

Abstract
Prostate cancer is a leading cause of cancer death in men due to the subset of cancers that progress to metastasis. Prostate cancers are thought to be hardwired to androgen receptor (AR) signaling, but AR-regulated changes in the prostate that facilitate metastasis remain poorly understood. We previously noted a marked reduction in Secreted protein, acidic and rich in cysteine-like 1 (SPARCL1) expression during invasive phases of androgen-induced prostate growth, suggesting that this may be a novel invasive program governed by AR. Herein, we show that SPARCL1 loss occurs concurrently with AR amplification or overexpression in patient based data. Mechanistically, we demonstrate that SPARCL1 expression is directly suppressed by androgen-induced AR activation and binding at the SPARCL1 locus via an epigenetic mechanism, and these events can be pharmacologically attenuated with either AR antagonists or HDAC inhibitors. We establish using the Hi-Myc model of prostate cancer that in Hi-Myc/Sparcl1−/− mice, SPARCL1 functions to suppress cancer formation. Moreover, metastatic progression of Myc-CaP orthotopic allografts is restricted by SPARCL1 in the tumor microenvironment. Specifically, we show that SPARCL1 both tethers to collagen in the extracellular matrix (ECM) and binds to the cell's cytoskeleton. SPARCL1 directly inhibits the assembly of focal adhesions thereby constraining the transmission of cell traction forces. Our findings establish a new insight into AR-regulated prostate epithelial movement and provide a novel framework whereby, SPARCL1 in the ECM microenvironment restricts tumor progression by regulating the initiation of the network of physical forces that may be required for metastatic-invasion of prostate cancer.
Keywords: SPARCL1, Hevin, Prostate, AR, Metastasis
Introduction
Despite progress in early detection and therapeutics, prostate cancer is a leading cause of cancer related mortality among men in the United States. AR signaling is integral to prostate cancer progression (1) and thus an increased understanding of AR modulated barriers that typically constrain prostate cancer progression is critical for further development of prognostic biomarkers and therapeutics. Using an AR-induced developmental system to model cancer progression (2, 3), we previously identified SPARCL1 (also called Hevin and SC1) as a potential novel AR-regulated gene. SPARCL1 is markedly downregulated during androgen-induced invasion during prostate development (4). Consistent with this, SPARCL1 expression also inversely correlates with prostate cancer aggressiveness; and its loss in clinically localized prostate cancer is a significant and independent prognostic factor of metastatic recurrence following surgery (4, 5). The mechanisms triggering SPARCL1 downregulation during physiologic or pathologic growth in the prostate are not known; however, the paralleled loss of SPARCL1 mRNA and protein suggests that SPARCL1 loss in many prostate cancers may be attributed to deregulation of SPARCL1 gene expression. Collectively, this implicates SPARCL1 as a potential AR-regulated gene.
While several functional studies support that SPARCL1 restricts tumor growth and progression (4, 6, 7), the role for SPARCL1 in prostate cancer remains poorly understood. The correlation between SPARCL1 loss and aggressiveness of clinically localized prostate cancer suggests that SPARCL1 may function as a barrier to tumor initiation and progression in the prostate (4). Consistent with this, overexpression of SPARCL1 in colon cancer cells suppressed growth of subcutaneous xenografts (6). While SPARCL1 has been shown to inhibit in vitro proliferation of colon cancer (6) and HeLa (8) cells, other studies support that SPARCL1 may not regulate cellular proliferation in the prostate (4, 7). Alternatively, SPARCL1 has been shown in multiple models to inhibit processes integral to both local and metastatic progression such as cancer cell adhesion, migration and invasion (4, 6, 7, 9, 10). Two recent reports demonstrate that SPARCL1 suppresses tumor nodule formation in visceral organs following intravenous injection (6, 7). While these studies collectively support that SPARCL1 constrains cancer growth; the precise role of SPARCL1 in the step-wise progression from prostate tumor initiation through localized progression has not been definitively examined in an autochthonous model. Thus it remains to be determined if SPARCL1 functions as a bona fide metastasis suppressor gene by limiting metastatic progression without affecting primary tumor growth or if SPARCL1 functions as a barrier to both localized and metastatic tumor progression in the prostate.
