

Abstract
Background
The application of molecular based diagnostics in sepsis has had limited success to date. Molecular community profiling methods have indicated that polymicrobial infections are more common than suggested by standard clinical culture. A molecular profiling approach was developed to investigate the propensity for polymicrobial infections in patients predicted to have bacterial sepsis.
Results
Disruption of blood cells with saponin and hypotonic shock enabled the recovery of microbial cells with no significant changes in microbial growth when compared to CFU/ml values immediately prior to the addition of saponin. DNA extraction included a cell-wall digestion step with both lysozyme and mutanolysin, which increased the recovery of terminal restriction fragments by 2.4 fold from diverse organisms. Efficiencies of recovery and limits of detection using Illumina sequencing of the 16S rRNA V3 region were determined for both viable cells and DNA using mock bacterial communities inoculated into whole blood. Bacteria from pre-defined communities could be recovered following lysis and removal of host cells with > 97 % recovery of total DNA present. Applying the molecular profiling methodology to three septic patients in the intensive care unit revealed microbial DNA from blood had consistent alignment with cultured organisms from the primary infection site providing evidence for a bloodstream infection in the absence of a clinical lab positive blood culture result in two of the three cases. In addition, the molecular profiling indicated greater diversity was present in the primary infection sample when compared to clinical diagnostic culture.
Conclusions
A method for analyzing bacterial DNA from whole blood was developed in order to characterize the bacterial DNA profile of sepsis infections. Preliminary results indicated that sepsis infections were polymicrobial in nature with the bacterial DNA recovered suggesting a more complex etiology when compared to blood culture data.
Keywords: Sepsis, Saponin, Blood, 16 s rRNA, Illumina, Polymicrobial, DNA Sequencing
Background
Sepsis is defined based on the clinical symptoms and signs of a systemic inflammatory response due to a microbial infection in sterile body sites or fluids [1]. Despite modern technologies and advances in health care, sepsis rates continue to climb and have more than doubled in the last ten years [2]. The socioeconomic costs associated with sepsis are high due to the increased need for hospital resources and long stays in the intensive care unit (ICU) [2]. In Canada, sepsis is one of the leading causes of in-hospital mortality with 10.9 % of hospital deaths in 2008–2009 being sepsis related [2]. Bacteria are implicated in the majority of sepsis infections with three independent studies reporting bacteremia, confirmed or suspected, in 63–74 % of all cases [3–5]. Currently, the “gold standard” of sepsis diagnostics is a confirmed bloodstream infection using blood culture. Microbial growth is monitored continuously and is detected using gas production. In order to identify the microbial growth, sub-culturing and biochemical testing is performed using clinical and laboratory standards institute (CLSI) guidelines [6]. The use of this diagnostic tool is time consuming and lacks sensitivity due to several inherent limitations such as a dependency on volume with efficacy decreasing as volume decreases, limited efficacy for slow-growing or fastidious organisms, and incubation times of 72–96 h [7]. These limitations allow an overall positivity of blood culture in the range of 30–40 % [7]. Since survival rates have been reported to decline with every hour without therapy, clinicians usually treat with broad-spectrum empiric antibiotics and then use any blood culture results obtained to tailor antimicrobial therapy [8].
There is a need for more rapid and comprehensive diagnostics for bloodstream infections in the management of sepsis. Nucleic acid technologies and proteomic approaches are being actively explored for sepsis diagnostics. Molecular testing on whole blood has capitalized on developments in PCR-based technology [9]. Broad-range assays, with primers targeting variable regions in the 16S rRNA or 18S/23S rRNA gene, have the greatest clinical applicability for sepsis diagnostics due to their short turnaround time and ability to directly detect any non-cultivable or cultivable pathogens [6, 9, 10].
To date, the application of nucleic acid technology in sepsis has focused on identifying 1–2 organisms from positive blood culture [11–16]. With the advent of next-generation sequencing technologies, there has been a shift in focus from singular pathogens causing illness to the concept of polymicrobial communities of organisms, pathogens and commensals in human infections. Despite this, the concept of a sepsis microbiota has not been explored and sepsis infections are considered to be monomicrobial with polymicrobial infections occurring in a minority of patients [17]. In order to better understand bloodstream infections in sepsis, we developed a method to optimize recovery microbial cells and DNA directly from blood in order to assess the bacterial DNA profile in sepsis patients using molecular community profiling methods of terminal restriction fragment length polymorphism (TRFLP) and paired-end Illumina sequencing. In this paper we outline the methods developed, their success in mock community studies and application to sepsis case studies.
Results
Impact of saponin on bacterial isolate viability
Whole blood is known to contain many substituents that can impair PCR including heme, leukocyte DNA, immunoglobulin G, hemoglobin and lactoferrin [18]. In this study, a method of lysing blood cells prior to DNA extraction was evaluated in order to improve the recovery of bacterial cells and DNA from small volumes of blood. Saponin, a plant metabolite, is known for its ability to lyse both red and white blood cells [19, 20]. Recent studies have shown that saponin at low concentrations (1 % w/v) does not impact microorganism growth [20–22]. In order to confirm the results from these studies, the ability of bacteria to grow directly in saponin culture was assessed.
Following incubation with the strains recovered from sepsis infections (Table 1), there were no significant changes in microbial growth when compared to CFU/ml values immediately prior to the addition of saponin (Fig. 1). Only Fusobacterium necrophorum had a 2-log drop in CFU/ml following treatment with saponin but the reduction was not significant (p = 0.2375, two-tailed t-test). The observed decrease in viability of F. necrophorum may result from its sensitivity to oxygen. Although incubations were carried out under anaerobic conditions, initial sample preparations were carried out with some exposure to ambient oxygen. These results demonstrated that bacteria were not affected by the presence of saponin.
Table 1.
Isolates used in synthetic community assessments
| Species | Source | Ward | Growth Conditions | Media |
|---|---|---|---|---|
| Enterobacter hormaechei | Peritoneal Fluid | FMC ICU | 5 % CO2 | CBA |
| Escherichia coli | Urine | FMC ICU | 5 % CO2 | MAC |
| Fusobacterium necrophorum | BAL | FMC ICU | Anaerobic | FAA |
| Micrococcus luteus | Whole blood | FMC ICU | 5 % CO2 | TSY |
| Neisseria flava | BAL | FMC ICU | 5 % CO2 | TSY |
| Prevotella melaninogenica | BAL | FMC ICU | Anaerobic | FAA |
| Staphylococcus aureus | Whole blood | FMC ED | 5 % CO2 | MSA |
| Staphylococcus epidermidis | Whole blood | FMC ICU | 5 % CO2 | MSA |
| Streptococcus intermedius | Abscess fluid | FMC ICU | 5 % CO2 | CNA |
| Streptococcus pneumoniae | Chest tube aspirate | FMC ICU | 5 % CO2 | CNA |
Fig. 1.
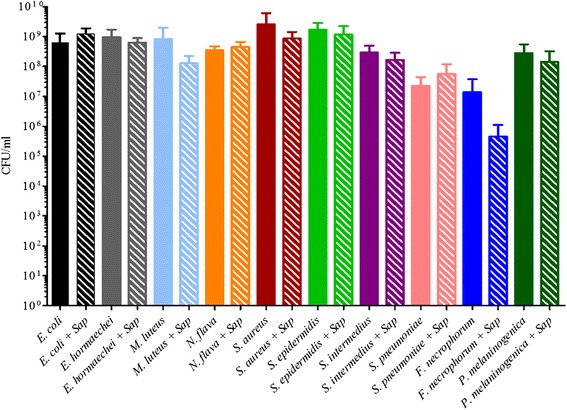
The addition of 0.85 % saponin to bacterial cells does not impact viability. The average CFU/ml and the standard deviation were plotted for each sample prior to and after the incubation with 0.85 % saponin (represented by “sap” in the figure label) for 1 h at room temperature either with ambient air or in an anaerobic chamber for F. necrophorum and P. melaninogenica. Each experiment was done in triplicate with samples recorded in duplicate. There were no statistically significant differences found in the viable cell count of the bacteria (two-tailed Students t-test, p > 0.05 % significance)
Optimization of DNA extraction from whole blood using enzymatic lysis
Our standard lab protocol included enzymatic digestion of the bacterial cell wall in a pretreatment step [23–25]. To evaluate if this was necessary in samples recovered from blood, a microbial community made from pooling all bacteria present on a Columbia Blood Agar (CBA) plate inoculated with bronchoalveolar lavage (BAL) fluid from a septic patient (ASN087) was spiked into blood and the bacterial DNA recovered using variations of our protocol were analyzed by terminal restriction fragment length polymorphism (TRFLP). Table 2 indicates the organisms represented in the culture pool. TRFLP analysis of the DNA recovered following treatment with no bacterial cell wall digesting enzymes had 7 principal T-RF peaks recovered (Fig. 2). The addition of lysozyme increased the recovery to 10 T-RFs whereas digestion with lysozyme and mutanolysin increased recovery to 24 T-RFs (Fig. 2). Based on this data, the DNA extraction method developed for use with whole blood samples included a cell-wall digestion step with both lysozyme and mutanolysin. Given the limitations of TRFLP in terms of providing a semi-quantitative assessment of highly concentrated culture pools it was only used for the initial method development. Further analysis was done using Illumina sequencing of PCR amplified 16S rDNA.
Table 2.
Organisms recovered from ASN087 BAL fluid cultivated on CBA and their respective T-RF cut size
| 16S Identification (HOMD) | T-RF Size (bp) | CFU/ml |
|---|---|---|
| Capnocytophaga sp | 86 | 102 |
| Prevotella melaninogenica | 99 | 105 |
| Lachnospiraceae [G-1] sp | 108 | 104 |
| Fusobacterium necrophorum | 194 | 105 |
| Bifidobacteriaceae [G-1] sp | 202 | 106 |
| Neisseria flava | 209 | 106 |
| Staphylococcus epidermidis | 234 | 103 |
| Neisseria flavescens | 370 | 104 |
| Neisseria elongata | 370 | 104 |
| Streptococcus mitis | 574 | 103 |
| Capnocytophaga sp | 584 | 104 |
| Streptococcus anginosus | 584 | 104 |
| Streptococcus constellatus | 584 | 105 |
Fig. 2.
