

Abstract
Watson-Crick base-pair changes, or single-nucleotide variants (SNV), have long been known as a source of mutations. However, the extent to which DNA structural variation, including duplication and deletion copy number variants (CNV) and copy number neutral inversions and translocations, contribute to human genome variation and disease has been appreciated only recently. Moreover, the potential complexity of structural variants (SV) was not envisioned; thus, the frequency of complex genomic rearrangements (CGR) and how such events form remained a mystery. The concept of genomic disorders, diseases due to genomic rearrangements and not sequence-based changes for which genomic architecture incite genomic instability, delineated a new category of conditions distinct from chromosomal syndromes and single-gene Mendelian diseases. Nevertheless, it is the mechanistic understanding of CNV/SV formation that has promoted further understanding of human biology and disease and provided insights into human genome and gene evolution.
Keywords: structural variant (SV), copy number variant (CNV), recombination, DNA replication, chromosome biology
Genome variation in evolution and disease
Structural variation (SV) refers to rearrangements of the DNA in a genome resulting in novel breakpoint junctional events. It can be copy number neutral (e.g. inversion or balanced translocation) or in a diploid genome result in copy number variants (CNV) at a given locus due to deletion, duplication, triplication, or further multiplication of one copy of a genomic segment on one chromosome of a homologous pair. The size can vary from as small as the size of an average exon (~100 – 200 bp) to as large as up to millions of base pairs (megabase or Mb) of DNA; the latter can be visible by microscopic techniques such as G-banded human chromosome analysis when > 5–10 Mb in size. SV and CNV require a break in the phosphodiediester backbone of the DNA and the formation of a novel joining thus generating a novel ‘joining point’ from discontinuous DNA sequences in the human reference haploid genome or a unique breakpoint junction.
Imagining our genomes as an encyclopedic book of life, analogized by 23 volumes with two copies each representing the 23 chromosome pairs, then whereas Watson-Crick base pair changes, or single nucleotide variants (SNVs), could be represented as typographical errors in letters of the alphabet, CNV would be represented by deletions of entire sentences, paragraphs, pages, or even large parts of an entire volume. CNV are ubiquitous in the human genome and account for more total differences between any two personal genomes, in terms of bases involved, than all the SNV combined [Conrad et al. 2010]. CNV have been associated with both Mendelian traits and common complex disease [Stankiewicz et al. 2010] and have played a major role in the evolution of genes and genomes [Carvalho et al. 2010]. However, a potential role for SV and CNV in environmentally mediated mutagenesis is only beginning to unfold (Table I).
Table I.
Structural Variation and Environmental Mutagenesis
Observations:
|
Questions:
|
Gene dosage and CNV
If one retrospectively reviews the scientific literature and examines where for the first time the concept of gene dosage was appreciated and experimentally demonstrated perhaps this is best found in the work of Albert Blakeslee and colleagues at the Cold Spring Harbor Laboratory [Blakeslee 1922]. Blakeslee showed in the jimson weed Datura stramonium that the observed plant phenotype was directly related to trisomy for each of the individual twelve chromosomes in this organism’s karyotype. Thus, phenotypic variation in Datura was due to changes in chromosome copy number – defining trisomy as one extra copy of an individual chromosome.
Perhaps the first direct observation of structural variation of the genome in association with trait manifestation was reported by Calvin Bridges in 1936 [Bridges 1936]. He showed that duplication of a locus in Drosophila melanogaster was responsible for the Bar phenotype which consisted of a narrower slit eye in the fruit fly. If the duplication was reverted the fly eye would return to the normal rounded shape. Moreover, rarely he could identify an ultraBar phenotype with a more narrowly slit eye and this was associated with a triplication at the Bar locus – “…it was found that the extra section observed in Bar was present still again, giving a thrice-repeated seriation in direct sequence”. Thus, these studies by Blakeslee and Bridges demonstrate the concept of gene dosage being related to trait manifestation and crystalized the idea that copy number variants (CNV) (i.e. duplication or triplication of a locus) in a diploid genome could represent mutational events important to phenotypic manifestations. Calvin Bridges argued that: “the respective shares attributable in the total effect to the genic balance change (i.e. gene dosage) and to the position-effect change seem to be at present a matter of taste.” Nevertheless, at the time it seems the ‘fly group’ was more enamored by position effects. Subsequent work in the fruitfly examining systematically both deletion (deficiencies) and duplication CNV constructed from translocation bearing strains concluded that: “…very few of the loci of Drosophila melanogaster can produce a dominant phenotype owing to dosage change” [Lindsley et al. 1972].
With the advent of molecular biology and genetic engineering tools, including recombinant DNA, Southern blotting and later the polymerase chain reaction (PCR), and molecular cytogenetics techniques such as fluorescence in situ hybridization or FISH, human genetics and molecular approaches led to some of the first elucidation of genic scale rearrangements in association with human disease. At the β-thalassemia locus, it was noted that a mild phenotype could result from duplication of the α-globin gene [Higgs et al. 1980]. Furthermore, the cloning of the opsin genes enabled the elucidation of the molecular genetic basis of red-green color blindness that in some individuals was associated with a rearrangement of these genes [Nathans et al. 1986]. Finally, even such common traits as hypertension were found to result in some families from a genomic rearrangement between the closely related genes encoding aldosterone synthase and 11 β-hydroxylase with the rearrangement fusing a different regulatory region resulting in mis-expression of a specific gene and causing glucocorticoid-remedial aldosteronism [Lifton et al. 1992]. Nevertheless, in each of these cases, the rearrangement occurred between closely related genes and thus, one could have envisioned interruption of the gene or disruption of the nearby regulatory region that contributed to trait manifestation.
One of the first gene dosage alterations in the human genome to be experimentally related to a clinical phenotype was an apparent increased copy number of the APP gene in association with an early-onset Alzheimer disease [Delabar et al. 1987]. In this study, quantitative Southern blotting was used to determine gene dosage and it was clearly demonstrated that the APP gene, which was mapped to chromosome 21, revealed increased APP gene dosage in the subject and it was comparable to that observed in genomic DNA from a patient with an abnormal 47, XY +21 karyotype in association with a Down syndrome phenotype. These observed gene dosage abnormalities were distinct from the molecular findings seen in genomic DNA from control individuals with normal 46, XX or 46, XY karyotypes. This was of particular interest given the known clinical association of Down syndrome with early onset dementia.
Moreover, in children with Down syndrome by clinical examination yet normal karyotype (46, XX or 46, XY) a submicroscopic duplication of one chromosome 21 homologue was revealed by molecular analyses showing increased APP gene dosage. This increase in gene dosage was demonstrated by Southern blotting that showed dosage differences of heterozygous alleles consistent with duplication on one chromosome homologue and three genomic copies of APP. Controls of both normal individuals and late-onset Alzheimer disease individuals showed two copies of APP. Nevertheless, two reports then argued against the role of APP duplication in Alzheimer disease. Each of these latter ‘negative data’ reports could not find evidence for duplication in the brains of patients with Alzheimer disease; however, only less than 10 patients with sporadic late onset dementia were studied [Podlisny et al. 1987; Tanzi et al. 1987]. Interestingly, 20 years later APP locus duplication was clearly demonstrated to cause Alzheimer disease with cerebral amyloid angiopathy in five families of French descent with autosomal dominant early-onset Alzheimer disease [Rovelet-Lecrux et al. 2006]. Thus, gene dosage as a molecular mechanism for chromosomal syndromes, Mendelian disease, and even complex traits began to emerge during a 20 year interval through studies on APP copy number variation in association with Alzheimer dementia. During this time interval studies on myelin disorders resulting in neurological disease robustly documented duplication CNV and gene dosage as a cause for disease phenotype, elucidated the concept of genomic disorders [Lupski 1998; Lupski 2009], and paved the way for understanding the mechanistic bases of CNV formation.
Myelin diseases of the nervous system, CNV and Genomic Disorders
Two major mechanisms for CNV formation in the human genome, nonallelic homologous recombination (NAHR) and the replication-based mechanisms FoSTeS/ MMBIR (Fork Stalling Template Switching/Microhomology Mediated Break Induced Replication), were elucidated primarily through studies of disorders of myelin in the peripheral nervous system (Charcot-Marie-Tooth disease; CMT1A MIM#118220) or the central nervous system (Pelizeaus-Merzbacher disease; PMD, MIM#312080) where copy number gain (e.g. duplication) of a dosage sensitive gene (PMP22 or PLP1, respectively) resulted in the disease (Figure 1). Recognition of the disease trait enabled ascertainment of mutational events in the population. Thus, the initial key experimental observation was that for both myelin disorders the disease resulted from a duplication of a genomic segment rather than a coding variant within a gene. The other important experimental observations came from mapping the breakpoint junctions of the duplication CNVs. At the CMT locus the duplication was almost always the same size with apparently identical breakpoint junctions that ‘clustered’ in the human genome documenting a recurrent mutational event [Lupski et al. 1991] (Figure 1). Clustering of breakpoint junctions was also noted for a chromosomal microdeletion syndrome [Greenberg et al. 1991; Guzzetta et al. 1992] known as the Smith-Magenis syndrome (SMS, MIM#182290) potentially mechanistically linking chromosomal aberrations with DNA rearrangements. In contrast, the gains at the PMD locus were of all different sizes and the breakpoint junction seemed distinct for each individual event; distinguished as nonrecurrent CNV [Inoue et al. 1999] (Figure 1).
Figure 1.

Mechanisms for CNV formation
The CMT1A duplication was independently identified in Antwerpen, Belgium and Houston, Texas and has become a paradigm to study CNV [Lupski et al. 1991; Raeymaekers et al. 1991] (Figure 2). The CMT1A duplication was shown to cause demyelinating CMT in about 70% of families and remarkably in 76–90% of sporadic demyelinating CMT, in the latter more frequently observed sporadic cases of the disease were caused by de novo mutation. The duplication conveyed the neuropathy trait by virtue of a gene dosage effect [Lupski et al. 1992].
Figure 2.