Herein, we delineate a specific AR-regulated pathway that facilitates prostate cancer progression. We demonstrate that direct AR binding at the SPARCL1 locus inhibited SPARCL1 expression through epigenetic modifications and that this could be pharmacologically modulated by either AR antagonists or HDAC inhibitors. In two independent patient based cohorts, we note that loss of SPARCL1 expression in the prostate significantly co-occurred with AR amplification or over expression. Using an animal model that recapitulates human prostate cancer progression, we demonstrate that SPARCL1 functions to suppress adenocarcinoma formation in the prostate. While temporal loss of SPARCL1 in invading epithelial buds has been shown to be necessary for prostate development (4), we show that constitutive absence of SPARCL1 did not lead to a hyperplastic phenotype. In the context of oncogenic activation such as c-MYC, SPARCL1 functioned to suppress tumor formation and limit metastatic progression. As a matricellular protein, SPARCL1 is secreted in the extracellular matrix (ECM). Mechanistically, we report that SPARCL1 in the ECM inhibited both biological and biophysical properties associated with cellular migration and invasion such as dynamics of cytoskeletal remodeling, focal adhesion assembly, cell stiffness, and cell traction forces. These data support the potential utility of targeting SPARCL1 and its associated pathways for therapeutic treatment to prevent local progression or to halt metastatic spread.
Materials and Methods
In vitro organ culture (4), UGE and UGM isolation (4), Quantitative real-time PCR (4), Johns Hopkins University prostate cancer anti-androgen therapy tissue microarray (11), Androgen gene regulation (12), Chromatin immunoprecipitation assay (12), Cell line methylation status (13, 14), Immunohistochemistry (4), Immunofluorescence (4), Cell proliferation (4), Live cell micromechanical methods (15-18), Fourier transform traction microscopy (17-20) and Statistical analysis (4) have been described previously and are detailed in the Supplementary Materials and Methods.
Pharmacological epigenetic modulation experiments
LNCaP, VCaP, 22RV1, and PC3 cells were treated with vehicle or 1μM 5-Aza-2’-deoxycytidine (Sigma-Aldrich) for 3 days. Similarly, LNCaP, VCaP, 22RV1, and PC3 cells were treated with 1-5nM Vorinostat (SelleckChem) or vehicle for 48 hours. Media with vehicle or Vorinostat was changed daily. LNCaP cells were treated with 1nM Panobinostat (SelleckChem) or vehicle for 24 hours.
Sparcl1 deficient mice and Hi-Myc mice
This protocol was approved by the Johns Hopkins University Animal Care and Use Committee. Sparcl1−/− 129/SvEv mice were gifted to our laboratory by Cagla Eroglu, PhD at Duke University (21). Sparcl1−/− 129/SvEv mice were backcrossed greater than seven generations to FVB/N. FVB-Tg(ARR2/Pbsn-MYC)7Key (Hi-Myc/FVB/N) were obtained from Jackson Labs. Sparcl1−/−FVB/N mice were crossed with Hi-Myc/FVB/N transgenic mice to obtain Sparcl1+/−/Hi-Myc mice. Sparcl1+/−/Hi-Myc were mated to generate the crosses used in this study. Prostates were harvested at 4.5 months (138 ± 2 days), formalin-fixed, paraffin embedded, and sectioned using standard methods. Two H&E stained slides 100μm apart were examined for evidence of adenocarcinoma by the study pathologist.
Orthotopic allografts
1×106 Myc-CaP cells in Matrigel were grafted into the anterior prostate of male WT and Sparcl1−/− FVB mice that were greater than 10 weeks in age. 1×106 Myc-CaP-Neo (B) or Myc-CaP–mSPARCL1 (A) cells in Matrigel were grafted into the anterior prostate of male WT FVB mice that were greater than 10 weeks in age. An Isoflurane gas anesthetic system (Caliper Life Sciences) was used for surgical anesthesia. A midline incision was made in the lower abdomen to externalize the seminal vesicles, bladder, and prostate. Following injection of 10μl of cells 1:1 in Matrigel into the anterior lobe of the prostate, the wound was closed with surgical sutures and surgical metal clips. Mice were monitored daily for distress. Mice were euthanized at day 21, necropsied and inspected for gross evidence of metastatic disease. Organs were removed, formalin-fixed, paraffin embedded, and sectioned using standard methods. H&E stained slides were examined for evidence of metastatic disease by the study pathologist.
