

Abstract
The Affymetrix Axiom genotyping standard and ‘best practice’ workflow for Linux and Mac users consists of three stand-alone executable programs (Affymetrix Power Tools) and an R package (SNPolisher). Currently, SNP analysis has to be performed in a step-by-step procedure. Manual intervention and/or programming skills by the user is required at each intermediate point, as Affymetrix Power Tools programs do not produce input files for the program next-in-line. An additional problem is that the output format of genotypes is not compatible with most analysis software currently available. AffyPipe solves all the above problems, by automating both standard and ‘best practice’ workflows for any species genotyped with the Axiom technology. AffyPipe does not require programming skills and performs all the steps necessary to obtain a final genotype file. Furthermore, users can directly edit SNP probes and export genotypes in PLINK format.
Availability and implementation: https://github.com/nicolazzie/AffyPipe.git.
Contact: ezequiel.nicolazzi@tecnoparco.org
1 INTRODUCTION
There are two widely used high-throughput SNP genotyping platforms: Illumina and Affymetrix. These producers differ not only in their genotyping technologies but also in the downstream software available to obtain genotype calls. Illumina has developed an integrated software, GenomeStudio®, with a graphical user interface (GUI) for visualization of the genotyping data from the assay platform. GenomeStudio® only runs on the Windows operating system (OS). Affymetrix provides a GUI software, Genotyping Console™, for Windows OS (http://www.affymetrix.com/estore/browse/level_seven_software_products_only.jsp?productId=131535). However, for Linux/Unix and Mac OS users, the Affymetrix genotyping workflow consists of a step-by-step procedure for which no GUI or automation is available. There are two types of workflow: the standard (http://www.affymetrix.com/support/downloads/manuals/axiom_genotyping_solution_analysis_guide.pdf) and the ‘best practice’ (http://www.affymetrix.com/support/downloads/manuals/axiom_best_practice_supplement_user_guide.pdf) workflow. The standard workflow consists of a four-step procedure, and is the basic workflow. The ‘best practice’ workflow includes an extra step for platewise quality control (QC).
In both workflows, the first three steps use a set of stand-alone executable programs, which were originally written in C++ (Affymetrix Power Tools; APTools), whereas the fourth step requires the use of the R programming language (http://www.R-project.org/) and a specific R package (SNPolisher). In addition, if the ‘best practice’ workflow is run, users must manually calculate two platewise statistics, excluding individuals in specific low-quality plates. A schematic overview of the process is shown in Figure 1. To carry out the analysis, executable programs, the SNPolisher R package and many input files for each of the steps have to be downloaded from the Affymetrix Web site, in addition to writing long command line instructions for each step.
Fig. 1.
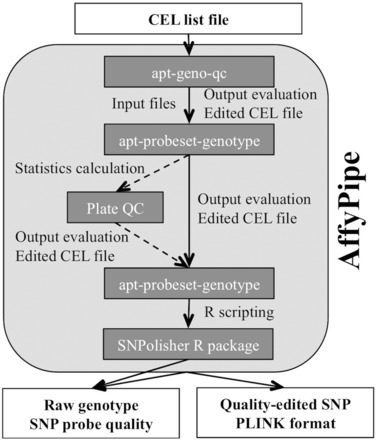
Schematic view of Affymetrix genotyping protocol for standard and ‘best practice’ (extra step in dashed line) workflows
The first phase of the analysis is sample QC, which creates a statistic (Dish-QC) to measure the signal across non-polymorphic loci. Affymetrix suggests retaining samples with a default Dish-QC >82%. Both, the evaluation of the output file and the creation of the new input file for the next program in the workflow (i.e. excluding samples that do not pass the threshold), have to be executed manually. In the second phase, genotyped samples with call rate (CR) lower than a default value of 97% are identified. Users then need to manually evaluate the output file and create a new input file, selecting the CR cutoff and excluding samples failing the CR criteria.
The ‘best practice’ workflow has an extra step for platewise QC. This step, however, is not automated and requires the manual calculation of two statistics, ‘Plate Pass Rate’ and ‘Average Plate Call Rate’, with default values 95 and 99%, respectively. Users then have to assess the quality of each plate and create a new input file for the next program.