The mechanism for generating the duplication CNV occurred through the misalignment of chromosomal loci at flanking low copy repeat (LCR) sequences and a subsequent homologous recombination between the nonallelic copies of these repeats from the two chromosomal homologues [Pentao et al. 1992] (Figure 2). This so called ‘nonallelic homologous recombination’ or NAHR [Stankiewicz et al. 2002] also was one of the first mechanisms elucidated for a host of disorders referred to as genomic disorders [Lupski 1998; Lupski 2009]. Interestingly, despite ~24 Kb of flanking ~99% identical sequence the crossovers showed a positional preference, or recombination hotspots, that were defined molecularly [Reiter et al. 1996; Timmerman et al. 1997; Lupski 2004]. The new mutation CMT1A duplication in male gametes was shown to occur at a frequency of 1.7×10−5 [Turner et al. 2008], and the frequency depended upon alleles at the PRDM9 locus, which encodes a protein that facilitates homologous recombination (HR) by recognizing a cis-acting recombination hotspot motif [Berg et al. 2010]. The mechanism also predicted a reciprocal deletion subsequently found to be responsible for a different clinical entity known as hereditary neuropathy with liability to pressure palsies (HNPP) [Chance et al. 1993; Chance et al. 1994] (Figure 2). Remarkably, in a recent study reporting genetic testing positive for disease associated variation in > 17,000 patients with neuropathy for whom physicians ordered CMT molecular testing the PMP22 duplication or deletion CNV were responsible for about 80% of the molecular diagnoses [DiVencenzo et al. 2015].
The key gene dosage hypothesis was supported by a multitude of experimental evidence. Initial genetic studies had documented an apparent more severe peripheral neuropathy phenotype in rare patients that were found to be homozygous for the CMT1A duplication [Lupski et al. 1991; Kaku et al. 1993]. Moreover, recent evidence identified the predicted CMT1A triplication and indeed that it also, as anticipated, conveyed a more severe phenotype [Liu et al. 2014a]. The recognition of PMP22 as the key dosage sensitive gene was initially suggested by its mapping within the duplication/deletion interval (Figure 2) and the identification of point mutations in CMT1A patients found to not have the CMT1A duplication [Roa et al. 1993]. This also was consistent with the idea that CMT1A duplications and some PMP22 missense mutations behaved as gain-of-function mutations whereas HNPP deletion mediated its phenotypic consequences through haploinsufficiency behaving as a loss-of-function mutation; the latter supported by the identification of frameshift mutations of PMP22 in non-deletion HNPP subjects [Nicholson et al. 1994]
For the SMS microdeletion syndrome, the breakpoints clustered in large LCRs of >200 Kb [Chen et al. 1997] and crossovers that occurred within the flanking repeats showed a positional preference consistent with a recombination hotspot [Bi et al. 2003] similar to those seen at the CMT1A/HNPP locus [Reiter et al. 1996; Timmerman et al. 1997]. These observations mechanistically suggested that a reciprocal microduplication syndrome may exist. Indeed, three years later, the reciprocal duplication was described [Potocki et al. 2000] (Figure 3) and the detailed clinical characteristics of the associated syndrome were elucidated seven years later [Potocki et al. 2007]. As the recognition of the NAHR mechanism was better appreciated many predicted reciprocal duplications to the well characterized microdeletion syndromes began to be discovered [Lupski 2009](please add Stankiewicz and Lupski 2002 here). Moreover, novel genomic disorders were discovered by an approach pioneered by Sharp and Eichler that used the segmental duplication architecture of the human genome to predict regions susceptible to NAHR mediated genomic instability [Sharp et al. 2006].
Figure 3.

The NAHR mechanism was clearly documented to be responsible for recurrent genomic rearrangements and chromosomal aberrations [Liu et al. 2012]. The recurrent genomic rearrangements use LCR (also called segmental duplications, SD) as substrates for NAHR [Dittwald et al. 2013a]. The mechanism can be responsible for deletions, duplications, inversions, as well as isochromosome formation through sister chromatid exchange utilizing inverted repeats as substrates [Barbouti et al. 2004]. Moreover, recurrent reciprocal translocations [t (8;12) (p23.1; p13.31), t (4;8) (p16.2; p23.1), t(4;11) (p16.2; p15.4)] [Ou et al. 2008] were documented to occur by NAHR [Liu et al. 2012].
Further studies of NAHR at the SMS/PTLS 17p11.2 locus noted a correlation between the frequency with which the rearrangements occurred utilizing LCR substrate pairs and the length of those flanking LCRs [Liu et al. 2011c]. This led to the concept of ectopic synapsis as a mediator of an ectopic crossing over. Consistent with this hypothesis were the following: i) the original observation of Calvin Bridges using polytene chromosomes and studies at the Bar locus wherein he showed that: “…synapses is disturbed” [Bridges 1936], ii) the observation that NAHR and AHR hotspots coincide [Lindsay et al. 2006], iii) the fact that the same PRDM9 hotspot motif is used for NAHR as for AHR (allelic homologous recombination) [Zhang et al. 2010] and iv) yeast synaptonemal complex mutants abolished ectopic homologous recombination or NAHR [Shinohara et al. 2013].
These locus-specific studies in humans were furthered complemented by computational analyses of patient-identified deletion and duplication CNV that occurred genomewide by NAHR as evidenced by recurrent and reciprocal events. Indeed, such in silico work supplemented by clinical laboratory studies on tens of thousands of patients demonstrated that there was a correlation between the NAHR de novo deletion frequency and the length of the LCR as well as the fraction matching or percent sequence identity whereas an inverse correlation existed with the distance between LCR substrates. The frequency of observed deletion/duplication events was also associated with significantly increased NAHR hotspots and furthermore the hotspot frequency density for the PRDM9 recognition motif also appeared to correlate with frequency [Dittwald et al. 2013b]. The latter computational finding is consistent with observations regarding duplication and triplication at the STS locus, one of the most frequent loci to undergo reciprocal CNV events, where the AHR/NAHR hotspot motif is embedded within a repeat unit of a microsatellite, making this region the highest density of the hotspot motif within the entire human genome [Liu et al. 2011a].
While the study of recurrent rearrangement events elucidated the NAHR mechanism, molecular studies of nonrecurrent rearrangements suggested a potentially different mechanism for CNV formation. Investigations of duplications at the PLP1 locus found in association with PMD revealed that the PLP1 duplications were of different sizes and encompassed distinct intervals of the human genome, but all those associated with disease contained the dosage-sensitive PLP1 gene. Thus, the distinguishing characteristics of the PMD-associated PLP1 duplication included its seemingly nonrecurrent nature, as well as the breakpoints occurring in different places of the human genome flanking the PLP1 gene (Figure 1). Furthermore, breakpoint junctions did not reveal a simple cutting and pasting of two DNA ends, but instead showed inserted segments of the genome in an apparent template driven manner, each apparently templated genomic interval separated by microhomology that was posited to potentially reflect priming of DNA replication. There was no recombination model that could explain these observations found at human CNV. Thus, a replicative mechanism for rearrangement was proposed that encompassed in the model microhomology to prime the replication and long distance template switches – FoSTeS or Fork Stalling Template Switching [Lee et al. 2007]. This latter mechanism was shown to cause CNV of all different sizes including potentially many megabases, both visible and submicroscopic chromosomal aberrations, as well as those of a size that encompassed only a single exon of a gene [Zhang et al. 2009].
The first observations of breakpoint junction sequences and recombination products suggesting long distance template switching after potential stalling of the replication fork (FoSTeS) were complemented by more detailed mechanistic modeling that included features embodied in the microhomology mediated break induced replication model (MMBIR) [Hastings et al. 2009]. The proposal was that a collapsed replication fork generated a single ended, double stranded DNA (seDNA) that was processed by extensive 5’ exonuclease degradation to expose a lengthy single stranded DNA with a 3’ overhang. This exposed 3’ end of a long flexible single stranded DNA could then scan through space and find a complementary sequence (i.e. microhomology) to prime the repair. The primer would be extended by a new low processivity DNA polymerase that can disassociate repeatedly (poor processivity). It would reform at different templates and complete replication resulting in an apparent breakpoint complexity.
From this model, it was easy to explain potential chromosome consequences of template switches during MMBIR. A duplication was explained by a sister chromatid template switch behind the position of fork collapse while a deletion would occur when a template switch occurred to a sister chromatid ahead of the position of the fork breakage. Template switch to a homologue of the wrong orientation resulted in an inversion while nonhomologous sequences or a separate chromosome would result in translocation. If the template switch occurred to sequences already duplicated this could lead to a triplication and if such template switch occurred in the same molecule behind the break a rolling circle mechanism could ensue with subsequent genomic amplification [Hastings et al. 2009]. MMBIR might also be utilized at telomeres where single-ended, double-stranded DNA might present for repair [Lowden et al. 2011; Yatsenko et al. 2012].
A replicative mechanism could drive evolution of genomes, chromosomes, and individual genes, the latter by shuffling exons via a template switch occurring in intronic sequences before and then after an individual exon. The FoSTeS/MMBIR replication based mechanisms (RBM) were shown to be associated also with an unusual form of observed complex genomic rearrangement (CGR) product defined as a complex triplication consisting of a duplication –inverted triplication – duplication (DUP-TRP/INV-DUP) [Carvalho et al. 2011]. Moreover evidence suggested that the polymerase involved was error prone in comparison to the intergenerational polymerases with apparently reduced fidelity causing an ‘error prone’ increase in point mutations or SNV around the breakpoint [Carvalho et al. 2013]. Thus, the properties of the involved polymerase began to emerge – it appeared to have reduced processivity, as it could switch templates over long distance and insert short templated sequences at the breakpoint junctions as it loaded on, extended, and then fell off and reloaded at a new template reflecting its reduced processivity, as well as show lower fidelity than intergenerational polymerases. For nonrecurrent rearrangements, those in which breakpoint junctions occur at distinct loci in each subject/patient, it was found that RBM appear to play a prominent role, particularly in CGR.
The MMBIR model predicted four potential outcomes that could be searched for experimentally. These included: 1) increased SNV mutagenesis concomitant with CGR, as has been observed with break induced replication (BIR), 2) copy number neutral absence of heterozygosity (AOH) when template switch occurred to a homologue versus a sister, 3) amplification of sequences to quadruplications and beyond by virtue of a rolling circle type mechanism and 4) conservative rather than semiconservative DNA replication. Experimental evidence for all four of these observations have now been recently obtained [Carvalho et al. 2013; Malkova et al. 2013; Saini et al. 2013; Wilson et al. 2013; Beck et al. 2015; Carvalho et al. 2015].
Triplication formation
Initial observations by Calvin Bridges in 1936 at the Bar locus identified a rare phenotype known as ultrabar with a more severe slit eye phenotype, and this was shown by polytene chromosome studies to be associated with triplication at the locus rather than the duplication associated with Bar (Figure 3). Thus, we sought to identify triplications at the CMT1A duplication locus with the potential possibility that such an event could lead to a more severe clinical phenotype. Moreover, we wanted to explore the frequency of triplication versus duplication given the initial observations by Charles Zeleny of triplication occurring in two out of every 14,000 flies with duplication [Zeleny 1921]. However, he could not determine the frequency of the de novo duplication mutational event because he did not find any evidence for the Bar locus trait in the 46,290 flies examined from his stocks in the collection of the fly room (Figure 4).
