the incorporation of nucleotides in a DNA molecule is recorded during synthesis for several thousand single template strands simultaneously (Eid et al., 2009). With the latest chemistry, this approach delivers reads >4 kb, enabling the assembly of larger repeat structures (Roberts et al., 2013). Additionally, amplification can be omitted. Since 2011, SMRT-based sequencing is commercially available from Pacific Biosciences of California. Their third-generation sequencer, PacBio RS II, generates to date up to 400 Mb per sequencing run.
Still, the advantages of third-generation sequencing come at a price. The accuracy of its LRs falls way behind those of short second-generation reads. Although current Illumina instruments offer a sequencing accuracy of 99% (Dohm et al., 2008), PacBio RS II achieves only 80–85% (Ono et al., 2013; Ross et al., 2013). Furthermore, the error model of both technologies differs. Although Illumina reads mainly contain miscalled bases with increasing frequency toward read ends, SMRT generates primarily insertions (10%) and deletions (5%) in a random pattern (Ross et al., 2013). Because SMRT uses circular templates, accuracy can be increased for shorter sequences (<1 kb). By sequencing each position multiple times, a circular consensus sequencing with an accuracy of 99% can be generated. However, this approach substantially decreases read length (Travers et al., 2010), erasing one of the major advantages of SMRT sequencing. In addition to this technical approach, two different methods for in silico correction of SMRT reads have been developed. (i) The hierarchical genome-assembly process (HGAP) uses shorter SMRT reads contained within longer reads to generate pre-assemblies and to calculate consensus sequences (Chin et al., 2013). (ii) PacBioToCA (Koren et al., 2012) and LSC (Au et al., 2012) use Illumina SRs in a hybrid approach to correct SMRT reads. These approaches result in higher quality LRs. Nevertheless, both approaches also have limitations. In the case of HGAP, a coverage of 80× to 100× has been recommended (Chin et al., 2013). This might not be an issue when targeting smaller, e.g. bacterial, genomes, but for larger, especially eukaryotic, genomes this would imply sequencing several hundred or thousands SMRT cells. Obviously, this increases the costs of the genome project substantially.
For a hybrid correction, as implemented by PacBioToCA and LSC, millions of SR to LR alignments have to be computed and processed. As these alignments have to tolerate error rates up to 20%, this can be a formidable computational challenge. These computational demands are usually met using massive parallelization on high-performance computers (HPCs) or computer grids, providing dozens or hundreds of computer nodes. Accordingly, LSC and PacBioToCA are designed to run on HPCs. PacBioToCA also works on computer clusters providing Sun grid engine (SGE) as a queuing system. Still, both require a large amount of memory during the correction process of large genomes. This can become a considerable limitation, as computing nodes in a grid are typically equipped only with limited memory. A second point that can determine the success of a genome project is the throughput of the correction method, i.e. the percentage of the bases in corrected reads that can be used in the assembly. The lower the throughput, the more material needs to be sequenced in the first place to achieve sufficient coverage for assembly after correction. As an example, PacBioToCA lost >40% when correcting sequences from Escherichia coli (Koren et al., 2012), which is a considerable loss of data. Ultimately, we expect that with the increasing use of SMRT sequencing, more genomes and transcriptomes with unusual features will be sequenced. Thus, a correction pipeline developed today should be flexible enough to be easily adopted to these new use cases. Although LSC was developed mainly for the correction of (human) transcriptomic data, PacBioToCA can handle different datasets, but is part of the Celera WGS pipeline and requires the installation of the complete package. Distributed computing is restricted to the now commercial SGE.
These limitations motivated us to implement a new SMRT sequencing correction pipeline. The goal was high flexibility such that the pipeline can (i) run on standard computers as well as computer grids and (ii) can be easily adapted to different use cases. Obviously, these objectives should not be at the cost of accuracy, length of corrected reads or throughput.
2 IMPLEMENTATION
2.1 Mapping—sensitive and trusted hybrid alignments
Because of the high rate and distinctive nature of SMRT sequencing errors, calculating hybrid alignments is challenging. Mapping of SRs with common alignment models and mapping programs either results in no or at best spurious alignments. For proovread, we thus devised a hybrid alignment scoring scheme based on the following assumptions: (i) The expected error rates for SMRT sequencing are ∼10% for insertions and up to 5% for deletions (Ono et al., 2013; Ross et al., 2013). Thus, the costs for gaps in the LR, which correspond to deletions, are about twice as high as for gaps in the SR, which represent insertion. (ii) Substitutions are comparatively rare (1%). This is reflected by a mismatch penalty of at least 10 times the cost of SR gaps. (iii) The distribution of SMRT sequencing errors is random. Hence, contrasting to biological scenarios, continuous insertions or deletions are less likely, resulting in higher costs for gap extension than for gap opening.
The implementation of the theoretical model strongly depends on the used mapping software. As default, proovread uses SHRiMP2 (David et al., 2011) for mapping. Its versatile interface allowed us to completely implement the hybrid scoring model with the following parameters: insertions are the most frequent errors and are penalized as gap open with −1. Deletions occur about half as often and are thus penalized with −2. Extensions for insertions and deletions are scored with −3 and −4, respectively. Mismatches are at least 10 times as rare, resulting in a penalty of −11 (Supplementary Table S1). All results presented here have been generated using these settings with SHRiMP2 version 2.2.3.
