

Abstract
Intraneuronal accumulation and extracellular deposition of amyloid beta (Aβ) protein continues to be implicated in the pathogenesis of Alzheimer’s disease (AD), be it familial in origin or sporadic in nature. Aβ is generated intracellularly following endocytosis of amyloid beta precursor protein (AβPP) and consequently factors that suppress AβPP internalization may decrease amyloidogenic processing of AβPP. Here we tested the hypothesis that caffeine decreases Aβ generation by suppressing AβPP internalization in primary cultured neurons. Caffeine concentration-dependently blocked LDL cholesterol internalization and a specific adenosine A3 receptor (A3R) antagonist as well as siRNA knockdown of A3Rs mimicked the effects of caffeine on neuronal internalization of LDL cholesterol. Further implicating A3Rs were findings that a specific A3R agonist increased neuronal internalization of LDL cholesterol. In addition, caffeine as well as siRNA knockdown of A3Rs blocked the ability of LDL cholesterol to increase Aβ levels. Furthermore, caffeine blocked LDL cholesterol-induced decreases in AβPP protein levels in neuronal plasma membranes, increased surface expression of AβPP on neurons, and the A3R antagonist as well as siRNA knockdown of A3Rs mimicked the effects of caffeine on AβPP surface expression. Moreover, the A3R agonist decreased neuronal surface expression of AβPP. Our findings suggest that caffeine exerts protective effects against amyloidogenic processing of AβPP at least in part by suppressing A3R-mediated internalization of AβPP.
Keywords: Caffeine, Adenosine A3 receptor, LDL cholesterol, Alzheimer’s disease, Amyloid-β precursor protein, Amyloid-β, Endocytosis
Introduction
Alzheimer’s disease (AD), the most common neurodegenerative disorder of old age, is characterized clinically by a progressive decline in cognitive function, and pathologically by loss of synaptic integrity and neurons, amyloid plaques composed of amyloid beta (Aβ) protein, and neuronal tangles composed of hyperphosphorylated tau [1, 2]. Brain deposition of Aβ, a proteolytic cleavage product of amyloid beta precursor protein (AβPP) by the beta-site APP cleavage enzyme 1 (BACE1) and γ-secretase, continues to be considered an important pathogenic factor of AD [1, 3]. Emerging evidence indicates that AβPP trafficking plays an important role in determining the extent to which AβPP is processed amyloidogenically [4, 5]. Internalized (trafficked) AβPP accumulates in endolysosomes wherein the acidic environment increases the activities of BACE-1 and γ-secretase and stimulates the amyloidogenic processing of AβPP [6–9]. Thus, factors that promote AβPP internalization and/or disturb endolysosome function may increase amyloidogenic processing of AβPP thus leading to increased AD pathogenesis. Alternatively, factors that prevent AβPP internalization may decrease amyloidogenic processing of AβPP and thus might decrease AD pathogenesis.
Elevated levels of plasma LDL cholesterol, independent of APOE genotypes, is a robust extrinsic factor that increases the risk of developing sporadic AD [10–14]. It has been shown that apoB, the exclusive apolipoprotein of LDL, co-localizes with cerebral Aβ in AD brain and in a transgenic mouse AD model, and that apoB levels are positively correlated with Aβ plaque abundance [15–17]. Others and we have shown that LDL receptors are highly expressed on neurons, that LDL receptors interact physically with AβPP, that LDL cholesterol affects AβPP trafficking [18–20], that LDL cholesterol is internalized via receptor-mediated endocytosis, and that this internalization process promotes AβPP internalization [4, 5, 20]. Mechanistically, we have shown that LDL cholesterol treatment promotes AβPP internalization and enhances amyloidogenesis [12]. Thus, LDL cholesterol endocytosis could promote AβPP internalization into neuronal endolysosomes and enhance amyloidogenesis.
Caffeine, the most commonly ingested psychoactive drug in the world, might be protective against AD pathogenesis [21–27]. Epidemiologically, caffeine ingestion has been correlated reciprocally with the prevalence and severity of AD [28–32]. In animal models, caffeine has been shown to prevent AD-like features as well as reverse the features once formed [33–37]. The mechanisms implicated in the protective actions of caffeine include blockage of adenosine A2A receptors [23, 37], activation of PKA signaling [34, 38], and decreased Aβ production through suppression of both beta- and gamma-secretases [34, 38]. Importantly, human, animal and in vitro studies all clearly show that these protective actions of caffeine occur at therapeutic concentrations easily obtainable through normal ingestion of food-based products.
The present studies were aimed to determine the extent to which and mechanisms whereby caffeine affects AβPP internalization and Aβ generation as induced by LDL cholesterol. In primary cultured neurons, we have described a novel mechanism whereby caffeine protects against Aβ generation. Specifically, we have demonstrated that caffeine suppresses LDL cholesterol-induced amyloidogenic processing of AβPP by blocking AβPP internalization via its actions on A3Rs.
Material and Methods
Primary cultures of rat cerebral cortical neurons
Primary cerebral cortical neurons were cultured from embryonic day 18 rats using a protocol approved by the University of North Dakota Animal Care and Use Committee adherent with the Guide for the Care and Use of Laboratory Animals (NIH publication number 80–23) [12].
Cultures of human neuroblastoma cells
Human neuroblastoma cells (SH-SY5Y) expressing wild type AβPP were kindly supplied by Dr. Norman Haughey (John Hopkins University). Cells were cultured in Eagle’s minimum essential medium (MEM) supplemented with 10% FCS, penicillin/streptomycin, nonessential amino acids, and sodium pyruvate (1 mM) at 37°C in 5% CO2/95% air. For the experiments, 4 × 106 cells were seeded on 60 mm2 dishes and cultured for 48 h. The cells were exposed to serum-free MEM for 24 h, then experimental treatments were performed in serum-free MEM.
LDL cholesterol internalization assay
Quantitative analysis of LDL cholesterol internalization in neurons was performed using a method as described previously, but with minor modifications [39]. Cells plated on glass-bottom 35-mm2 tissue culture dishes were pretreated with various concentrations of drugs for 24 hours prior to addition of 1 μg/ml DiI-labeled LDL cholesterol (Kalein Biomedical) for 30 min at 37°C. Cells were washed with an acid wash solution (0.2 M acetic acid, 0.5 M NaCl, pH 2.8) at 4°C for 10 min and then washed with ice-cold PBS for 5 min to remove surface-bound LDL cholesterol. Cells were fixed in 4% paraformaldehyde and images were taken with a confocal laser-scanning microscope (Olympus). All experiments were performed in triplicate. The average integrated intensity of DiI-LDL cholesterol signal per cell was calculated for each well using ImageJ software.