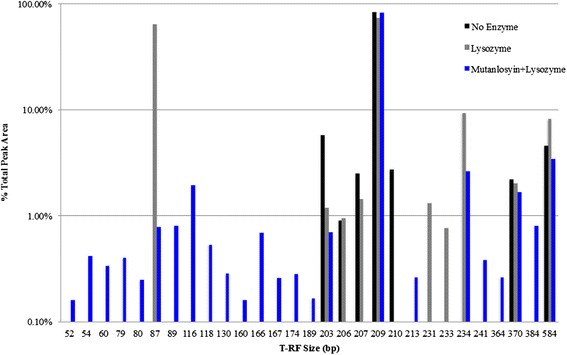
Enzymatic digestion requirement for bacterial DNA extraction of whole blood. Whole blood was spiked with a CBA plate pool from ASN087 BAL sample. Following a treatment with saponin, the resulting pellet was either digested with no enzyme (black), lysozyme (grey), or lysozyme and mutanolysin (blue). More T-RFs were present when enzymatic digestion was done with lysozyme (10, grey) and lysozyme plus mutanolysin (24, blue) identified, respectively, when compared to no enzymatic treatment with only 7 T-RFs present
Saponin treatment of blood spiked with synthetic communities
In order to determine if our method could recover total microbial abundance from a defined community, a mock community of organisms typically recovered from sepsis infections was used. The applicability of the saponin blood-treatment method was assessed in both cultivation-dependent and -independent approaches. The mock community was spiked into blood collected from healthy donors in vacutainers to parallel the collection from actual septic patients enrolled in the study. The addition of saponin to lyse red blood cells and washing to remove debris from the lysis of the human cells on bacterial DNA recovery of the synthetic community (Table 1) was examined at different bacterial concentrations. 16S rRNA community profiling as described was used to assess recovery. The limit of detection and percent recovery is summarized in Table 3. Following the addition of saponin and no washes, the percent recovery of synthetic community organisms was above 80 % for 9 out of the 10 organisms in SC1 and SC3 but only 4 out of 10 for SC5 (Table 3). The treatment with saponin and one wash had highly variable results with percent recovery above 80 % for 8 of the 10 organisms in SC1, 6 out of 10 for SC3 and 1 out 10 for SC5 (Table 3). After two washes, 5 of the 10 organisms could be recovered from all SCs regardless of concentration (Table 3). In order to examine the effects of competition in the recovery of the bacteria, each organism was spiked into whole blood independently. Recovery of each organism was optimal using saponin alone with the notable exception of N. flava (Fig. 3). Anaerobic organisms, F. necrophorum and P. melaninogenica, were recovered under all treatment conditions when cultivated alone and under anaerobic conditions (Fig. 3).
Table 3.
Percent recovery and limit of detection for mock community spiked into whole blood
| Percent recovery (%) | Limit of detection (CFU/ml) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Saponin alone | Saponin + 1 Wash | Saponin + 2 washes | Saponin alone | Saponin +1 wash | Saponin + 2 washes | |||||||
| Organism | SC1 | SC3 | SC5 | SC1 | SC3 | SC5 | SC1 | SC3 | SC5 | |||
| E. coli | 82.8 | 100 | 73.7 | 24.8 | 0 | 0 | 32.3 | 51.7 | 15.5 | 102 | 104 | 103 |
| E. hormaechei | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 10 | 10 | 10 |
| N. flava | 100 | 100 | 23.4 | 100 | 100 | 75.6 | 100 | 100 | 37.8 | 105 | 104 | 104 |
| M. luteus | 100 | 68.7 | 8.0 | 100 | 6.9 | 0 | 20.1 | 0 | 0 | 102 | 10 | 10 |
| S. epidermidis | 100 | 100 | 100 | 100 | 100 | 39.6 | 100 | 100 | 100 | 10 | 10 | 10 |
| S. aureus | 100 | 100 | 39.3 | 100 | 100 | 31.7 | 100 | 100 | 100 | 10 | 10 | 10 |
| S. intermedius | 100 | 100 | 100 | 100 | 100 | 19.9 | 100 | 100 | 100 | 10 | 10 | 10 |
| S. pneumoniae | 100 | 100 | 0 | 96.9 | 100 | 0 | 100 | 100 | 100 | 102 | 102 | 10 |
| F. necrophorum a | 0 | 100 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 102 | 103 | 104 |
| P. melaninogenica a | 100 | 100 | 0 | 100 | 0 | 0 | 0 | 0 | 0 | 102 | 102 | 102 |
aBased on recovery alone (Fig. 3) not in SC
Fig. 3.
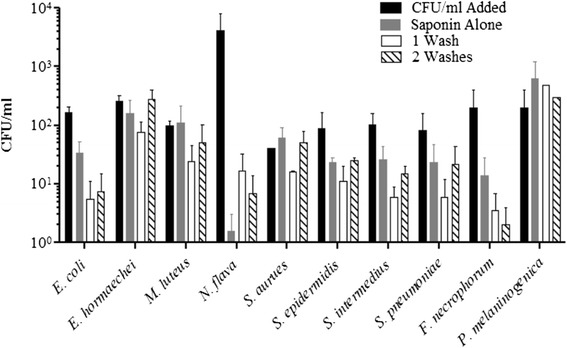
Limit of detection for synthetic communities of bacteria spiked into whole blood. The CFU/ml of each bacterium in the community was determined prior to blood spiking (solid black bars). The CFU/ml of bacteria recovered after each step in the saponin-blood treatment protocol was determined; addition of 0.85 % saponin with no further washes (solid grey bars), addition of saponin at 0.85 % with the addition of a 1 ml sterile double distilled water wash (diagonal lined bars), or addition of saponin at 0.85 % with two washes with 1 ml sterile double distilled water (solid white bars). Each organism was spiked into whole blood alone to verify the limit of detection observed in Table 3 (D)
Bacterial DNA extracted from each community prior to treatment and after all blood spiking conditions was analyzed. Sequence reads that did not map to the bacterial 16S rRNA reference library resulting from primer cross-reactivity to human DNA [26, 27] were removed. A total of 210 OTUs were identified. OTUs were filtered to remove OTUs representing less than 0.1 % total abundance, resulting in 32 OTUs representing 97 % or greater of the total amplified bacterial DNA present. The top 3 OTUs in all samples were those representing the genera Streptococcus, Staphylococcus, and Enterobacter (Fig. 4). The family Enterobacteriaceae OTU was also prevalent in all samples (Fig. 4). Micrococcus DNA could only be identified at the order level, Actinomycetales (Fig. 4). DNA from 9 of the 10 community organisms was recovered from all treatments with Prevotella, Micrococcus and Neisseria OTUs present at scant levels (Table 4). Fusobacterium DNA could not be recovered from SC5 when treated with saponin and with saponin plus two washes (Table 4). Other DNA represented OTUs that did not match DNA from the organisms in the synthetic community. It was present from 0.1 % in the SC1 communities up to 8 % in the SC5 communities (Table 4).
Fig. 4.
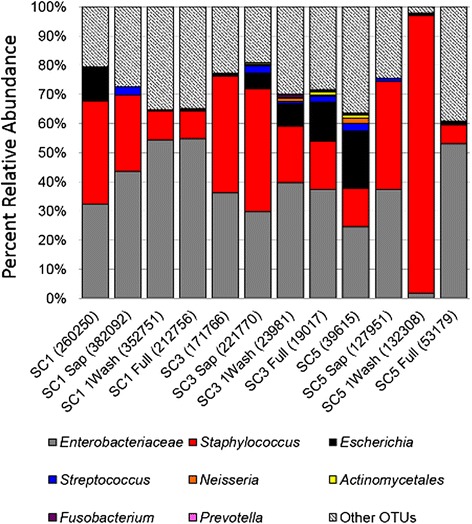
OTU abundance of 16S rRNA Illumina sequenced DNA from synthetic communities. Taxonomic summaries for the synthetic community samples after each step in the saponin blood-treatment protocol were compared. Each bar represents the total PCR amplified DNA sequenced for the sample and the relative abundance of each OTU in the molecular profile. The representative sequence for each OTU was aligned to both the NCBI and HOMD 16S databases in order to determine what synthetic community organism they represented. E. hormaechei sequences were represented by the Enterobacteriaceae OTU, S. aureus was represented by the Staphylococcus OTU, K. pneumoniae by the Klebsiella OTU, E. coli by the Escherichia OTU, S. pneumoniae and S. intermedius by the Streptococcus OTU, N. flava by the Neisseria OTU, M. luteus by the Actinomycetales OTU, F. necrophorum by the Fusobacterium OTU, and P. melaninogenica by the Prevotella OTU. All OTUs with sequence alignments that could not be correlated to the bacteria spiked into the synthetic community were combined into “Other OTUs”, which accounted for 20–40 % of the total OTU abundance
Table 4.