Figure 4.
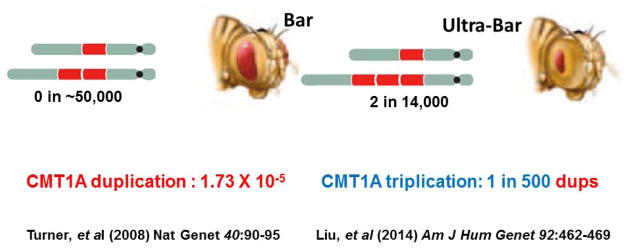
With the implementation of clinical testing for the CMT1A duplication in patient populations, and the determination of CNV using an MLPA (multiplex ligation dependent probe amplification) assay, this MLPA assay could be calibrated with array CGH to distinguish duplication from triplication at the CMT1A locus. Systematic studies applying the MLPA assay in patients with neuropathy occurring during a six year interval, 2007 to 2012, in a clinical diagnostic lab ensued. Triplications were identified in about 1 in every 550 copy number gains at the CMT1A duplication locus [Liu et al. 2014a]. This was approximately 100 times more frequent than the de novo CMT1A duplication as measured by single sperm PCR assay (2×10−5) [Turner et al. 2008] (Figure 4). Thus, the duplication goes to triplication at a higher mutational frequency than the rate at which de novo duplication is formed. Further work suggested that increased mutational frequency occurred because of the fact that one can proceed from duplication to triplication by virtue of many more NAHR substrate mechanism choices (e.g. a larger genomic interval segmentally duplicated on one allele) [Liu et al. 2014a]. At the STS locus 1/20 recurrent gains observed in a clinical population were found to be triplications [Liu et al. 2011a], potentially reflecting both more substrate choices and increased concentration of the HR hotspot motif. However, the dup-to-trp frequency may be overestimated at the STS locus because the prevalence may be higher than the incidence for this X-linked event that may not manifest a phenotype affecting fitness in carrier females. Interestingly, NAHR occurs meiotically, thus to generate triplication it takes more than one generation and occurs from a duplication substrate.
Triplication does not always occur by a simple NAHR and duplication proceeding to triplication. Early on we identified a novel complex genomic rearrangement product that consisted of a triplication embedded within and surrounded by duplications in which the triplicated genomic fragment was in an inverted orientation. This DUP-TRP/INV-DUP CGR was shown to occur by a two-step template switching mechanism and thus, it became clear that replicative mechanisms allow for a diversity of genomic rearrangement products [Carvalho et al. 2011]. Moreover, the triplicated segments in inverted orientation results often in the generation of a new gene by virtue of using the reverse complement strand to make a novel fusion gene. Like the potential for exon shuffling, this latter observation again links FoSTeS/MMBIR to an important evolutionary change – new gene formation.
Complex Genomic Rearrangements – further complexities to mutagenesis
The RBM (FoSTeS/MMBIR) also explained some of the very complex genomic rearrangements observed to occur somatically in association with cancers. This phenomena was initially described from whole genome sequencing studies of ~800 cancers and referred to as “chromothripsis” – that was proposed to result from a shattering process and then a gluing together potentially by nonhomologous end joining [Stephens et al. 2011]. Similar complex chromosomal rearrangement products were also elucidated in patients with developmental disabilities as constitutional mutational events. They were identified and further characterized by using high resolution array CGH and SNP chips in combination with breakpoint junction analyses. These CGR were generated in the germline, or postzygotically during early embryogenesis, and thus were found in constitutional genomes rather than occurring somatically in cancer genomes. Such complex DNA rearrangement processes were proposed to occur by FoSTeS/MMBIR particularly because such a mechanism could readily explain the observations of triplicated regions and short insertions, as well as microhomology, at breakpoint junctions [Liu et al. 2011b], and higher order amplifications as well as copy number neutral SV including inversions. The latter was referred to as chromoanasynthesis to emphasize the potential mechanism for formation of the chromothripsis phenomenon [Maher et al. 2012]. Yet a third mechanism, referred to as chromoanogenesis, postulated that the chromosome was separated from the nucleus of the cell into a mini-cell for the replicative repair of shattered chromosomes to result in the apparent observed chromothripsis phenomena [Crasta et al. 2012; Holland et al. 2012].
An additional observation from mutational studies of cancer genomes consisted of multiple point mutations, or single nucleotide variants (SNV), found as mutational patches in the human genome. This phenomena was termed kataegis in which there was apparent mutational showers of clustered mutations around the genome in association with breast cancers [Nik-Zainal et al. 2012]. The mechanism by which such mutational showers were proposed to occur was that patchy single stranded DNA was mutagenic for SNV. Recently, a unifying mechanism of apparent BIR/MMBIR that could potentially explain both chromothripsis and kaetegis was proposed from studies in yeast. This novel mechanism postulated a migrating bubble during break induced replication, driven by PIF1 helicase of polymerase delta, promotes recombination-coupled DNA synthesis via bubble migration [Saini et al. 2013; Wilson et al. 2013; Sakofsky et al. 2014]. This mechanism was shown to result in conservative DNA synthesis [Malkova et al. 2013] rather than semiconservative DNA replication as originally described by Meselson and Stall [Meselson et al. 1958] and further supports the MMBIR model.
Postzygotic SV Mutagenesis and Transmission Genetics
Mutations that occur in somatic cells can lead to cancer whereas those that occur in germ cells can cause a sporadic genetic disease trait and then be transmitted to the next generation according to Mendelian Laws of inheritance. Recent studies suggest that new mutations may occur postzygotically more frequently than previously thought. Such postzygotic mutagenesis results in mosaicism – the presence of two or more cell lines within an organism [Lupski 2013]. Some mutations that convey embryonic lethality can only exist in the organism in a mosaic state. Chromosomal mosaicism in humans was recognized as early as the 1960s [Hirschhorn et al. 1960]. With the advent of genomewide assays the ability to detect both chromosomal mosaicism [Cheung et al. 2007; Pham et al. 2014] and mosaicism for small genic and even exonic CNV was enabled [Zhang et al. 2009; Boone et al. 2010].
For humans during development a single cell undergoes about 1016 mitoses during the process of becoming a multicellular adult organism. Thus, mutational mechanisms that occur mitotically, such as the replication-based mechanisms FoSTeS/MMBIR, may feature prominently in postzygotic mutagenesis. Consistent with this notion, passage number is a major contributor to structural variation in mouse iPS cells, and the majority of the induced CNVs show breakpoint junction characteristics that are hallmarks of RBM [Liu et al. 2014b]. If the SV mutation occurs early during embryogenesis, it may also be present in the developing germ cells and result in gonadal mosaicism; the developing child may not have disease, but as a subsequent parent may be at risk for transmitting the disease. Parental somatic mosaicism may be under recognized, and it can influence recurrence risk for a genomic disorder [Campbell et al. 2014a]. New mutations causing sporadic genetic disease are more likely to have occurred on the paternally inherited chromosome – risk of new mutation disease increases with paternal age [Kong et al. 2012]. However, the new mutation that occurs on the maternally inherited chromosome is at a higher risk of recurrence. Probabilistic modeling explains the broken symmetry of transmission genetics. Moreover, somatic mosaicism together with sexual differences in gametogenesis could potentially explain a considerable fraction of unexpected recurrences of X-linked recessive disease [Campbell et al. 2014b].
Mirror Traits
Mirror traits refer to those that appear at the opposite ends of a spectrum and represent phenotypic extremes of a bell-shaped curve signifying the distribution of trait manifestation in a population; importantly, the opposing traits are associated with reciprocal mutations (e.g. duplication versus deletion). The concept emerged from studies of genomic disorders. Patients with Smith-Magenis syndrome associated with del17p11.2 were anecdotally noted to be overweight with high BMI while those with Potocki-Lupski syndrome due to dup17p11.2 were observed to be underweight. Mouse models for these syndromes were constructed by chromosome engineering and indeed the deletion mice were overweight in a mixed genetic background as well as pure bred backgrounds, whereas the mice with duplication were underweight [Walz et al. 2003; Walz et al. 2006; Ricard et al. 2010] (Figure 5a). Moreover, whereas the deletion syndrome animal model mice, like patients, were found to be prone to obesity and manifest metabolic syndrome; the duplication mice were protected from diet-induced obesity when fed a ‘super-sizing’ high fat diet revealing gene X environmental interactions at a CNV locus [Lacaria et al. 2012a; Lacaria et al. 2012b] (Figure 5b). Even complex behaviors, as exemplified by ‘licking behavior’ in mice could be shown to objectively and quantitatively manifest as mirror traits in these del/dup CNV animal models [Heck et al. 2012]. This was of interest given that the PTLS duplication was initially reported in association with autism in some patients [Potocki et al. 2000; Potocki et al. 2007] and objectively found in 11 of 15 subjects tested [Treadwell-Deering et al. 2010]. Moreover, in the PTLS animal model the objectively assessed CNV-based autistic like traits could be mitigated by enriched rearing [Lacaria et al. 2012b].
Figure 5.

In humans, studies at the 1q21.2 and 16p11.2 loci revealed mirror traits for head size; whereas 1q21.1 deletion was associated with small head size or microcephaly, 1q21.1 duplication were shown to have macrocephaly [Brunetti-Pierri et al. 2008] (Figure 5c). Interestingly, the mirror trait is expressed in a reciprocal manner at the 16p11.2 locus: deletion associated with macrocephaly and microcephaly found with duplication [Shinawi et al. 2010] (Figure 5c). These findings were quite remarkable in light of a prominent theory regarding evolution of the social brain that posited that autism and schizophrenia represented opposing phenotypic extremes of normal human behavior [Crespi et al. 2010]. Interestingly, del16p11.2 was identified in association with autism [Weiss et al. 2008] while dup16p11.2 was found with schizophrenia [McCarthy et al. 2009; Shinawi et al. 2010]. The reciprocal is observed at 1q21.1; deletion is associated with schizophrenia [International Schizophrenia 2008; Lupski 2008; Stefansson et al. 2008], duplication tends towards autism [Figure 5c]
Thus, CNV and mirror traits may have an important function during evolution enabling extension of phenotypes at either end of a spectrum by variation at a single locus [Figure 5d]. Mirror extreme BMI phenotypes were also shown to occur in association with duplication versus deletion CNV at the 16p11.2 locus [Jacquemont et al. 2011]. Furthermore, overexpression of a human transcript for a single gene, KCTD13 that maps within the 16p11.2 del/dup critical genomic interval, in zebrafish embryos identified it as the sole message capable of inducing microcephaly. Reduced expression of the same gene yielded a microcephaly phenotype recapitulating the mirror trait observations at the 16p11.2 locus [Golzio et al. 2012].
