

Abstract
Summary: STAMP is a graphical software package that provides statistical hypothesis tests and exploratory plots for analysing taxonomic and functional profiles. It supports tests for comparing pairs of samples or samples organized into two or more treatment groups. Effect sizes and confidence intervals are provided to allow critical assessment of the biological relevancy of test results. A user-friendly graphical interface permits easy exploration of statistical results and generation of publication-quality plots.
Availability and implementation: STAMP is licensed under the GNU GPL. Python source code and binaries are available from our website at: http://kiwi.cs.dal.ca/Software/STAMP
Contact: donovan.parks@gmail.com
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Taxonomic and functional profiles arise in many areas of the biological sciences. Statistical hypothesis tests can be used to identify features (e.g. taxa or metabolic pathways) that differ significantly between pairs of profiles or between sets of profiles organized into two or more groups (e.g. sick versus healthy). Here we introduce STAMP v2, a tool that provides extensive hypothesis testing, exploratory plots, effect size measures and confidence intervals for aiding in the identification of biologically relevant differences. We illustrate the use of STAMP on two microbial datasets: (i) taxonomic profiles from coalbed methane (CBM) communities and (ii) functional profiles from photosynthetic and non-photosynthetic Cyanobacteria.
2 FEATURES
The original release of STAMP (Parks and Beiko, 2010) was limited to comparing a single pair of taxonomic or functional profiles. This release adds statistical tests and plots for assessing differences between two or more treatment groups along with increased compatibility with popular bioinformatic software:
Input data: STAMP can process functional and taxonomic profiles produced by QIIME (Caporaso et al., 2010), PICRUSt (Langille et al., 2013), MG-RAST (Meyer et al., 2008), IMG/M (Markowitz et al., 2008) and RITA (MacDonald et al., 2012). Custom profiles can also be specified as a tab-separated values file. STAMP can process input files containing hundreds of samples spanning thousands of features with a standard desktop computer.
Statistical hypothesis tests: Welch’s t-test and White’s non-parametric t-test (White et al., 2009) are provided for comparing profiles organized into two groups. STAMP implements the ANOVA and Kruskal–Wallis H-test for comparing three or more groups of profiles. Statistically significant features can be further examined with post hoc tests (e.g. Tukey–Kramer) to determine which groups of profiles differ from each other.
Effect size and confidence intervals: Widely used effect size measures are provided for all statistical tests to aid in determining features with biologically relevant differences between groups. Two-group tests use the difference in mean proportion effect size measure along with Welch’s confidence intervals. The eta-squared effect size measure is used when considering multiple groups.
Filtering of features: A feature can be filtered based on its P-value, effect size or prevalence within a group of profiles, to create plots focused on features likely to be biologically relevant. Specific subsets of features can also be manually filtered.
Plots: Numerous publication-quality plots can be produced using STAMP. Principal component analysis (PCA; e.g. Fig. 1a) plots, bar plots (e.g. Supplementary Fig. S1), box-and-whisker plots (e.g. Fig. 1b), scatter plots and heat maps permit an initial exploratory analysis of profiles. Extended error bar plots (e.g. Fig. 1c) provide a single figure indicating statistically significant features along with the P-values, effect sizes and confidence intervals.
Fig. 1.