Relative abundance of OTUs recovered from synthetic communities spiked into whole blood
| OTU taxonomic ID | SC1 | SC1 Sap | SC1 1Wash | SC1 Full | SC3 | SC3 Sap | SC3 1Wash | SC3 Full | SC5 | SC5 Sap | SC5 1Wash | SC5 Full |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Enterobacteriaceae (E. hormaechei) | 32.22 % | 43.58 % | 54.37 % | 54.78 % | 36.15 % | 29.76 % | 39.65 % | 37.26 % | 24.60 % | 37.40 % | 1.94 % | 53.01 % |
| Staphylococcus (S. aureus, S. epidermidis) | 35.59 % | 26.11 % | 9.76 % | 9.40 % | 40.03 % | 42.14 % | 19.45 % | 16.69 % | 13.03 % | 36.95 % | 94.93 % | 6.54 % |
| Escherichia (E. coli) | 10.82 % | 0.01 % | 0.33 % | 0.26 % | 0.90 % | 5.36 % | 7.56 % | 13.30 % | 19.67 % | 0.03 % | 0.70 % | 1.00 % |
| Streptococcus (S. pneumoniae, S. intermedius) | 0.30 % | 2.98 % | 0.17 % | 0.24 % | 0.00 % | 2.56 % | 0.80 % | 2.08 % | 2.72 % | 1.27 % | 0.16 % | 0.19 % |
| Neisseria (N. flava) | 0.13 % | 0.00 % | 0.06 % | 0.10 % | 0.00 % | 0.24 % | 1.20 % | 0.36 % | 1.83 % | 0.00 % | 0.03 % | 0.01 % |
| Actinomycetales (M. luteus) | 0.01 % | 0.01 % | 0.02 % | 0.12 % | 0.00 % | 0.55 % | 0.28 % | 1.09 % | 0.95 % | 0.00 % | 0.12 % | 0.07 % |
| Fusobacterium (F. necrophorum) | 0.12 % | 0.00 % | 0.05 % | 0.13 % | 0.00 % | 0.29 % | 0.75 % | 0.40 % | 0.40 % | 0.00 % | 0.02 % | 0.00 % |
| Prevotella (P. melaninogenica) | 0.25 % | 0.00 % | 0.01 % | 0.14 % | 0.00 % | 0.02 % | 0.33 % | 0.44 % | 0.31 % | 0.01 % | 0.01 % | 0.02 % |
| Other OTUs | 20.56 % | 27.32 % | 35.23 % | 34.84 % | 22.90 % | 19.08 % | 29.98 % | 28.39 % | 36.47 % | 24.34 % | 2.09 % | 39.15 % |
DNA profiling of healthy donor blood
In order to parallel the sepsis population cohort, whole blood from 12 healthy adults (age 38–73 years, median 43 years) was subjected to the same DNA extraction and sequencing protocols. The rationale was to determine if there was bacterial DNA in healthy donor blood or determine if there was a source of contamination in the processing of whole blood and DNA extraction. Of these 12 healthy donor blood samples, one failed to amplify in the initial PCR and one had less than 50 sequences amplified and were removed from further analysis. The remaining 10 samples had a minimum sequencing depth of 772 sequences per sample and a maximum of 33,133 sequences per sample with a median of 6174 sequences per sample. A total of 285,067 sequences were identified and aligned to 519 OTUs representing 105 taxonomic groups. In addition, PCR was performed on all the reagents and buffers used in the saponin blood-treatment and the DNA extraction. Of these, only PBS gave a positive signal and was included in the analysis as well as two negative template controls (NTC). The DNA amplified from the 10 healthy controls had similar taxonomic profiles (Fig. 5a). In all samples, the top OTUs were Enterobacteriaceae, Staphylococcus, and Escherichia representing 54.7 % up to 96.3 % of the OTU diversity in each sample. Lower levels of Streptococcus DNA were also present and ranged from 0.32 % to 4.17 % (Fig. 5a). The majority of the remaining OTUs did not represent human-associated bacteria.
Fig. 5.
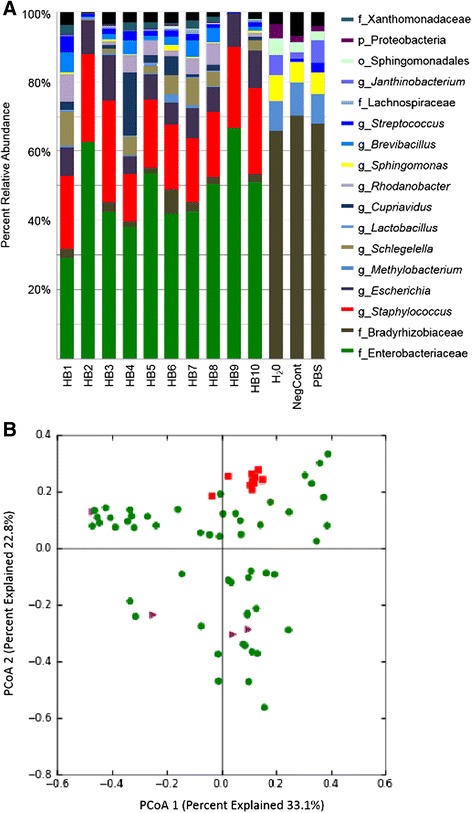
The bacteria DNA profiles of healthy blood. Whole blood was collected from 10 adult donors that worked in a health care setting but were healthy at the time of sampling. Two negative template controls (NTC) and sterile PBS were included for comparison. Taxonomic summaries for the blood samples were compared. Each bar represents the total DNA sequenced for the sample and the percent relative abundance of each OTU identified (a). The bacterial DNA profile profiles of the HB samples were similar to each other but distinct from the NTCs and PBS samples. The letter in front of each taxonomic group indicates the level of taxonomic depth with p__ representing phyla, f __representing family, o__ representing order, and g__ representing genus (a). Principal coordinates analysis (PCoA), based on weighted UniFrac, indicated the healthy blood samples (red) clustered separately from septic blood samples (green) (b)
Since the taxonomic identify of the OTUs in the health samples could also represent potential pathogens in a clinical setting, these taxonomic profiles were compared to a large cohort of whole blood samples collected from ICU patients. The results of the septic patient cohort will be discussed in a separate manuscript. Principal coordinates analysis (PCoA) of weighted unifrac metrics [28–30] was used to visualize the relationships between samples and indicated that the HB samples clustered separately from the sepsis samples (Fig. 5b). Permutational analysis of variance (PERMANOVA) was done to determine if these differences were statistically significant. This test was chosen, as it assumes no distribution and allows for the comparison of categorical factors such as sample type [31]. The PERMANOVA analyses supported the PCoA demonstrating that the healthy blood controls had taxonomic profiles that were significantly different from those seen in septic patients (p = 0.001).
Case studies applying the saponin treatment to whole blood from septic ICU patients
Having established the method, we next set out to apply it to clinical samples collected from adult ICU patients. Three septic patient samples were characterized to evaluate how the bacterial DNA profiles could be interpreted in a clinical context. The first case involved a septic pneumonia patient, ASN165. Chest tube (CT) aspirate fluid was collected on Day 1 and 3 and whole blood was collected on Day 3 of the patient’s ICU stay. In-depth culture of the CT fluid recovered S. pneumoniae at 105 CFU/ml (Fig. 6). Illumina sequencing of the 16S rRNA V3 region resulted in over 155,000 reads for Day 1 and Day 3 CT fluid. The genera Streptococcus represented 99.99 % OTU abundance on Day 1 and Day 3 (Fig. 6, Day 1 not shown). The OTU representative sequence for the most prevalent Streptococcus OTU showed alignments to the S. mitis/pneumoniae group 16S rRNA. Day 3 whole blood was treated with saponin prior to in-depth culture. Partial 16S rRNA sequencing of recovered isolates indicated Streptococcus species, S. vestibularis and Actinomycetes species were present at less than 10 CFU/ml (Fig. 6). Molecular profiling of saponin treated whole blood collected on Day 3 resulted in 431 reads representing 10 OTUs at greater than 1 % of the relative DNA abundance. Diagnostic lab results for the blood culture results were negative on Day 1 and Day 3. Sputum culture indicated growth of S. pneumoniae on Day 1.
Fig. 6.
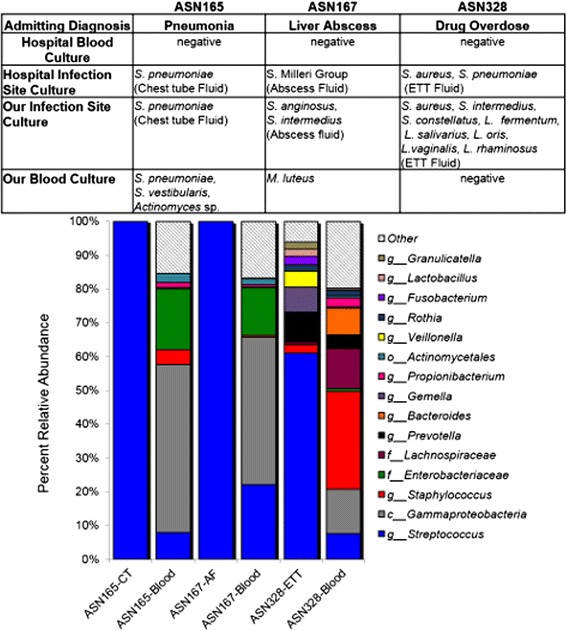
Bacterial DNA profiles of case studies from septic ICU patients. Taxonomic summaries of the bacterial DNA extracted from the primary infection sample and saponin treated whole blood from each case study patient. Each bar represents the combined results from two separate experiments with each PCR sample amplified in triplicate. The size of the bar indicated the percent relative abundance with the taxonomic identification labeled for each major group. The letter in front of each taxonomic group indicates the level of taxonomic depth with p__ representing phyla, f __representing family, o__ representing order, and g__ representing genus. The table indicated the comparison of the clinical diagnostic culture results to our culture results
The second case study, ASN167, was a 37-year-old patient admitted to ICU with a pyogenic liver abscess related sepsis. Abscess drainage fluid and whole blood were obtained. In-depth culture of the abscess fluid indicated two members of the Streptococcus anginosus/milleri group (SMG); S. anginosus and S. intermedius, were present at 105 CFU/ml (Fig. 6). Clinical lab culture of the abscess fluid also indicated heavy SMG growth. Molecular profiling of abscess fluid identified the genera Streptococcus representing 99.99 % of the total OTU abundance (Fig. 6). The OTU representative sequence for the most prevalent Streptococcus OTU showed alignments to the SMG group 16S rRNA. Saponin treated whole blood was cultured and Micrococcus luteus was recovered at less than 10 CFU/ml. Molecular profiling of saponin treated blood from ASN167 was done for Day 1 of ICU admission (Fig. 6). There were 4434 reads that matched to 9 OTUs above 1 % of the relative DNA abundance. Clinical laboratory blood culture results for ASN167 were positive for Streptococcus milleri Group three days prior to ICU admission. Upon ICU admission, blood culture results remained negative after 72 h of cultivation and may reflect rapid response to antibiotic therapy.