Clinical Genomics – assaying structural variant mutagenesis
Clinical genomics is perhaps best defined as the implementation and application of human genome information and personal variation to the practice of clinical medicine. The development of such genomewide assays could not have taken place until a reference human genome was established [Lander et al. 2001; International Human Genome Sequencing 2004] that enabled a scaffold for probes allowing interrogation of specific genomic intervals for variation (CNV) that might be associated with and explain disease pathology [Stankiewicz et al. 2010]. Previous work had established the utility of clinical cytogenetics initially with the elucidation of disorders based on an alteration in the chromosome number, such as trisomy 21 linked to Down syndrome, with subsequent microscopically visible deletions, duplications, and translocations, both balanced and unbalanced, being resolved and associated with disease pathology as the resolution of human chromosomes was technically improved (Table II). However, until de novo submicroscopic CNV could be robustly identified, their role in sporadic diseases was not generally appreciated [Lupski 2007b].
Table II.
Brief history of human genome analysis
| 1956 | human chromosomes number = 46 |
| 1959 & 1960s | abnormalities in chromosome number,
|
| 1970s | banding techniques, detect subtle aberrations of chromosome structure |
| 1981 | 1st microdeletion Sx described in Kleberg Cytogenetics Laboratory;
|
| 1980s | fluorescence in situ hybridization or FISH; limit to small genomic interval |
| 1990s | *telomere FISH – simultaneously assay chromosome ends |
| 21st century The postgenomic era | Array CGH scan entire genome for submicroscopic CNV Personal genome sequence (WGS and WES) |
demarcates a conceptual shift in thinking from single locus to multi locus testing
As the mechanisms for copy number variation were more clearly elucidated [Figure 63] and human genome instability better understood, these led to informed design of genomic arrays that could be applied in the clinic. Such genomewide arrays were able to detect CNV of relatively small sizes (< 10 Kb) including a design that focused the interrogating oligonucleotides into every exon of known OMIM (give URL; http://www-ncbi-nlm-nih-gov.ezproxyhost.library.tmc.edu/omim/) genes and could readily detect exon dropouts whereby a single exon would be removed or potentially shuffled around the genome. CNV studies in patients over a period of one decade at Baylor College of Medicine resulted in over 66,000 clinical arrays that were signed-out. These clinical genomic studies documented a number of principles and lessons learnt that could be gleaned from the personal genomes of these patient population studies (Figure 6). Major improvements that enabled more genomic diagnoses included; i) the addition of more oligonucleotide probes to arrays enabling higher resolution of the assay and the ability to detect smaller genomic changes, ii) the addition of ‘exonic probes’ to resolve intragenic and even single exon changes [Boone et al. 2010; Boone et al. 2013], iii) combining the abilities to obtain CGH data and SNP data in a single platform [Wiszniewska et al. 2014], and iv) mechanistic informed designs that accounted for predicted regions of genomic instability [Dittwald et al. 2013b].
Figure 6.

However, it would take several more years for the development of next generation multiple parallel sequencing to detect rare variant SNV potentially associated with human disease traits [Gonzaga-Jauregui et al. 2012]. Such implementation was brought about, as in the case for CNV analysis, by again having a reference human genome sequence with which to compare sequence variation (SNV) genomic data generated from individuals. But it also required reduction in the cost of generating genomic information – heralded by the advent of next generation sequencing - to enable the generation of diploid sequence information of an individual’s personal genome. The first genome to be sequenced by next generation sequencing was that of James D. Watson, and this publication occurred 55 years after the elucidation of the Watson-Crick model of DNA [Wheeler et al. 2008]. From these Whole Genome Sequencing (WGS) studies, it became apparent that personal genomes have tremendous variation. There were millions of bases that showed no match to the human haploid reference sequence. Both SNV and CNV vary a lot, and the allele frequency spectrum of CNV revealed smaller CNV, particularly less than 1000 base pairs were more frequent. The amount of variation made it challenging to interpret what was relevant variation with respect to clinical medicine as well as to the personal health of that individual. Variation of African personal genomes was greater in frequency than found in Caucasian genomes [Schuster et al. 2010].
With the ability to perform rare variant detection, WGS of an individual with a specific recessive Mendelian neuropathy was performed to determine if the causative variation could be identified. Indeed, compound heterozygous variant alleles at the SH3TC2 locus were identified [Lupski et al. 2010] – the power of personal genome sequencing was revealed to me and indeed became personal. I had studied CMT neuropathy, a disease that segregates as an autosomal recessive trait in my family, for over 25 years but never knew the molecular basis of my own disease until my genome was sequenced! Subsequent analyses and further whole exome sequencing (WES) documented that one of those SH3TC2 alleles was actually more complex than originally thought and identified by the initial sequencing technology that was utilized [Lupski et al. 2013]. Additional studies of this genome enabled the development of tools for assessing structural variations and towards the establishment of a reference diploid human genome [English et al. 2015]
The next milestone for potential clinical implementation was to see if specific variation could be identified and how it might enable an approach to therapy that had not been considered previously. Here a set of fraternal twins with L-DOPA-responsive dystonia was studied by WGS, and remarkably variation in the sepiapterin reductase locus SPR was identified [Bainbridge et al. 2011] This enabled a new therapy to be tried that would not only treat the defective dopamine pathway but also the serotonergic pathway by supplementation with 5HT. Indeed, the patients seemed to respond better clinically to this additional supplementation than without it [Bainbridge et al. 2011]. With these WGS and WES studies demonstrating proof of principal, and exome capture for whole exome sequencing being more robustly developed, it now enabled sequencing to be done at a cost enabling clinical implementation. At Baylor College of Medicine this began in November 2011 and approximately 230 clinical WES are now performed per month, with the slope of the curve ever increasing, suggesting future widespread implementation of this clinical technology [Yang et al. 2013; The Deciphering Developmental Disorders 2014; Wright et al. 2014; Yang et al. 2014].
As was found with the implementation of clinical arrays, once large numbers of patients were studied, the genomic sequencing revealed novel findings regarding the genetic architecture of human disease. In a pilot study of 250 cases, a remarkable frequency of 25% diagnostic yield was made; 62 molecular diagnoses in 250 cases. These studies confirmed and further demonstrated the remarkable genetic heterogeneity of a number of clinical phenotypes including Cornelia De Lange and Noonan syndromes. Most interestingly, four subjects were found to have two mutations at two Mendelian loci that could explain a diagnostic dilemma by virtue of a blended phenotype where the patient had features of each syndrome blended together. Subsequent work on the next 2,000 clinical exomes confirmed the observation from pilot studies that approximately 4–5% of patients with diagnostic dilemmas that were referred for WES had two molecular diagnoses resulting in a blended phenotype. Moreover, rare genetic events for molecular diagnoses, including a high frequency of de novo mutation, many novel variants, mosaicism, and uniparental disomy, as well as combinations of SNV + CNV [Bayer et al. 2014] at a recessive disease locus, were identified in patients [Yang et al. 2014].
Environmentally Induced SV Mutagenesis
As clinical genomics permeates medical research and eventually clinical practice, Homo sapiens become not only the ‘model organism’ of choice, but rather are perhaps better characterized as a pioneer organism. Medical research seeks to understand disease and to identify what genetic and environmental exposures result in perturbations of biological systems and networks resulting in susceptibility to disease. Human genetics and genomics experiments are data rich allowing derivation of models testable in model organisms. Delineating the mechanisms for SV/CNV mutagenesis has helped provide insights into environmentally induced SV mutagenesis.
Initial genetic screens in yeast for gene mutations that cause an increase in rearrangements (CNV/SV) utilized a gene dosage assay to screen for spontaneous duplication in a single gene [Payen et al. 2008]. Surprisingly, this screen did not identify any genes affecting genetic recombination, but instead found only a gene important to DNA replication – the pol32 gene encoding a subunit of DNA polymerase delta! [Payen et al. 2008]. Microhomologies were found at breakpoint junctions, and the duplication CNV occurred in the apparent absence of the NHEJ pathway. Interestingly, Pol32 had been previously shown to be involved in break-induced replication (BIR) [Lydeard et al. 2007]. Thus, long distance template switching, microhomology mediated junctional events, the involvement of DNA replicative processes, and the lack of classical recombination pathways including NHEJ – all predicted by models (FoSTeS/MMBIR) formulated based on experimental observations in human subjects with genomic disorders – were also recapitulated in the yeast model organism.
Moreover, exposure to compounds that cause replication stress, in this case aphidicolin, which directly inhibits DNA polymerase function and can lead to stalled replication forks, increases de novo CNV formation. Incredibly, breakpoint junction analyses of these fork stall-induced CNV reveal microhomologies at the junctions [Arlt et al. 2009]. Such induced CNV in human cells were also observed with exposure to hydroxyurea [Arlt et al. 2011]. These environmental exposure induced events, apparently mediated by a FoSTeS/MMBIR replicative mechanism, occurred in the absence of the NHEJ pathway in mouse Xrcc4−/− cells and included both CGR and chromothripsis/chromoanasynthesis/chromoanagenesis like structural changes [Arlt et al. 2012].
These mechanistic studies document that chemical exposures (aphidicolin and hydroxyurea) and ionizing radiation [Arlt et al. 2014] can induce SV mutagenesis. However, studies are needed to establish background rates for SV mutagenesis in humans somatic cells and understand what exposures might influence these rates and potentially result in further susceptibility to cancer. In addition, it is necessary to establish background levels of variability in germline mutation rates genome-wide for CNV and SNV and identify environmental agents that influence these rates and heritable disease [Yauk et al. 2013].
CNV and SV in evolution, health and disease
Mutational studies in genomic disorders delineated the role of CNV in disease, experimentally defined recurrent versus nonrecurrent events, documented the role of new mutation in sporadic traits, demonstrated the increased frequency of de novo mutation for CNV versus SNV, and even derived mechanisms for SV mutagenesis (NAHR, FoSTeS/MMBIR)(Figures 3 and 7). In the ‘post-genomic era’ and with the application of genome-wide assays, the extent of SV/CNV in the human genome became apparent [Iafrate et al. 2004; Sebat et al. 2004; Redon et al. 2006; Conrad et al. 2010; Mills et al. 2011], and insights into human evolution [Dumas et al. 2007; Lupski 2007a; Perry et al. 2007; Dumas et al. 2012] and environmentally mediated SV mutagenesis [Arlt et al. 2012] began to be elucidated. Thus, SV mutagenesis is important to genome biology, gene and genome evolution, human biology, and to trait manifestations including susceptibility to both rare and common disease. The ability to measure genetic variation (SNV, CNV/SV) genome-wide enables one to contemplate measuring background rates of intergenerational mutagenesis and examine the potential influences of environmental exposures on these rates [Yauk et al. 2015].