RNA interference
A3R expression levels were knocked down with specific siRNAs at a final concentration of 60 nM (Invitrogen); negative siRNAs (Invitrogen) were used as controls. Before siRNA transfection, fresh Neurobasal media was added to cultured neurons plated for 10 days. The transfection cocktail containing 300 μl of transfection buffer (SignaGen), 12 μl of siRNA stock (15 μM) for each target protein, and 9 μl of GenMute™ reagent was added carefully to each dish along with 1 ml of media. After incubation (37°C, 5% CO2) for 5 h, the transfection media was replaced with fresh Neurobasal media, and neurons were treated with LDL cholesterol for 3 days. Knockdown efficiency was measured by immunoblotting as described below.
Immunoblotting
Total cell lysates and plasma membrane fractions were prepared using a Plasma Membrane Protein Extraction kit (Bio-Rad). Protein concentrations were determined with a DC protein assay (Bio-Rad). Equal amounts of proteins (50 μg) were separated by SDS-PAGE (12% gel) and, following transfer, polyvinylidene difluoride membranes were incubated overnight at 4°C with antibodies against N-terminal AβPP (Milipore) and A3R (Alomone Lab); β-actin (Abcam) was used as a gel loading control. Blots were developed with enhanced chemiluminescence, and bands were visualized and analyzed by LabWorks 4.5 software on a UVP Bioimaging System (Upland). Quantification of results was performed by densitometry and the results were analyzed as total integrated densitometric volume values (arbitrary units).
Surface immunostaining
Neurons were fixed with 4% paraformaldehyde for 10 min, washed with PBS, blocked with 5% goat serum, and incubated overnight at 4°C with a primary antibody against N-terminal AβPP (Milipore). After washing with PBS, neurons were incubated with fluorescence-conjugated secondary antibody (Invitrogen). Neurons were examined by confocal microscopy (Olympus). The average integrated signal intensity per cell was calculated (ImageJ software). Controls for immunostaining specificity included staining neurons with primary antibodies without fluorescence-conjugated secondary antibodies (background controls), and staining neurons with only secondary antibodies.
Immunostaining for AβPP and endosomes
Neurons were fixed with 4% paraformaldehyde for 10 min followed by cold methanol (−20°C) for 10 min. The cells were then washed with PBS, blocked with 5% goat serum, and incubated overnight at 4°C with primary antibodies targeting early endosome antigen-1 (EEA1, 1:500, rabbit polyclonal, Santa Cruz), and N-termianl AβPP (1:500, Milipore). After washing with PBS, neurons were incubated with corresponding fluorescence-conjugated secondary antibodies including Alexa 488-conjugated goat anti-mouse antibodies (Invitrogen) and Alexa 546-conjugated goat anti-rabbit antibodies (Invitrogen). Neurons were examined by confocal microscopy (Olympus). Controls for immunostaining specificity included staining neurons with primary antibodies without fluorescence-conjugated secondary antibodies (background controls), and staining neurons with only secondary antibodies; these controls helped eliminate auto-fluorescence in each channel and bleed-through (crossover) between channels.
Quantification of Aβ levels
Aβ levels were quantified using human/rat Aβ1–40 and Aβ1–42 ELISA kits as per the manufacturer’s protocol (Wako). Media from cultured neurons were collected, diluted 1:4 with standard diluent buffer, and each sample was analyzed in duplicate. Protein levels from neurons in each dish were determined by a DC protein assay (Bio-Rad). Aβ levels were normalized to total protein content in each sample.
Quantitative RT-PCR measurement of AβPP mRNA
Total RNA was extracted with TRIzol-Reagent (Invitrogen) and levels were determined spectrophotometrically. Reverse transcription reactions were carried out using a SuperScript® III First-Strand Synthesis supermix (Invitrogen). The primers for BACE-1 and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were as follows: f: 5′-CGGACAGCATCGATTCTGCG -3′ and r: 5′- CTCTCTCGGTGCTTGGCTTC -3′ for AβPP; f: 5′-TGCACCACCAACTGCTTAG-3′ and r: 5′-GGATGCAGGGATGATGTTC-3′ for GAPDH. Samples were run with our iCycler IQ™ Multicolor Real-Time PCR Detection System (Bio-Rad) that monitors fluorescence as a direct indication of PCR product [40]. All samples were run in triplicate and the averaged values were used for the relative quantification of gene expression. AβPP mRNA expression levels were calculated as the ratio of their expression compared with that of GAPDH.
Measurement of neuronal cell injury
Neuronal cell injury was quantitatively assessed by the measurement of lactate dehydrogenase (LDH), released from damage or destroyed cells, in the extracellular fluid after completion of the experiment (Sigma). An aliquot of bathing media was combined with NADH and pyruvate solutions. LDH activity is proportional to the rate of pyruvate loss, which was assayed by absorbance change using a microplate reader (Molecular Device). Data were expressed as percentages of the control samples.
Statistical analysis
All data were expressed as means and SEM. Statistical significance between two groups was analyzed with a Student’s t-test, and statistical significance among multiple groups was analyzed with one-way ANOVA plus a Tukey post-hoc test. P < 0.05 was considered to be statistically significant.
Results
Caffeine prevented LDL cholesterol internalization
We first determined the extent to which caffeine affected LDL cholesterol internalization using primary cultured neurons. DiI-labeled LDL cholesterol was rapidly taken up by neurons and the internalization reached maximal levels by 2 hrs (data not shown). When neurons were pretreated with caffeine (0–200 μM) for 24 hrs prior to adding DiI-labeled LDL cholesterol for 30 min, internalization of DiI-labeled LDL cholesterol accumulation was decreased in a concentration-dependent and statistically significant (P < 0.001) manner (Figure 1A).
Figure 1. Caffeine blocked LDL cholesterol internalization.