The final case study was ASN328, a 26-year-old male patient admitted to ICU with a drug overdose and suspected aspirtation pneumonia. In-depth culture of the ETT fluid indicated the patient had S. aureus at 103 CFU/ml, S. intermedius and S. constellatus at 102 CFU/ml, as well as five species of Lactobacillus (L. fermentum, salivarius, oris, vaginalis, rhamnosus) at 102 CFU/ml. The diagnostic lab report indicated S. aureus present moderately and heavy S. pneumoniae. Molecular profiling of the ET fluid resulted in 10 OTUs at 1 % or higher relative DNA abundance. The most abundant OTU matched to the genera Streptococcus that represented 64.3 % of the relative DNA diversity (Fig. 6). The OTU representative sequence for the most prevalent Streptococcus OTUs showed alignments to the S. salivarius/vestibulrius group (25.5 %), the S. mitis/pneumonia group (23.7 %), and the SMG group (11.9 %). Molecular profiling of whole blood from ASN328 resulted in 3003 reads that fell into 16 OTUs representing 1 % or greater of the relative DNA diversity (Fig. 6). Sequences match to the genera Staphylococcus were the most abundant at 30.7 % (Fig. 6). Blood culture obtained from ASN328 on the same day was negative.
Discussion
Currently, there is no “gold-standard” for DNA extraction from clinical samples. Many molecular studies have chosen DNA extraction methods without proper validation or rationale and used extraction protocols based on commercially available kits [6, 32–39] that have not been carefully evaluated in clinical samples [40]. As such, we developed our own comprehensive DNA extraction protocol for this study.
Our study demonstrated that the addition of the two enzymatic lysis agents, lysozyme and mutanolysin, optimized the recovery of polymicrobial DNA (Fig. 2). These results were comparable to those of Yuan et al., 2012 that also found that a lytic enzyme cocktail of containing lysozyme, mutanolysin and lysostaphin (a Staphylococcal specific pentaglycin cleaving enzyme) consistently lysed cells of different species more effectively than lysozyme alone [40]. The addition of lysostaphin was deemed unnecessary given the results of the mock community demonstrated that Staphylococcus DNA was recovered at high abundance under all conditions (Fig. 3). The DNA extraction process included the addition of Proteinase K and RNase A to eliminate any PCR inhibitors present in the preparations and separate the DNA from organic components [40, 41]. In the final portion of the DNA extraction a column-based purification was employed to remove any remaining contaminants from the DNA preparation.
Our method used saponin at a concentration of 0.85 % mixed directly with 1-2 ml of whole blood. The use of detergents in sepsis diagnostics has been evaluated for blood culture media since the 1990s when it was shown to improve the recovery of fungal organisms, coagulase-negative Staphylococcus and Pseudomonas species while reducing the incidence of false-positive results [42]. Saponin interacts with cellular membrane components including phospholipids and sterols thereby resulting in the lysis of both red and white blood cells [19, 20]. In accordance with previous studies, no significant loss of bacterial growth across a panel of bacteria ranging from highly ubiquitous organisms (E. coli, S. epidermidis), fastidious organisms (S. intermedius, pneumoniae), and anaerobic organisms (P. melaninogenica and F. necrophorum, Fig. 1) was observed [19–22]. TRFLP results of the culture pools spiked into whole blood subsequently treated with saponin prior to DNA extraction suggested that our DNA extraction protocol could recover polymicrobial DNA from saponin-treated whole blood. There were limitations to these results that restricted our ability to assess microbial recovery. Namely, the TRFLP data only provided a semi-quantitative assessment and the culture pools were highly concentrated. As such, mock community of organisms recovered from sepsis infections (Table 1) was used in subsequent experiments assessing the applicability of our method in a cultivation-dependent as well as cultivation-independent approach.
Our results indicated a concentration dependent effect on the recovery of bacteria (Table 3). Gram-negative bacteria E. coli and N. flava had the poorest recovery in the mixed communities with higher inoculum required to recovery these organisms compared to the other bacteria (Table 3). Other culture-independent studies have reported under-representation with Gram-negatives including E. coli for unknown reasons [40]. In order to determine if there was a competitive effect, each organism was spiked individually into blood and the recovery was assessed (Fig. 3). The same phenomenon was observed with 1–2 log drops in E. coli and 3–4 log drops in N. flava CFU following treatment. Our findings suggest the inhibition of growth was likely due to bacteriostatic and bactericidal effects of human blood cells and plasma [43]. The use of sodium polyanethole sulfonate (SPS) in clinical culture media is often used to circumvent the bacteriostatic/bactericidal effects [43]. SPS is also a strong PCR inhibitor [41] and we were unable to use SPS-treated samples for molecular profiling (data not shown). Overall, the recovery of a panel of organisms spiked into whole following the saponin treatment with or without hypotonic washes suggested that saponin treatment could be used to recover bacterial cells directly from whole blood without compromising microbial viability.
Illumina based profiling of the DNA recovered from the mock communities indicated the saponin lysis followed by two washes with DNase/RNase free water resulted in the most representative bacterial DNA profile (Fig. 4). All of the mock community organisms were identified but at varying levels of sequence resolution ranging from M. luteus identified as Actinomycetales, the E. hormaechei identified as Enterobacteriaceae, and the remaining organisms identified at the genera level (Table 4). The composition of the bacterial DNA profiles paralleled the culture-based composition.
In developing a molecular profiling strategy, there were several limitations that needed to be addressed. The first was the primer cross-reactivity with human DNA. Non-specific amplification of human DNA with universal 16S primers has been well documented [26, 27, 44, 45]. However, the abundance of human DNA often represented a large portion of the amplified sequences in whole blood likely reflecting the low ratio of bacterial to host DNA in these samples. The proportion of amplified human DNA increased as the concentration of bacteria decreased. Nevertheless, these sequences were easily removed from the taxonomic profile thereby permitting analysis of the bacterial components of the DNA profiles. The use of other 16S rRNA primer sets was considered but the paired-end Illumina V3 region was selected as it had better taxonomic resolution and longer length when compared to other regions [46, 47].
The bacterial DNA amplified from healthy control samples was not unexpected as our PCR approach was exquisitely sensitive and the universal 16S primers allow for the amplification of any DNA from a bacterial source [48]. Contamination from reagents and the environment (laboratory and hospital) is a common problem in PCR using universal bacterial gene probes. The bacterial DNA in the taxonomic profiles from the control samples were represented by OTUs for Enterobacteriaceae, Escherichia, and Staphylococcus (Fig. 5a). These OTUs were not recovered from the NTS or PBS control samples suggesting PCR reagents were likely not the source of this bacterial DNA (Fig. 5a). The DNA present in these samples was interpreted as a mixture skin and environment contamination as a result of the way in which the blood samples were collected. The samples were peripheral blood draws into vacutainers. Since this was not done in a hospital setting, the same procedures used for blood culture to minimize skin contamination were not employed [49]. Further, studies have indicated that antisepsis at the skin cannot completely prevent contamination since 20 % of the skin bacteria are located in deep layers of the skin or in structures that the surface antiseptics do not penetrate [49]. In comparison, our septic patient blood samples were collected mainly from central lines. Further, the skin was clean using a sterile isopropyl alcohol swab. These swabs are effective at killing bacteria but do not degrade bacterial DNA [50]. As such, DNA from skin microbiota may have been introduced during the venipuncture of the skin. This could account for the abundance of Staphylococcus DNA recovered as the Staphylococcus are considered as part of the skin microbiota [51]. Although the Enterobacteriaceae are not commonly associated with the skin microbiota, a study assessing the hands of health care workers recovered Enterobacter species as well as other Gram-negative bacteria, not taxonomically identified, in addition to Staphylococcus species from individuals in which there was documented skin damage [52]. Overall, the DNA representing human-associated taxonomic groups was interpreted as representative of skin microbiota contaminating the venipuncture. We also compared the ten healthy control samples to a cohort of blood samples collected from 62 ICU patients with sepsis. The taxonomic profiles of the septic patient blood samples clustered separately and were statistically different from all the healthy control samples (Fig. 5b). Attempts to quantitate the bacterial load in the healthy control samples, using Real time-PCR, were unsuccessful due to the cross-reactivity of the 16S primers to human DNA in these samples (data not shown). As such, the total abundance of the bacterial DNA in the healthy control samples was unknown. Nevertheless, knowledge of this type of contamination in the whole blood samples indicated that caution was needed for the interpretation of the Staphylococcus, Enterobacteriaceae, and Escherichia OTUs present in the clinical sample’s molecular profiles. These OTUs were not interpreted as significant unless there was clinical evidence to support the presence of bacterial DNA represented by these OTUs in the patients.