Results
Androgen suppresses Sparcl1 expression during prostate development
We previously reported a marked suppression of Sparcl1 gene expression during invasive phases of androgen induced prostate development and regeneration in vivo (4). To determine if androgen signaling mediates Sparcl1 gene repression, we examined Sparcl1 expression during prostate development in response to 5α-dihydrotestosterone (DHT). DHT treatment of androgen naïve urogenital sinus (UGS) specifically suppressed Sparcl1 expression in the invading epithelium (UGE) compared to mesenchyme (UGM) (Fig. 1A-C). As SPARCL1 has been shown to inhibit epithelial bud outgrowth (4), these results suggest that androgen facilitates epithelial bud invasion by suppressing Sparcl1 gene expression.
Figure 1.

Androgen represses SPARCL1 expression in prostate cancer. A, B, Sparcl1 expression in (A) UGE and (B) UGM (n≥3) of DHT treated UGS. C, WT male UGS were examined by IHC for SPARCL1 (n=3; 400× magnification). D, Co-occurrence of SPARCL1 loss and AR amplification or overexpression in MSKCC cohort analyzed from www.cbioportal.com and Mayo Clinic cohort. E, F, SPARCL1 and KLK3 expression as determined by qRT-PCR in (E) LNCaP and (F) VCaP cells in 10% FCS with vehicle, in 10% androgen depleted FCS (C/D serum), and 10% C/D serum with 100nM DHT (n≥3). G, SPARCL1 expression as determined by qRT-PCR in LNCaP cells treated with vehicle or 50μM MDV3100 (n=3). H, TMA containing localized tumor samples from controls (n=18) who did not receive therapy prior to radical prostatectomy (NT) and cases (n=46) who were treated with a luteinizing hormone-releasing hormone agonist (anti-androgen therapy) before sampling at radical prostatectomy was examined for SPARCL1 expression by IHC. I, Schematic of a potential AR binding site in a putative promoter/enhancer region of SPARCL1 located in the intron 3’ to the transcriptional start site (TSS). Black bars and numbers underneath represent genomic location in relation to the TSS amplified by qRT-PCR primers following ChIP of AR. J, ChIP of AR in LNCaP cells treated with vehicle or 100nM DHT (n≥3). Statistical analysis performed by One-way ANOVA with Tukey's Multiple Comparison Test (A, E, F), Student's t test (G), Student's t test with Welsh's Correction (H), and Two-way ANOVA (J) (mean ± SEM; *P≤0.05, **P≤0.01, ****P≤0.0001).
Androgen suppresses SPARCL1 expression in prostate cancer
We previously reported that SPARCL1 expression inversely correlates with Gleason grade with the most pronounced loss seen in metastatic lesions (4). Herein, we postulated that, similar to prostate development, androgens also repress SPARCL1 expression in prostate cancer. In canonical androgen signaling, androgens regulate gene transcription through AR. Analysis of human gene expression data showed that AR gene amplification and overexpression occurred concurrently with the loss of SPARCL1 expression with an Odds Ratio of 9.23 (95% CI: 3.92-21.74; P=0.000001 by Fisher's exact test) (22-24) (Fig. 1D). We validated this finding in a prospectively-designed study of high-risk men who underwent radical prostatectomy at the Mayo Clinic (4, 25, 26). We found that the loss of SPARCL1 expression co-occurred with overexpression of AR with an Odds Ratio of 2.78 (95% CI: 1.59 to 4.90; P=0.0001485 by Fisher's exact test) (Fig. 1D). To determine if androgen directly suppressed SPARCL1 gene expression, we assayed for SPARCL1 expression in response to androgen depletion. Androgen was removed by culturing androgen responsive prostate cancer cell lines, LNCaP and VCaP, in media with charcoal:dextran stripped fetal bovine serum (C/D serum). Compared with cells grown with serum, LNCaP and VCaP cells grown with C/D serum showed a significant increase in SPARCL1 gene expression (Fig. 1E, F). SPARCL1 gene expression in turn decreased with the addition of DHT while KLK3 (PSA), a known androgen-induced gene, was increased with similar kinetics (Fig. 1E, F). Furthermore, chemical inhibition of AR with MDV3100/Enzalutamide, an AR antagonist approved for the treatment of metastatic prostate cancer, significantly increased SPARCL1 gene expression in prostate cancer cells in the presence of androgen (Fig. 1G). Consistent with these results, men treated with anti-androgen therapy (ADT) prior to radical prostatectomy (RP) for localized prostate cancer (n=46) had significantly higher SPARCL1 expression in their prostate at RP by IHC compared to men who did not have ADT (n=18) (Fig. 1H). SPARCL1 expression was also increased in an additional cohort of men treated with ADT compared to untreated men (Supplementary Fig. S1A) (27). Complementary to data in humans, treatment of castrated mice with DHT decreased SPARCL1 levels in the prostate (Supplementary Fig. S1B). Collectively, these results demonstrate that androgen functions through AR to suppress SPARCL1 gene expression in prostate cancer.
