

Abstract
Motivation: MicroRNAs (miRNAs) play crucial roles in complex cellular networks by binding to the messenger RNAs (mRNAs) of protein coding genes. It has been found that miRNA regulation is often condition-specific. A number of computational approaches have been developed to identify miRNA activity specific to a condition of interest using gene expression data. However, most of the methods only use the data in a single condition, and thus, the activity discovered may not be unique to the condition of interest. Additionally, these methods are based on statistical associations between the gene expression levels of miRNAs and mRNAs, so they may not be able to reveal real gene regulatory relationships, which are causal relationships.
Results: We propose a novel method to infer condition-specific miRNA activity by considering (i) the difference between the regulatory behavior that an miRNA has in the condition of interest and its behavior in the other conditions; (ii) the causal semantics of miRNA–mRNA relationships. The method is applied to the epithelial–mesenchymal transition (EMT) and multi-class cancer (MCC) datasets. The validation by the results of transfection experiments shows that our approach is effective in discovering significant miRNA–mRNA interactions. Functional and pathway analysis and literature validation indicate that the identified active miRNAs are closely associated with the specific biological processes, diseases and pathways. More detailed analysis of the activity of the active miRNAs implies that some active miRNAs show different regulation types in different conditions, but some have the same regulation types and their activity only differs in different conditions in the strengths of regulation.
Availability and implementation: The R and Matlab scripts are in the Supplementary materials.
Contact: jiuyong.li@unisa.edu.au
Supplementary information: Supplementary data are available at Bioinformatics online.
1 BACKGROUND
MicroRNAs (miRNAs) are a family of short non-coding RNA molecules (usually 19–25 nt) that regulate gene expression via the full degradation of the target messenger RNA (mRNA) transcript or the translational repression of it (Bartel, 2009). miRNAs have been found to be involved in most biological processes, including developmental timing, cell proliferation, metabolism, differentiation, apoptosis, stress responses, cellular signaling and even various human cancers (Ambros, 2003; Bartel, 2004; Du and Zamore, 2007).
miRNA target prediction is a vital step toward the understanding of miRNA activity. Because experimental methods are limited by their low efficiency and high cost, computational approaches have become a key alternative for predicting miRNA activity. Several tools have been developed to identify miRNA targets, such as MicroCosm (Griffiths-Jones et al., 2008), PicTar (Krek et al., 2005), TargetScan (Friedman et al., 2009) and miRanda (Betel et al., 2008). However, the predictions are based on sequence complementarity and/or structural stability of the putative duplex and thus have a high rate of false positives and false negatives (Rajewsky, 2006). Furthermore, sequence data are static and they do not change in different conditions or at different times. Thus, from sequence data alone we are unable to identify the effect of miRNAs on their targets’ expression in specific biological conditions, while miRNA regulation or activity is often condition-specific (Le and Bar-Joseph, 2013).
Some recent work has combined sequence data and gene expression data to infer miRNA activity. Cheng and Li (2008) employed an enrichment score used by the Gene Set Enrichment Analysis (Subramanian et al., 2005) to infer miRNA activity. They identified the activity enhancement of miRNAs in miRNA-transfected HeLa cells. Madden et al. (2010) combined correspondence analysis, between group analysis and co-inertia analysis to detect miRNA activity using microarray datasets. They produced a ranked list of miRNAs associated with a specific splitting in the samples, by combining miRNA target predictions with gene expression levels. Volinia et al. (2010) proposed T-REX to build miRNA activity map. They used the effect of miRNAs over their targets for detecting miRNA activity with mRNA expression profiles. Some tools also have been developed, including miReduce (Sood et al., 2006), Sylamer (van Dongen et al., 2008), BIRTA (Zacher et al., 2012), DIANA-mirExTra (Alexiou et al., 2010), mirAct (Liang et al., 2011), miTEA (Steinfeld et al., 2013) and cWords (Rasmussen et al., 2013), to infer miRNA activity. The first three are stand-alone applications and the last four provide online services.
Although these methods were successfully applied to infer condition-specific miRNA activity, most of them only look at a specific condition (e.g. cancer), without considering the difference in miRNA activity between conditions. Therefore, the miRNA activity found based on the information in one condition of interest may contain regulatory relationships that are not unique to the condition. Moreover, when considering only one specific condition, the number of samples that can be used is smaller, worsening the over-fitting problem with high-dimensional gene expression data.
Additionally, most existing methods use statistical correlations or associations to identify miRNA–mRNA interactions. However, associations may not reveal gene regulatory relationships that are indeed causal relationships. For example, the expression levels of an miRNA and a gene can be strongly correlated, but the correlation may not indicate a regulatory relationship between the miRNA and the gene because the strong correlation may be the consequence of the regulation of a common regulator of them.
To address the above limitations, in this article, we propose a novel approach to discovering condition-specific miRNA activity.