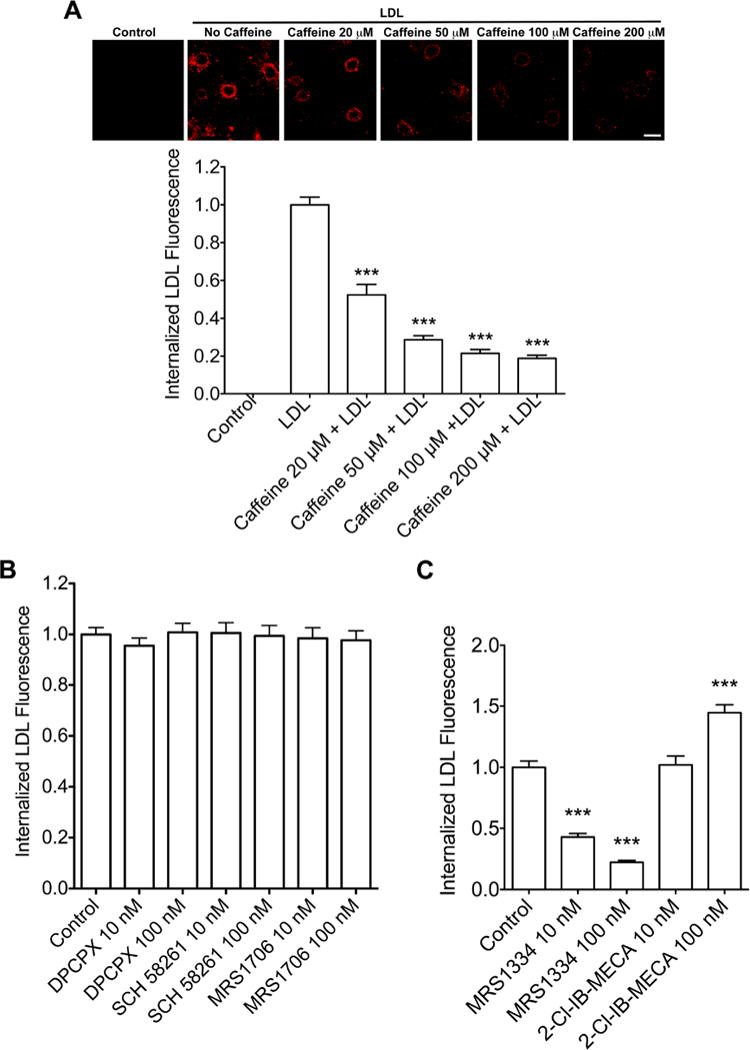
(A) In a receptor-mediated endocytosis assay, DiI-LDL-cholesterol was rapidly (30 min) internalized by neurons. Caffeine pretreatment for 24 h blocked LDL-cholesterol internalization in a concentration-dependent manner (n = 45, ***P < 0.001). Bar = 10 μm. (B) Neuronal internalization of DiI-LDL cholesterol was not affected by blocking (pretreatment for 24 h) adenosine A1R with DPCPX, A2AR with SCH 58261, or A2BR with MRS1706 (n = 41, P > 0.05). (C) Neuronal internalization of DiI-LDL cholesterol was attenuated significantly by blocking (pretreatment for 24 h) adenosine A3R with MRS1334 (n = 52, ***P < 0.001). Activation (pretreatment for 24 h) of adenosine A3R with 2-Cl-IB-MECA enhanced significantly neuronal internalization of DiI-LDL cholesterol (n = 49, ***P < 0.001).
Pharmacologically, caffeine concentrations in the μM range can block all four subtypes of adenosine receptors (A1R, A2AR, A2BR and A3R) [41]. Thus, we determined next which subtype(s) of adenosine receptors was(were) involved in neuronal internalization of LDL cholesterol. Using adenosine receptor subtype specific antagonists at two concentrations each (10 and 100 nM), we tested the ability of DPCPX (for A1Rs), SCH58261 (for A2ARs), MRS1706 (for A2BRs), and MRS1334 (for A3Rs) to block LDL cholesterol internalization. We found that the specific A3R antagonist MRS1334 at 10 and 100 nM concentrations decreased significantly (P < 0.001) neuronal internalization of DiI-LDL cholesterol (Figure 1C). In contrast, none of the other adenosine receptor antagonists tested, at either concentration, produced statistically significant changes in Dil-LDL cholesterol internalization (Figure 1B). We then tested the effects of the specific A3R agonist 2-Cl-IB-MECA on Dil-LDL cholesterol internalization, and found that pretreatment of neurons with the A3R agonist at 100 nM, but not at 10 nM, increased significantly (P < 0.001) DiI-LDL cholesterol internalization (Figure 1C).
To confirm our pharmacological findings, we knocked-down protein expression levels of A3Rs using an RNA interference approach (Figure 2A) and found that siRNA knockdown of A3Rs decreased significantly (P < 0.001) neuronal internalization of DiI-LDL cholesterol (Figure 2B). Collectively, our findings suggest strongly that the actions of caffeine on LDL cholesterol internalization were mediated through A3R-mediated actions.
Figure 2. A3R knockdown attenuated LDL cholesterol internalization.
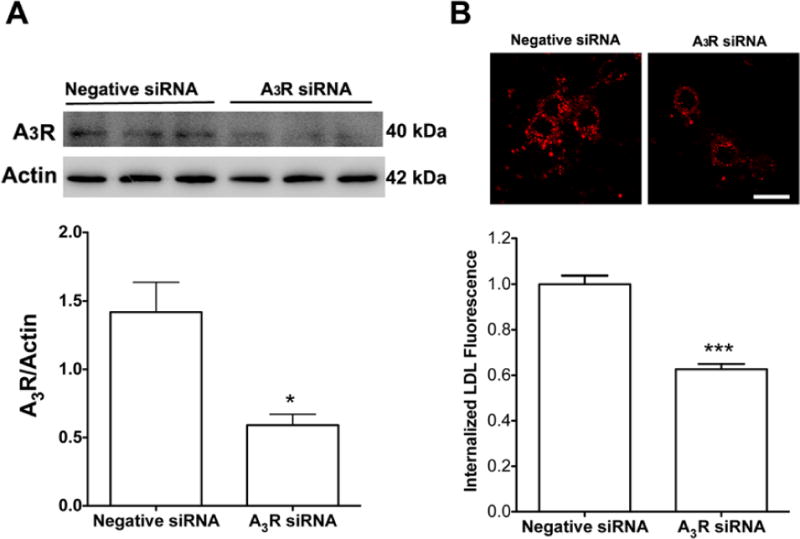
(A) Immunoblotting data showed that a specific A3R siRNA decreased significantly A3R expression (n = 6, *P < 0.05). (B) A3R knockdown decreased significantly DiI-LDL cholesterol internalization (n = 82, ***P < 0.001). Bar=10 μm.
Caffeine suppressed LDL cholesterol-induced Aβ generation
Next we determined the extent to which caffeine affected Aβ generation as induced by LDL cholesterol, an extrinsic factor that promotes AβPP internalization and enhances amyloidogenesis [12]. For these studies we chose to use 200 μM of caffeine because at this concentration we observed maximal effectiveness in blocking LDL cholesterol internalization. However, before conducting these studies, we first demonstrated that caffeine treatment (200 μM up to 4 days) did not induce neurotoxicity as indicated by a LDH releasing assay (99.9 ± 3.6 in control vs. 101.7 ± 3.5 in caffeine treatment, n=6); a finding that is consistent with that reported by others [42, 43]. We found that caffeine pretreatment (200 μM for 24 h) blocked significantly LDL cholesterol (50 μg/ml for 3 days)-induced increases in levels of Aβ1–40 and Aβ1–42 (Figure 3A). We have replicated this experiment in AβPP over-expressing SH-SY5Y cells, and the findings were essentially the same (Figure 3B). Furthermore, we demonstrated that caffeine treatment alone decreased significantly basal levels of Aβ1–40 and Aβ1–42 in AβPP over-expressing SH-SY5Y cells (Figure 3B). Because of our findings on the involvement of A3Rs in regulating LDL cholesterol internalization, next we determined the extent to which siRNA knockdown of A3Rs affected LDL cholesterol-induced increases in Aβ levels. We found that A3R knockdown decreased significantly LDL cholesterol-induced increases in Aβ1–40 but not Aβ1–42 (Figure 3C).
Figure 3. Caffeine blocked LDL cholesterol-induced elevated levels of Aβ.