In the mock community samples, the PCR amplified DNA was distributed into 203 OTUs sequenced thereby overestimating the diversity in these mock communities consisting of 8 bacterial genera. The over-estimation of diversity in next-generation sequencing studies has been well documented [53]. In particular, the use of heuristic methods for OTU clustering overestimates the number of groups. With our methods optimized to recover all microbial DNA, there was also a risk of amplifying any environmental DNA present during the blood collection, the DNA extraction procedure, and the PCR set-up. Other studies have also reported recovery of DNA not correlated to the mock community, and found higher rates of contamination in samples with lower bacterial DNA [54]. Our study exhibited the same phenomenon as the mock communities containing the lowest CFU/ml of bacteria resulted in the greatest abundance of OTUs that could not be correlated to the original mock community. Although there was still over-estimation of diversity, the mean proportion of OTUs that could not be taxonomically assigned to the mock community organisms was 26.7 %. These OTUs were considered contaminant DNA that were minor components of the taxonomic profile yet when the template DNA became limiting, their relative abundance was increased. For future clinical diagnostic application it will be important to minimize contamination from reagents.
Despite some limitations in the mock communities, the Illumina molecular profiling of saponin treated blood was successfully applied to three ICU case studies. In the case of ASN165, the molecular profiling data indicated that this patient had S. pneumoniae pneumonia that progressed to a bloodstream infection (Fig. 6). The culturing of saponin treated blood and the molecular profiling method provided supporting evidence for a S. pneumoniae infection with a positive blood culture and a dominant Streptococcus OTU alignment to the S. mitis/pneumonia group. The clinical diagnostic laboratory blood culture was negative for any organism despite our recovery of Streptococcus bacteria and DNA from blood collected the same day as the blood culture. For patient ASN167, the clinical data suggested that the patient no longer had bacteremia, however, the molecular profiling data suggested Streptococcus bacteria or bacterial products were still present in the bloodstream (Fig. 6). This would not be unexpected as abscess formation provides a reservoir of bacteria that can continuously be shed into the bloodstream [9]. The molecular profiling data indicated that the patient still had detectable Streptococcus DNA signal despite a negative clinical diagnostic blood culture. For ASN328 the molecular profiling data and in-depth culture data suggested a more complex infection aetiology when compared to the clinical diagnostic results. The presence of several Lactobacillus species, Prevotella, and Fusobacterium in the ETT fluid from this patient suggested that there was an aspiration event in addition to the Streptococcus and Staphylococcus infection (Fig. 6). Molecular profiling also detected Rothia, Prevotella, Granulicatella, Veillonella, Gemella, and Fusobacterium OTUs that have also been recovered in studies on chronic airway infections [24, 25]. The blood molecular profile for ASN328 shared several OTUs with the ETT fluid (Fig. 5) suggesting that there was possible contribution of these bacteria or their DNA to a bloodstream infection despite the negative blood culture results.
Polymicrobial DNA profiles were identified in all three patients using a non-targeted molecular profiling methods. In addition, some of the OTUs identified in the blood molecular profile were the same genus as bacteria cultivated from either the primary infection sample or the blood sample (Fig. 6). There were varying levels of sequencing depth between samples despite normalizing the DNA concentration used in the PCR reactions. This may reflect varying levels of bacterial DNA template present in each sample. The mock community analysis suggested that as the level of bacteremia decreased the proportion of amplified human DNA indicating that the issue was associated with the ratio of bacterial to human DNA. As such, samples with lower number of reads reflected a higher ratio of human to bacterial DNA. Since this was a ratio-based issue, the use of larger blood volumes was not predicted to circumvent these limitations. Nevertheless, the removal of these DNA sequences from the taxonomic profile enabled the analysis of the remaining, low proportion, bacterial DNA in the case study samples. Based on this preliminary data, we concluded that the use of a saponin-mediated blood cells lysis combined with a robust DNA extraction method could be used to enable the successful application of an Illumina molecular profiling approach to identify bacterial infection in the blood and primary infection samples of sepsis patients.
Conclusions
The paired-end Illumina 16S rRNA community profiling of bacteria has been successfully applied to human clinical samples to provide a more robust evaluation of polymicrobial infections [47, 55–57]. To our knowledge, this was the first study in which paired-end Illumina 16S rRNA gene community profiling was done on whole blood. The results of this study indicate that a saponin blood pre-treatment lysis steps combined with the paired-end Illumina sequencing enabled molecular-profiling of small volumes of whole blood. Blood culture based assessments indicate that the incidence of polymicrobial sepsis is low ranging from 10–20 % [17, 58]. However, the limited results from these case studies suggest that a molecular approach may enable improved detection of polymicrobial infections. The application of sensitive molecular methods to clinical samples can identify more organisms in samples when compared to clinical diagnostics, which is selective for specific organisms. It is important to consider that DNA based methods indicate the presence of DNA rather than viable organisms and positive results should be interpreted accordingly. Whether or not they represent viable organisms in the blood, a positive signal indicates the presence of bacterial products in the blood, which would contribute to systemic inflammation. Future work will involve application of this protocol to subsets of adult and paediatric sepsis patients to further verify the utility of this method.
Methods
Isolates and culture conditions
Isolates were obtained from clinical specimens as outlined in Table 1. Isolates were maintained at -80C in 10 % skim milk for long term storage. Samples were grown on solid media including Columbia blood agar (CBA), Colombia CNA agar (CNA), mannitol salt agar (MSA), and trypticase soy agar supplemented with 3 % yeast extract (TSY), all from BD Diagnostics, Canada as well as, fastidious anaerobic agar (FAA, Neogen Acumedia, USA). Difibrinated sheep’s blood (Dalynn Biologics, Canada) was added to a 5 % final volume for CBA, CNA, and FAA. Isolates were cultivated at 37 °C with 5 % CO2 or in a Ruskinn Concept 400 anaerobic chamber (Ruskinn, Bridgend, UK) with 80 % N, 10 % CO2 10 % H2.
Sample collection and processing
Approval for this study was obtained from the Conjoint Health Research Ethics Board of the University of Calgary. All human samples were collected following the guidelines outlined in the Canadian of Health Research, Natural Sciences and Engineering Research Council of Canada, and Social Sciences and Humanities Research Council of Canada, Tri-Council Policy Statement: Ethical Conduct for Research Involving Humans, December 2010. Written informed consent was obtained for all patients, or their proxy decision maker, enrolled in the study. Patients of 18 years of age or older admitted to the ICU of the Foothills Medical Center meeting the published criteria for systemic inflammatory response and clinical suspicion of sepsis within the first 24 h of ICU admission were eligible for the study [1, 59, 60]. Clinical and laboratory data was collected daily during the ICU stay of the patient and mortality was tracked until hospital discharge. Control individuals were adults above 30 years of age who worked in a health care setting but at the time collected had no major autoimmune disorders, no symptoms of illness, were not on anti-inflammatories for the prior 7 days, and were otherwise healthy (no colds/fever/chills/respiratory symptoms) in the prior 7 days with a normal respiratory rate and temperature measured as supportive data. Collection was done as part of the Critical Care Epidemiologic and Biologic Tissue Resource (CCEPTR). Approval for CCEPTR was granted by the Conjoint Health Research Ethics Board of the University of Calgary with the Ethics ID for the study E-22236 on April 7, 2009. Whole blood was collected from an existing arterial line, central line or venous line with a maximum volume of 4 ml in sterile K2EDTA spray coated vacutainers (BD Diagnostics). Biologic samples included bronchoalveolar lavage (BAL) fluid, endotracheal tube (ETT) fluid and abscess drainage fluid (AF) collected in sterile containers.
Biological fluid samples were first sheared to an even consistency using a 1 ml tuberculin slip-tip syringe. Ten-fold serial dilutions were prepared as needed. Samples were plated using 100 μl of sample per solid media type and incubated as indicated above.
Bacterial viability with Saponin
To evaluate whether saponin impacted bacterial viability, in vitro experiments were carried out in which bacterial strains recovered from sepsis infections (Table 1) were co-incubated with 0.85 % saponin at concentrations ranging from 101 to 109 CFU/ml for 60 min. Whole blood was treated with 0.85 % saponin (Sigma-Aldrich, USA) final volume at room temperature for 15 min to lyse red blood cells. Following treatment, the blood was centrifuged at 20,800 rcf for 15 min to remove lysed cells in the supernatant. The supernatant was removed and the remaining cells were washed 1-3x with 1 ml sterile DNase/RNase free double distilled water (Life Technologies, Burlington, ON, Burlington, ON). This involved re-suspending the pellet in the sterile water and vortexing to ensure homogeneity prior to centrifugation. After each wash the cells were spun at 20,800 rcf for 15 min and the supernatant was removed. Cells were suspended in 500 μl sterile PBS for storage prior to DNA extraction or in 500 μl BHI broth for cultivation.
DNA extraction from whole blood
To each 500 μl sample of saponin treated blood, 50 μl of Lysozyme (100 mg/ml, Sigma-Aldrich, Oakville, ON), 20 μl of Mutanolysin (10U/μl, Sigma-Aldrich, Oakville, ON), and 20 μl of RNase A (10 mg/ml, Life Technologies, Burlington, ON) were added and incubated overnight at 37 °C. Following this, 50 μl of 25 % sodium dodecyl sulphate (SDS, Sigma-Aldrich, Oakville, ON), 50 μl of 20 mg/ml proteinase K (Invitrogen, Life Technologies, Burlington, ON), and 100 μl 5 M NaCl were added. The mixture was incubated at 65 °C for 1–2 h. Cellular debris was pelleted by centrifugation at 20,800 rcf for 10 min. The supernatant was then treated with one standard phenol-chloroform-isoamyl (25:24:1, Life Technologies, Burlington, ON) alcohol extraction. The DNA in the aqueous layer was transferred to a Zymo DNA Clean & Concentrator™-25 (Zymo Research, Irvine, CA) column containing 200 μl of ChIP DNA Binding Buffer (Zymo Research, Irvine, CA). The column was spun for 1 min at 20,800 rcf and the flow-through was discarded. Wash buffer was added at 500 μl twice to the column with a 1 min, 20,800 rcf centrifugation and discard of flow-through in between each wash. A final 1 min, 20,800 rcf centrifugation step was done to ensure the column was completely dry and free of any ethanol carry-over from the wash buffer. Pre-warmed DNase/RNase free deionized and UV irradiated water was used to elute the DNA with 50 μl added per column. DNA was quantified using a Nanodrop 2000c Spectrophotometer.