Figure 7.

Replication-based mechanisms (FoSTeS/MMBIR) feature prominently in upstream CNV formation (Figure 7), whereas downstream mechanisms resulting in phenotypic consequences of SV may be mediated by gene-dosage effects of a single [Lalani et al. 2014] or multiple genes [Carvalho et al. 2014] or by breakpoint junctions that disrupt a gene, alter its regulation, or creat a novel gene fusion (Figure 7). A novel consequence of reciprocal CNV and gene dosage is the concept of mirror traits. Other diseases will be defined, gene function delineated, and novel genetic mechanisms for both rare and common/complex disorders elucidated by CNV studies [Lupski et al. 2013; Riccardi et al. 2013; Trivellin et al. 2014; Wu et al. 2015].
The elucidation of CNV and the concepts of i) genomic disorders, ii) gene dosage in trait manifestation, and iii) new mutation CNV and sporadic traits, have helped bridge the gaps in our knowledge of mutations causing disease and our understanding between single gene Mendelizing disease traits and chromosomal syndromes. Furthermore, the seemingly disparate categories of disease consisting of chromosomal syndromes, Mendelian disease and common/complex traits with our understanding of genomic disorders can now all be thought of in the context of genome variation rather than mutation at a single locus (Lupski et al 2011). Driven by the human genome project and technical innovations in genomics, the field of human and medical genetics has been opened up to exploration of the underlying genetic architecture of disease. The Clan Genomics hypothesis, which posits that recently arising mutations in you or recent ancestors in your Clan, rather than population specific and prevalent variation, can be tested for its relevance to disease trait manifestation [Lupski et al. 2011]. As such, avoiding environments with chemical exposures that might cause SV mutagenesis, including in germ cells and during early embryologic development, and avoiding the introduction of compounds into the environment that might stimulate SV mutagenesis, seems prudent.
2014 EMGS AWARD.
The Environmental Mutagenesis and Genomics Society conferred this award to Dr. James R. Lupski for his seminal contributions that have significantly increased our understanding of mutational mechanisms and human disease. These contributions span basic DNA chemistry to ground-breaking applications of genomic technologies. Dr. Lupski’s most recent studies focus on genomic instability and rearrangements, disease-related mutations and copy number variants (CNVs), and the technological advances in whole genome and exome sequence analysis of Mendelian disease. His contributions to elucidating mechanisms that cause human mutations, as well linking those mutations to diseases, are unparalleled. His research has led him to develop the concept of genomic disorders, which refers to syndromes that originate not from point mutations that affect the function of specific genes, but from duplications and deletions of genomic regions.
Acknowledgments
I thank all of the patients and families who volunteered to participate and collaborate in our research efforts and my colleagues Claudia Carvalho, Pengfei Liu, Pawel Stankiewicz and Tamar Harel for critical reviews. I also am grateful for the generous support of the National Institutes of Neurological Disorders and Stroke (NINDS), the National Institute of Child Health and Development (NICHD), the National Eye Institute (NEI), the National Human Genome Research Institute (NHGRI), the Muscular Dystrophy Association, the March of Dimes, the Smith-Magenis Syndrome Research Foundation (SMSRF), the Parents and Researchers Interested in Smith-Magenis Syndrome (PRISMS), and the CMT Association. Current laboratory support includes NINDS (R01NS058529) and NHGRI (U54HG006542). I also thank the Environmental Mutagenesis and Genomics Society for their 2014 Annual Award and for honoring the Lupski laboratory in such a manner.
References
- Arlt MF, Mulle JG, Schaibley VM, Ragland RL, Durkin SG, Warren ST, Glover TW. Replication stress induces genome-wide copy number changes in human cells that resemble polymorphic and pathogenic variants. Am J Hum Genet. 2009;84:339–350. doi: 10.1016/j.ajhg.2009.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arlt MF, Ozdemir AC, Birkeland SR, Wilson TE, Glover TW. Hydroxyurea induces de novo copy number variants in human cells. Proc Natl Acad Sci U S A. 2011;108:17360–17365. doi: 10.1073/pnas.1109272108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arlt MF, Rajendran S, Birkeland SR, Wilson TE, Glover TW. De novo CNV formation in mouse embryonic stem cells occurs in the absence of Xrcc4-dependent nonhomologous end joining. PLoS Genet. 2012;8:e1002981. doi: 10.1371/journal.pgen.1002981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arlt MF, Rajendran S, Birkeland SR, Wilson TE, Glover TW. Copy number variants are produced in response to low-dose ionizing radiation in cultured cells. Environ Mol Mutagen. 2014;55:103–113. doi: 10.1002/em.21840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bainbridge MN, Wiszniewski W, Murdock DR, Friedman J, Gonzaga-Jauregui C, Newsham I, Reid JG, Fink JK, Morgan MB, Gingras MC, et al. Whole-genome sequencing for optimized patient management. Sci Transl Med. 2011;3:87re83. doi: 10.1126/scitranslmed.3002243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbouti A, Stankiewicz P, Nusbaum C, Cuomo C, Cook A, Hoglund M, Johansson B, Hagemeijer A, Park SS, Mitelman F, et al. The breakpoint region of the most common isochromosome, i(17q), in human neoplasia is characterized by a complex genomic architecture with large, palindromic, low-copy repeats. Am J Hum Genet. 2004;74:1–10. doi: 10.1086/380648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer DK, Martinez CA, Sorte HS, Forbes LR, Demmler-Harrison GJ, Hanson IC, Pearson NM, Noroski LM, Zaki SR, Bellini WJ, et al. Vaccine-associated varicella and rubella infections in severe combined immunodeficiency with isolated CD4 lymphocytopenia and mutations in IL7R detected by tandem whole exome sequencing and chromosomal microarray. Clin Exp Immunol. 2014;178:459–469. doi: 10.1111/cei.12421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck CR, Carvalho CMB, Banser L, Gambin T, Stubbolo D, Yuan B, Sperle K, McCahan SM, Henneke M, Seeman P, et al. Complex genomic rearrangements at the PLP1 locus includes triplication and quadruplication. PLoS Genet. 2015 doi: 10.1371/journal.pgen.1005050. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg IL, Neumann R, Lam KW, Sarbajna S, Odenthal-Hesse L, May CA, Jeffreys AJ. PRDM9 variation strongly influences recombination hot-spot activity and meiotic instability in humans. Nat Genet. 2010;42:859–863. doi: 10.1038/ng.658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi W, Park SS, Shaw CJ, Withers MA, Patel PI, Lupski JR. Reciprocal crossovers and a positional preference for strand exchange in recombination events resulting in deletion or duplication of chromosome 17p11.2. Am J Hum Genet. 2003;73:1302–1315. doi: 10.1086/379979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakeslee AF. Variations in datura due to changes in chromosome number. The American Naturalist. 1922;642:16–31. [Google Scholar]
- Boone PM, Bacino CA, Shaw CA, Eng PA, Hixson PM, Pursley AN, Kang SH, Yang Y, Wiszniewska J, Nowakowska BA, et al. Detection of clinically relevant exonic copy-number changes by array CGH. Hum Mutat. 2010;31:1326–1342. doi: 10.1002/humu.21360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boone PM, Campbell IM, Baggett BC, Soens ZT, Rao MM, Hixson PM, Patel A, Bi W, Cheung SW, Lalani SR, et al. Deletions of recessive disease genes: CNV contribution to carrier states and disease-causing alleles. Genome Res. 2013;23:1383–1394. doi: 10.1101/gr.156075.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges CB. The Bar “Gene” a Duplication. Science. 1936;83:210–211. doi: 10.1126/science.83.2148.210. [DOI] [PubMed] [Google Scholar]
- Brunetti-Pierri N, Berg JS, Scaglia F, Belmont J, Bacino CA, Sahoo T, Lalani SR, Graham B, Lee B, Shinawi M, et al. Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat Genet. 2008;40:1466–1471. doi: 10.1038/ng.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell IM, Stewart JR, James RA, Lupski JR, Stankiewicz P, Olofsson P, Shaw CA. Parent of origin, mosaicism, and recurrence risk: probabilistic modeling explains the broken symmetry of transmission genetics. Am J Hum Genet. 2014a;95:345–359. doi: 10.1016/j.ajhg.2014.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell IM, Yuan B, Robberecht C, Pfundt R, Szafranski P, McEntagart ME, Nagamani SC, Erez A, Bartnik M, Wisniowiecka-Kowalnik B, et al. Parental somatic mosaicism is underrecognized and influences recurrence risk of genomic disorders. Am J Hum Genet. 2014b;95:173–182. doi: 10.1016/j.ajhg.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho CM, Pehlivan D, Ramocki MB, Fang P, Alleva B, Franco LM, Belmont JW, Hastings PJ, Lupski JR. Replicative mechanisms for CNV formation are error prone. Nat Genet. 2013;45:1319–1326. doi: 10.1038/ng.2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho CM, Pfundt R, King DA, Lindsay SJ, Zuccherato LW, Macville MVE, Liu P, Johnson D, Stankiewicz P, Brown CW, et al. Absence of heterozygosity due to template switching during replicative rearrangements. American Journal of Human Genetics. 2015 doi: 10.1016/j.ajhg.2015.01.021. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho CM, Ramocki MB, Pehlivan D, Franco LM, Gonzaga-Jauregui C, Fang P, McCall A, Pivnick EK, Hines-Dowell S, Seaver LH, et al. Inverted genomic segments and complex triplication rearrangements are mediated by inverted repeats in the human genome. Nat Genet. 2011;43:1074–1081. doi: 10.1038/ng.944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho CM, Vasanth S, Shinawi M, Russell C, Ramocki MB, Brown CW, Graakjaer J, Skytte AB, Vianna-Morgante AM, Krepischi AC, et al. Dosage changes of a segment at 17p13.1 lead to intellectual disability and microcephaly as a result of complex genetic interaction of multiple genes. Am J Hum Genet. 2014;95:565–578. doi: 10.1016/j.ajhg.2014.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho CM, Zhang F, Lupski JR. Evolution in health and medicine Sackler colloquium: Genomic disorders: a window into human gene and genome evolution. Proc Natl Acad Sci U S A. 2010;107(Suppl 1):1765–1771. doi: 10.1073/pnas.0906222107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chance PF, Abbas N, Lensch MW, Pentao L, Roa BB, Patel PI, Lupski JR. Two autosomal dominant neuropathies result from reciprocal DNA duplication/deletion of a region on chromosome 17. Hum Mol Genet. 1994;3:223–228. doi: 10.1093/hmg/3.2.223. [DOI] [PubMed] [Google Scholar]