To determine if AR directly suppresses SPARCL1 gene expression, we examined AR binding at the SPARCL1 locus. Androgen binding to AR induces AR recruitment to a conserved 15 base-pair palindromic androgen response element (ARE) on target genes (28, 29). In silico analyses of previously published chromatin immunoprecipitation coupled with massively parallel sequencing (ChIP-Seq) data demonstrated that synthetic androgen (R1881) induced AR binding at the SPARCL1 locus in LNCaP and VCaP cells (Supplementary Fig. S2A, B) (30). ChIP-Seq data also demonstrated that AR bound to the SPARCL1 locus in human prostate tissue (30). Similar to many AR target genes, this SPARCL1 locus contained a potential non-canonical ARE (NC-ARE) with 75% homology to the canonical ARE (28) encompassing a putative promoter/enhancer region (Fig. 1I). We validated these results using targeted ChIP of AR in LNCaP, an AR responsive prostate cancer cell line (Fig. 1J). When treated with DHT, androgen induced AR binding within the region encompassing the NC-ARE in SPARCL1, but not in a control upstream region. Dynamics of AR binding at SPARCL1 were similar to other known AR targets such as PSA and TMPRSS2 (Supplementary Fig. S2C, D). These data, taken together support the conclusion that androgen directly suppresses SPARCL1 expression through AR binding at the SPARCL1 locus.
AR mediates SPARCL1 suppression through modulation of histone acetylation
How AR negatively regulates transcriptional activation by direct DNA binding is not fully understood. Because suppressors of tumor and metastatic progression can be epigenetically repressed in cancer (31, 32), and aberrant epigenetic modifications such as DNA methylation and histone acetylation often occur in prostate cancer (33), we considered if SPARCL1 expression is governed by epigenetic modifications at the SPARCL1 locus. In order to assess the role of promoter hypermethylation on SPARCL1 expression, we treated prostate cancer cell lines with a DNA methyltransferase inhibitor (5-Aza-2’-deoxycytidine). 5-Aza-2’-deoxycytidine treatment did not increase SPARCL1 expression (Fig. 2A) but did increase expression of the control gene GSTP1 (Supplementary Fig. S2E), which is known to be epigenetically repressed by promoter DNA hypermethylation in prostate cancer cells. Notably, CpG methylation patterns at the SPARCL1 locus in cancer cells were comparable to that of benign primary prostate epithelial cells (PrEC) (Fig. 2B). These findings are consistent with prior studies in cancers of other origins (9, 34), and indicate that promoter CpG methylation is not primarily responsible for the observed suppression of SPARCL1 in prostate cells.
Figure 2.
