

Abstract
Summary: Single nucleotide variations (SNVs) located within a reading frame can result in single amino acid polymorphisms (SAPs), leading to alteration of the corresponding amino acid sequence as well as function of a protein. Accurate detection of SAPs is an important issue in proteomic analysis at the experimental and bioinformatic level. Herein, we present sapFinder, an R software package, for detection of the variant peptides based on tandem mass spectrometry (MS/MS)-based proteomics data. This package automates the construction of variation-associated databases from public SNV repositories or sample-specific next-generation sequencing (NGS) data and the identification of SAPs through database searching, post-processing and generation of HTML-based report with visualized interface.
Availability and implementation: sapFinder is implemented as a Bioconductor package in R. The package and the vignette can be downloaded at http://bioconductor.org/packages/devel/bioc/html/sapFinder.html and are provided under a GPL-2 license.
Contact: siqiliu@genomics.cn
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Recent advances in next-generation sequencing(NGS) technologies, such as whole-genome sequencing and total RNA sequencing (RNA-Seq), have yielded large volumes of genetic variation data (1000 Genomes Project Consortium, 2012; Peng et al., 2012). Among these variations, single nucleotide variations (SNVs) are recognized as one of the most common type of genetic variants in human genome. The SNVs that are located within protein-coding regions can cause the changes in the corresponding amino acids, which could result in change of protein functions and further influence particular physiological or pathological traits in individuals. Detection of the proteins containing single amino acid polymorphisms (SAPs) derived from SNVs provides valuable information in studying the functional significance of the genetic variations. Tandem mass spectrometry (MS/MS)-based proteomics, especially shotgun proteomics, is a powerful means to detect SAPs on a large scale (Sheynkman et al., 2014; Wang et al., 2014). A common strategy for peptide and protein identification is database searching by comparing experimental MS/MS spectra against theoretical mass spectra derived from a reference protein sequence database; however, this strategy fails to identify the varied amino acid sequences that do not exist in the reference database. Several researchers have addressed this problem by developing new searching approaches. For instance, one approach is developed by MASCOT by using error-tolerant searching (Creasy and Cottrell, 2002) or by X!Tandem by conducting the refinement search (Craig and Beavis, 2004). As this approach allows exhaustive test of all amino acid substitutions and greatly expands search space, statistical significance for the variant identifications is not easily evaluated (Creasy and Cottrell, 2002). Another approach is based on the construction of a database that includes SAPs found within SNVs or cancer mutation repositories, such as dbSNP (Sherry et al., 2001), COSMIC (Forbes et al., 2011) or sample-specific NGS data (Li et al., 2011; Sheynkman et al., 2014; Wang et al., 2014). The SAPs are detected through MS searches against the SAP-containing databases. A technical bottleneck in this approach is how to effectively and conveniently integrate diverse sources of sequence variation information into an MS-searchable protein database. Wang et al. developed customProDB, an R package that generates customized databases from RNA-Seq data or public SNV repositories (Wang and Zhang, 2013). To define a SAP site, we could not only depend on the variant peptide database but also need to search the variant peptides based on MS/MS spectra. Unfortunately, CustomProDB does not possess such function. SysPIMP is a Web-based platform for identifying human disease-related mutant sequences using X!Tandem as the search engine (Xi et al., 2009). SysPIMP collects human disease-related mutant sequences from the Online Mendelian Inheritance in Man (Hamosh et al., 2005), Protein Mutant Database (Kawabata et al., 1999) and SwissProt database (Boeckmann et al., 2003); however, it does not incorporate sample-specific NGS data, and its Web-based nature prevents analysis of large files and scale-up.
Herein, we describe sapFinder, a program that can use data from public repositories of genetic variants or sample-specific NGS data to automate the generation of variant-containing databases and thereby enable detection of SAP-containing peptides. The program also allows for efficient MS/MS data searching, post-processing and report generation from HTML-based format.
2 METHODS AND IMPLEMENTATION
As illustrated in Figure 1, the workflow for identifying canonical and variant peptides based on shotgun proteomics data is broadly divided into four steps.
Fig. 1.
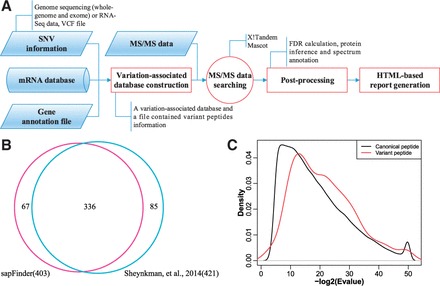
(A) Schematic overview of the sapFinder package. (B) A Venn diagram illustrating the overlap of identified variant peptides between sapFinder and Sheynkman et al. study. (C) Search score distribution of variant and canonical peptides
2.1 Construction of variation-associated database
sapFinder was used to construct a variation-associated database. The inputs contained three files: a variant call format (VCF) file, which was either generated from a Binary Alignment/Map (BAM) file using SNV calling tools such as SAMtools (Li et al., 2009) or the Genome Analysis Toolkit (McKenna et al., 2010) or was directly downloaded from dbSNP or COSMIC; a gene annotation file downloaded from the University of California, Santa Cruz (UCSC) table browser; and a FASTA format mRNA sequences file, which was also downloaded from UCSC table browser. Based on the three files, SAPs at the protein level were obtained by identifying SNVs and translating them into protein. Correspondingly, all possible tryptic peptides derived from two possible missed cleavage sites and including the SAP were extracted from the protein variations. Only peptides with more than five amino acid residues were accepted. The FASTA headers for the peptides were prefixed with ‘VAR’ as to distinguish from canonical protein entries. The SAP-containing sequences were appended to the reference protein sequences (FASTA format). In addition, reverse sequences were appended to the original forward sequences to act as decoys in false discovery rate (FDR) estimation (Elias and Gygi, 2007).