To identify miRNA activity that is specific to a condition of interest, our method exploits the difference between the regulatory behavior of miRNAs in the condition of interest and in the other condition. We divide matched samples of miRNA and mRNA expression into groups according to sample conditions, e.g. cancer and normal. Then miRNA–mRNA causal interactions are examined using each group of samples, respectively, but only those interactions showing significant difference in their strengths in different conditions (called significant causal interactions) are retained. These significant causal interactions are then used to find out active miRNAs with respect to the condition of interest, i.e. miRNAs that have significantly different causal interactions with mRNAs in different conditions. The significant causal interactions associated with an active miRNA are the condition-specific activity of this miRNA.
To capture the causal semantics of miRNA–mRNA regulatory relationships, in the above procedure, we use IDA (Maathuis et al., 2009, 2010), a causal inference method, to estimate the strengths of miRNA–mRNA interactions. With observational data, IDA simulates an intervention process (e.g. a gene knockdown experiment) and predicts the causal effects of the intervention. It is proved to be an effective method for predicting the causal regulatory effects that an miRNA has on an mRNA (Le et al., 2013).
To validate the proposed method, we apply it to two gene expression datasets: epithelial–mesenchymal transition (EMT) and multi-class cancer (MCC), respectively. The identified miRNA activity is validated by using the miRNA transfection experiments data, as well as by functional analysis, pathway analysis and the information from literature. The results show that the proposed method can effectively infer condition-specific miRNA activity.
2 METHODS
2.1 Overview of the proposed method
As illustrated in Figure 1, the method comprises the following steps:
Data preparation. Given the matched miRNA and mRNA expression profiles, a list of differentially expressed miRNAs and mRNAs are identified. The expression profiles of the differentially expressed miRNAs and mRNAs are then split into sample groups according to the conditions (phenotypes) of the samples. In each condition, the miRNA and mRNA samples are matched and integrated into one dataset (matrix) as an input of the next step.
Using IDA to learn a causal structure and to calculate the causal effects of each miRNA on each mRNA. This is done for each condition separately. To overcome the over-fitting problem of high-dimensional data, we use bootstrapping to improve the stability of the estimation of causal effects.
Extracting significant miRNA–mRNA causal interactions. Kolmogorov–Smirnov (KS) test is used to evaluate the significance of the difference of the causal effects of an miRNA–mRNA causal interaction in different conditions. miRNA target binding information is used as a constraint in this step for extracting significant causal interactions, and an interaction that passes the KS test and is implied by the target binding information is selected as a significant miRNA–mRNA causal interaction.
Detecting active miRNAs and condition-specific miRNA activity. For each miRNA, the difference of its significant causal interactions across all conditions is assessed using the KS test. If the test result is significant for the miRNA, then it is an active miRNA with respect to the condition of interest, and its activity in the condition (significant causal interactions with the mRNAs) is considered specific to the condition.
Fig. 1.
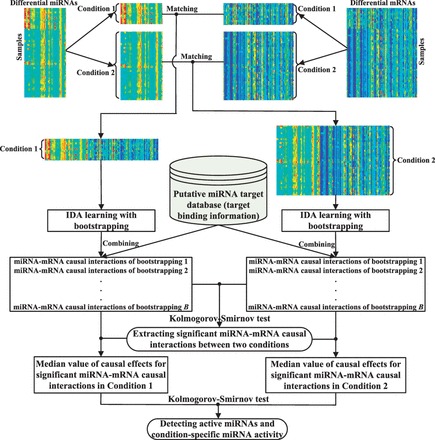
A flowchart of the proposed method. The expression profiles of differentially expressed miRNA and mRNAs are firstly split according to the conditions (phenotypes) of the samples, and in each condition the matched miRNA and mRNA expression samples are integrated. Then, we identify miRNA–mRNA causal relationships with IDA learning by combining target binding information, in each condition, respectively. Bootstrapping is used to improve the result of IDA with high-dimensional data. KS test is then conducted to identify significant miRNA–mRNA causal interactions. To determine if an miRNA is an active miRNA, in each condition, we obtain the median value of all causal effects obtained in all the B runs of bootstrapping for each significant miRNA–mRNA causal interaction associated with this miRNA, and use KS test to evaluate the significance of the difference of this miRNA’s activity in the two conditions
In the following, we will present the key steps in detail.
2.2 Causal inference with IDA
The application of IDA (Maathuis et al., 2009, 2010) to matched miRNA and mRNA expression data can be divided into two steps: (i) learning a causal structure from expression data, and (ii) calculating causal effects (Le et al., 2013).
In step (i), the expression levels of miRNAs and mRNAs are represented by a set of random variables. The PC algorithm (Spirtes et al., 2000) is used to learn the causal structure of the variables in the form of a directed acyclic graph (DAG), where the nodes represent the random variables (miRNAs or mRNAs) and the edges denote causal relationships between these variables.
The PC algorithm is based on conditional dependence tests. Because different DAGs may encode the same conditional independencies in a given dataset, the output of the PC algorithm is an equivalence class of DAGs, which can be uniquely described by a completed partially directed acyclic graph (CPDAG) (Maathuis et al., 2009). Learning a CPDAG from high-dimensional data is computational expensive, and we need to select an efficient conditional independent test to implement it. The PC algorithm with partial correlation test (Kalisch and Bühlmann, 2007) is proved to be uniformly consistent in the high-dimensional context, and thus, we can use it to learn causal structures with gene expression data in inferring gene causal regulatory networks. In this article, we use the R-package pcalg (Kalisch et al., 2012), which implements the PC algorithm with partial correlation test and set the significant level of the conditional independence test α = 0.01.