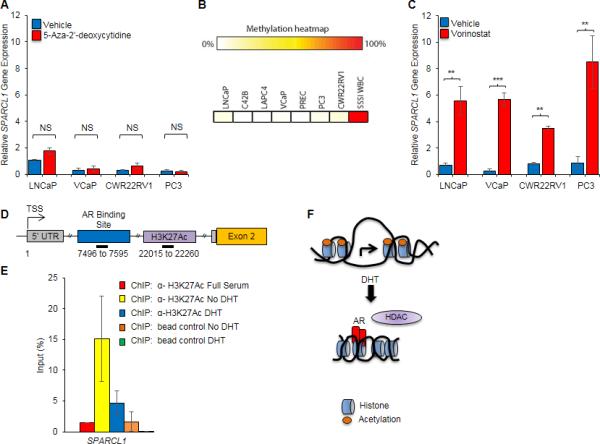
Androgen mediated chromatin remodeling at the SPARCL1 locus. A, SPARCL1 expression as determined by qRT-PCR in LNCaP, VCaP, CWR22RV1, and PC3 cells treated with vehicle or 1 μM 5-Aza-2’-deoxycytidine (DNA methyltransferase inhibitor) for 3 days (n=3). B, Methylation heatmap of SPARCL1 locus in prostate cancer cell lines (LNCaP, C42B, LAPC4, VCaP, PC3, CWR22RV1), primary prostate cells (PREC), and positive control (SSSI WBC). C, SPARCL1 expression as determined by qRT-PCR in LNCaP, VCaP, CWR22RV1 and PC3 cells treated with vehicle or Vorinostat (HDAC inhibitor) for 48 hours (n=3). D, Schematic of a potential acetylation site in the putative promoter/enhancer region of SPARCL1. Black bars and numbers underneath represent genomic location in relation to the TSS amplified by qRT-PCR primers following ChIP of AR or H3K27Ac. E, ChIP of H3K27Ac in LNCaP cells treated with vehicle or 100nM DHT (n≥3). F, Schematic of Androgen mediated suppression of SPARCL1 expression. Statistical analyses performed by Student's t test (A and C) (mean ± SEM; NS=not significant, **P≤0.01, ***P≤0.001).
Another important mechanism involved in gene regulation is chromatin remodeling through histone acetylation. Histone acetylation leads to formation of open chromatin, which thereby allows for transcriptional activation. In the absence of acetylation, transcription is hindered due to formation of closed chromatin conformations. HDACs remove acetyl groups from ε-N-acetyl lysine (K) in histones to regulate gene transcription. Inhibition of HDACs with Vorinostat, an HDAC inhibitor currently in clinical trials for the treatment of metastatic prostate cancer, or a broad spectrum HDAC inhibitor, Panobinostat, significantly increased SPARCL1 expression, even in the presence of androgen, suggesting that HDACs function downstream of AR binding to the SPARCL1 locus (Fig. 2C and Supplementary Fig. S2F). In androgen-independent prostate cancer cells (PC3), HDAC inhibition also increased SPARCL1 expression (Fig. 2C). In addition, ChIP-Seq data from ENCODE and analyzed through the UCSC genome browser (hg19) supports that a region downstream of the NC-ARE at the SPARCL1 locus is enriched for the Histone H3 acetyl Lys27 (H3K27Ac) marks (35), which are active transcription marks often found at transcriptional promoters and/or enhancers (36) (Fig. 2D). In the absence of androgen, ChIP for H3K27Ac demonstrated that this region in SPARCL1 was acetylated (Fig. 2E). Upon androgen stimulation, H3K27Ac was lost at the SPARCL1 locus (Fig. 2E and Supplementary Fig. S2G). Collectively, these results are consistent with a model of androgen-induced AR binding and HDAC-mediated deacetylation at the SPARCL1 locus thereby functioning to suppress SPARCL1 expression (Fig. 2F).