Additionally, proovread is able to directly digest mapping data provided in SAM format (Li et al., 2009), leaving the mapping procedure entirely up to the user. As an alternative to SHRiMP2, Bowtie2 (Langmead and Salzberg, 2012) is supported as an experimental feature. However, corrections using Bowtie2 lagged behind owing to a limited set of parameters regarding scoring and sensitivity. LR and SR data can be provided in either FASTA or FASTQ format. Usage of trimmed (e.g. sickle https://github.com/najoshi/sickle) or corrected SRs [e.g. Quake (Kelley et al., 2010)] is recommended as it increases correction accuracy.
To distinguish valid from spurious alignments, the obtained mapping results require further evaluation. In general, the number of reported alignments differs strongly between different regions of the reference sequences. Repetitive regions tend to collect a vast amount of SRs contrary to non-repetitive regions with a high local error rate, which might not trigger alignment computation at all. In addition, the alignment score distribution reported by the mapping program fluctuates along the references with the varying amount of errors. We therefore assess length normalized scores in a localized context. For this purpose, LRs are internally represented by a consecutive series of small bins. Each SR mapping is allocated to a bin by the position of its center. The size of the bins is a trade-off based on two considerations: ideally, the range for local context comprises a single site and all used SRs are of identical length. Only then, all alignments of a single bin will contain the exact same amount of reference errors and carry unambiguously comparable scores. However, with median SR lengths ranging from 75 to 150 bp at an operating coverage of 50×, the rough estimate for a valid alignment is only about one for every other site. At a bin size of 20 bp, we gather a sufficient amount of alignments per bin for comparison while maintaining a local context for proper score assessment for our E.coli dataset (Supplementary Figs S6–S8).
Only the highest scoring alignments of each bin, not the overall highest scoring alignments, up to the specified coverage cutoff are considered for the next step—the calculation of the consensus sequence. The default coverage cutoff, in congruence with a recommended SR coverage of >50×, is 40. It is dynamically adjusted according to the current SR sampling rate as described in Section 2.4.
2.2 Consensus call with quality computation and chimera detection
To compute the consensus, a matrix with one column per nucleotide is composed for each LR. This matrix is filled with SR bases according to the alignment information obtained from the mapper (Fig. 1). Empty cells represent insertions on the LR; multiple nucleotides within one cell indicate deletions. The gap favoring scoring scheme, which is needed to accurately model SMRT sequencing errors, is prone to incorrectly introduce gaps close to the end of an alignment. Thus, it is crucial that alignments with gaps close to their ends are trimmed during matrix initialization. The consensus is computed by a majority vote in each column. If no SR bases are available, the original LR sequence is kept. The support of the called base is used as confidence criterion and converted to a phred mimicking quality score (Supplementary Equation S2, Supplementary Table S2).
Fig. 1.
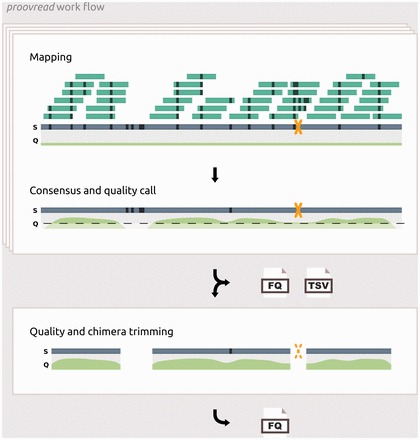
Generic work flow of the proovread correction pipeline. Box 1: Simplified representation of the correction-by-short-read-consensus approach; short reads (dark green bars) are mapped onto an erroneous and chimeric (yellow X) long read (blue bar); black strokes indicate sequencing errors and non-matching alignment positions. Regions with an unusually high amount of errors prevent short read mappings. Base call qualities are represented below the long read (light green curve). During consensus generation, the majority of errors are removed and possible chimeric break points are identified. New qualities scores are inferred from coverage and the composition of the consensus at each position. Processed reads and chimera annotations are written to files. Box 2: The resulting reads are trimmed using a quality cutoff and the chimera annotations generating proovread’s primary output: high-accuracy long reads
More than 1% of raw SMRT single pass reads are chimeric (Fichot and Norman, 2013). The majority of these reads emerges through ‘misligation’ of LRs during SMRT cell library construction. The location of the fusion within the chimeric read is defined as chimeric break point. Here, adjacent sequences derive from different locations of the sequenced reference. Thus, SRs only align to one side of the break point with high identity. The second part of the alignment is enriched for gaps and mismatches. In combination with a score threshold alignments of reads largely overlapping break points are highly unlikely. However, the sensitive mapping parameters facilitate break point overlapping read ends. Therefore, break points are hard to localize considering only per base coverage. For detection of possible break points, proovread therefore considers the per bin coverage generated during the consensus call in the finishing correction cycle. The bin coverage is obtained by computing the total number of bases of all reads assigned to one bin. Only reads centered close to a break point contribute to the coverage of the according bin, whereas partially break point overlapping reads do not. Hence, in contrast to per base coverage, local decreases in bin coverage serve as strong indicators for break points. By default, an occurrence of two to five consecutive bins with <20% of the specified coverage cutoff triggers advanced break point verification. For longer stretches of low coverage bins, chimera detection becomes obsolete. These stretches presuppose low overall per base coverage leading to quality-based trimming regardless of the result of the chimera detection. We analyze the error pattern in the alignments overlapping with the break point using an entropy-based approach (Fig. 2). For that, all alignments located within four bins up- and downstream of the low coverage bins are considered and divided into two populations: reads placed upstream of the center of the low coverage region and reads placed downstream, respectively. From these alignments, one overall and two flank-specific consensus matrices are generated. For each column of each matrix, a Shannon entropy (Supplementary Equation S1) value is computed, resulting in three values per site: entropy of upstream reads (HU), entropy of downstream reads (HD) and total entropy (HT). For comparison of flank-specific entropy and overall entropy, the difference (ΔH) of the overall entropy (HT) and the larger value of the flank-specific entropies (HU and HD) are computed. At a true break point, an accumulation of positive ΔH values distinguishes contradicting signals on both side from random noise or generic low coverage. proovread determines a chimera score for a break point location by dividing the number of positive scoring ΔH values by the total number of signal containing columns. Chimera coordinates and scores are stored to a chimera annotation file and subsequently used during trimming. By default, a chimera break point is confirmed at a minimum score of 0.01.