In step (ii), we simulate the controlled experiments with do-calculus (Judea, 2000), to estimate the causal effect that each miRNA has on an mRNA. Given a DAG, do-calculus can estimate the causal effect of a node on any other node in the DAG from observational data. For an miRNA–mRNA causal interaction, we calculate the causal effect ef(miRNA, mRNA) based on each of the DAGs represented by the CPDAG learnt by the PC algorithm, respectively. We then use the minimum absolute value of the obtained causal effects as the final result of this step, to get a lower bound on the estimated strength of miRNA–miRNA causal interaction. For example, ef(miRNA, mRNA) ∈ {0.75, 0.55, −0.7, 0.65}, then the final result ef (miRNA, mRNA) is 0.55. It suggests that the causal effect of the miRNA on the target gene is at least 0.55. Details of how the causal effects are calculated are out of the scope of this article and interested readers are referred to (Le et al., 2013; Maathuis et al., 2009, 2010) for more information.
Unstable estimation caused by the small number of samples is a challenge to the proposed method, and the problem may get more serious with high-dimensional gene expression data. To tackle this problem, we use a bootstrapping strategy. The above described IDA procedure is carried out in each run of the bootstrapping, and all the results will be used in the next step for identifying significant miRNA–mRNA causal interactions.
2.3 Identifying significant miRNA–mRNA interactions
As mentioned previously, to identify condition-specific miRNA activity, significant miRNA–mRNA causal interactions, i.e. those that vary in their strengths in the condition of interest and the other conditions should be the focus of the examination. The interactions that do not change much across different conditions are not unique to the condition of interest.
To evaluate the significance of a miRNA–mRNA causal interaction, we compare its causal effect, ef, calculated in the two different conditions using a two-sample KS statistic test. The KS test can assess whether the distribution of ef in the samples of one condition is significantly shifted compared with the distribution in the samples of the other condition. We choose to use KS test because it has the following advantages: (i) it is non-parametric and hence does not rely on any assumptions about the distribution of the changes of causal effects; (ii) it does not rely on arbitrary thresholds; and (iii) it measures significant shifts between the entire distribution rather than just comparing the tails.
Suppose that is the empirical cumulative distribution function (cdf) of ef in the two groups of samples, where j ∈ {1, 2}, B is the number of bootstrapping runs and I is the indicator function whose value is 1 when and 0 otherwise. Then the KS test is the maximum difference (D) between the two groups of samples in value of the cdfs, i.e. , where is the supremum of the set of difference.
We use the Matlab function kstest2 to calculate the KS test statistic and the asymptotic P-value [adjusted by Benjamini–Hochberg (BH) method] of each miRNA–mRNA causal interaction. The miRNA–mRNA causal interaction with adjusted P < 0.05 is regarded as a significant miRNA–mRNA causal interaction between the two conditions.
In the implementation, before conducting the KS test, for each miRNA–mRNA causal interaction, we check if the mRNA is a predicted target of the miRNA by using miRNA target binding information, and the interaction is undergoing the KS test only if the interaction is confirmed by the target binding information.
2.4 Inferring condition-specific miRNA activity
Generally, a single significant miRNA–mRNA interaction only shows the partial activity of the miRNA regarding the condition of interest, as the miRNA may be involved in multiple significant miRNA–mRNA interactions. Thus, to obtain a complete picture of the condition-specific activity of an miRNA we need to investigate all the significant interactions in which the miRNA is involved. Note that our definition of an active miRNA is specific to the condition of interest. The overall causal effect that the active miRNA has on all the mRNAs significantly interacting with it must have changed significantly between the condition of interest and the other condition.
To infer such condition-specific active miRNAs or their activity, firstly, for each identified significant miRNA–mRNA causal interaction, we find out the median value of its causal effects calculated during the B times of bootstrapping, in each condition, respectively.
Then for each miRNA, we examine the difference of the distributions of the median causal effects of all its associated significant causal interactions in the condition of interest and the other condition, using the KS test. We also use the Matlab function kstest2 to calculate the KS test statistic and the asymptotic P-value (adjusted by BH method) for the miRNA. If the adjusted P is <0.05, then this miRNA is regarded as an active miRNA specific to the condition of interest.
3 RESULTS
3.1 Data sources and preparation
To demonstrate our method, we apply it to the matched miRNA and mRNA expression profiles from the EMT and MCC datasets.
The miRNA expression profiles of EMT are from Søkilde et al. (2011) (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE26375). They were profiled from the 60 cancer cell lines of the drug screening panel of human cancer cell lines at the National Cancer Institute (NCI-60). The mRNA expression profiles of EMT for NCI-60 were obtained from ArrayExpress (http://www.ebi.ac.uk/arrayexpress, accession number E-GEOD-5720). Samples of the EMT data categorized as epithelial (11 samples) and mesenchymal (36 samples) were used for this work.