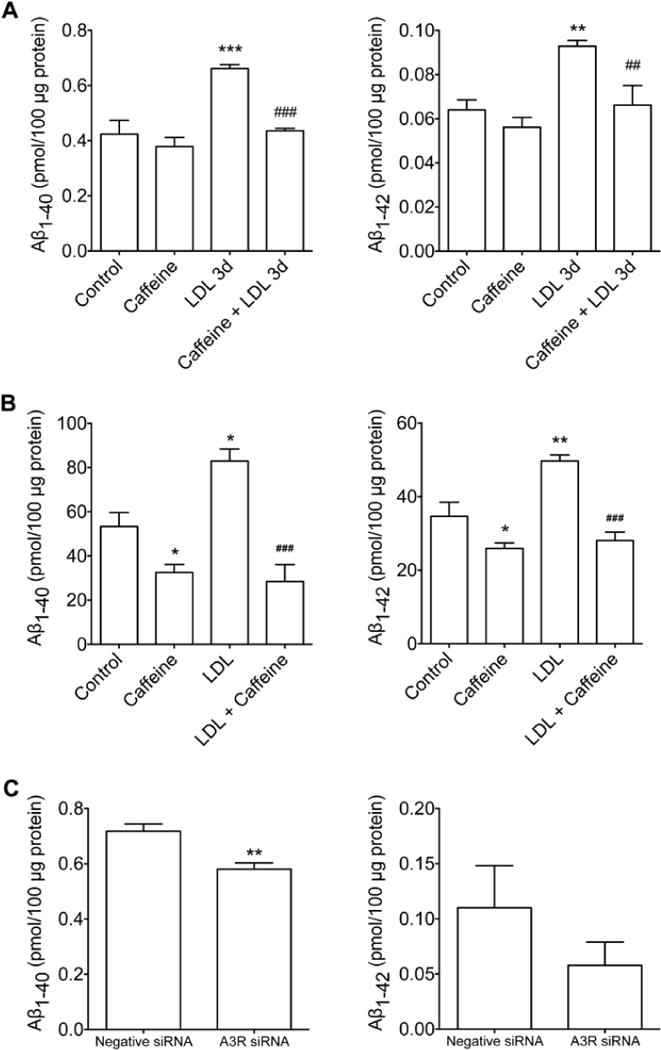
(A) In primary cultured neurons, pretreatment with caffeine (200 μM for 24 h) blocked increased levels of Aβ1–40 and Aβ1–42 as induced by LDL cholesterol treatment (50 μg/ml) for 3 days (n = 4; **P < 0.01 vs. Control; ***P < 0.001 vs. Control; #P < 0.05 vs. LDL 3d). (B) In AβPP over-expressing SH-SY5Y cells, pretreatment with caffeine decreased significantly basal levels of Aβ1–40 and Aβ1–42 and blocked increased levels of Aβ1–40 and Aβ1–42 as induced by LDL cholesterol treatment (50 μg/ml) for 2 days (n = 6; *P<0.05 vs. Control; **P<0.01 vs. Control; ###P < 0.001 vs. LDL). (C) In primary cultured neurons, siRNA knockdown of A3R decreased LDL cholesterol (50 μg/ml, for 3 days) induced increased levels of Aβ1–40 and Aβ1–42 (n = 4; **P < 0.01). n = 4; **P < 0.01).
Caffeine suppressed AβPP internalization
Given the above results, we determined next the extent to which caffeine affected AβPP internalization and plasma membrane expression levels of AβPP. We found that LDL cholesterol treatment (50 μg/ml for 30 min) did not affect total expression levels of AβPP protein, but LDL cholesterol decreased significantly protein levels of AβPP in plasma membrane fractions; these decreased levels were attenuated significantly by caffeine (200 μM for 24 hrs) pretreatment (Figure 4A). For this experiment, plasma membrane protein Na+/K+ ATPase was used as a control and we found that neither LDL nor caffeine affected significantly protein levels of Na+/K+ ATPase in plasma membrane fractions or in total lysates.
Figure 4. Caffeine suppressed AβPP internalization.
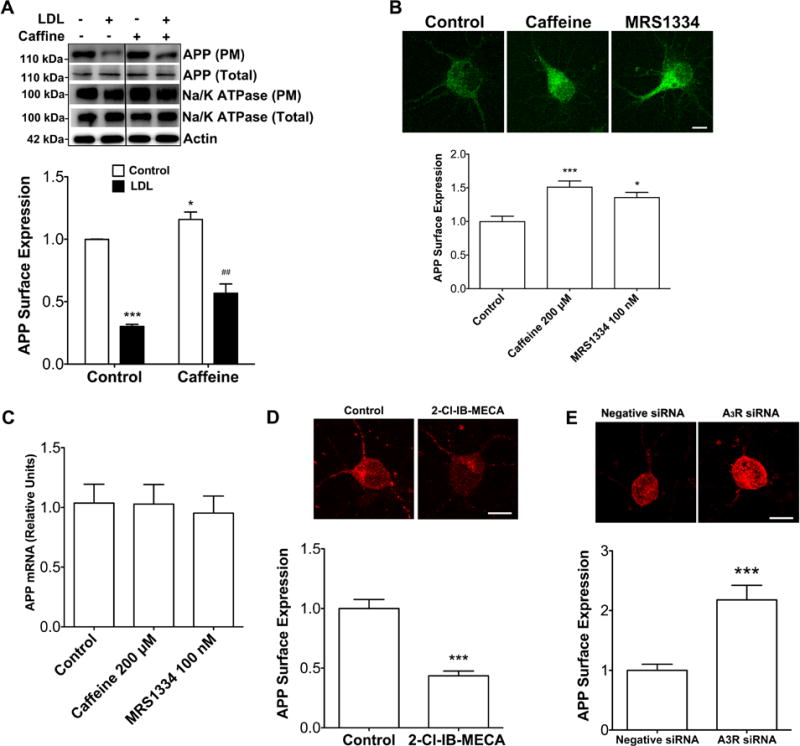
(A) LDL cholesterol (50 μg/ml) treatment for 30 min did not affect AβPP protein levels in total cell lysates, but decreased significantly AβPP protein levels in plasma membrane fractions, and such effects were attenuated significantly by caffeine (200 μM) treatment for 24 h (n = 4, ***P < 0.001 vs. Control, ##P < 0.01 vs. LDL). Neither LDL nor caffeine affected significantly protein levels of Na/K ATPase in plasma membrane fractions or in total lysates. In addition, caffeine treatment alone increased significantly AβPP protein levels in plasma membrane fractions (n = 4, *P < 0.05 vs. Control). (B) Caffeine (200 μM) treatment for 24 h increased significantly surface expression levels of AβPP (n = 23, ***P < 0.001). Similar to caffeine, blocking adenosine A3Rs with MRS1334 (100 nM for 24 h) increased significantly surface expression levels of AβPP (n = 20, *P < 0.05). Bar=10 μm. (C) Neither caffeine nor A3R antagonist treatment affected AβPP mRNA levels. (D) Activation of A3Rs with 2-Cl-IB-MECA (100 nM for 24 h) decreased significantly surface expression levels of AβPP (n = 23, ***P < 0.001). Bar = 10 μm. (E) A3R knockdown increased significantly surface expression levels of AβPP (n = 21, ***P < 0.001). Bar = 10 μm
Furthermore, we found that caffeine (200 μM for 24 hrs) pretreatment alone increased significantly AβPP levels in plasma membrane fractions (Figure 4A). To confirm these latter findings, we determined the extent to which caffeine affected AβPP trafficking using a surface immunostaining approach. Consistent with our immunoblotting findings, caffeine (200 μM for 24 hrs) pretreatment (Figure 4B) and the specific A3R antagonist MRS1334 (100 nM for 24 hrs) (Figure 4B) increased significantly surface expression levels of AβPP. To exclude the possibility that such increased AβPP expression may be due to modifications at the transcriptional level, we quantified AβPP mRNA and we demonstrated that neither caffeine nor A3R antagonist affected AβPP mRNA levels (Figure 4C). In contrast to the A3R antagonist, the specific A3R agonist 2-Cl-IB-MECA (100 nM for 24 hrs) decreased significantly surface expression levels of AβPP (Figure 4D). To confirm our pharmacological findings, we knocked down protein expression levels of A3Rs using an RNA interference approach and found that A3R knockdown increased significantly surface expression levels of AβPP (Figure 4E).