Optimization of DNA extraction and purification
In order to validate the DNA extraction, whole blood was collected in K2EDTA vacutainers and spiked with a mixed bacterial community recovered on CBA from a septic pneumonia BAL fluid sample from an ICU patient (Table 2). This plate pool represented bacteria recovered at or greater than 103 CFU/ml. All bacteria recovered on the CBA plate were resuspended in 1 ml of sterile 10 % skim milk and stored at −80 °C. Aliquots of 500 μl of blood were spiked with 50 μl of the culture pool and subjected to a 0.85 % saponin treatment. DNA was extracted from the resulting blood pellet in one of three ways; with no enzymatic lysis, lysozyme alone, and lysozyme plus mutanolysin. The RNAase and proteinase K treatments were the same for all three samples. The supernatant was then treated with one standard phenol-chloroform-isoamyl (25:24:1, Life Technologies, Burlington, ON) alcohol extraction. The DNA in the aqueous layer was transferred to a Zymo DNA Clean & Concentrator™-25 (Zymo Research, Irvine, CA) column containing 200 μl of ChIP DNA Binding Buffer (Zymo Research, Irvine, CA). The column was spun for 1 min at 20,800 rcf and the flow-through was discarded. Wash buffer was added at 500 μl twice to the column with a 1 min, 20,800 rcf centrifugation and discard of flow-through in between each wash. A final 1 min, 20,800 rcf centrifugation step was done to ensure the column was completely dry and free of any ethanol carry-over from the wash buffer. Pre-warmed DNase/RNase free deionized and UV irradiated water was used to elute the DNA with 50 μl added per column. DNA was quantified using a Nanodrop 2000c Spectrophotometer.
Partial 16S rDNA sequencing
Grown colonies of bacteria were distinguished using colony morphology and identification was done on 10 % chelex (Bio-Rad, USA) boiled colony preps [61] using PCR with the universal 16S rDNA primers 8 F (5'AGAGTTTGATCCTGGCTCAG3') and 926R (5'CCGTCAATTCCTTTRAGTTT3'). PCR cycle conditions were initial denaturation at 94 °C for 1 min, 32 cycles of denaturation at 94 °C for 1 min, annealing for 1 min at 57 °C and extension at 72 °C for 1 min followed with a final extension at 72 °C for 10 min. PCR products were sequenced unidirectionally from the 8 F primer at Beckman Coulter Genomics (Danvers, USA) and the sequences were aligned to curated ribosomal sequence databases including HOMD (human oral microbiome database, www.homd.org), and Greengenes (greengenes.lbl.gov/cgi-bin/nph-index.cgi).
Terminal restriction fragment length polymorphism
Molecular analysis was done using terminal restriction fragment length polymorphism (TRFLP) as outlined by Sibley et al. [25]. Briefly, a portion of the 16S rRNA gene was PCR amplified using 8 F (5'AGAGTTTGATCCTGGCTCAG3') and 926r (5'CCGTCAATTCCTTTRAGTTT3'). The 8 F primer was fluorescently tagged with 6-FAM at the 5′ end. PCR conditions were the same as those outlined above. PCR products were concentrated and purified using Zymo DNA Clean and Concentrator™ columns (Zymo Research) then digested overnight with CfoI (10U, Sigma-Alrdich). Fragment analysis was done at the UCDNA core facility (University of Calgary, Canada). Analysis was done using GeneMapper 4.0 (Applied Biosystems, Life Technologies) and the percent of total peak area for each fragment size was calculated.
16S rRNA gene bacterial community profiling with paired-end Illumina
PCR and sequencing was done following Bartram et al. (2011) with some modifications. PCR amplification of the V3 region was done using the primers 341 F (5′CCTACGGGAGGCAGCAG3′) and 518R (5′ATTACCGCGGCTGCTGG3′). Primer modifications included the addition of Illumina multiplexing, bridge amplification and sequencing regions [47]. Reverse primers were barcoded to allow multiple processing of samples. For whole blood samples, 200–300 ng of DNA was PCR amplified whereas 50-100 ng of DNA was used for primary infection site samples. The PCR cycle consisted of an initial denaturation at 94 °C for 2 min followed by 30 cycles of denaturation at 94 °C for 30 s, annealing at 50 °C for 30 s, extension at 72 °C for 30 s followed by a final extension at 72 °C for 10 min. Each sample was amplified in triplicate and the pooled PCR reactions were run on a 2 % agarose gel. PCR products were excised from the gel and purified using the QIAquick gel extraction kit (Qiagen, Netherlands) following manufacturer guidelines. The resulting PCR products were sequenced using the Illumina MiSeq personal sequencer (Illumina Incorporated, USA) at the McMaster Genomics Facility, Ontario, Canada. After sequencing, image analysis, base calling, and error estimation were completed using the Illumina Analysis Pipeline (version 2.6) [47]. The sequencing data was processed with custom, in-house Perl scripts [62]. Initial sequence processing was carried out with Cutadapt [63] and paired-end sequences aligned and consensus generated using PANDAseq [64]. Operational taxonomic units (OTUs) clustering at a threshold of 97 % sequence similarity was carried out using AbundantOTU+ [65]. The taxonomic identification was assigned using the Ribosomal Database Project classifier [66] using the Greengenes reference database, February 4th 2011 release [67] as a training set. QIIME computational analysis pipeline was used for community analysis (alpha and beta diversity) [68]. β-diversity was used as a measure to examine differences between samples. Both weighted and un-weighted UniFrac distance and clustering of the samples was done and visualised using principal coordinate analysis (PCoA) [28, 30]. PCoA plots were visualized using KiNG version 2.21 visualization software [69].
Synthetic community experiments
Ten bacterial strains recovered from sepsis cases (bloodstream or organ site infections (Table 1) were used to generate a mixed community. Colonies from each sample were suspended in sterile M9 salts + 0.1 % cysteine to a McFarland 2 unit. Viability in saponin was assessed by incubating 10−1 through to 10−8 dilutions of each isolate with 0.85 % saponin for 1 h at room temperature. Colony forming units (CFU) were determined prior to and after treatment as an average of duplicate plates. Communities of 100 μl of each strain diluted at a 10−1, 10−3, or 10−5 dilution were mixed together resulting in the three communities SC1, SC3, and SC5 respectively. Human whole blood was collected in K2EDTA vacutainers and 20 μl of each synthetic community was added to 500 μl of whole blood. Saponin was added at a 0.85 % final volume and incubated for 30 min at room temperature. Three treatments with saponin were performed; addition of 0.85 % saponin with no further washes, addition of saponin at 0.85 % with the addition of a 1 ml sterile double distilled water wash, or addition of saponin at 0.85 % with two washes with 1 ml sterile double distilled water. All recovered cells were resuspended in BHI and plated in duplicate on MSA, CNA, MAC, TSY, and FAA using 100 μl per plate prior to and after addition to blood. In addition, 10 μl of each strain alone was diluted and spiked into 250 μl of 0.85 % saponin treated whole blood, incubated for 30 min at room temperature and plated on their respective media type (Table 1). DNA was also extracted from all communities for TRFLP and 16S rRNA gene community profiling. These experiments were done in triplicate with three different samples of human blood done on different days.
Acknowledgements
Many members of the Alberta Sepsis Network facilitated the sample collection and the clinical data collection for the case studies portion of the study. Included are the leaders of the ASN project, Drs. Chip Doig, Paul Kubes, and Ari Joffe, the manager and co-ordinator of the CCEPTR resource, Dr. Brent Winston and Joseé Wong for the sample collection, Dean Yergens for the collection of clinical data for this study and the research nurses and assistants who identified and enrolled patients with the ICU and ED at Foothills Medical Centre and the Alberta Children’s Hospital including Janice Hammond, Linda Knox, Christine Skinner, Dori-Ann Martin, Claudia Maki, Stacey Ruddell, and Dan Lane. The Alberta Heritage Foundation for Medical Research (now Alberta Innovates Health Solutions) funded Alberta Sepsis Network, a five-year team grant awarded to Drs. Paul Kubes, Ari Joffe and Christopher Doig, provided funding for this project.
Abbreviations
- AF
Abscess fluid
- BAL
Bronchoalveolar lavage
- BHI
Brain-heart infusion
- BHIco
Brain-heart infusion with colistin and oxolinic acid
- BLAST
Basic local alignment search tool
- CBA
Colombia blood agar
- CCEPTR
Critical care epidemiologic and biologic tissue bank resource
- CNA
Colombia CNA agar
- CT
Chest tube fluid
- ETT
Endotracheal tube fluid
- FAA
Fastidious anaerobe agar
- HOMD
Human oral microbiome database
- MAC
MacConkey agar
- MSA
Mannitol-salt agar
- OTU
Operational taxonomic unit
- QIIME
Quantitative insights into microbial ecology
- SC
Synthetic community
- SMG
Streptococcus milleri group
- T-RF
Terminal restriction fragment
- TRFLP
Terminal restriction fragment length polymorphism
- TSY
Tryptic soy yeast
Footnotes
Competing interests
None of the authors had competing interests to declare at the time of submission.
Authors’ contributions
MFC carried out all of the experiments presented in this manuscript. MFC also drafted the manuscript. MGS conceived of the study and performed the molecular sequencing analysis. JMC contributed to the coordination of the study and helped draft the manuscript. All authors read and approved of the final manuscript.
Contributor Information
MMP Faria, Phone: 905-525-9140, Email: mmfaria@ucalgary.ca.
JM Conly, Email: john.conly@albertahealthservices.ca.
MG Surette, Email: surette@mcmaster.ca.