The miRNA expression profiles of MCC were obtained from Lu et al. (2005) (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE2564). The mRNA expression profiles of MCC are from Ramaswamy et al. (2001) (http://www.broad.mit.edu/cancer/pub/migcm). Samples of the MCC data classified as normal (21 samples) and tumor (67 samples) were used in this work.
We perform differential gene expression analysis on the gene expression profiles to identify differentially expressed miRNAs and mRNAs between the two conditions in each dataset using the limma package (Smyth, 2005) of Bioconductor. Genes with >10 missing values are removed. As a result, 46 probes of miRNAs and 1612 probes of mRNAs for the EMT dataset and 66 probes of miRNAs and 1318 probes of mRNAs for the MCC dataset are identified to be differentially expressed at a significant level (adjusted P < 0.05, adjusted by BH method). The detailed results of the differentially expressed miRNAs and mRNAs are in Supplementary Material 1.
We use the putative miRNA target information in MicroCosm v5 (Griffiths-Jones et al., 2008) as the constraint when identifying significant miRNA–mRNA causal interactions (Fig. 1). Note that the putative target information is an independent component in our method, and any database of miRNA target information can be used. We choose MicroCosm to illustrate the method.
The number of bootstrapping, B, is set to 100.
3.2 Validations by transfection experiment result
In this section, we validate identified significant miRNA–mRNA interactions using the transfection experimental data from (Khan et al., 2009), and the data included in the transfection data are listed in Supplementary Material 2. The 18 unique miRNAs overlap with 11 and 12 miRNAs in the EMT and MCC datasets, respectively, which enables the validation of the significant interactions involving these miRNAs. Differentially expressed genes between the control and miRNA transfected samples are considered as targets of the miRNA, and are used as the ground truth to validate the predicted miRNA targets (significant miRNA–mRNA interactions). In the transfection experiment, mRNA differential expression levels are calculated by comparing mRNA expression levels between transfection and control samples via log2 fold change (LFC). The larger the absolute value of the LFC is, the more significant the mRNA differential expression level is. The commonly used fold change (FC) cutoffs are 1.5 and 2.0 (Dalman et al., 2012), so we use the logarithm of the FC cutoffs and round them to 0.5 and 1.0 in this work. The following validations are done using the differentially expressed genes (ground truth) obtained with both LFCs, respectively.
3.2.1 Validation in comparison with MicroCosm
To assess how well the proposed method enriches the putative miRNA target information (MicroCosm), we compare the performance of the method with MicroCosm. For each dataset, we retrieve all the target genes of each miRNA predicted by the method and calculate the percentage of confirmed targets. As shown in Figure 2, our method overall performs better than MicroCosm in most miRNAs in both datasets. When the LFC cutoff is set to 1.0, our method also performs better than MicroCosm in terms of the number of validated targets (see Supplementary Fig. S1 in Supplementary Material 3). The results suggest that the method significantly enriches the putative target information used in the model. We also conceive a cumulative hypergeometric (HG) test to assess the statistical significance of the number of validated miRNA–mRNA interactions in our method. We found that the number of validated miRNA–mRNA interactions is statistically significant (P < 0.05) in both the EMT and MCC datasets (see Supplementary Table S1 in Supplementary Material 3 for details).
Fig. 2.
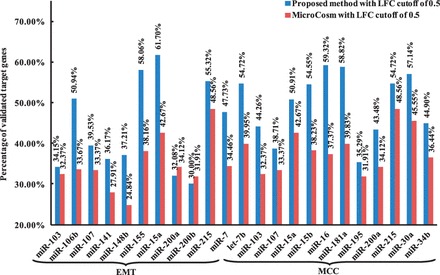
The percentage of confirmed target genes identified by using the proposed method and MicroCosm in the EMT and MCC dataset (LFC = 0.5)
3.2.2 Validation in comparison with non–condition-specific approach
To evaluate the effectiveness of the condition-specific approach, we compare the performance of the proposed method with its non–condition-specific variant that does not consider sample categories. The non–condition-specific approach will not split the dataset into different conditions, and it simply applies IDA to the whole dataset to enrich the putative target information.
We extract the top 10 and 20 predicted targets for all the transfected miRNAs in the EMT and MCC datasets (11 for EMT and 12 in MCC) and compare the total number of validated targets for the two approaches. Figure 3 shows that the proposed condition-specific approach predicts more confirmed targets than that by the non–condition-specific approach in all cases (Top10-EMT, Top20-EMT, Top10-MCC and Top20-MCC). When the LFC cutoff in transfection experiments is set to 1.0, the proposed method also performs better than non–condition-specific method in terms of the number of validated targets (see Supplementary Fig. S2 in Supplementary Material 3).
Fig. 3.
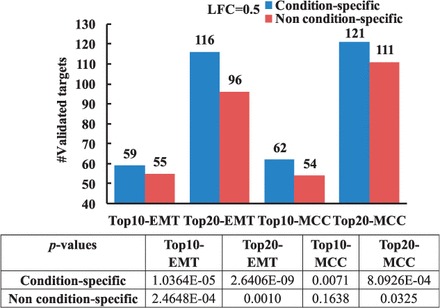
Comparison between condition-specific and non–condition-specific analyses in terms of the number of validated targets (LFC = 0.5). P-values of validated targets are calculated using cumulative HG test
3.2.3 Validation in comparison with the cases using correlation methods
To show the advantage of causal inference, in this section, we replace step (2) of our proposed method (see Section 2.1) with each of the five correlation methods, Pearson, Spearman, Kendall, Lasso and Elastic-net (called the correlation methods), and compare their results with the results obtained by the proposed method in terms of the number of validated miRNA targets.