Fig. 2.
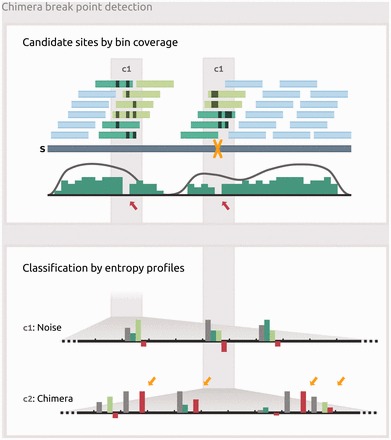
Detection of chimera break points. Box 1: Mapping of short reads (short light blue and green bars) onto pre-corrected long read (blue bar) containing a chimeric break point (yellow X); black strokes indicate non-matching alignment positions. While not detectable by per-base coverage (gray curve), break point candidate sites (c1, c2) can be inferred from decreases in bin coverage (green curve, red arrows). Reads overlapping candidate sites from the right hand side (dark green bars) and reads overlapping from the left hand side (light green bars) are considered for further classification of each individual site. Box 2: Entropy profiles derived from the short read alignment matrix at the two candidate sites. Each position comprises four entropy values: HT calculated from the entire read set (gray bar), HU for the reads placed upstream (dark green bar), HD for the reads placed downstream (light green bar) and ΔH (red bar). ΔH is the difference of HT and the greater value of HU and HD. An accumulation of positive ΔH values (yellow arrows) is observed at true chimera locations
2.3 Quality and chimera trimming
The reads obtained from the consensus step are considered untrimmed corrected LRs. These reads are returned in FASTQ format, with consensus phred scores ASCII encoded in the quality line. These data comprise high-accuracy regions as well as uncorrected regions and unprocessed chimeras. Using a window-based quality filter, we identify and trim low-quality regions from these reads. Chimeric reads are cut according to annotations. The trimming procedure can easily be rerun on the untrimmed corrected reads to adjust strictness according to the users needs.
2.4 Iterative correction
To entirely cover a set of erroneous LR with SR alignments, the level of sensitivity has to match the regions with the locally highest error rates, even though most regions exhibit much lower rates. Mapping at the required level of sensitivity, especially on large-scale data, is computationally demanding and time consuming. Therefore, proovread provides an iterative procedure for mapping and correcting with successively increasing sensitivity (Fig. 3). It consists of three pre-correction and one finishing cycle. During pre-corrections, only subsets of the SRs are used, by default 20, 30 and 50%, respectively. These subsets are generated through systematic sampling such that samples from successive cycles complement each other.
Fig. 3.
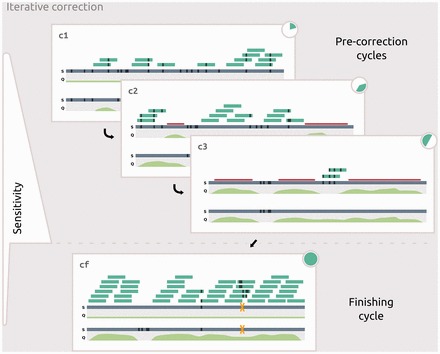
Iterative correction by short read consensus. Short reads (dark green bars) are mapped onto an erroneous long read (blue bar) in four iterations. During pre-correction cycles (c1–c3) subreads are mapped with increasing sensitivity and with complementary subsamples of SR (Pie charts: 20, 30 and 50%). After each cycle a consensus is generated. Sufficiently covered regions are masked before the next iteration, reducing effective reference size for computationally more expensive mappings at increased sensitivity. In the final iteration (cf, 100%), all available short read data are mapped onto unmasked, pre-corrected long reads at high specificity, resulting in a high-accuracy consensus with trusted qualities and chimera annotations
In the first cycle, the SR subset is mapped at a moderate level of sensitivity and thus at high speed. Subsequently, LRs are pre-corrected and regions with sufficient SR support, by default regions with a minimum per base coverage of five, are identified and masked. As a result, the effective search space is substantially reduced before the next cycles. As masking is limited, unmasked edges of pre-corrected regions can act as seeds for mappings during the next cycle, facilitating extensions of masked regions. The process is repeated in cycle two and three, with larger and complementary SR subsamples at increased sensitivity. We tuned parameters such that on average >80% of the reference is masked after the first two cycles. Consequently, mapping at highest sensitivity during cycle three is limited to remaining error-enriched islands. Finally, the entire SR set is mapped at high specificity to the unmasked pre-corrected LRs to merge and refine all previously performed corrections. The time benefit of this iterative approach far out-weights the computational overhead generated by repeated mapping and correction. In summation, proovread combines high sensitivity with low run time.