Sparcl1 is dispensable for prostate development
Prostate development occurs in an undifferentiated UGS when androgens induce proliferation and invasion of UGE into the surrounding mesenchyme to form epithelial prostate buds. Prior studies support that discrete loss of SPARCL1 expression within the invading epithelial bud tip is required for bud migration and invasion during bud elongation (4) (Fig. 1C). It is not known, however, if SPARCL1 expression elsewhere in the UGE is necessary to restrict appendicular patterning. When compared to WT male UGS in organ culture, Sparcl1−/− UGS had similar spatial patterning, length and number of buds (Fig. 3A-C). Analogous to early development, SPARCL1 was also dispensable for adult prostate maturation. Sparcl1−/− adult prostates were grossly indistinguishable from WT prostates as measured by appearance and weight (Fig. 3D, E). Histological and immunohistological analyses of Sparcl1−/− prostates ≥12 months (n=20) demonstrated comparable gland architecture including basal (p63) and luminal (CK8) cell number (Fig. 3F) and ratio (Fig. 3G) as WT. Consistent with prior studies demonstrating that SPARCL1 does not regulate cellular proliferation in the prostate (4, 7), Sparcl1−/− prostates did not have elevated proliferation as measured by Ki67 (Fig. 3F). Consistent with this, Sparcl1−/− prostates did not have histological evidence of hyperplasia, cancer precursor lesions [murine prostatic intraepithelial neoplasia (mPIN)] or invasive prostate adenocarcinoma formation (Fig. 3D, E, F). These data demonstrate that, while the temporal loss of SPARCL1 has been shown to be necessary for bud migration and invasion (4), its constitutive absence does not disrupt early prostate development or adult maturation and is not sufficient to initiate prostate carcinogenesis. This is consistent with the notion that while SPARCL1 loss is necessary to release a “brake” to physiological prostate development, its loss is not sufficient to activate a corresponding “acceleration” switch.
Figure 3.
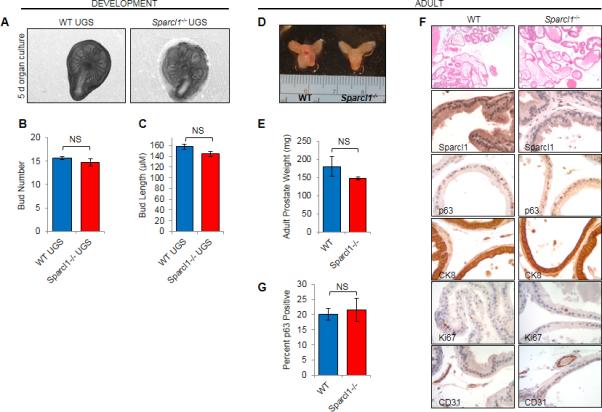
Sparcl1 is not necessary for prostate development. A-C, Bud number and length determined from photomicrographs of WT and Sparcl1−/− male UGS (n≥3) cultured in vitro for 5 days. D-E, Adult (8.5 months) prostates from WT and Sparcl1−/− were weighed (n≥5). F-G, WT and Sparcl1−/− prostates were examined by H&E (n=20 at ≥12 months; 40× magnification). Expression of p63, CK8 and Ki67 were examined in WT and Sparcl1−/− prostates by IHC (n≥3; 400× magnification). Statistical analyses performed by Student's t test (mean ± SEM; NS=not significant).
SPARCL1 functions to suppress adenocarcinoma formation in the prostate
In contrast to prostate cancer initiation, compelling data suggest that SPARCL1 functions as a tumor suppressor by restricting local invasion (6, 9). To test the hypothesis that SPARCL1 inhibits local invasion driving the transition between cancer pre-cursor lesions and invasive adenocarcinoma, we modeled dynamics of human prostate cancer in mice. MYC is one of the most commonly amplified and overexpressed oncogenes in human prostate cancer. Prostate specific overexpression of MYC is a widely used mouse model of prostate cancer that recapitulates human disease progression. Hi-Myc transgenic mice develop murine prostatic intraepithelial neoplasia (mPIN) lesions as early as 2 weeks that progress to locally invasive adenocarcinoma of the prostate by 6 months (37). Consistent with human prostate cancer (Fig. 4A), SPARCL1 loss correlates with tumor progression in Hi-Myc mice (Fig. 4B, C) with the most pronounced loss observed in Myc-CaP cells (38), a spontaneously immortalized cell line derived from a Hi-Myc prostate. To determine the role of SPARCL1 in cancer progression, we examined prostate carcinogenesis in Hi-Myc mice with heterozygous (Hi-Myc/Sparcl1+/−) and homozygous (Hi-Myc/Sparcl1−/−) genetic deletions of Sparcl1. Consistent with prior studies demonstrating that SPARCL1 does not restrict cellular proliferation in prostate cells (4, 7), prostate weight was comparable between Hi-Myc and Hi-Myc/Sparcl1−/− mice at 4.5 months (Fig. 4D). However, pathological examination demonstrated that homozygous loss of Sparcl1 in Hi-Myc mice led to a significant increase (by over 50%) in invasive adenocarcinoma incidence at 4.5 months (Fig. 4E, F). An intermediate phenotype was seen with the loss of one copy of Sparcl1 with an increase in incidence of nearly 20% over Hi-Myc mice at 4.5 months (Fig. 4E). These results support that SPARCL1 restricts the transition from pre-cancerous lesions to invasive carcinoma.