We extract the top 10 and 20 predicted targets for all transfected miRNAs in the EMT and MCC datasets (11 in EMT and 12 in MCC), and compare the total number of validated targets obtained by different methods. As illustrated in Figure 4, our method performs better than all the correlation methods in all cases (Top10-EMT, Top20-EMT, Top10-MCC and Top20-MCC). When the LFC cutoff is set to 1.0, the proposed method also outperforms all the five correlation methods in the number of validated targets (see Supplementary Fig. S3 in Supplementary Material 3).
Fig. 4.
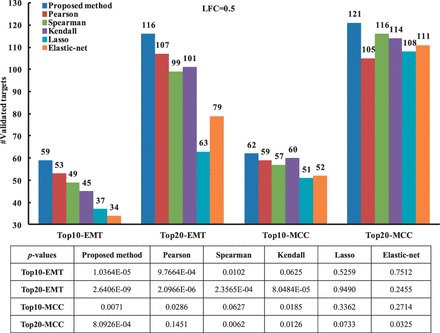
Comparison between the proposed method and the correlation methods in terms of the number of confirmed targets (LFC = 0.5). P-values of validated targets are calculated using cumulative HG test
3.3 Inferring condition-specific active miRNAs
Using our method, we have identified 18 and 41 active miRNAs in the EMT and MCC datasets, respectively. The identified miRNA activity with the KS tests and the box plots of causal effects of the active miRNAs in both datasets are provided in Supplementary Material 4.
3.3.1 Causal effects vs. correlations for inferring condition-specific active miRNAs
To show the effectiveness of using causal effects as the measure of the strength of miRNA–mRNA interactions, we also use the five correlation methods (Pearson, Spearman, Kendall, Lasso and Elastic-net) for detecting condition-specific miRNA activity. That is, in Step (2) of our method (Section 2.1), instead of using IDA, we use one of the correlation methods to compute the strength of an interaction, and the obtained values instead of casual effects are used in the next two steps.
Because no benchmarks are available, to compare the performance of each method in inferring condition-specific miRNA activity, we use the number of identified active miRNAs out of the differentially expressed miRNAs as the criterion. If a method identifies the largest number of condition-specific active miRNAs, the method performs the best. As shown in Table 1, our method significantly outperforms all the five correlation methods in detecting active miRNAs, suggesting that causal effect is a useful measure to detect active miRNAs. The results of condition-specific miRNA activity using the five correlation methods are provided in Supplementary Material 5.
Table 1.
Comparing the number of active miRNAs found by the proposed method and the five correlation methods
| Dataset | Proposed method | Pearson | Spearman | Kendall | Lasso | Elastic-net |
|---|---|---|---|---|---|---|
| EMT | 18 | 6 | 2 | 2 | 0 | 0 |
| MCC | 41 | 38 | 26 | 24 | 1 | 5 |
3.3.2 Comparing with existing methods in inferring condition-specific active miRNAs
We evaluate the performance of our method by comparing it with other five existing methods: DIANA-mirExTra (Alexiou et al., 2010), Sylamer (van Dongen et al., 2008), MIR (Cheng and Li, 2008), miReduce (Sood et al., 2006) and cWords (Rasmussen et al., 2013). Similarly, an miRNA with P < 0.05 is regarded as an active miRNA. We also use the number of identified active miRNAs out of the differentially expressed miRNAs as the criterion. As illustrated in Table 2, for the EMT dataset, our method is comparable with cWords and performs better than the other four existing methods. For the MCC dataset, our method outperforms all the five existing methods. The results of condition-specific active miRNAs (P < 0.05) using the five existing methods are provided in Supplementary Material 6.
Table 2.
Comparing the number of active miRNAs found by the proposed method and the five existing methods
| Dataset | Proposed method | DIANA- mirExTra | Sylamer | MIR | miReduce | cWords |
|---|---|---|---|---|---|---|
| EMT | 18 | 5 | 10 | 13 | 3 | 18 |
| MCC | 41 | 35 | 20 | 10 | 0 | 6 |
3.4 Validation of active miRNAs
We use the TAM (Lu et al., 2010) software to conduct the functional analysis of the active miRNAs found by our method. Significant biological functions and associated diseases are identified for an active miRNA with the adjusted P-value (adjusted by BH method) of 0.05. The analysis of the molecular pathways that the active miRNAs are potentially involved is performed with mirPath (Vlachos et al., 2012), and TarBase 6.0 (Vergoulis et al., 2012) is regarded as a reference database to mine significantly enriched pathways.
For the EMT dataset, the 47 samples are closely related to nine human cancer cell lines, Breast, Cardiovascular Nervous System, Colon, Leukemia, Lung, Melanoma, Ovarian, Prostate and Renal. Here, we only discuss EMT in the biological functions and diseases associated with these nine human cancer cell lines.