To further determine whether caffeine and/or the A3R antagonist decreased AβPP internalization into endosomes, we performed double staining for AβPP and early endosome antigen 1 (EEA1). We demonstrated that LDL treatment (50 μg/ml for 3 days) enlarged endosomes and increased the co-localization of AβPP with endosomes, and that these effects were attenuated by either pretreatment with caffeine (200 μM for 24 hrs) or pretreatment with the specific A3R antagonist MRS1334 (100 nM for 24 hrs). Together, our findings suggest that A3Rs are involved in caffeine-induced suppression of AβPP internalization.
Discussion
Findings from a large number of epidemiological and experimental studies indicate that caffeine, the world’s most used psychoactive drug, is protective against behavioral and pathological features of AD [21–23]. However, efforts are ongoing to determine the mechanisms underlying caffeine’s protective effects against AD. Here, we focused our studies to determine the extent to which and mechanisms by which caffeine exerts its protective effects against LDL cholesterol-induced amyloidogenesis by suppressing AβPP internalization with a focus on the role of specific subtypes of adenosine receptors.
The pathogenesis of sporadic AD, the major form of AD, is believed to result from complex interactions between nutritional, environmental, epigenetic and genetic factors [3]. Among those factors that contribute to the development of sporadic AD, elevated levels of circulating LDL cholesterol, independent of APOE genotypes, have been robustly linked to enhanced amyloidogenic processing of AβPP [10–13]. Although apoB, the exclusive apolipoprotein of LDL, is not normally found in brain [44], it has been shown that apoB is present in AD brain [15], co-localizes with cerebral Aβ in AD brain and in a transgenic mouse AD model [15–17], and that apoB level is positively correlated with Aβ plaque abundance. Thus, a compromised blood brain barrier (BBB), an early pathological feature of sporadic AD that precedes brain deposition of Aβ [45], may allow peripheral apoB-containing LDL cholesterol to enter into brain parenchyma and contribute to the pathogenesis of AD.
We have shown that elevated levels of LDL cholesterol, the essential lipoprotein transporting circulating cholesterol in the blood, (1) induces BBB leakage and increases brain levels of apoB [11, 46], (2) disturbs neuronal endolysosome structure and function – another early pathological features of sporadic AD [47], and (3) promotes the development of pathological hallmarks of AD including disrupted synaptic integrity, brain deposition of Aβ, and tau pathology [11]. Furthermore, we demonstrated that LDL cholesterol treatment promoted AβPP internalization, enhanced BACE-1 activity, and increased amyloidogenic processing of AβPP in endolysosomes of primary cultured neurons [12]. Collectively, our findings suggest that elevated levels of LDL cholesterol, when it enters brain parenchyma via a leaky BBB, are internalized by neurons via receptor-mediated endocytosis. Because some LDLRs including LRP1 and LRP10 have been shown to interact with AβPP and affect AβPP trafficking [18–20], LDL cholesterol endocytosis could promote AβPP internalization into neuronal endolysosomes thus enhancing amyloidogenesis. Such a notion is consistent with the concept that amyloidogenic processing of AβPP occurs predominantly within endolysosomes, where the acidic environment is optimum for activities of BACE-1 and γ-secretase [4, 5].
Substantial evidence from human epidemiological studies and from experimental studies conducted in animals and cultured cell models indicate that caffeine decreases Aβ levels and protects against the onset and severity of AD [22, 24–26, 28, 30, 31, 33, 34, 38]. Furthermore, some studies have shown that caffeine can reverse behavioral and pathological features of AD [34, 38]. Less clear, however, are the mechanisms by which caffeine exerts these protective effects.
Previously, caffeine was reported to inhibit endocytosis [48, 49] and at mM concentrations it affected exocytosis via calcium-dependent mechanisms [50, 51]. Thus, caffeine might affect AβPP trafficking and subsequent amyloidogenic processing. Here, we showed that μM concentrations of caffeine inhibited LDL cholesterol internalization. This basic biological effect might be of significance to the pathogenesis of AD because amyloidogenic processing of AβPP occurs predominantly within endolysosomes after AβPP is internalized, and because LDLRs interact with AβPP and affect AβPP trafficking [18–20]. Indeed, we have shown that LDL cholesterol treatments promote AβPP internalization and enhance amyloidogenic processing of AβPP within the endolysosome pathway [12]. It therefore follows that caffeine, by blocking LDL cholesterol internalization, could suppress LDL cholesterol-induced AβPP internalization thus suppressing LDL cholesterol-induced amyloidogenic processing of AβPP in endolysosomes. Supporting such a notion, we demonstrated that caffeine blocked LDL-cholesterol-induced increases in Aβ levels, decreases in protein levels of AβPP on neuronal plasma membranes, and increases in AβPP internalization in endosomes. Furthermore, we demonstrated that caffeine pretreatment, in the absence of LDL cholesterol, increased AβPP protein expression levels on neuronal cell surfaces and decreased basal levels of Aβ. Such findings suggest that caffeine can suppress AβPP internalization independently of LDL cholesterol and/or LDLRs. Thus, caffeine might affect AβPP internalization and subsequent amyloidogenic processing both in the absence and presence of LDLR activation. Indeed, caffeine at μM concentrations has been shown to decrease significantly Aβ levels in APP-swe over-expressing N2a cells [34].