2.5 Configuration and customization
proovread is highly transparent and flexible with regard to parameter settings and tuning. It comprises a comprehensive optional configuration file that allows tracing and modification of the parameters down to the core algorithms (Supplementary Listing 5). The settings include scoring schemes, binning, masking, iteration procedure and post-processing. The default setup is optimized for genomic data. During the initial pre-correction cycles, for example, only subsamples of the provided SRs are used. For data with uniform coverage distribution, this optimization significantly reduces run time without any loss in overall correction efficiency. However, on data with uneven coverage distribution, e.g. derived from RNA-Seq experiments, the setup can be improved. To sufficiently cover low abundance transcripts, increasing the subsample ratios is required. Such data-specific adjustments can easily be achieved using a modified configuration file (Supplementary Listing 5). Similarly, with appropriate parameter settings, proovread can be tuned to correct data with entirely different characteristics, e.g. obtained from newly emerging LR sequencing technologies.
2.6 Scalability and parallelization
The advantages of SMRT LR sequencing render the technology useful for a variety of sequencing projects, ranging from specific studies on microbial genomes, viruses and plastids to large-scale sequencing of eukaryotic genomes. The varying amount of data demands high flexibility in scalability and parallelization of the processing software. proovread provides several optimizations for this purpose. By design, LRs are corrected independently from each other and as a consequence, instances of proovread can operate on packages of LR data at adaptable size without affecting correction results (Supplementary Fig. S1). The memory footprint of the pipeline increases linearly with the amount of supplied LR bases (Supplementary Fig. S2). The amount of SR data, however, does not affect memory consumption, as our approach does not require the generation of SR indexes or kmer spectra. Therefore, by tuning the package size, the memory requirements of proovread can easily be adjusted to meet available hardware, regardless of the scale of the project. Thus, proovread can run efficiently on high-memory multi-core machines as well as distributed cluster architectures with nodes of limited capabilities or even desktop PCs. Queuing engines like SGE (commercially available from http://www.univa.com/products/grid-engine.php) or Slurm (Jette et al., 2002) can be used to distribute individual jobs. Additionally, the number and size of temporary files are strongly reduced. The SR subsamples are generated on-the-fly from the user-supplied input data. Systematic sampling is realized with the program SeqChunker, which is included in proovread. SeqChunker splits FASTA/FASTQ files into a user-defined number of chunks of similar size. Using simple arithmetics, e.g. return only the first chunk from a block of three chunks, different and in particular complementary read sets can be generated. A high efficiency is accomplished through low-level copy processes for chunk core regions, while actual parsing is limited to chunk boundaries, where exact record offsets are determined. The output of SeqChunker is directly fed to the mapper rendering storage of subsample files obsolete. The primary output of the mapper is also redirected and parsed directly. We determine location, score and offset information of each alignment and use this information to populate an alignment index. In the index structure, alignments are assigned to bins and ranked by score. Only records up to the given coverage cutoff are kept for each bin. An alignment will only be written to disk if the according bin has not been completely filled or if it outscores at least one other alignment in the bin. In the latter case, the record of the alignment ranked last will be deleted from the index. This way, while in the beginning almost all alignments will be stored to file, the further the mapping progresses, the fewer unnecessary alignments will have to be stored. The maximum size of the index structure is determined by the number of records per bin and the total number of bins, which is directly dependent on chunk size. After completion of the mapping process, the index is used to extract all relevant alignments and generate a sorted alignment file without having to run a computationally more expensive sort process on an actual file. These optimizations decrease the required disk space as well as the read and write operations several fold.
3 MATERIALS AND METHODS
3.1 Datasets and preparation
All LRs used with correction software are filtered subreads derived from PacBio’s SMRT-Portal analysis suite, with adapters trimmed and reads split accordingly. Here, we give an overview of the test datasets; more detailed information is provided in the Supplementary Material (Supplementary Table S3). The genomic LR sequences were obtained from the PacBio DevNet (http://www.pacb.com/devnet/).
As bacterial dataset, E.coli genomic sequences (available from PacBio DevNet as ‘E.coli K12 MG1655 Resequencing’) of ∼100 Mb and an N50 of 4082 bp were analyzed. These data were also used as benchmark by Koren et al. (2012). For the correction, a subsample of a 100 bp Illumina library (SRA ERX002508) was used. The reference was the K12 strain of E.coli (Assembly GCA_000005845.1).