As illustrated in Figure 5, 9 of the 18 active miRNAs identified using our method are significantly associated with EMT, and 11 miRNAs are closely related to Breast Neoplasms. As shown in Figure 6, of the 18 active miRNAs, most of them are significantly enriched in the top five KEGG pathways, including Pathways in cancer, Chronic myeloid leukemia, Cell cycle, HTLV-I infection and Colorectal cancer.
Fig. 5.
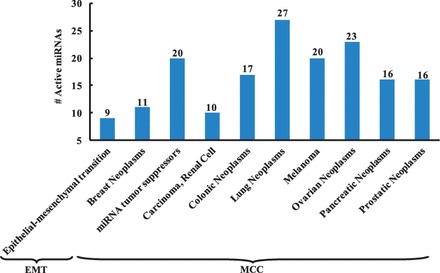
Functional analysis of active miRNAs. The results are generated by TAM, and significant biological functions and associated diseases are identified with a P-value cutoff of 0.05
Fig. 6.
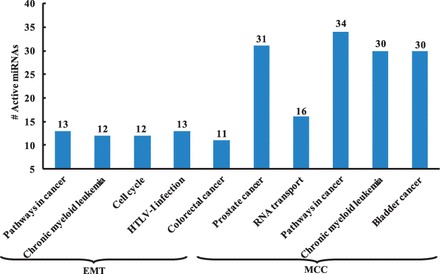
Pathway analysis of active miRNAs. The results are generated by mirPath, the top five KEGG pathways are listed for both datasets
Our method has found a number of literature-confirmed active miRNAs in EMT, including four members (miR-141, miR-200a, miR-200c and miR-429) of the miR-200 family and two members (miR-192 and miR-215) of the miR-192 family. Previous studies (Gregory et al., 2008; Mongroo and Rustgi, 2010) have revealed that members of the miR-200 family play a critical role in the suppression of EMT, tumor cell adhesion, migration, invasion and metastasis and may have therapeutic implications for the treatment of metastatic and drug-resistant tumors. The miR-200 family and miR-192 family are critical mediators of p53-regulated EMT (Kim et al., 2011).
The MCC samples are closely associated with 11 human cancer lines, including Bladder, Breast, Colon, Lung, Melanoma, Mesothelioma, Ovarian, Pancreas, Prostate, Renal and Uterus. In the results, of the 41 miRNAs indentified to be active between the normal and tumor samples, 20 miRNAs are shown to be significantly associated with miRNA tumor suppressors in Figure 5. Furthermore, many active miRNAs are significantly associated with Carcinoma of Renal Cell, Colonic Neoplasms, Lung Neoplasms, Melanoma, Ovarian Neoplasms, Pancreatic Neoplasms and Prostatic Neoplasms.
The pathway analysis indicates that more than half of the 41 active miRNAs are significantly enriched in the top five KEGG pathways including Prostate cancer, Pathways in cancer, Chronic myeloid leukemia and Bladder cancer, except RNA transport (Fig. 6).
Our method has also identified that six members (let-7a, let-7b, let-7c, let-7d, let-7f and let-7g) of the let-7 family, two members (miR-181a and miR-181c) of the miR-181 family and two members (miR-29a and miR-29c) of the miR-29 family are active in the process of tumor. Recent research (Boyerinas et al., 2010) has found out that let-7 and its family members are highly conserved across species in sequence and function, and misregulation of the let-7 family leads to a less differentiated cellular state and the development of cell-based diseases such as cancer. The miR-181 family has been demonstrated to play an important role in occurrence and progression of malignant tumors such as lung cancer, pancreatic cancer, prostate cancer and breast cancer (Zhu et al., 2012). The miR-29 family has also been shown to be silenced or downregulated in many different types of cancer and have subsequently been attributed predominantly tumor-suppressing properties (Schmitt et al., 2013).
3.5 Condition-specific miRNA activity
To understand the types of regulation of active miRNAs on their targets in different conditions, we compare the number of positive and negative effect of each active miRNA on their targets. With our method, a positive (negative) causal effect indicates upregulation (downregulation) of the miRNA on its interacting mRNA. If the number of negative effects of an active miRNA on its targets is more than that of positive effects on its targets in one condition, the active miRNA dominantly downregulates its target genes in the condition, and vice versa. When an active miRNA has the same number of negative effects and positive effects on its targets in a condition, the regulation type of the active miRNA is uncertain in the condition.
As shown in Table 3, most active miRNAs identified using our method (13 of 18) downregulate their target genes in class E (epithelial), but most active miRNAs (10 of 18) upregulate their targets in class M (mesenchymal). Most active miRNAs (11 of 18) have different regulation types between E and M. For the MCC dataset, most active miRNAs (30 of 41) upregulate their target genes in class N (normal), but most active miRNAs (23 of 41) downregulate their targets in class T (tumor). In total, 17 active miRNAs have different regulation types between N and T.
Table 3.