The next phase requires the genotyping step to be rerun for the samples passing the quality criteria. In the last phase, the SNPolisher R package is used to evaluate the quality of the signal and to classify each SNP probe into six classes: ‘PolyHighResolution’, ‘MonoHighResolution’, ‘NoMinorHomozygote’, ‘OffTargetVariant’, ‘CallRateBelowThreshold’ and ‘Other’. The Affymetrix workflow provides genotypes, irrespective of their SNP probe quality, in SNP-probe by row format, with a 0/1/2/−1 coding, for homozygotes ‘B’ and ‘A’ (0 and 2, respectively), heterozygotes (1) and missing genotypes (−1).
Although the use of the APTools does not require any C++ programming skills, just basic command line use, the use of SNPolisher requires at least a basic knowledge of R. Therefore, the lay user cannot easily run the full analysis. Currently, a Linux or Linux-based environment is commonly used to handle large datasets, therefore a pipeline that integrates the analyses steps into a single application would be of general use. AffyPipe is a Python-based pipeline that addresses the need to automate the whole workflow and hence reduce the time, effort and programming skills needed to obtain genotype calls. AffyPipe allows both standard and ‘best practice’ workflows to be run, and produces edited final files in PLINK format (Purcell et al., 2007), a standard and widely used format for genetic analyses.
Although this pipeline was built originally for the Water Buffalo Axiom 90 K assay (Iamartino et al., 2013), it is able to handle any species genotyped with the Affymetrix Axiom technology.
2 METHODS
AffyPipe pipeline links APTools, intermediate steps and SNPolisher software into a single integrated application by automatically evaluating output files from each step and creating the input file(s) for the program next-in-line. AffyPipe was programmed entirely in python, using python default libraries. Although the use of more advanced python libraries could have increased the efficiency of this pipeline, default libraries were used to ensure portability and ease of installation and use, even for inexperienced users. Running the pipeline is achieved using simple command line execution, and does not require knowledge of any of the programming languages used (python, C++ or R). The pipeline is flexible and includes a large range of options. The input file used contains the list of CEL files, which are the standard Affymetrix raw intensity files, to be analyzed, and a simplified parameter file. This parameter file is the key to extend the capabilities of the AffyPipe tool to any of the species genotyped with the Axiom technology, by specifying three array- and species-specific variables: the prefix of the array type library files, their release number and the name of the annotation file for the desired species.
AffyPipe provides a script (createcelfile.sh) to help users to automatically create the list of CEL files. AffyPipe can directly edit and export genotypes and map information into PLINK format (Purcell et al., 2007), by selecting the desired probe classes (default classes are ‘PolyHighResolution’, ‘MonoHighResolution’ and ‘NoMinorHomozygote’), and the ‘best’ probe for each SNP. Best probes are directly identified by SNPolisher R package scripts, and are coded as ‘1’ in the ‘BestProbeset’ field of ‘Ps.performance.txt’ output file.
Source code, full documentation of the process and requirements, in terms of proprietary software to be obtained from Affymetrix Web site, are provided in the readme file at https://github.com/nicolazzie/AffyPipe.git.
AffyPipe can be used for any species genotyped with the Axiom technology and has been successfully tested on Buffalo, Human Exome 319 and EUR arrays (NCBI GEO, http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GPL18760; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE52691).
ACKNOWLEDGEMENTS
The authors acknowledge the BuffaloSNP international cooperation project Italy–Brazil (ID 16978 and Ref N. AGRO-09) and The International Water Buffalo Genome Consortium for test datasets. Authors wish also to acknowledge the two anonymous reviewers that provided good suggestions to improve this tool further.
Funding: Italian Ministry of Education, University and Research, project GenHome (D.M. 505/Ric); and the European Union's Seventh Framework Programme, project Gene2Farm (G.A. 289592).
Conflicts of interest: none declared.
REFERENCES
- Iamartino D, et al. The Buffalo Genome and the Application of Genomics in Animal Management and Improvement. Buffalo Bull. 2013;32:151–158. [Google Scholar]
- Purcell S, et al. PLINK: a toolset for whole-genome association and population-based linkage analysis. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]