- Chance PF, Alderson MK, Leppig KA, Lensch MW, Matsunami N, Smith B, Swanson PD, Odelberg SJ, Disteche CM, Bird TD. DNA deletion associated with hereditary neuropathy with liability to pressure palsies. Cell. 1993;72:143–151. doi: 10.1016/0092-8674(93)90058-x. [DOI] [PubMed] [Google Scholar]
- Chen KS, Manian P, Koeuth T, Potocki L, Zhao Q, Chinault AC, Lee CC, Lupski JR. Homologous recombination of a flanking repeat gene cluster is a mechanism for a common contiguous gene deletion syndrome. Nat Genet. 1997;17:154–163. doi: 10.1038/ng1097-154. [DOI] [PubMed] [Google Scholar]
- Cheung SW, Shaw CA, Scott DA, Patel A, Sahoo T, Bacino CA, Pursley A, Li J, Erickson R, Gropman AL, et al. Microarray-based CGH detects chromosomal mosaicism not revealed by conventional cytogenetics. Am J Med Genet A. 2007;143A:1679–1686. doi: 10.1002/ajmg.a.31740. [DOI] [PubMed] [Google Scholar]
- Conrad DF, Bird C, Blackburne B, Lindsay S, Mamanova L, Lee C, Turner DJ, Hurles ME. Mutation spectrum revealed by breakpoint sequencing of human germline CNVs. Nat Genet. 2010;42:385–391. doi: 10.1038/ng.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crasta K, Ganem NJ, Dagher R, Lantermann AB, Ivanova EV, Pan Y, Nezi L, Protopopov A, Chowdhury D, Pellman D. DNA breaks and chromosome pulverization from errors in mitosis. Nature. 2012;482:53–58. doi: 10.1038/nature10802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespi B, Stead P, Elliot M. Evolution in health and medicine Sackler colloquium: Comparative genomics of autism and schizophrenia. Proc Natl Acad Sci U S A. 2010;107(Suppl 1):1736–1741. doi: 10.1073/pnas.0906080106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delabar JM, Goldgaber D, Lamour Y, Nicole A, Huret JL, de Grouchy J, Brown P, Gajdusek DC, Sinet PM. Beta amyloid gene duplication in Alzheimer’s disease and karyotypically normal Down syndrome. Science. 1987;235:1390–1392. doi: 10.1126/science.2950593. [DOI] [PubMed] [Google Scholar]
- Dittwald P, Gambin T, Gonzaga-Jauregui C, Carvalho CM, Lupski JR, Stankiewicz P, Gambin A. Inverted low-copy repeats and genome instability--a genome-wide analysis. Hum Mutat. 2013a;34:210–220. doi: 10.1002/humu.22217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittwald P, Gambin T, Szafranski P, Li J, Amato S, Divon MY, Rodriguez Rojas LX, Elton LE, Scott DA, Schaaf CP, et al. NAHR-mediated copy-number variants in a clinical population: mechanistic insights into both genomic disorders and Mendelizing traits. Genome Res. 2013b;23:1395–1409. doi: 10.1101/gr.152454.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiVencenzo C, Elzinga CD, Medeiros AC, Karbassi I, Jones JR, Evans MC, Braastad CD, Bishop CM, Jaremko M, Wang Z, et al. The allelic spectrum of Charcot-Marie-Tooth disease in over 17,000 individuals with neuropathy. Mol Genet Genomic Med. 2015;2:522–529. doi: 10.1002/mgg3.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumas L, Kim YH, Karimpour-Fard A, Cox M, Hopkins J, Pollack JR, Sikela JM. Gene copy number variation spanning 60 million years of human and primate evolution. Genome Res. 2007;17:1266–1277. doi: 10.1101/gr.6557307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumas LJ, O’Bleness MS, Davis JM, Dickens CM, Anderson N, Keeney JG, Jackson J, Sikela M, Raznahan A, Giedd J, et al. DUF1220-domain copy number implicated in human brain-size pathology and evolution. Am J Hum Genet. 2012;91:444–454. doi: 10.1016/j.ajhg.2012.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English AC, Salerno WJ, Hampton OA, Gonzaga-Jauregui C, Ambreth S, Ritter DI, Beck CR, Davis CF, Dahdouli M, Ma S, et al. Assessing structural variation in a personal genome. BMC Genomics. 2015 doi: 10.1186/s12864-015-1479-3. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golzio C, Willer J, Talkowski ME, Oh EC, Taniguchi Y, Jacquemont S, Reymond A, Sun M, Sawa A, Gusella JF, et al. KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature. 2012;485:363–367. doi: 10.1038/nature11091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzaga-Jauregui C, Lupski JR, Gibbs RA. Human genome sequencing in health and disease. Annu Rev Med. 2012;63:35–61. doi: 10.1146/annurev-med-051010-162644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg F, Guzzetta V, Montes de Oca-Luna R, Magenis RE, Smith AC, Richter SF, Kondo I, Dobyns WB, Patel PI, Lupski JR. Molecular analysis of the Smith-Magenis syndrome: a possible contiguous-gene syndrome associated with del(17)(p11.2) Am J Hum Genet. 1991;49:1207–1218. [PMC free article] [PubMed] [Google Scholar]
- Guzzetta V, Franco B, Trask BJ, Zhang H, Saucedo-Cardenas O, Montes de Oca-Luna R, Greenberg F, Chinault AC, Lupski JR, Patel PI. Somatic cell hybrids, sequence-tagged sites, simple repeat polymorphisms, and yeast artificial chromosomes for physical and genetic mapping of proximal 17p. Genomics. 1992;13:551–559. doi: 10.1016/0888-7543(92)90124-b. [DOI] [PubMed] [Google Scholar]
- Hastings PJ, Ira G, Lupski JR. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 2009;5:e1000327. doi: 10.1371/journal.pgen.1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heck DH, Gu W, Cao Y, Qi S, Lacaria M, Lupski JR. Opposing phenotypes in mice with Smith-Magenis deletion and Potocki-Lupski duplication syndromes suggest gene dosage effects on fluid consumption behavior. Am J Med Genet A. 2012;158A:2807–2814. doi: 10.1002/ajmg.a.35601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgs DR, Old JM, Pressley L, Clegg JB, Weatherall DJ. A novel alpha-globin gene arrangement in man. Nature. 1980;284:632–635. doi: 10.1038/284632a0. [DOI] [PubMed] [Google Scholar]
- Hirschhorn K, Decker WH, Cooper HL. Human intersex with chromosome mosaicism of type XY/XO. Report of a case. N Engl J Med. 1960;263:1044–1048. doi: 10.1056/NEJM196011242632102. [DOI] [PubMed] [Google Scholar]
- Holland AJ, Cleveland DW. Chromoanagenesis and cancer: mechanisms and consequences of localized, complex chromosomal rearrangements. Nat Med. 2012;18:1630–1638. doi: 10.1038/nm.2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, Lee C. Detection of large-scale variation in the human genome. Nat Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- Inoue K, Osaka H, Imaizumi K, Nezu A, Takanashi J, Arii J, Murayama K, Ono J, Kikawa Y, Mito T, et al. Proteolipid protein gene duplications causing Pelizaeus-Merzbacher disease: molecular mechanism and phenotypic manifestations. Ann Neurol. 1999;45:624–632. [PubMed] [Google Scholar]
- International Human Genome Sequencing C. Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–945. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- International Schizophrenia C. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–241. doi: 10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemont S, Reymond A, Zufferey F, Harewood L, Walters RG, Kutalik Z, Martinet D, Shen Y, Valsesia A, Beckmann ND, et al. Mirror extreme BMI phenotypes associated with gene dosage at the chromosome 16p11.2 locus. Nature. 2011;478:97–102. doi: 10.1038/nature10406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaku DA, Parry GJ, Malamut R, Lupski JR, Garcia CA. Nerve conduction studies in Charcot-Marie-Tooth polyneuropathy associated with a segmental duplication of chromosome 17. Neurology. 1993;43:1806–1808. doi: 10.1212/wnl.43.9.1806. [DOI] [PubMed] [Google Scholar]
- Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, Gudjonsson SA, Sigurdsson A, Jonasdottir A, Jonasdottir A, et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature. 2012;488:471–475. doi: 10.1038/nature11396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacaria M, Saha P, Potocki L, Bi W, Yan J, Girirajan S, Burns B, Elsea S, Walz K, Chan L, et al. A duplication CNV that conveys traits reciprocal to metabolic syndrome and protects against diet-induced obesity in mice and men. PLoS Genet. 2012a;8:e1002713. doi: 10.1371/journal.pgen.1002713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacaria M, Spencer C, Gu W, Paylor R, Lupski JR. Enriched rearing improves behavioral responses of an animal model for CNV-based autistic-like traits. Hum Mol Genet. 2012b;21:3083–3096. doi: 10.1093/hmg/dds124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalani SR, Zhang J, Schaaf CP, Brown CW, Magoulas P, Tsai AC, El-Gharbawy A, Wierenga KJ, Bartholomew D, Fong CT, et al. Mutations in PURA Cause Profound Neonatal Hypotonia, Seizures, and Encephalopathy in 5q31.3 Microdeletion Syndrome. Am J Hum Genet. 2014;95:579–583. doi: 10.1016/j.ajhg.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Lee JA, Carvalho CM, Lupski JR. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 2007;131:1235–1247. doi: 10.1016/j.cell.2007.11.037. [DOI] [PubMed] [Google Scholar]
- Lifton RP, Dluhy RG, Powers M, Rich GM, Cook S, Ulick S, Lalouel JM. A chimaeric 11 beta-hydroxylase/aldosterone synthase gene causes glucocorticoid-remediable aldosteronism and human hypertension. Nature. 1992;355:262–265. doi: 10.1038/355262a0. [DOI] [PubMed] [Google Scholar]