Caffeine, at μM concentrations, can block all four subtypes of adenosine receptors, while at higher and potentially toxic concentrations it inhibits cAMP phosphodiesterase activity and increases the release of calcium from intracellular stores [41]. Importantly, activation of adenosine receptors (A1R and A2AR) has been implicated previously in the pathogenesis of AD [52–54], and blockage of A2AR with caffeine has been shown to suppress Aβ generation [34, 38] and protect against Aβ-induced neurotoxicity [55]. Here we showed that of the four subtypes of adenosine receptors studied only A3Rs affected neuronal internalization of LDL cholesterol; a specific A3R antagonist decreased and a specific A3R agonist enhanced neuronal internalization of LDL. Consistent with these pharmacological findings, we found that siRNA knockdown of A3Rs decreased significantly neuronal internalization of LDL cholesterol. Collectively, our findings suggest that A3Rs play an importance role in regulating neuronal internalization of LDL cholesterol. Furthermore, we found that siRNA knockdown of A3Rs decreased LDL cholesterol-induced increases in Aβ levels. Of mechanistic importance, we demonstrated that the A3R antagonist as well as A3R knockdown increased significantly surface expression levels of AβPP, whereas the specific A3R agonist decreased significantly surface expression levels of AβPP. In addition, we demonstrated that A3R blockage attenuated LDL-induced increased accumulation of AβPP in endosomes. Thus, similar to caffeine, A3R blockage could suppress AβPP internalization thus suppressing amyloidogenesis.
In summary, we have described here a novel mechanism whereby caffeine protects against Aβ generation. This mechanism includes suppression of LDL cholesterol-enhanced amyloidogenic processing of AβPP by blocking AβPP internalization via its actions on A3Rs. Further elucidation of the underlying signaling events may provide insight into the pathogenesis of sporadic AD and may lead to new effective therapeutic strategies against this devastating neurodegenerative disease.
Figure 5. Caffeine suppresses AβPP accumulation in endosomes.
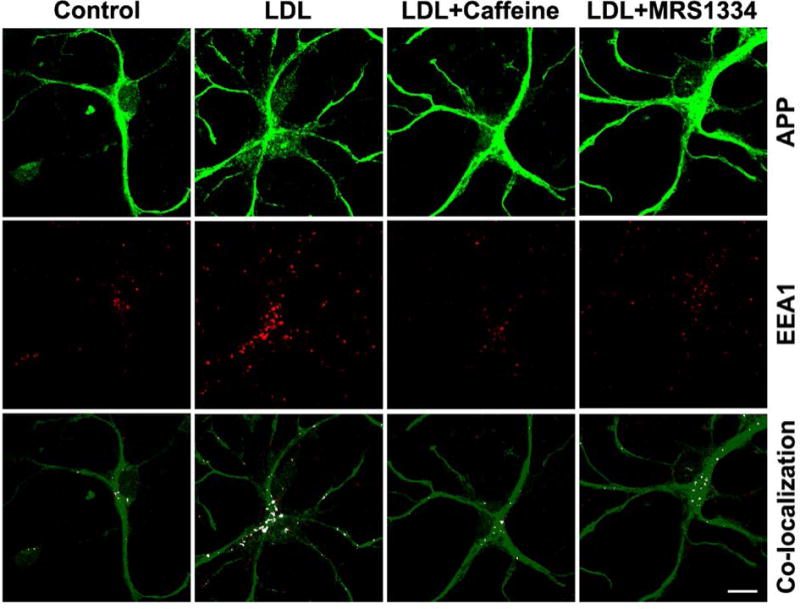
LDL treatment (50 μg/ml for 3 days) enlarged endosomes and increased co-localization of AβPP with endosomes (EEA1) as compared with controls. Such effects were attenuated by either pretreatment with caffeine (200 μM for 24 hrs) or pretreatment with the specific A3R antagonist MRS1334 (100 nM for 24 hrs).
Acknowledgments
Supported by P30GM103329, R01MH100972, and R01MH105329.
Footnotes
The authors declare no competing financial interests
References
- 1.Goate A, Hardy J. Twenty years of Alzheimer’s disease-causing mutations. J Neurochem. 2012;120(Suppl 1):3–8. doi: 10.1111/j.1471-4159.2011.07575.x. [DOI] [PubMed] [Google Scholar]
- 2.Holtzman DM, Morris JC, Goate AM. Alzheimer’s disease: the challenge of the second century. Sci Transl Med. 2011;3:77sr71. doi: 10.1126/scitranslmed.3002369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reitz C, Brayne C, Mayeux R. Epidemiology of Alzheimer disease. Nat Rev Neurol. 2011;7:137–152. doi: 10.1038/nrneurol.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rajendran L, Annaert W. Membrane trafficking pathways in Alzheimer’s disease. Traffic. 2012;13:759–770. doi: 10.1111/j.1600-0854.2012.01332.x. [DOI] [PubMed] [Google Scholar]
- 5.Morel E, Chamoun Z, Lasiecka ZM, Chan RB, Williamson RL, Vetanovetz C, Dall’Armi C, Simoes S, Point Du Jour KS, McCabe BD, et al. Phosphatidylinositol-3-phosphate regulates sorting and processing of amyloid precursor protein through the endosomal system. Nat Commun. 2013;4:2250. doi: 10.1038/ncomms3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nixon RA. Endosome function and dysfunction in Alzheimer’s disease and other neurodegenerative diseases. Neurobiol Aging. 2005;26:373–382. doi: 10.1016/j.neurobiolaging.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 7.Rajendran L, Schneider A, Schlechtingen G, Weidlich S, Ries J, Braxmeier T, Schwille P, Schulz JB, Schroeder C, Simons M, et al. Efficient inhibition of the Alzheimer’s disease beta-secretase by membrane targeting. Science. 2008;320:520–523. doi: 10.1126/science.1156609. [DOI] [PubMed] [Google Scholar]
- 8.Sannerud R, Declerck I, Peric A, Raemaekers T, Menendez G, Zhou L, Veerle B, Coen K, Munck S, De Strooper B, et al. ADP ribosylation factor 6 (ARF6) controls amyloid precursor protein (APP) processing by mediating the endosomal sorting of BACE1. Proc Natl Acad Sci U S A. 2011;108:E559–568. doi: 10.1073/pnas.1100745108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shimizu H, Tosaki A, Kaneko K, Hisano T, Sakurai T, Nukina N. Crystal structure of an active form of BACE1, an enzyme responsible for amyloid beta protein production. Mol Cell Biol. 2008;28:3663–3671. doi: 10.1128/MCB.02185-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Solomon A, Kivipelto M, Wolozin B, Zhou J, Whitmer RA. Midlife serum cholesterol and increased risk of Alzheimer’s and vascular dementia three decades later. Dement Geriatr Cogn Disord. 2009;28:75–80. doi: 10.1159/000231980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen X, Wagener JF, Morgan DH, Hui L, Ghribi O, Geiger JD. Endolysosome Mechanisms Associated with Alzheimer’s Disease-like Pathology in Rabbits Ingesting Cholesterol-Enriched Diet. J Alzheimers Dis. 2010;22:1289–1303. doi: 10.3233/JAD-2010-101323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hui L, Chen X, Geiger JD. Endolysosome involvement in LDL cholesterol-induced Alzheimer’s disease-like pathology in primary cultured neurons. Life Sci. 2012;91:1159–1168. doi: 10.1016/j.lfs.2012.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reed B, Villeneuve S, Mack W, DeCarli C, Chui HC, Jagust W. Associations between serum cholesterol levels and cerebral amyloidosis. JAMA Neurol. 2014;71:195–200. doi: 10.1001/jamaneurol.2013.5390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lesser GT, Beeri MS, Schmeidler J, Purohit DP, Haroutunian V. Cholesterol and LDL relate to neuritic plaques and to APOE4 presence but not to neurofibrillary tangles. Curr Alzheimer Res. 2011;8:303–312. doi: 10.2174/156720511795563755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Namba Y, Tsuchiya H, Ikeda K. Apolipoprotein B immunoreactivity in senile plaque and vascular amyloids and neurofibrillary tangles in the brains of patients with Alzheimer’s disease. Neurosci Lett. 1992;134:264–266. doi: 10.1016/0304-3940(92)90531-b. [DOI] [PubMed] [Google Scholar]