As first eukaryotic genomic dataset, ∼126 Mb of Arabidopsis thaliana (available from PacBio DevNet as ‘Arabidopsis P5C3’; N50 8109 bp) were corrected with 100 bp Illumina sequences (SRA SRX158552). The TAIR10 assembly was used as reference (Assembly GCA_000001735.1).
The second genomic dataset was 393 Mb from a human sequencing project (available from PacBio DevNet as ‘H. sapiens 10× Sequence Coverage with PacBio data’; N50 9938 bp). These sequences were corrected with SRs sampled for a 50× coverage from the 1000 genomes project (SRA SRX246904, SRX246905, SRX246906, SRX246907, SRX247361 and SRX247362) leading to a 50× coverage. The reference sequence was the human hg19 assembly (Assembly GCA_000001405.12).
The applicability of proovread for the correction of transcriptomic data was tested with the human brain transcriptome set used by Au et al. (2012) (available from http://www.stanford.edu/∼kinfai/human_cerebellum_PacBioLR.zip). It contained 138 Mb RNA-seq LRs with a N50 of 972 bp. SRs from the human Map 2.0 project (SRA ERX011200 and ERX011186) were used for correction. The human hg19 assembly was used as reference (Assembly GCA_000001405.12).
Before correction all SRs were quality trimmed using sickle (https://github.com/najoshi/sickle). The normalization of the SRs for the human genome was achieved using the normalize-by-median.py script of the khmer package (Brown et al., 2012).
3.2 Correction programs and parameters
We evaluated the correction efficiency of proovread (v1.01) in comparison with the existing pipelines PacBioToCA (v7.0) and LSC (v.0.3.1). The majority of the corrections were performed on a HPC offering 32 CPU cores and 192 GB memory. The correction of the genomic human dataset was performed in a computer grid offering single nodes with 4 CPU cores and 8 GB memory. The queuing system Slurm was used to distribute jobs. The results were assessed in terms of correction accuracy, total throughput, N50, run time and required memory. The required memory and CPU time were monitored for each correction run by a custom script. LSC and PacBioToCA were run with 32 threads. proovread was run in eight independent processes using four mapping threads. The upper bounds for memory requirements were estimated by summing up the eight most memory intensive subtasks.
3.3 Quality assessment procedure
The quality of the correction results was assessed by an evaluation script (available on request from the authors). During the assessment the corrected LRs were remapped onto the reference sequence by GMAP (Wu and Watanabe, 2005). All LR sequences without a global alignment were realigned using exonerate v2.2.0 (Slater and Birney, 2005). Genomic LRs were aligned using the affine:bestfit model to assure global alignments to the reference. Exonerate does not offer global alignment with intron modeling; therefore, transcriptomic LRs were aligned with the model est2genome and reduced gap costs to maximize the length of the local hits. All LR nucleotides without an alignment were considered as erroneous positions. Only reads with a unique mapping were used for the accuracy estimation. Reads were classified as chimeric by GMAP. Reads without an initial GMAP alignment were not considered for the accuracy assessment. The proportions of these ambiguous read placements for all corrections were low, with the exception of the A.thaliana correction with up to 17% for LSC (Supplementary Table S4). The overall throughput and the N50 values for each correction method were calculated for the complete sequence set returned by the programs, respectively, regardless of their mapping result.
3.4 Estimation of the required SR coverage
The SR coverage has a major effect on mapping run time and correction efficiency. We empirically determined the optimal SR coverage for proovread using the E.coli dataset. We selected random subsets of the SRs yielding an expected coverage of 10×, 20×, 35×, 50×, 75×, 100× and 200×, with larger sets including the preceding smaller set. The correction result of the 50× set was only slightly improved by increasing the expected coverage (Supplementary Fig. S3 and S4). Indeed, the memory consumption was saturated by a higher expected coverage (Supplementary Fig. S5). Therefore, we used a 50× as a good trade-off between run time and accuracy, which is in agreement with Koren et al. (2012).
4 RESULTS
Today, the major application of the PacBio RS II is the sequencing of bacterial genomes. Therefore, we used an E.coli dataset as first test case (Table 1). Mapping of the corrected reads onto the E.coli reference genome revealed accuracies of 99.98% for proovread, followed by 99.93% (PacBioToCA) and 88.79% (LSC). LSC correction resulted in a N50 of 4158 bp, which was higher than in the uncorrected reads, as LSC returns only LRs with SR mapping and omitted other LRs. Therefore, the starting N50 for LSC was in fact 4450 bp. Proovread-corrected reads had an N50 of 2147 bp and PacBioToCA of 1639 bp. In matters of throughput, proovread recovered 81% of the input, whereas LSC and PacBioToCA returned 76 and 73%, respectively. The required run time was shortest for PacBioToCA (2.6 h), while proovread took 7× longer, and LSC was most time-consuming (25× of PacBioToCA). The memory consumption was in the same range for LSC and proovread (18.4 and 19.6 GB), whereas PacBioToCA required more than twice as much (44 GB). This differs from the originally published 2.1 GB (Koren et al., 2012), as here PacBioToCA ran multi-threaded.
Table 1.