- Lindsay SJ, Khajavi M, Lupski JR, Hurles ME. A chromosomal rearrangement hotspot can be identified from population genetic variation and is coincident with a hotspot for allelic recombination. Am J Hum Genet. 2006;79:890–902. doi: 10.1086/508709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsley DL, Sandler L, Baker BS, Carpenter AT, Denell RE, Hall JC, Jacobs PA, Miklos GL, Davis BK, Gethmann RC, et al. Segmental aneuploidy and the genetic gross structure of the Drosophila genome. Genetics. 1972;71:157–184. doi: 10.1093/genetics/71.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Carvalho CM, Hastings PJ, Lupski JR. Mechanisms for recurrent and complex human genomic rearrangements. Curr Opin Genet Dev. 2012;22:211–220. doi: 10.1016/j.gde.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Erez A, Nagamani SC, Bi W, Carvalho CM, Simmons AD, Wiszniewska J, Fang P, Eng PA, Cooper ML, et al. Copy number gain at Xp22.31 includes complex duplication rearrangements and recurrent triplications. Hum Mol Genet. 2011a;20:1975–1988. doi: 10.1093/hmg/ddr078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Erez A, Nagamani SC, Dhar SU, Kolodziejska KE, Dharmadhikari AV, Cooper ML, Wiszniewska J, Zhang F, Withers MA, et al. Chromosome catastrophes involve replication mechanisms generating complex genomic rearrangements. Cell. 2011b;146:889–903. doi: 10.1016/j.cell.2011.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Gelowani V, Zhang F, Drory VE, Ben-Shachar S, Roney E, Medeiros AC, Moore RJ, DiVincenzo C, Burnette WB, et al. Mechanism, prevalence, and more severe neuropathy phenotype of the Charcot-Marie-Tooth type 1A triplication. Am J Hum Genet. 2014a;94:462–469. doi: 10.1016/j.ajhg.2014.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Kaplan A, Yuan B, Hanna JH, Lupski JR, Reiner O. Passage number is a major contributor to genomic structural variations in mouse iPSCs. Stem Cells. 2014b;32:2657–2667. doi: 10.1002/stem.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Lacaria M, Zhang F, Withers M, Hastings PJ, Lupski JR. Frequency of nonallelic homologous recombination is correlated with length of homology: evidence that ectopic synapsis precedes ectopic crossing-over. Am J Hum Genet. 2011c;89:580–588. doi: 10.1016/j.ajhg.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowden MR, Flibotte S, Moerman DG, Ahmed S. DNA synthesis generates terminal duplications that seal end-to-end chromosome fusions. Science. 2011;332:468–471. doi: 10.1126/science.1199022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR. Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998;14:417–422. doi: 10.1016/s0168-9525(98)01555-8. [DOI] [PubMed] [Google Scholar]
- Lupski JR. Hotspots of homologous recombination in the human genome: not all homologous sequences are equal. Genome Biol. 2004;5:242. doi: 10.1186/gb-2004-5-10-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR. An evolution revolution provides further revelation. Bioessays. 2007a;29:1182–1184. doi: 10.1002/bies.20686. [DOI] [PubMed] [Google Scholar]
- Lupski JR. Genomic rearrangements and sporadic disease. Nat Genet. 2007b;39:S43–47. doi: 10.1038/ng2084. [DOI] [PubMed] [Google Scholar]
- Lupski JR. Schizophrenia: Incriminating genomic evidence. Nature. 2008;455:178–179. doi: 10.1038/455178a. [DOI] [PubMed] [Google Scholar]
- Lupski JR. Genomic disorders ten years on. Genome Med. 2009;1:42. doi: 10.1186/gm42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR. Genetics. Genome mosaicism--one human, multiple genomes. Science. 2013;341:358–359. doi: 10.1126/science.1239503. [DOI] [PubMed] [Google Scholar]
- Lupski JR, Belmont JW, Boerwinkle E, Gibbs RA. Clan genomics and the complex architecture of human disease. Cell. 2011;147:32–43. doi: 10.1016/j.cell.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR, de Oca-Luna RM, Slaugenhaupt S, Pentao L, Guzzetta V, Trask BJ, Saucedo-Cardenas O, Barker DF, Killian JM, Garcia CA, et al. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell. 1991;66:219–232. doi: 10.1016/0092-8674(91)90613-4. [DOI] [PubMed] [Google Scholar]
- Lupski JR, Gonzaga-Jauregui C, Yang Y, Bainbridge MN, Jhangiani S, Buhay CJ, Kovar CL, Wang M, Hawes AC, Reid JG, et al. Exome sequencing resolves apparent incidental findings and reveals further complexity of SH3TC2 variant alleles causing Charcot-Marie-Tooth neuropathy. Genome Med. 2013;5:57. doi: 10.1186/gm461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR, Reid JG, Gonzaga-Jauregui C, Rio Deiros D, Chen DC, Nazareth L, Bainbridge M, Dinh H, Jing C, Wheeler DA, et al. Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathy. N Engl J Med. 2010;362:1181–1191. doi: 10.1056/NEJMoa0908094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR, Wise CA, Kuwano A, Pentao L, Parke JT, Glaze DG, Ledbetter DH, Greenberg F, Patel PI. Gene dosage is a mechanism for Charcot-Marie-Tooth disease type 1A. Nat Genet. 1992;1:29–33. doi: 10.1038/ng0492-29. [DOI] [PubMed] [Google Scholar]
- Lydeard JR, Jain S, Yamaguchi M, Haber JE. Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature. 2007;448:820–823. doi: 10.1038/nature06047. [DOI] [PubMed] [Google Scholar]
- Maher CA, Wilson RK. Chromothripsis and human disease: piecing together the shattering process. Cell. 2012;148:29–32. doi: 10.1016/j.cell.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkova A, Ira G. Break-induced replication: functions and molecular mechanism. Curr Opin Genet Dev. 2013;23:271–279. doi: 10.1016/j.gde.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy SE, Makarov V, Kirov G, Addington AM, McClellan J, Yoon S, Perkins DO, Dickel DE, Kusenda M, Krastoshevsky O, et al. Microduplications of 16p11.2 are associated with schizophrenia. Nat Genet. 2009;41:1223–1227. doi: 10.1038/ng.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meselson M, Stahl FW. The replication of DNA. Cold Spring Harb Symp Quant Biol. 1958;23:9–12. doi: 10.1101/sqb.1958.023.01.004. [DOI] [PubMed] [Google Scholar]
- Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C, Abyzov A, Yoon SC, Ye K, Cheetham RK, et al. Mapping copy number variation by population-scale genome sequencing. Nature. 2011;470:59–65. doi: 10.1038/nature09708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathans J, Piantanida TP, Eddy RL, Shows TB, Hogness DS. Molecular genetics of inherited variation in human color vision. Science. 1986;232:203–210. doi: 10.1126/science.3485310. [DOI] [PubMed] [Google Scholar]
- Nicholson GA, Valentijn LJ, Cherryson AK, Kennerson ML, Bragg TL, DeKroon RM, Ross DA, Pollard JD, McLeod JG, Bolhuis PA, et al. A frame shift mutation in the PMP22 gene in hereditary neuropathy with liability to pressure palsies. Nat Genet. 1994;6:263–266. doi: 10.1038/ng0394-263. [DOI] [PubMed] [Google Scholar]
- Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, Jones D, Hinton J, Marshall J, Stebbings LA, et al. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012;149:979–993. doi: 10.1016/j.cell.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou Z, Berg JS, Yonath H, Enciso VB, Miller DT, Picker J, Lenzi T, Keegan CE, Sutton VR, Belmont J, et al. Microduplications of 22q11.2 are frequently inherited and are associated with variable phenotypes. Genet Med. 2008;10:267–277. doi: 10.1097/GIM.0b013e31816b64c2. [DOI] [PubMed] [Google Scholar]
- Payen C, Koszul R, Dujon B, Fischer G. Segmental duplications arise from Pol32-dependent repair of broken forks through two alternative replication-based mechanisms. PLoS Genet. 2008;4:e1000175. doi: 10.1371/journal.pgen.1000175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pentao L, Wise CA, Chinault AC, Patel PI, Lupski JR. Charcot-Marie-Tooth type 1A duplication appears to arise from recombination at repeat sequences flanking the 1.5 Mb monomer unit. Nat Genet. 1992;2:292–300. doi: 10.1038/ng1292-292. [DOI] [PubMed] [Google Scholar]
- Perry GH, Dominy NJ, Claw KG, Lee AS, Fiegler H, Redon R, Werner J, Villanea FA, Mountain JL, Misra R, et al. Diet and the evolution of human amylase gene copy number variation. Nat Genet. 2007;39:1256–1260. doi: 10.1038/ng2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham J, Shaw C, Pursley A, Hixson P, Sampath S, Roney E, Gambin T, Kang SH, Bi W, Lalani S, et al. Somatic mosaicism detected by exon-targeted, high-resolution aCGH in 10,362 consecutive cases. Eur J Hum Genet. 2014;22:969–978. doi: 10.1038/ejhg.2013.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podlisny MB, Lee G, Selkoe DJ. Gene dosage of the amyloid beta precursor protein in Alzheimer’s disease. Science. 1987;238:669–671. doi: 10.1126/science.2960019. [DOI] [PubMed] [Google Scholar]
- Potocki L, Bi W, Treadwell-Deering D, Carvalho CM, Eifert A, Friedman EM, Glaze D, Krull K, Lee JA, Lewis RA, et al. Characterization of Potocki-Lupski syndrome (dup(17)(p11.2p11.2)) and delineation of a dosage-sensitive critical interval that can convey an autism phenotype. Am J Hum Genet. 2007;80:633–649. doi: 10.1086/512864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potocki L, Chen KS, Park SS, Osterholm DE, Withers MA, Kimonis V, Summers AM, Meschino WS, Anyane-Yeboa K, Kashork CD, et al. Molecular mechanism for duplication 17p11.2- the homologous recombination reciprocal of the Smith-Magenis microdeletion. Nat Genet. 2000;24:84–87. doi: 10.1038/71743. [DOI] [PubMed] [Google Scholar]
- Raeymaekers P, Timmerman V, Nelis E, De Jonghe P, Hoogendijk JE, Baas F, Barker DF, Martin JJ, De Visser M, Bolhuis PA, et al. Duplication in chromosome 17p11.2 in Charcot-Marie-Tooth neuropathy type 1a (CMT 1a). The HMSN Collaborative Research Group. Neuromuscul Disord. 1991;1:93–97. doi: 10.1016/0960-8966(91)90055-w. [DOI] [PubMed] [Google Scholar]
- Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter LT, Murakami T, Koeuth T, Pentao L, Muzny DM, Gibbs RA, Lupski JR. A recombination hotspot responsible for two inherited peripheral neuropathies is located near a mariner transposon-like element. Nat Genet. 1996;12:288–297. doi: 10.1038/ng0396-288. [DOI] [PubMed] [Google Scholar]
- Ricard G, Molina J, Chrast J, Gu W, Gheldof N, Pradervand S, Schutz F, Young JI, Lupski JR, Reymond A, Walz K. Phenotypic consequences of copy number variation: insights from Smith-Magenis and Potocki-Lupski syndrome mouse models. PLoS Biol. 2010;8:e1000543. doi: 10.1371/journal.pbio.1000543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccardi VM, Lupski JR. Duplications, deletions, and single-nucleotide variations: the complexity of genetic arithmetic. Genet Med. 2013;15:172–173. doi: 10.1038/gim.2012.124. [DOI] [PubMed] [Google Scholar]
- Roa BB, Garcia CA, Suter U, Kulpa DA, Wise CA, Mueller J, Welcher AA, Snipes GJ, Shooter EM, Patel PI, Lupski JR. Charcot-Marie-Tooth disease type 1A. Association with a spontaneous point mutation in the PMP22 gene. N Engl J Med. 1993;329:96–101. doi: 10.1056/NEJM199307083290205. [DOI] [PubMed] [Google Scholar]
- Rovelet-Lecrux A, Hannequin D, Raux G, Le Meur N, Laquerriere A, Vital A, Dumanchin C, Feuillette S, Brice A, Vercelletto M, et al. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet. 2006;38:24–26. doi: 10.1038/ng1718. [DOI] [PubMed] [Google Scholar]
- Saini N, Ramakrishnan S, Elango R, Ayyar S, Zhang Y, Deem A, Ira G, Haber JE, Lobachev KS, Malkova A. Migrating bubble during break-induced replication drives conservative DNA synthesis. Nature. 2013;502:389–392. doi: 10.1038/nature12584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakofsky CJ, Roberts SA, Malc E, Mieczkowski PA, Resnick MA, Gordenin DA, Malkova A. Break-induced replication is a source of mutation clusters underlying kataegis. Cell Rep. 2014;7:1640–1648. doi: 10.1016/j.celrep.2014.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster SC, Miller W, Ratan A, Tomsho LP, Giardine B, Kasson LR, Harris RS, Petersen DC, Zhao F, Qi J, et al. Complete Khoisan and Bantu genomes from southern Africa. Nature. 2010;463:943–947. doi: 10.1038/nature08795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, Maner S, Massa H, Walker M, Chi M, et al. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–528. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- Sharp AJ, Hansen S, Selzer RR, Cheng Z, Regan R, Hurst JA, Stewart H, Price SM, Blair E, Hennekam RC, et al. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat Genet. 2006;38:1038–1042. doi: 10.1038/ng1862. [DOI] [PubMed] [Google Scholar]
- Shinawi M, Liu P, Kang SH, Shen J, Belmont JW, Scott DA, Probst FJ, Craigen WJ, Graham BH, Pursley A, et al. Recurrent reciprocal 16p11.2 rearrangements associated with global developmental delay, behavioural problems, dysmorphism, epilepsy, and abnormal head size. J Med Genet. 2010;47:332–341. doi: 10.1136/jmg.2009.073015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara M, Shinohara A. Multiple pathways suppress non-allelic homologous recombination during meiosis in Saccharomyces cerevisiae. PLoS One. 2013;8:e63144. doi: 10.1371/journal.pone.0063144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankiewicz P, Lupski JR. Genome architecture, rearrangements and genomic disorders. Trends Genet. 2002;18:74–82. doi: 10.1016/s0168-9525(02)02592-1. [DOI] [PubMed] [Google Scholar]
- Stankiewicz P, Lupski JR. Structural variation in the human genome and its role in disease. Annu Rev Med. 2010;61:437–455. doi: 10.1146/annurev-med-100708-204735. [DOI] [PubMed] [Google Scholar]
- Stefansson H, Rujescu D, Cichon S, Pietilainen OP, Ingason A, Steinberg S, Fossdal R, Sigurdsson E, Sigmundsson T, Buizer-Voskamp JE, et al. Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–236. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144:27–40. doi: 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanzi RE, Bird ED, Latt SA, Neve RL. The amyloid beta protein gene is not duplicated in brains from patients with Alzheimer’s disease. Science. 1987;238:666–669. doi: 10.1126/science.2890207. [DOI] [PubMed] [Google Scholar]
- The Deciphering Developmental Disorders S. Large-scale discovery of novel genetic causes of developmental disorders. Nature. 2014 doi: 10.1038/nature14135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmerman V, Rautenstrauss B, Reiter LT, Koeuth T, Lofgren A, Liehr T, Nelis E, Bathke KD, De Jonghe P, Grehl H, et al. Detection of the CMT1A/HNPP recombination hotspot in unrelated patients of European descent. J Med Genet. 1997;34:43–49. doi: 10.1136/jmg.34.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treadwell-Deering DE, Powell MP, Potocki L. Cognitive and behavioral characterization of the Potocki-Lupski syndrome (duplication 17p11.2) J Dev Behav Pediatr. 2010;31:137–143. doi: 10.1097/DBP.0b013e3181cda67e. [DOI] [PubMed] [Google Scholar]
- Trivellin G, Daly AF, Faucz FR, Yuan B, Rostomyan L, Larco DO, Schernthaner-Reiter MH, Szarek E, Leal LF, Caberg JH, et al. Gigantism and acromegaly due to Xq26 microduplications and GPR101 mutation. N Engl J Med. 2014;371:2363–2374. doi: 10.1056/NEJMoa1408028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner DJ, Miretti M, Rajan D, Fiegler H, Carter NP, Blayney ML, Beck S, Hurles ME. Germline rates of de novo meiotic deletions and duplications causing several genomic disorders. Nat Genet. 2008;40:90–95. doi: 10.1038/ng.2007.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walz K, Caratini-Rivera S, Bi W, Fonseca P, Mansouri DL, Lynch J, Vogel H, Noebels JL, Bradley A, Lupski JR. Modeling del(17)(p11.2p11.2) and dup(17)(p11.2p11.2) contiguous gene syndromes by chromosome engineering in mice: phenotypic consequences of gene dosage imbalance. Mol Cell Biol. 2003;23:3646–3655. doi: 10.1128/MCB.23.10.3646-3655.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walz K, Paylor R, Yan J, Bi W, Lupski JR. Rai1 duplication causes physical and behavioral phenotypes in a mouse model of dup(17)(p11.2p11.2) J Clin Invest. 2006;116:3035–3041. doi: 10.1172/JCI28953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R, Saemundsen E, Stefansson H, Ferreira MA, Green T, et al. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med. 2008;358:667–675. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- Wheeler DA, Srinivasan M, Egholm M, Shen Y, Chen L, McGuire A, He W, Chen YJ, Makhijani V, Roth GT, et al. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–876. doi: 10.1038/nature06884. [DOI] [PubMed] [Google Scholar]
- Wilson MA, Kwon Y, Xu Y, Chung WH, Chi P, Niu H, Mayle R, Chen X, Malkova A, Sung P, Ira G. Pif1 helicase and Poldelta promote recombination-coupled DNA synthesis via bubble migration. Nature. 2013;502:393–396. doi: 10.1038/nature12585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiszniewska J, Bi W, Shaw C, Stankiewicz P, Kang SH, Pursley AN, Lalani S, Hixson P, Gambin T, Tsai CH, et al. Combined array CGH plus SNP genome analyses in a single assay for optimized clinical testing. Eur J Hum Genet. 2014;22:79–87. doi: 10.1038/ejhg.2013.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright CF, Fitzgerald TW, Jones WD, Clayton S, McRae JF, van Kogelenberg M, King DA, Ambridge K, Barrett DM, Bayzetinova T, et al. Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data. Lancet. 2014 doi: 10.1016/S0140-6736(14)61705-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu N, Ming X, Xiao J, Wu Z, Chen X, Shinawi M, Shen Y, Yu G, Liu J, Xie H, et al. TBX6 null variants and a common hypomorphic allele in congenital scoliosis. N Engl J Med. 2015;372:341–350. doi: 10.1056/NEJMoa1406829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, Braxton A, Beuten J, Xia F, Niu Z, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med. 2013;369:1502–1511. doi: 10.1056/NEJMoa1306555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Muzny DM, Xia F, Niu Z, Person R, Ding Y, Ward P, Braxton A, Wang M, Buhay C, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA. 2014;312:1870–1879. doi: 10.1001/jama.2014.14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatsenko SA, Hixson P, Roney EK, Scott DA, Schaaf CP, Ng YT, Palmer R, Fisher RB, Patel A, Cheung SW, Lupski JR. Human subtelomeric copy number gains suggest a DNA replication mechanism for formation: beyond breakage-fusion-bridge for telomere stabilization. Hum Genet. 2012;131:1895–1910. doi: 10.1007/s00439-012-1216-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yauk CL, Aardema M, van Benthem J, Bishop JB, DeMarini DM, Dubrova YE, Honma M, Lupski JR, Marchetti F, Meistrich ML, et al. Approaches for identifying germ cell mutagens: Report of the 2013 IEWGT workshop on germ cell assays. Mutation Research: Genetic Toxicology & Environmental Genetic Toxicology. 2015 doi: 10.1016/j.mrgentox.2015.01.008. In press. [DOI] [PubMed] [Google Scholar]
- Yauk CL, Lucas Argueso J, Auerbach SS, Awadalla P, Davis SR, Demarini DM, Douglas GR, Dubrova YE, Elespuru RK, Glover TW, et al. Harnessing genomics to identify environmental determinants of heritable disease. Mutat Res. 2013;752:6–9. doi: 10.1016/j.mrrev.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeleny C. The direction and frequency of mutation in the bar-eye series of multiple allelomorphs of Drosophila. Journal of Experimental Zoology. 1921;34:202–233. [Google Scholar]
- Zhang F, Khajavi M, Connolly AM, Towne CF, Batish SD, Lupski JR. The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat Genet. 2009;41:849–853. doi: 10.1038/ng.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Potocki L, Sampson JB, Liu P, Sanchez-Valle A, Robbins-Furman P, Navarro AD, Wheeler PG, Spence JE, Brasington CK, et al. Identification of uncommon recurrent Potocki-Lupski syndrome-associated duplications and the distribution of rearrangement types and mechanisms in PTLS. Am J Hum Genet. 2010;86:462–470. doi: 10.1016/j.ajhg.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]