References
- 1.Levy MM, Fink MP, Marshall JC, Abraham E, Angus D, Cook D, et al. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit Care Med. 2003;31(4):1250–6. doi: 10.1097/01.CCM.0000050454.01978.3B. [DOI] [PubMed] [Google Scholar]
- 2.Husak L, Marcuzzi A, Herring J, Wen E, Yin L, Capan DD, et al. National analysis of sepsis hospitalizations and factors contributing to sepsis in-hospital mortality in Canada. Healthc Q. 2010;13(Spec No):35–41. doi: 10.12927/hcq.2010.21963. [DOI] [PubMed] [Google Scholar]
- 3.Brun-Buisson C, Doyon F, Carlet J. Bacteremia and severe sepsis in adults: a multicenter prospective survey in ICUs and wards of 24 hospitals. French Bacteremia-Sepsis Study Group. Am J Respir Crit Care Med. 1996;154(3 Pt 1):617–24. doi: 10.1164/ajrccm.154.3.8810595. [DOI] [PubMed] [Google Scholar]
- 4.Pittet D, Thievent B, Wenzel RP, Li N, Auckenthaler R, Suter PM. Bedside prediction of mortality from bacteremic sepsis. A dynamic analysis of ICU patients. Am J Respir Crit Care Med. 1996;153(2):684–93. doi: 10.1164/ajrccm.153.2.8564118. [DOI] [PubMed] [Google Scholar]
- 5.Valles J. [Bacteremias in intensive care] Enferm Infecc Microbiol Clin. 1997;15(Suppl 3):8–13. [PubMed] [Google Scholar]
- 6.Kaleta EJ, Clark AE, Johnson DR, Gamage DC, Wysocki VH, Cherkaoui A, et al. Use of PCR coupled with electrospray ionization mass spectrometry for rapid identification of bacterial and yeast bloodstream pathogens from blood culture bottles. J Clin Microbiol. 2011;49(1):345–53. doi: 10.1128/JCM.00936-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Afshari A, Schrenzel J, Ieven M, Harbarth S. Bench-to-bedside review: rapid molecular diagnostics for bloodstream infection - a new frontier? Crit Care. 2012;16(3):222. doi: 10.1186/cc11202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rivers E, Nguyen B, Havstad S, Ressler J, Muzzin A, Knoblich B, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med. 2001;345(19):1368–77. doi: 10.1056/NEJMoa010307. [DOI] [PubMed] [Google Scholar]
- 9.Mancini N, Carletti S, Ghidoli N, Cichero P, Burioni R, Clementi M. The era of molecular and other non-culture-based methods in diagnosis of sepsis. Clin Microbiol Rev. 2010;23(1):235–51. doi: 10.1128/CMR.00043-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fenollar F, Raoult D. Molecular diagnosis of bloodstream infections caused by non-cultivable bacteria. Int J Antimicrob Agents. 2007;30(Suppl 1):S7–15. doi: 10.1016/j.ijantimicag.2007.06.024. [DOI] [PubMed] [Google Scholar]
- 11.Tuttle MS, Mostow E, Mukherjee P, Hu FZ, Melton-Kreft R, Ehrlich GD, et al. Characterization of bacterial communities in venous insufficiency wounds by use of conventional culture and molecular diagnostic methods. J Clin Microbiol. 2011;49(11):3812–9. doi: 10.1128/JCM.00847-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haag H, Locher F, Nolte O. Molecular diagnosis of microbial aetiologies using SepsiTest (TM) in the daily routine of a diagnostic laboratory. Diagn Microbiol Infect Dis. 2013;76(4):413–8. doi: 10.1016/j.diagmicrobio.2013.04.027. [DOI] [PubMed] [Google Scholar]
- 13.Avolio M, Diamante P, Modolo ML, De Rosa R, Stano P, Camporese A. Direct molecular detection of pathogens in blood as specific rule-in diagnostic biomarker in patients with presumed sepsis - our experience on a heterogeneous cohort of patients with signs of infective SIRS. Shock. 2014;42:86–92. doi: 10.1097/SHK.0000000000000191. [DOI] [PubMed] [Google Scholar]
- 14.Burdino E, Ruggiero T, Allice T, Milia MG, Gregori G, Milano R, et al. Combination of conventional blood cultures and the SeptiFast molecular test in patients with suspected sepsis for the identification of bloodstream pathogens. Diagn Microbiol Infect Dis. 2014;79(3):287–92. doi: 10.1016/j.diagmicrobio.2014.03.018. [DOI] [PubMed] [Google Scholar]
- 15.Liesenfeld O, Lehman L, Hunfeld KP, Kost G. Molecular diagnosis of sepsis: new aspects and recent developments. Eur J Microbiol Immunol. 2014;4(1):1–25. doi: 10.1556/EuJMI.4.2014.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang L, Gowardman J, Morrison M, Krause L, Playford EG, Rickard CM. Molecular investigation of bacterial communities on intravascular catheters: no longer just Staphylococcus. Eur J Clin Microbiol Infect Dis. 2014;33(7):1189–98. doi: 10.1007/s10096-014-2058-2. [DOI] [PubMed] [Google Scholar]
- 17.Ngo JT, Parkins MD, Gregson DB, Pitout JD, Ross T, Church DL, et al. Population-based assessment of the incidence, risk factors, and outcomes of anaerobic bloodstream infections. Infection. 2013;41(1):41–8. doi: 10.1007/s15010-012-0389-4. [DOI] [PubMed] [Google Scholar]
- 18.Abu al-Soud W, Radstrom P. Purification and characterization of PCR-inhibitory components in blood cells. J Clin Microbiol. 2001;39(2):485–93. doi: 10.1128/JCM.39.2.485-493.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lupetti A, Barnini S, Castagna B, Capria AL, Nibbering PH. Rapid identification and antimicrobial susceptibility profiling of Gram-positive cocci in blood cultures with the Vitek 2 system. Eur J Clin Microbiol Infect Dis. 2010;29(1):89–95. doi: 10.1007/s10096-009-0825-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Doorne H, van der Tuuk Adriani WP, van de Ven LI, Bosch EH, de Natris T, Smit Sibinga CT. Saponin, an inhibitory agent of carbon dioxide production by white cells: its use in the microbiologic examination of blood components in an automated bacterial culture system. Transfusion. 1998;38(11–12):1090–6. doi: 10.1046/j.1537-2995.1998.38111299056321.x. [DOI] [PubMed] [Google Scholar]
- 21.Meex C, Neuville F, Descy J, Huynen P, Hayette MP, De Mol P, et al. Direct identification of bacteria from BacT/ALERT anaerobic positive blood cultures by MALDI-TOF MS: MALDI Sepsityper kit versus an in-house saponin method for bacterial extraction. J Med Microbiol. 2012;61(Pt 11):1511–6. doi: 10.1099/jmm.0.044750-0. [DOI] [PubMed] [Google Scholar]
- 22.Lupetti A, Barnini S, Morici P, Ghelardi E, Nibbering PH, Campa M. Saponin promotes rapid identification and antimicrobial susceptibility profiling of Gram-positive and Gram-negative bacteria in blood cultures with the Vitek 2 system. Eur J Clin Microbiol Infect Dis. 2013;32(4):493–502. doi: 10.1007/s10096-012-1762-z. [DOI] [PubMed] [Google Scholar]
- 23.Sibley CD, Church DL, Surette MG, Dowd SE, Parkins MD. Pyrosequencing reveals the complex polymicrobial nature of invasive pyogenic infections: microbial constituents of empyema, liver abscess, and intracerebral abscess. Eur J Clin Microbiol Infect Dis. 2012;31(10):2679–91. doi: 10.1007/s10096-012-1614-x. [DOI] [PubMed] [Google Scholar]
- 24.Sibley CD, Grinwis ME, Field TR, Eshaghurshan CS, Faria MM, Dowd SE, et al. Culture enriched molecular profiling of the cystic fibrosis airway microbiome. PLoS One. 2011;6(7):e22702. doi: 10.1371/journal.pone.0022702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sibley CD, Parkins MD, Rabin HR, Duan K, Norgaard JC, Surette MG. A polymicrobial perspective of pulmonary infections exposes an enigmatic pathogen in cystic fibrosis patients. Proc Natl Acad Sci U S A. 2008;105(39):15070–5. doi: 10.1073/pnas.0804326105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Vlaminck I, Khush KK, Strehl C, Kohli B, Luikart H, Neff NF, et al. Temporal response of the human virome to immunosuppression and antiviral therapy. Cell. 2013;155(5):1178–87. doi: 10.1016/j.cell.2013.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kommedal O, Simmon K, Karaca D, Langeland N, Wiker HG. Dual priming oligonucleotides for broad-range amplification of the bacterial 16S rRNA gene directly from human clinical specimens. J Clin Microbiol. 2012;50(4):1289–94. doi: 10.1128/JCM.06269-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lozupone C, Hamady M, Knight R. UniFrac--an online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinformatics. 2006;7:371. doi: 10.1186/1471-2105-7-371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hamady M, Knight R. Microbial community profiling for human microbiome projects: tools, techniques, and challenges. Genome Res. 2009;19(7):1141–52. doi: 10.1101/gr.085464.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lozupone C, Lladser ME, Knights D, Stombaugh J, Knight R. UniFrac: an effective distance metric for microbial community comparison. ISME J. 2011;5(2):169–72. doi: 10.1038/ismej.2010.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Navas-Molina JA, Peralta-Sanchez JM, Gonzalez A, McMurdie PJ, Vazquez-Baeza Y, Xu ZJ, et al. Advancing our understanding of the human microbiome using QIIME. Method Enzymol. 2013;531:371–444. doi: 10.1016/B978-0-12-407863-5.00019-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schabereiter-Gurtner C, Nehr M, Apfalter P, Makristathis A, Rotter ML, Hirschl AM. Evaluation of a protocol for molecular broad-range diagnosis of culture-negative bacterial infections in clinical routine diagnosis. J Appl Microbiol. 2008;104(4):1228–37. doi: 10.1111/j.1365-2672.2007.03648.x. [DOI] [PubMed] [Google Scholar]