- 16.Takechi R, Galloway S, Pallebage-Gamarallage M, Wellington C, Johnsen R, Mamo JC. Three-dimensional colocalization analysis of plasma-derived apolipoprotein B with amyloid plaques in APP/PS1 transgenic mice. Histochem Cell Biol. 2009;131:661–666. doi: 10.1007/s00418-009-0567-3. [DOI] [PubMed] [Google Scholar]
- 17.Takechi R, Galloway S, Pallebage-Gamarallage MM, Wellington CL, Johnsen RD, Dhaliwal SS, Mamo JC. Differential effects of dietary fatty acids on the cerebral distribution of plasma-derived apo B lipoproteins with amyloid-beta. Br J Nutr. 2010;103:652–662. doi: 10.1017/S0007114509992194. [DOI] [PubMed] [Google Scholar]
- 18.Brodeur J, Theriault C, Lessard-Beaudoin M, Marcil A, Dahan S, Lavoie C. LDLR-related protein 10 (LRP10) regulates amyloid precursor protein (APP) trafficking and processing: evidence for a role in Alzheimer’s disease. Mol Neurodegener. 2012;7:31. doi: 10.1186/1750-1326-7-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoon IS, Chen E, Busse T, Repetto E, Lakshmana MK, Koo EH, Kang DE. Low-density lipoprotein receptor-related protein promotes amyloid precursor protein trafficking to lipid rafts in the endocytic pathway. FASEB J. 2007;21:2742–2752. doi: 10.1096/fj.07-8114com. [DOI] [PubMed] [Google Scholar]
- 20.Jiang S, Li Y, Zhang X, Bu G, Xu H, Zhang YW. Trafficking regulation of proteins in Alzheimer’s disease. Mol Neurodegener. 2014;9:6. doi: 10.1186/1750-1326-9-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cao C, Loewenstein DA, Lin X, Zhang C, Wang L, Duara R, Wu Y, Giannini A, Bai G, Cai J, et al. High Blood caffeine levels in MCI linked to lack of progression to dementia. J Alzheimers Dis. 2012;30:559–572. doi: 10.3233/JAD-2012-111781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cao C, Cirrito JR, Lin X, Wang L, Verges DK, Dickson A, Mamcarz M, Zhang C, Mori T, Arendash GW, et al. Caffeine suppresses amyloid-beta levels in plasma and brain of Alzheimer’s disease transgenic mice. J Alzheimers Dis. 2009;17:681–697. doi: 10.3233/JAD-2009-1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Flaten V, Laurent C, Coelho JE, Sandau U, Batalha VL, Burnouf S, Hamdane M, Humez S, Boison D, Lopes LV, et al. From epidemiology to pathophysiology: what about caffeine in Alzheimer’s disease? Biochem Soc Trans. 2014;42:587–592. doi: 10.1042/BST20130229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arendash GW, Cao C. Caffeine and coffee as therapeutics against Alzheimer’s disease. J Alzheimers Dis. 2010;20(Suppl 1):S117–126. doi: 10.3233/JAD-2010-091249. [DOI] [PubMed] [Google Scholar]
- 25.Eskelinen MH, Kivipelto M. Caffeine as a protective factor in dementia and Alzheimer’s disease. J Alzheimers Dis. 2010;20(Suppl 1):S167–174. doi: 10.3233/JAD-2010-1404. [DOI] [PubMed] [Google Scholar]
- 26.Wostyn P, Van Dam D, Audenaert K, De Deyn PP. Increased Cerebrospinal Fluid Production as a Possible Mechanism Underlying Caffeine’s Protective Effect against Alzheimer’s Disease. Int J Alzheimers Dis. 2011;2011:617420. doi: 10.4061/2011/617420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carman AJ, Dacks PA, Lane RF, Shineman DW, Fillit HM. Current evidence for the use of coffee and caffeine to prevent age-related cognitive decline and Alzheimer’s disease. J Nutr Health Aging. 2014;18:383–392. doi: 10.1007/s12603-014-0021-7. [DOI] [PubMed] [Google Scholar]
- 28.Ritchie K, Carriere I, de Mendonca A, Portet F, Dartigues JF, Rouaud O, Barberger-Gateau P, Ancelin ML. The neuroprotective effects of caffeine: a prospective population study (the Three City Study) Neurology. 2007;69:536–545. doi: 10.1212/01.wnl.0000266670.35219.0c. [DOI] [PubMed] [Google Scholar]
- 29.Santos C, Costa J, Santos J, Vaz-Carneiro A, Lunet N. Caffeine intake and dementia: systematic review and meta-analysis. J Alzheimers Dis. 2010;20(Suppl 1):S187–204. doi: 10.3233/JAD-2010-091387. [DOI] [PubMed] [Google Scholar]
- 30.Santos C, Lunet N, Azevedo A, de Mendonca A, Ritchie K, Barros H. Caffeine intake is associated with a lower risk of cognitive decline: a cohort study from Portugal. J Alzheimers Dis. 2010;20(Suppl 1):S175–185. doi: 10.3233/JAD-2010-091303. [DOI] [PubMed] [Google Scholar]
- 31.Gelber RP, Petrovitch H, Masaki KH, Ross GW, White LR. Coffee intake in midlife and risk of dementia and its neuropathologic correlates. J Alzheimers Dis. 2011;23:607–615. doi: 10.3233/JAD-2010-101428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eskelinen MH, Ngandu T, Tuomilehto J, Soininen H, Kivipelto M. Midlife coffee and tea drinking and the risk of late-life dementia: a population-based CAIDE study. J Alzheimers Dis. 2009;16:85–91. doi: 10.3233/JAD-2009-0920. [DOI] [PubMed] [Google Scholar]
- 33.Arendash GW, Mori T, Cao C, Mamcarz M, Runfeldt M, Dickson A, Rezai-Zadeh K, Tane J, Citron BA, Lin X, et al. Caffeine reverses cognitive impairment and decreases brain amyloid-beta levels in aged Alzheimer’s disease mice. J Alzheimers Dis. 2009;17:661–680. doi: 10.3233/JAD-2009-1087. [DOI] [PubMed] [Google Scholar]