The number of active miRNAs showing downregulation (Down), upregulation (Up) and different-regulation (Dif) in EMT and MCC dataset
| Dataset | #Down in E(N) | #Up in E(N) | #Down in M(T) | #Up in M(T) | #Dif in both conditions |
|---|---|---|---|---|---|
| EMT | 13 | 5 | 8 | 10 | 11 |
| MCC | 7 | 30 | 23 | 17 | 17 |
Note: In the MCC dataset, 4 active miRNAs have uncertain regulation type in N and 1 in T. E:Epithelial, N, Normal; M, Mesenchymal; T, Tumor.
The results indicate that in each sample condition there is a dominant regulation type. The results also show that some active miRNAs behave differently in different conditions in the types of regulation (up or down), but some active miRNAs have the same regulation type in different conditions and the difference across conditions is just the strengths of their regulation. If we only look at the whole dataset without considering the difference between conditions, we may miss the interactions in a specific condition (e.g. cancer).
4 CONCLUSIONS
miRNAs have been regarded as the main regulators at the post-transcriptional level. Identifying the targets of miRNAs is a fundamental task in predicting miRNA functions. Great efforts have been made to elucidate miRNA functions and regulatory mechanism. One stream of the research is focused on miRNA activity specific to a condition of interest. However, most of the studies only use samples obtained in the specific condition, without examining the difference of miRNA behavior in the specific condition and the other conditions, thus the miRNA activity discovered may not be unique to the specific condition. Furthermore, most computational methods only use associations or correlations in predicting miRNA–mRNA regulation while the regulation is in fact causal relationships.
In this study, we have proposed an alternative method to reveal ‘truly’ condition-specific miRNA activity with the consideration of the causal semantics of miRNA–mRNA relationships.
We have applied our method to the EMT and MCC datasets. The validation with transfection experiment data illustrates that our method is more efficient than MicroCosm v5 in identifying the miRNA targets, and considering the difference across different sample conditions improves the number of validated interactions.
The comparison with five correlation methods demonstrates that causal effects provide a better measure than correlations in modeling the strengths of miRNA–mRNA interactions, leading to more effective discovery of active miRNAs.
As the main aim of the article is to identify condition-specific active miRNAs and their activity, we conduct function and pathway analysis of the active miRNAs detected using our method. The results have shown that a significant number of the identified active miRNAs are closely related to the biological functions associated with the conditions of samples in the EMT and MCC datasets, and play a vital role in the potential pathogenesis of complex diseases. Furthermore, to understand the activity of the active miRNAs, we investigate how these miRNAs behave differently in different conditions. It was found out that some active miRNAs show different regulation types in different conditions and some active miRNAs have the same regulation types and their activity only differs in different conditions in terms of the strengths of regulation.
In conclusion, the validation and analysis results indicate that the proposed method can be an effective method to detect condition-specific miRNA activity.
Funding: This work has been partially supported by the Applied Basic Research Foundation of Science and Technology of Yunnan Province (No: 2013FD038), the Australian Research Council Discovery grant DP130104090 and the Science Research Foundation for Youth Scholars of Dali University (No: KYQN201203).
Conflict of Interest: none declared.
Supplementary Material
REFERENCES
- Ambros V. MicroRNA pathways in flies and worms: growth, death, fat, stress, and timing. Cell. 2003;113:673–676. doi: 10.1016/s0092-8674(03)00428-8. [DOI] [PubMed] [Google Scholar]
- Alexiou P, et al. The DIANA-mirExTra web server: from gene expression data to microRNA function. PLoS One. 2010;5:e9171. doi: 10.1371/journal.pone.0009171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betel D, et al. The microRNA.org resource: targets and expression. Nucleic Acids Res. 2008;36:D149–D153. doi: 10.1093/nar/gkm995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyerinas B, et al. The role of let-7 in cell differentiation and cancer. Endocr. Relat. Cancer. 2010;17:F19–F36. doi: 10.1677/ERC-09-0184. [DOI] [PubMed] [Google Scholar]
- Cheng C, Li LM. Inferring microRNA activities by combining gene expression with microRNA target prediction. PLoS One. 2008;3:e1989. doi: 10.1371/journal.pone.0001989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalman MR, et al. Fold change and p-value cutoffs significantly alter microarray interpretations. BMC Bioinformatics. 2012;13:S11. doi: 10.1186/1471-2105-13-S2-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du T, Zamore PD. Beginning to understand microRNA function. Cell Res. 2007;17:661–663. doi: 10.1038/cr.2007.67. [DOI] [PubMed] [Google Scholar]
- Friedman RC, et al. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory PA, et al. MicroRNAs as regulators of epithelial-mesenchymal transition. Cell Cycle. 2008;7:3112–3118. doi: 10.4161/cc.7.20.6851. [DOI] [PubMed] [Google Scholar]