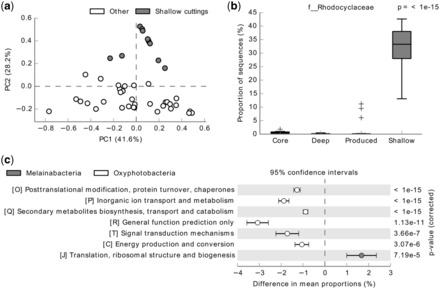
Example outputs from STAMP. (a) PCA plot comparing class-level taxonomic profiles of 44 CBM communities sampled from shallow core cuttings or other (drilled cores, deep core cuttings, produced waters) niches within the coalbed environment. (b) Box-and-whisker plot illustrating Rhodocyclaceae taxa are only present in appreciable numbers within communities sampled from shallow core cuttings. (c) COG categories differing significantly between Melainabacteria and Oxyphotobacteria genomes with an effect size ≥0.75%
3 CBM COMMUNITIES
The metabolic activity of microbial communities has been implicated as a major source of methane in many CBM reservoirs. Here we use STAMP to examine the taxonomic profiles of 44 CBM communities sampled from drilled cores, shallow (<1000 mbs) and deep (≥1000 mbs) core cuttings, and produced waters (Supplementary Methods; An et al., 2013). A PCA plot indicates that communities from shallow core cuttings are relatively distinct (Fig. 1a). Specifically, the Rhodocyclaceae and Comamonadaceae families were found to be overrepresented in these communities (Fig. 1b; Supplementary Fig. S1). A PCA plot coloured by the company performing the drilling reveals secondary clustering of shallow core cuttings indicating that the difference between CBM samples may be the result of secondary factors such as collection protocols or geography as opposed to different niches within the CBM environment (Supplementary Fig. S2). A clear example is the five Nexen samples (three from deep core cuttings, two from produced waters), which are the only samples containing an appreciable percentage of taxa from the Propionigenium and Halomonas genera (Supplementary Fig. S3).
4 MELAINABACTERIA GENOMES
The Melainabacteria are a recently discovered and highly diverse group of bacteria that form a sister class within (Soo et al., 2014) or phylum to (Di Rienzi et al., 2013) the Cyanobacteria. Here we use STAMP to compare COG profiles (Supplementary Methods) of the non-photosynthetic Melainabacteria with the Oxyphotobacteria, the class name proposed by Soo et al. to describe photosynthetic cyanobacteria. Several COG categories were found to differ significantly between these groups (Fig. 1c) indicating that at a broad scale these groups are metabolically distinct from each other. Examining individual COG categories in detail indicates that the Melainabacteria contains relatively few genes assigned to categories O, P and Q compared with named orders within the Oxyphotobacteria (Supplementary Fig. S4). Significantly different COGs within categories P and Q associated with photosynthesis were previously identified with STAMP (Soo et al., 2014).
Funding: D.H.P. is supported by the Natural Sciences and Engineering Research Council of Canada. G.W.T. and P.H. are supported by a Discovery Outstanding Researcher Award (DORA) and Queen Elizabeth II Fellowship from the Australian Research Council, grants DP120103498 and DP1093175, respectively. R.G.B. acknowledges the support of Genome Atlantic, the Canada Foundation for Innovation and the Canada Research Chairs program.
Conflict of interest: none declared.
Supplementary Material
REFERENCES
- An D, et al. Metagenomics of hydrocarbon resource environments indicates aerobic taxa and genes to be unexpectedly common. Environ. Sci. Technol. 2013;47:10708–10717. doi: 10.1021/es4020184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Rienzi SC, et al. The human gut and groundwater harbor non-phtosynthetic bacteria nelonging to a new candidate phylum sibling to Cyanobacteria. eLife. 2013;2:e01102. doi: 10.7554/eLife.01102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langille MGI, et al. Predicitive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013;31:814–821. doi: 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald NJ, et al. Rapid identification of high-confidence taxonomic assignments for metagenomic data. Nucleic Acids Res. 2012;40:e111. doi: 10.1093/nar/gks335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowitz VM, et al. IMG/M: a data management and analysis system for metagenomes. Nucleic Acids Res. 2008;36:D534–D538. doi: 10.1093/nar/gkm869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer F, et al. The metagenomics RAST server—a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics. 2008;9:386. doi: 10.1186/1471-2105-9-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks DH, Beiko RG. Identifying biologically relevant differences between metagenomic communities. Bioinformatics. 2010;26:715–721. doi: 10.1093/bioinformatics/btq041. [DOI] [PubMed] [Google Scholar]
- Soo RM, et al. An expanded genomic representation of the phylum Cyanobacteria. Genome Biol. Evol. 2014;6:1031–1045. doi: 10.1093/gbe/evu073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JR, et al. Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput. Biol. 2009;5:e1000352. doi: 10.1371/journal.pcbi.1000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.