- 34.Arendash GW, Schleif W, Rezai-Zadeh K, Jackson EK, Zacharia LC, Cracchiolo JR, Shippy D, Tan J. Caffeine protects Alzheimer’s mice against cognitive impairment and reduces brain beta-amyloid production. Neuroscience. 2006;142:941–952. doi: 10.1016/j.neuroscience.2006.07.021. [DOI] [PubMed] [Google Scholar]
- 35.Laurent C, Eddarkaoui S, Derisbourg M, Leboucher A, Demeyer D, Carrier S, Schneider M, Hamdane M, Muller CE, Buee L, et al. Beneficial effects of caffeine in a transgenic model of Alzheimer’s disease-like tau pathology. Neurobiol Aging. 2014 doi: 10.1016/j.neurobiolaging.2014.03.027. [DOI] [PubMed] [Google Scholar]
- 36.Han K, Jia N, Li J, Yang L, Min LQ. Chronic caffeine treatment reverses memory impairment and the expression of brain BNDF and TrkB in the PS1/APP double transgenic mouse model of Alzheimer’s disease. Mol Med Rep. 2013;8:737–740. doi: 10.3892/mmr.2013.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Espinosa J, Rocha A, Nunes F, Costa MS, Schein V, Kazlauckas V, Kalinine E, Souza DO, Cunha RA, Porciuncula LO. Caffeine consumption prevents memory impairment, neuronal damage, and adenosine A2A receptors upregulation in the hippocampus of a rat model of sporadic dementia. J Alzheimers Dis. 2013;34:509–518. doi: 10.3233/JAD-111982. [DOI] [PubMed] [Google Scholar]
- 38.Arendash GW, Mori T, Cao C, Mamcarz M, Runfeldt M, Dickson A, Rezai-Zadeh K, Tan J, Citron BA, Lin X, et al. Caffeine Reverses Cognitive Impairment and Decreases Brain Amyloid-beta Levels in Aged Alzheimer’s Disease Mice. J Alzheimers Dis. 2009;17:661–680. doi: 10.3233/JAD-2009-1087. [DOI] [PubMed] [Google Scholar]
- 39.Vaslin A, Puyal J, Borsello T, Clarke PG. Excitotoxicity-related endocytosis in cortical neurons. J Neurochem. 2007;102:789–800. doi: 10.1111/j.1471-4159.2007.04564.x. [DOI] [PubMed] [Google Scholar]
- 40.Chen X, Lan X, Roche I, Liu R, Geiger JD. Caffeine protects against MPTP-induced blood-brain barrier dysfunction in mouse striatum. J Neurochem. 2008 doi: 10.1111/j.1471-4159.2008.05697.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fredholm BB, Battig K, Holmen J, Nehlig A, Zvartau EE. Actions of caffeine in the brain with special reference to factors that contribute to its widespread use. Pharmacol Rev. 1999;51:83–133. [PubMed] [Google Scholar]
- 42.Kang SH, Lee YA, Won SJ, Rhee KH, Gwag BJ. Caffeine-induced neuronal death in neonatal rat brain and cortical cell cultures. Neuroreport. 2002;13:1945–1950. doi: 10.1097/00001756-200210280-00023. [DOI] [PubMed] [Google Scholar]
- 43.Fligner CL, Opheim KE. Caffeine and its dimethylxanthine metabolites in two cases of caffeine overdose: a cause of falsely elevated theophylline concentrations in serum. J Anal Toxicol. 1988;12:339–343. doi: 10.1093/jat/12.6.339. [DOI] [PubMed] [Google Scholar]
- 44.Pitas RE, Boyles JK, Lee SH, Hui D, Weisgraber KH. Lipoproteins and their receptors in the central nervous system. Characterization of the lipoproteins in cerebrospinal fluid and identification of apolipoprotein B,E(LDL) receptors in the brain. J Biol Chem. 1987;262:14352–14360. [PubMed] [Google Scholar]
- 45.Ujiie M, Dickstein DL, Carlow DA, Jefferies WA. Blood-brain barrier permeability precedes senile plaque formation in an Alzheimer disease model. Microcirculation. 2003;10:463–470. doi: 10.1038/sj.mn.7800212. [DOI] [PubMed] [Google Scholar]
- 46.Chen X, Gawryluk JW, Wagener JF, Ghribi O, Geiger JD. Caffeine blocks disruption of blood brain barrier in a rabbit model of Alzheimer’s disease. J Neuroinflammation. 2008;5:12. doi: 10.1186/1742-2094-5-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, Nixon RA. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer’s disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am J Pathol. 2000;157:277–286. doi: 10.1016/s0002-9440(10)64538-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aubry L, Klein G, Satre M. Endo-lysosomal acidification in Dictyostelium discoideum amoebae. Effects of two endocytosis inhibitors: caffeine and cycloheximide. Eur J Cell Biol. 1993;61:225–228. [PubMed] [Google Scholar]
- 49.Gonzalez C, Klein G, Satre M. Caffeine, an inhibitor of endocytosis in Dictyostelium discoideum amoebae. J Cell Physiol. 1990;144:408–415. doi: 10.1002/jcp.1041440307. [DOI] [PubMed] [Google Scholar]
- 50.Kang G, Holz GG. Amplification of exocytosis by Ca2+-induced Ca2+ release in INS-1 pancreatic beta cells. J Physiol. 2003;546:175–189. doi: 10.1113/jphysiol.2002.029959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Krizaj D, Bao JX, Schmitz Y, Witkovsky P, Copenhagen DR. Caffeine-sensitive calcium stores regulate synaptic transmission from retinal rod photoreceptors. J Neurosci. 1999;19:7249–7261. doi: 10.1523/JNEUROSCI.19-17-07249.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wei CJ, Li W, Chen JF. Normal and abnormal functions of adenosine receptors in the central nervous system revealed by genetic knockout studies. Biochim Biophys Acta. 2011;1808:1358–1379. doi: 10.1016/j.bbamem.2010.12.018. [DOI] [PubMed] [Google Scholar]
- 53.Gomes CV, Kaster MP, Tome AR, Agostinho PM, Cunha RA. Adenosine receptors and brain diseases: neuroprotection and neurodegeneration. Biochim Biophys Acta. 2011;1808:1380–1399. doi: 10.1016/j.bbamem.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 54.Marques S, Batalha VL, Lopes LV, Outeiro TF. Modulating Alzheimer’s disease through caffeine: a putative link to epigenetics. J Alzheimers Dis. 2011;24(Suppl 2):161–171. doi: 10.3233/JAD-2011-110032. [DOI] [PubMed] [Google Scholar]
- 55.Dall’Igna OP, Porciuncula LO, Souza DO, Cunha RA, Lara DR. Neuroprotection by caffeine and adenosine A2A receptor blockade of beta-amyloid neurotoxicity. Br J Pharmacol. 2003;138:1207–1209. doi: 10.1038/sj.bjp.0705185. [DOI] [PMC free article] [PubMed] [Google Scholar]