2.2 MS/MS data searching
X!Tandem can recognize the varied MS/MS data formats in database searching, such as DTA, PKL or mgf (Craig and Beavis, 2004). In this article, rTANDEM, an R encapsulation of X!Tandem (Fournier et al., 2014), was adopted to search the variation-associated databases against tandem mass spectra. Alternatively, sapFinder also accepts Mascot dat file as input.
2.3 Post-processing
X!Tandem Parser (Muth et al., 2010) was used to extract the information of peptide and protein identification from the rTANDEM result. Taken into consideration the high FDR risk for variant peptide identification, a refined FDR estimation approach for these identifications was used in our workflow (Li et al., 2011). After PSMs filtration based on a specified FDR threshold, the Occam’s razor approach (Nesvizhskii et al., 2003) was adopted to deal with degenerated canonical peptides, by finding a minimum subset of proteins that are covered by all of the identified canonical peptides, and to export two txt format files containing the identification results of either peptides or proteins.
2.4 HTML-based report generation
sapFinder outputs an HTML-based interactive report, which contained quality control plots, annotated spectra and identification information of variant peptides and canonical peptides.
3 APPLICATION
To evaluate the utility of sapFinder, a published dataset was processed using our pipeline (Sheynkman et al., 2014). MS/MS spectra files and SNVs listed in the VCF file in the dataset, the mRNA database and the gene annotation file downloaded from UCSC table browser were treated as the input for sapFinder. We used similar MS database search parameters as in the original paper. The canonical and variant peptides were filtered with a threshold of 1% FDR. As shown in Figure 1B, 403 variant peptides were identified and ∼80% of them were also identified in the original study, and a density distribution plot of their E-value distributions was compared in Figure 1C. As also found in the Sheynkman et al. paper, the distributions reveal that there is a slight shift to higher score distributions for the variant peptides as compared with that for canonical peptides, suggesting that the variant peptide identifications as a group have a similar or lower FDR as compared with the canonical peptide identifications. The HTML-based report can be found in Supplementary Data. These results demonstrate the utility of sapFinder in detecting genetic variations at the protein level.
Funding: We acknowledge the support from the State Key Development Program for Basic Research of China–973 Program (NO. 2010CB912703, 2013CB945204); the Program of International S&T Cooperation (2014DFB30020); Guangdong Provincial Engineering Laboratory for Proteomics; Shenzhen Key Laboratory of Transomics Biotechnologies (NO.CXB201108250096A); ShenZhen Engineering Laboratory for Proteomics.
Conflict of interest: none declared.
Supplementary Material
REFERENCES
- Boeckmann B, et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003;31:365–370. doi: 10.1093/nar/gkg095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig R, Beavis RC. TANDEM: matching proteins with tandem mass spectra. Bioinformatics. 2004;20:1466–1467. doi: 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- Creasy DM, Cottrell JS. Error tolerant searching of uninterpreted tandem mass spectrometry data. Proteomics. 2002;2:1426–1434. doi: 10.1002/1615-9861(200210)2:10<1426::AID-PROT1426>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- Forbes SA, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39:D945–D950. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier F, et al. rTANDEM, an R/Bioconductor package for MS/MS protein identification. Bioinformatics. 2014 doi: 10.1093/bioinformatics/btu178. [DOI] [PubMed] [Google Scholar]
- 1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamosh A, et al. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005;33:D514–D517. doi: 10.1093/nar/gki033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabata T, et al. The protein mutant database. Nucleic Acids Res. 1999;27:355–357. doi: 10.1093/nar/27.1.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, et al. A bioinformatics workflow for variant peptide detection in shotgun proteomics. Mol. Cell. Proteomics. 2011;10 doi: 10.1074/mcp.M110.006536. M110 006536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muth T, et al. XTandem Parser: an open-source library to parse and analyse X!Tandem MS/MS search results. Proteomics. 2010;10:1522–1524. doi: 10.1002/pmic.200900759. [DOI] [PubMed] [Google Scholar]
- Nesvizhskii AI, et al. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 2003;75:4646–4658. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- Peng Z, et al. Comprehensive analysis of RNA-Seq data reveals extensive RNA editing in a human transcriptome. Nat. Biotechnol. 2012;30:253–260. doi: 10.1038/nbt.2122. [DOI] [PubMed] [Google Scholar]
- Sherry ST, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheynkman GM, et al. Large-scale mass spectrometric detection of variant peptides resulting from nonsynonymous nucleotide differences. J. Proteome Res. 2014;13:228–240. doi: 10.1021/pr4009207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, et al. Omics evidence: single nucleotide variants transmissions on chromosome 20 in liver cancer cell lines. J. Proteome Res. 2014;13:200–211. doi: 10.1021/pr400899b. [DOI] [PubMed] [Google Scholar]
- Wang X, Zhang B. customProDB: an R package to generate customized protein databases from RNA-Seq data for proteomics search. Bioinformatics. 2013;29:3235–3237. doi: 10.1093/bioinformatics/btt543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi H, et al. SysPIMP: the web-based systematical platform for identifying human disease-related mutated sequences from mass spectrometry. Nucleic Acids Res. 2009;37:D913–D920. doi: 10.1093/nar/gkn848. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.