Benchmark correction results for proovread, PacBioToCA and LSC: Ec—Escherichia coli, At—Arabidopsis thaliana, Hsg—Homo sapiens genome, Hst—Homo sapiens transcriptome
| Program | Accuracy (%) |
N50 (bp) |
Throughput (%) |
Run time (h or d) |
Memory (GB) |
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ec | At | Hsg | Hst | Ec | At | Hsg | Hst | Ec | At | Hsg | Hst | Ec | At | Hsg | Hst | Ec | At | Hsg | Hst | |
| Uncorrected | 4082 | 8109 | 9938 | 1234 | ||||||||||||||||
| proovread def. | 99.98 | 98.48 | 98.90 | 99.87 | 2147 | 2714 | 2219 | 821 | 81 | 79 | 88 | 48 | 19.3 h | 173 d | 2288 d | 84 h | 19.6 | 43.6 | 40.1 | 8.1 |
| proovread norm. | 99.10 | 1327 | 80 | 1009 d | 33.6 | |||||||||||||||
| proovread adapt. | 99.88 | 730 | 70 | 282 h | 9.9 | |||||||||||||||
| PacBioToCA | 99.93 | 97.44 | 98.67 | 1639 | 1528 | 193 | 73 | 46 | 29 | 2.6 h | 20 d | 49 h | 44.0 | 27.5 | 45.4 | |||||
| LSC | 88.79 | 95.43 | 4158 | 908 | 76 | 60 | 66.2 h | 137 h | 18.4 | 18.4 | ||||||||||
With their outstanding length, SMRT sequencing reads will be of increasing use for the sequencing of eukaryotic genomes. To evaluate the performance of proovread on this type of data, we used the comparably small genome of A.thaliana as a second test case (Table 1). For this dataset, correction with LSC exceeded the maximum available memory (192 GB) and therefore did not finish. proovread and PacBioToCA achieved a high correction accuracy of 98.48 and 97.44%, respectively. Starting with an N50 of 8109 bp in the raw reads, proovread returned an N50 of 2714 bp and PacBioToCA of 1528 bp. The throughput was 79% for proovread and 46% for PacBioToCA. The latter required a run time of 481 h with proovread 8.7× longer. Concerning the memory consumption, proovread required 43.6 GB and PacBioToCA 27.5 GB.
To analyze the influence of the size and the complexity of the sequenced genome on the correction result, we used the human genome as third test case (Table 1). For run time reasons, we corrected this set on a cluster infrastructure. Unfortunately, the available grid provided only Slurm as queuing system, whereas PacBioToCA is implemented only for SGE. Furthermore, and typically for larger grids, each node offers only four CPU cores and 8 GB memory. Both do not fulfill the hardware requirements of LSC and a stand-alone PacBioToCA correction. Therefore, only the performance of proovread was analyzed for this LR dataset. Here, we examined two different SR datasets: first, a 50× coverage SR sample and second, a digitally normalized version of these data. The normalization process required 37 GB memory and took 49 h. The correction accuracy for the non-normalized and the normalized SR set was 98.9 and 99.1%, with an N50 of 2219 and 1327 bp, respectively. The throughput was 88.4% for the complete SR set and 79.5% for the normalized dataset. The total run time was shorter for the normalized set (1009 days versus 2288 days) and the required memory was similar for both cases (33.6 and 29.3 GB).
In addition to genomics studies, SMRT sequencing is frequently used for transcriptome analyses. With its LRs, chances are high that a whole transcript is sequenced in a single read, thereby avoiding the assembly process. To evaluate the applicability of proovread for this type of data, we used human brain transcriptomic data as a final test case (Table 1). This dataset was previously used to assess the performance of LSC (Au et al., 2012), although here we used a more recent version of the program (0.3.1). proovread was run with two different parameter settings: (i) the default settings and (ii) a modified set tailored-made for transcriptomic data (see Section 2). Indeed, the correction accuracy was highest with the default and modified proovread settings (99.87 and 99.88%). PacBioToCA and LSC performed worse (98.67 and 95.43%). Starting with an N50 of 972 bp, LSC returned the highest N50 (908 bp) followed by default proovread (821 bp), modified proovread (730 bp) and PacBioToCA (193 bp). Modified proovread resulted in the highest throughput (69.5% of input data), followed by LSC (59.6%), proovread with default settings (47.5%) and PacBioToCA (29%). The shortest run time was required by PacBioToCA (49.1 h). LSC took 1.35× longer, while standard proovread ran 1.71× longer. Modified proovread required the longest run time (5.75×). In contrast, proovread needed <10 GB memory (8.1 and 9.9 GB for default and modified settings), while LSC needed almost twice as much (18.4 GB) and PacBioToCA needed most memory (45.4 GB).
5 DISCUSSION
proovread is designed to correct erroneous LRs sequenced by SMRT with high-quality SR data as generated by Illumina sequencers. Our benchmarks revealed that proovread is well suited for the correction of microbial and eukaryotic as well as genomic and transcriptomic data.