- Griffiths-Jones S, et al. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36:D154–D158. doi: 10.1093/nar/gkm952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judea P. Causality: Models, Reasoning, and Inference. New York, USA: Cambridge University Press; 2000. [Google Scholar]
- Kalisch M, Bühlmann P. Estimating high-dimensional directed acyclic graphs with the PC-algorithm. J. Mach. Learn. Res. 2007;8:613–636. [Google Scholar]
- Kalisch M, et al. Causal inference using graphical models with the R Package pcalg. J. Stat. Softw. 2012;47:1–26. [Google Scholar]
- Khan AA, et al. Transfection of small RNAs globally perturbs gene regulation by endogenous microRNAs. Nat. Biotechnol. 2009;27:549–555. doi: 10.1038/nbt.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T, et al. p53 regulates epithelial-mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J. Exp. Med. 2011;208:875–883. doi: 10.1084/jem.20110235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krek A, et al. Combinatorial microRNA target predictions. Nat. Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- Le HS, Bar-Joseph Z. Integrating sequence, expression and interaction data to determine condition-specific miRNA regulation. Bioinformatics. 2013;29:i89–i97. doi: 10.1093/bioinformatics/btt231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le TD, et al. Inferring microRNA-mRNA causal regulatory relationships from expression data. Bioinformatics. 2013;29:765–771. doi: 10.1093/bioinformatics/btt048. [DOI] [PubMed] [Google Scholar]
- Liang Z, et al. mirAct: a web tool for evaluating microRNA activity based on gene expression data. Nucleic Acids Res. 2011;39:W139–W144. doi: 10.1093/nar/gkr351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- Lu M, et al. TAM: a method for enrichment and depletion analysis of a microRNA category in a list of microRNAs. BMC Bioinformatics. 2010;11:419. doi: 10.1186/1471-2105-11-419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maathuis HM, et al. Estimating high-dimensional intervention effects from observational data. Ann. Stat. 2009;37:3133–3164. [Google Scholar]
- Maathuis HM, et al. Predicting causal effects in large-scale systems from observational data. Nat. Methods. 2010;7:247–249. doi: 10.1038/nmeth0410-247. [DOI] [PubMed] [Google Scholar]
- Madden SF, et al. Detecting microRNA activity from gene expression data. BMC Bioinformatics. 2010;11:257. doi: 10.1186/1471-2105-11-257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mongroo PS, Rustgi AK. The role of the miR-200 family in epithelial-mesenchymal transition. Cancer Biol. Ther. 2010;10:219–222. doi: 10.4161/cbt.10.6312548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajewsky N. microRNA target predictions in animals. Nat. Genet. 2006;38:S8–S13. doi: 10.1038/ng1798. [DOI] [PubMed] [Google Scholar]
- Ramaswamy S, et al. Multiclass cancer diagnosis using tumor gene expression signatures. Proc. Natl Acad. Sci. USA. 2001;98:15149–15154. doi: 10.1073/pnas.211566398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SH, et al. cWords–systematic microRNA regulatory motif discovery from mRNA expression data. Silence. 2013;4:2. doi: 10.1186/1758-907X-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt MJ, et al. MiRNA-29: a microRNA family with tumor-suppressing and immune-modulating properties. Curr. Mol. Med. 2013;13:572–585. doi: 10.2174/1566524011313040009. [DOI] [PubMed] [Google Scholar]
- Smyth GK. Limma: linear models for microarray data. In: Gentleman R, editor. Bioinformatics and Computational Biology Solutions using R and Bioconductor. New York: Springer; 2005. pp. 397–420. [Google Scholar]
- Sood P, et al. Cell-type-specific signatures of microRNA on target mRNA expression. Proc. Natl Acad. Sci. USA. 2006;103:2746–2751. doi: 10.1073/pnas.0511045103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Søkilde R, et al. Global microRNA analysis of the NCI-60 cancer cell panel. Mol. Cancer Ther. 2011;10:375–384. doi: 10.1158/1535-7163.MCT-10-0605. [DOI] [PubMed] [Google Scholar]
- Spirtes P, et al. Causation, Prediction, and Search. 2nd edn. Cambridge, MA: MIT Press; 2000. [Google Scholar]
- Steinfeld I. miRNA target enrichment analysis reveals directly active miRNAs in health and disease. Nucleic Acids Res. 2013;41:e45. doi: 10.1093/nar/gks1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dongen S, et al. Detecting microRNA binding and siRNA off-target effects from expression data. Nat. Methods. 2008;5:1023–1025. doi: 10.1038/nmeth.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergoulis T, et al. TarBase 6.0: capturing the exponential growth of miRNA targets with experimental support. Nucleic Acids Res. 2012;40:D222–D229. doi: 10.1093/nar/gkr1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlachos IS, et al. DIANA miRPath v.2.0: investigating the combinatorial effect of microRNAs in pathways. Nucleic Acids Res. 2012;40:W498–W504. doi: 10.1093/nar/gks494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volinia S, et al. Identification of microRNA activity by Targets’ Reverse Expression. Bioinformatics. 2010;26:91–97. doi: 10.1093/bioinformatics/btp598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacher B, et al. Joint Bayesian inference of condition-specific miRNA and transcription factor activities from combined gene and microRNA expression data. Bioinformatics. 2012;28:1714–1720. doi: 10.1093/bioinformatics/bts257. [DOI] [PubMed] [Google Scholar]
- Zhu YK, et al. Advances in research on miR-181 family members and malignant tumors. Tumor. 2012;32:837–841. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.