- 33.Reier-Nilsen T, Farstad T, Nakstad B, Lauvrak V, Steinbakk M. Comparison of broad range 16S rDNA PCR and conventional blood culture for diagnosis of sepsis in the newborn: a case control study. BMC Pediatr. 2009;9:5. doi: 10.1186/1471-2431-9-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wiesinger-Mayr H, Jordana-Lluch E, Martro E, Schoenthaler S, Noehammer C. Establishment of a semi-automated pathogen DNA isolation from whole blood and comparison with commercially available kits. J Microbiol Methods. 2011;85(3):206–13. doi: 10.1016/j.mimet.2011.03.003. [DOI] [PubMed] [Google Scholar]
- 35.Vondracek M, Sartipy U, Aufwerber E, Julander I, Lindblom D, Westling K. 16S rDNA sequencing of valve tissue improves microbiological diagnosis in surgically treated patients with infective endocarditis. J Infect. 2011;62(6):472–8. doi: 10.1016/j.jinf.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 36.Wallet F, Nseir S, Baumann L, Herwegh S, Sendid B, Boulo M, et al. Preliminary clinical study using a multiplex real-time PCR test for the detection of bacterial and fungal DNA directly in blood. Clin Microbiol Infect. 2010;16(6):774–9. doi: 10.1111/j.1469-0691.2009.02940.x. [DOI] [PubMed] [Google Scholar]
- 37.Westh H, Lisby G, Breysse F, Boddinghaus B, Chomarat M, Gant V, et al. Multiplex real-time PCR and blood culture for identification of bloodstream pathogens in patients with suspected sepsis. Clin Microbiol Infect. 2009;15(6):544–51. doi: 10.1111/j.1469-0691.2009.02736.x. [DOI] [PubMed] [Google Scholar]
- 38.Tsalik EL, Jones D, Nicholson B, Waring L, Liesenfeld O, Park LP, et al. Multiplex PCR to diagnose bloodstream infections in patients admitted from the emergency department with sepsis. J Clin Microbiol. 2010;48(1):26–33. doi: 10.1128/JCM.01447-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Palka-Santini M, Cleven BE, Eichinger L, Kronke M, Krut O. Large scale multiplex PCR improves pathogen detection by DNA microarrays. BMC Microbiol. 2009;9:1. doi: 10.1186/1471-2180-9-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yuan S, Cohen DB, Ravel J, Abdo Z, Forney LJ. Evaluation of methods for the extraction and purification of DNA from the human microbiome. PLoS One. 2012;7(3):e33865. doi: 10.1371/journal.pone.0033865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fredricks DN, Relman DA. Improved amplification of microbial DNA from blood cultures by removal of the PCR inhibitor sodium polyanetholesulfonate. J Clin Microbiol. 1998;36(10):2810–6. doi: 10.1128/jcm.36.10.2810-2816.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Murray PR, Spizzo AW, Niles AC. Clinical comparison of the recoveries of bloodstream pathogens in Septi-Chek brain heart infusion broth with saponin, Septi-Chek tryptic soy broth, and the isolator lysis-centrifugation system. J Clin Microbiol. 1991;29(5):901–5. doi: 10.1128/jcm.29.5.901-905.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Palarasah Y, Skjoedt MO, Vitved L, Andersen TE, Skjoedt K, Koch C. Sodium polyanethole sulfonate as an inhibitor of activation of complement function in blood culture systems. J Clin Microbiol. 2010;48(3):908–14. doi: 10.1128/JCM.01985-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Edwards U, Rogall T, Blocker H, Emde M, Bottger EC. Isolation and direct complete nucleotide determination of entire genes. Characterization of a gene coding for 16S ribosomal RNA. Nucleic Acids Res. 1989;17(19):7843–53. doi: 10.1093/nar/17.19.7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bosshard PP, Zbinden R, Altwegg M. Turicibacter sanguinis gen. nov., sp. nov., a novel anaerobic, Gram-positive bacterium. Int J Syst Evol Microbiol. 2002;52(Pt 4):1263–6. doi: 10.1099/00207713-52-4-1263. [DOI] [PubMed] [Google Scholar]
- 46.Huse SM, Dethlefsen L, Huber JA, Mark Welch D, Relman DA, Sogin ML. Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing. PLoS Genet. 2008;4(11):e1000255. doi: 10.1371/journal.pgen.1000255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bartram AK, Lynch MD, Stearns JC, Moreno-Hagelsieb G, Neufeld JD. Generation of multimillion-sequence 16S rRNA gene libraries from complex microbial communities by assembling paired-end illumina reads. Appl Environ Microbiol. 2011;77(11):3846–52. doi: 10.1128/AEM.02772-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Corless CE, Guiver M, Borrow R, Edwards-Jones V, Kaczmarski EB, Fox AJ. Contamination and sensitivity issues with a real-time universal 16S rRNA PCR. J Clin Microbiol. 2000;38(5):1747–52. doi: 10.1128/jcm.38.5.1747-1752.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Calfee DP, Farr BM. Comparison of four antiseptic preparations for skin in the prevention of contamination of percutaneously drawn blood cultures: a randomized trial. J Clin Microbiol. 2002;40(5):1660–5. doi: 10.1128/JCM.40.5.1660-1665.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McDonnell G, Russell AD. Antiseptics and disinfectants: activity, action, and resistance. Clin Microbiol Rev. 1999;12(1):147–79. doi: 10.1128/cmr.12.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rosenthal M, Goldberg D, Aiello A, Larson E, Foxman B. Skin microbiota: microbial community structure and its potential association with health and disease. Infect Genet Evol. 2011;11(5):839–48. doi: 10.1016/j.meegid.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rocha LA, Borges LFDE, Gontijo PP. Changes in hands microbiota associated with skin damage because of hand hygiene procedures on the health care workers. Am J Infect Control. 2009;37(2):155–9. doi: 10.1016/j.ajic.2008.04.251. [DOI] [PubMed] [Google Scholar]
- 53.Sun DL, Jiang X, Wu QL, Zhou NY. Intragenomic heterogeneity of 16S rRNA genes causes overestimation of prokaryotic diversity. Appl Environ Microbiol. 2013;79(19):5962–9. doi: 10.1128/AEM.01282-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Willner D, Daly J, Whiley D, Grimwood K, Wainwright CE, Hugenholtz P. Comparison of DNA extraction methods for microbial community profiling with an application to pediatric bronchoalveolar lavage samples. PLoS One. 2012;7(4):e34605. doi: 10.1371/journal.pone.0034605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maughan H, Wang PW, Diaz Caballero J, Fung P, Gong Y, Donaldson SL, et al. Analysis of the cystic fibrosis lung microbiota via serial Illumina sequencing of bacterial 16S rRNA hypervariable regions. PLoS One. 2012;7(10):e45791. doi: 10.1371/journal.pone.0045791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012;6(8):1621–4. doi: 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stearns JC, Davidson CJ, McKeon S, Whelan FJ, Fontes ME, Schryvers AB, et al. Culture and molecular-based profiles show shifts in bacterial communities of the upper respiratory tract that occur with age. ISME J. 2015;9(5):1268. doi: 10.1038/ismej.2015.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Laupland KB, Gregson DB, Zygun DA, Doig CJ, Mortis G, Church DL. Severe bloodstream infections: a population-based assessment. Crit Care Med. 2004;32(4):992–7. doi: 10.1097/01.CCM.0000119424.31648.1E. [DOI] [PubMed] [Google Scholar]
- 59.Dellinger RP, Levy MM, Carlet JM, Bion J, Parker MM, Jaeschke R, et al. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock: 2008. Crit Care Med. 2008;36(1):296–327. doi: 10.1097/01.CCM.0000298158.12101.41. [DOI] [PubMed] [Google Scholar]
- 60.Dellinger RP, Levy MM, Rhodes A, Annane D, Gerlach H, Opal SM, et al. Surviving sepsis campaign: international guidelines for management of severe sepsis and septic shock: 2012. Crit Care Med. 2013;41(2):580–637. doi: 10.1097/CCM.0b013e31827e83af. [DOI] [PubMed] [Google Scholar]
- 61.Grinwis ME, Sibley CD, Parkins MD, Eshaghurshan CS, Rabin HR, Surette MG. Characterization of Streptococcus milleri group isolates from expectorated sputum of adult patients with cystic fibrosis. J Clin Microbiol. 2010;48(2):395–401. doi: 10.1128/JCM.01807-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Whelan FJ, Verschoor CP, Stearns JC, Rossi L, Luinstra K, Loeb M, et al. The loss of topography in the microbial communities of the upper respiratory tract in the elderly. Ann Am Thorac Soc. 2014;11(4):513–21. doi: 10.1513/AnnalsATS.201310-351OC. [DOI] [PubMed] [Google Scholar]
- 63.Martin M. Cutadapt removes adapter sequences from high-throughput seqeuncing reads. EMBnet J. 2011;17:10. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 64.Masella AP, Bartram AK, Truszkowski JM, Brown DG, Neufeld JD. PANDAseq: paired-end assembler for illumina sequences. BMC Bioinformatics. 2012;13:31. doi: 10.1186/1471-2105-13-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ye Y. Identification and quantification of abundant species from pyrosequences of 16S rRNA by consensus alignment. 2010 IEEE International Conference on Bioinformatics and Medicine 2010, BIBM:153–157. [DOI] [PMC free article] [PubMed]
- 66.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73(16):5261–7. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72(7):5069–72. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335–6. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen VB, Davis IW, Richardson DC. KING (Kinemage, Next Generation): a versatile interactive molecular and scientific visualization program. Protein Sci. 2009;18(11):2403–9. doi: 10.1002/pro.250. [DOI] [PMC free article] [PubMed] [Google Scholar]