Arguably, the most prominent characteristic of a correction pipeline is the accuracy of the corrected reads. With accuracies of >99% in almost all test cases, proovread and PacBioToCA clearly outperformed LSC. The latter achieved only <90% for genomic and 95% for transcriptomic reads, as LSC omits trimming of corrected reads, which results in reads that are only partially corrected. Obviously, the overall accuracy of these reads will be low. When comparing accuracies, it has to be taken into account that all corrected reads that could not be mapped onto the reference or were identified as chimeric were classified as ambiguous and not considered. In general, their amount was small. Only for the A.thaliana set, it exceeds 6.5% for all programs, as here the reference and the sequenced strain differ (Supplementary Table S4). Still, if these reads were included, the accuracy of all programs would decrease. This effect would be smallest on proovread, as it generated fewer of these ambiguous reads than PacBioToCA and LSC.
Still, accuracy is only one criterion for the evaluation of an LR correction pipeline. A key advantage of SMRT sequencing is the length of the resulting reads. Therefore, the correction process shortens the reads as few as possible. When comparing the N50 of the corrected reads, LSC resulted in the longest reads. This does not come as a surprise taking into account that LSC does not trim corrected reads (see above). Still, we think that trimming is an important feature, as it not only avoids the inclusion of poorly corrected regions in the following analyses but also enables the correction of chimeric reads. Therefore, both, proovread and PacBioToCA, trim corrected reads. Still the N50 of proovread-corrected reads was in all test cases considerably higher than for PacBioToCA. To give the user maximum flexibility, proovread also reports the untrimmed corrected reads. Furthermore, the trimming step is independent of the correction, thereby enabling the user to easily optimize the trimming parameters for the given dataset.
Finally, during the correction process, regions and reads can be rejected, decreasing the throughput of the pipeline. This factor might be neglectable if initially a high coverage of the sequenced material was provided. In the case of larger, e.g. eukaryotic, genomes, this is usually too expensive, resulting in lower coverage. Here, a decrease in throughput could have a strong impact on the further steps of the projects, especially the assembly. In the worst case, costly re-sequencing might be needed. Thus, minimizing the rejection of reads is an important objective of a correction pipeline. proovread corrected in almost all cases ≥80% of the input data, while LSC and PacBioToCA generated dramatically smaller throughput. In the case of the Homo sapiens transcriptome and the A.thaliana genome PacBioToCA omitted ≫50% of the LRs. Thus, >1.6× raw reads would be needed for the same amount of corrected reads. Taken together, proovread is able to correct larger percentages with higher accuracy, leading to longer reads than previous tools.
Apart from read length and accuracy, in practice particularly the horizontal coverage of the reference is of high importance. SMRT sequencing has little to no bias toward nucleotide composition (Loomis et al., 2013; Shin et al., 2013). In contrast, especially with early chemistry, Illumina had various issues with bias-free sequencing of GC-rich molecules (Aird et al., 2011). As a consequence, SR data for hybrid correction has to be chosen with care. Any biases in the SRs are to some extent transferred onto the LRs, thus, impairing correction quality and potentially eliminating specific SMRT sequencing advantages.
A general issue of the consensus correction method is the potential loss of single nucleotide variants. The haplotype of the corrected LRs is determined by the SR dataset. Nevertheless, Carneiro et al. (2012) describe the recovery of the original variants using the uncorrected LRs given a sufficient coverage.
When designing proovread it was one goal to achieve a maximum of independence from the existing computer infrastructure. Therefore, the correction can be performed in a single process, which requires a maximum of memory, or can be split into smaller chunks, each needing less memory. In addition, the amount of SRs is unlimited, as proovread does not require an indexing of the SR data. This allows the parallel execution of independent proovread processes on computers with limited memory and CPU configuration. Indeed, the memory footprint of a single proovread process is smaller in comparison to the other programs. If the available memory is not sufficient, the package size can be lowered to fit the available memory. Admittedly, this increases the run time of proovread. Still, we think that this can be overcome by clusters of comparably cheap machines. Here, each machine corrects only a fraction of the reads. Contrarily, LSC requires large memory and does not benefit from running in a grid infrastructure. PacBioToCA allows the parallel execution in a grid system, but is restricted to the SGE queuing system. Moreover, it requires up to 48 GB memory on a local computer, which requires large server systems. Thus, proovread does not dictate the architecture of the computer system.
This idea of flexibility is also encoded in the correction process itself. This starts with the mapping of the reads. Currently, we support SHRiMP2 as default mapper and Bowtie2 as experimental option. As we can not foresee the development of new mappers, proovread can also work directly on user-provided mapping files. Next, the proovread iterative correction is highly configurable. A user can modify the number of iterations and thereby decrease the overall run time by performing fewer iterations. Obviously, this might affect the correction accuracy and therefore has to be considered carefully. Finally, the correction and the filtering steps are independent from each other. Without filtering, the maximum of the input read length can be preserved. In contrast with a strict filtering, only highly accurate positions will be returned. Furthermore, this separation enables repeating the filtering step without rerunning the time-consuming correction.
The idea to correct errors in LRs from SMRT sequencing with Illumina SRs has been implemented before by LSC and PacBioToCA. But, both have strong demands on hardware as well as software infrastructure. If either can not be met, correction can not be performed. This will make following analyses like an assembly challenging if possible at all. Contrarily, proovread can be easily adapted to the available resources. It handles the correction of an E.coli genome on a laptop as well as of a human genome on a HPC cluster. This in-built flexibility also enables the adaptation to and optimization for different datasets as generated in genomic and transcriptomic projects. Finally, correction with proovread delivered more, more accurate and longer reads. Thus, proovread is well suited for the correction of LRs irrespective of the target of sequencing and regardless of the computational resources.
Supplementary Material
ACKNOWLEDGEMENT
We would like to thank the ‘Leibniz Rechenzentrum’ and the ‘Data Intensive Academic Grid’ for providing us with the infrastructure for evaluation and benchmarking of the programs, Henri van de Geest, Wageningen UR, Plant Research International, Netherlands for testing of proovread and Jessika D’Lorge-Harnisch for proofreading the manuscript.
Funding: The research leading to these results has received funding from the European Research Council under the European Union's Seventh Framework Programme (FP/20010-2015)/ERC Grant Agreement number 250194-Carnivorom.
Conflicts of interest: F.F. is a share holder of Pacific Biosciences.
REFERENCES
- Aird D, et al. Analyzing and minimizing pcr amplification bias in illumina sequencing libraries. Genome Biol. 2011;12:R18. doi: 10.1186/gb-2011-12-2-r18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au KF, et al. Improving pacbio long read accuracy by short read alignment. PLoS One. 2012;7:e46679. doi: 10.1371/journal.pone.0046679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CT, et al. 2012. A reference-free algorithm for computational normalization of shotgun sequencing data. ( http://arxiv.org/abs/1203.4802) [Google Scholar]
- Carneiro MO, et al. Pacific biosciences sequencing technology for genotyping and variation discovery in human data. BMC Genomics. 2012;13:375. doi: 10.1186/1471-2164-13-375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin C-S, et al. Nonhybrid, finished microbial genome assemblies from long-read smrt sequencing data. Nat. Methods. 2013;10:563–569. doi: 10.1038/nmeth.2474. [DOI] [PubMed] [Google Scholar]
- David M, et al. Shrimp2: sensitive yet practical short read mapping. Bioinformatics. 2011;27:1011–1012. doi: 10.1093/bioinformatics/btr046. [DOI] [PubMed] [Google Scholar]
- Dohm JC, et al. Substantial biases in ultra-short read data sets from high-throughput dna sequencing. Nucleic Acids Res. 2008;36:e105. doi: 10.1093/nar/gkn425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid J, et al. Real-time dna sequencing from single polymerase molecules. Science. 2009;323:133–138. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- Fichot EB, Norman RS. Microbial phylogenetic profiling with the pacific biosciences sequencing platform. Microbiome. 2013;1:10. doi: 10.1186/2049-2618-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnerre S, et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl Acad. Sci. USA. 2011;108:1513–1518. doi: 10.1073/pnas.1017351108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jette MA, et al. In Lecture Notes in Computer Science: Proceedings of Job Scheduling Strategies for Parallel Processing (JSSPP) 2003. Berlin, Germany: Springer-Verlag; 2002. Slurm: simple linux utility for resource management; pp. 44–60. [Google Scholar]
- Kelley DR, et al. Quake: quality-aware detection and correction of sequencing errors. Genome Biol. 2010;11:R116. doi: 10.1186/gb-2010-11-11-r116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S, et al. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat. Biotechnol. 2012;30:693–700. doi: 10.1038/nbt.2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. The sequence alignment/map format and samtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010;20:265–272. doi: 10.1101/gr.097261.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomis EW, et al. Sequencing the unsequenceable: expanded cgg-repeat alleles of the fragile x gene. Genome Res. 2013;23:121–128. doi: 10.1101/gr.141705.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JR, et al. Aggressive assembly of pyrosequencing reads with mates. Bioinformatics. 2008;24:2818–2824. doi: 10.1093/bioinformatics/btn548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers EW, et al. A whole-genome assembly of drosophila. Science. 2000;287:2196–2204. doi: 10.1126/science.287.5461.2196. [DOI] [PubMed] [Google Scholar]
- Ono Y, et al. Pbsim: Pacbio reads simulator–toward accurate genome assembly. Bioinformatics. 2013;29:119–121. doi: 10.1093/bioinformatics/bts649. [DOI] [PubMed] [Google Scholar]
- Roberts RJ, et al. The advantages of smrt sequencing. Genome Biol. 2013;14:405. doi: 10.1186/gb-2013-14-7-405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross MG, et al. Characterizing and measuring bias in sequence data. Genome Biol. 2013;14:R51. doi: 10.1186/gb-2013-14-5-r51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin SC, et al. Advantages of single-molecule real-time sequencing in high-gc content genomes. PLoS One. 2013;8:e68824. doi: 10.1371/journal.pone.0068824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater GSC, Birney E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics. 2005;6:31. doi: 10.1186/1471-2105-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers KJ, et al. A flexible and efficient template format for circular consensus sequencing and snp detection. Nucleic Acids Res. 2010;38:e159. doi: 10.1093/nar/gkq543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TD, Watanabe CK. Gmap: a genomic mapping and alignment program for mrna and est sequences. Bioinformatics. 2005;21:1859–1875. doi: 10.1093/bioinformatics/bti310. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.