Figure 4.

SPARCL1 suppresses adenocarcinoma formation in the prostate. A, Representative image of SPARCL1 expression examined by IHC in human benign adjacent (1), PIN (2), and adenocarcinoma (3) in the prostate. B, Representative images of SPARCL1 expression examined by IHC in benign adjacent, PIN, and prostate cancer from Hi-Myc mice at 6 months of age (400×). C, Sparcl1 expression examined by qRT-PCR in WT murine prostates at 5 months (n=3), Hi-Myc murine prostates at 4.5 (n=3) and 6 months (n=3), and Myc-CaP cells. D, Prostates from 4.5 month old WT (n=6), Sparcl1−/− (n=8), Hi-Myc (n=5), Hi-Myc/Sparcl1+/− (n=8) and Hi-Myc/Sparcl1−/− (n=11) mice were weighed. E, F, Prostates from 4.5 month old WT (n=7), Sparcl1−/− (n=10), Hi-Myc (n=60), Hi-Myc/Sparcl1+/− (n=10) and Hi-Myc/Sparcl1−/− (n=11) mice were examined by H & E by the study pathologist (100× magnification). G, SPARCL1 expression in Myc-CaP-EV stable clones (A and B) and Myc-CaP-mSPARCL1 clones (A and B) as determined by immunoblotting. H, In vitro proliferation of Myc-CaP-EV and Myc-CaP-mSPARCL1 stable clones as measured by MTT assay. I, J, Orthotopic allografts of Myc-CaP-EV (B) and Myc-CaP-mSPARCL1 (A) at 21 days (n≥4). Statistical analyses performed by Oneway ANOVA (C), Chi-squared test (F), and Student's t test (J) (mean ± SEM; *P≤0.05; **P≤0.01).
As SPARCL1 expression is inversely associated with cancer aggressiveness as measured by Gleason grade, we tested the hypothesis that SPARCL1 restricts tumor progression. To examine this, Myc-CaP cells overexpressing murine SPARCL1 (Myc-CaP-mSPARCL1) or empty-vector (Myc-CaP-EV) were orthotopically allografted into WT prostates (Fig. 4G and Supplementary Fig. S4A). While in vitro proliferation was not significantly different between Myc-CaP-EV and Myc-CaP-mSPARCL1 cells, Myc-CaP-mSPARCL1 allografts were significantly smaller compared to Myc-CaP-EV allografts (Fig. 4H-J). Collectively, these results suggest that SPARCL1 functions as a barrier to tumor formation and that its loss contributes to tumor progression.
SPARCL1 in the tumor microenvironment suppresses metastatic progression in the prostate
As a matricellular protein, SPARCL1 is secreted into the ECM and regulates cellular function. We postulated that if SPARCL1 inhibited tumor invasion, then prostate cancer cells would be more invasive in Sparcl1−/− prostates compared to WT prostates. To examine the role of SPARCL1 as a matricellular protein in the tumor microenvironment, Myc-CaP cells, which do not express SPARCL1 (Fig. 4C), were orthotopically allografted into the prostates of WT and Sparcl1−/− mice. Myc-CaP tumor size (measured by GU weight) and proliferation (measured by Ki67) were comparable between WT and Sparcl1−/− mice (Fig. 5A, B and Supplementary Fig. S3B) suggesting that SPARCL1 in the local tumor environment secreted by adjacent benign cells was not sufficient to restrict tumor size. While Hi-Myc mice only very rarely develop metastases (37, 39), Myc-CaP cells (38) metastasize to regional and distant lymphatic tissues when grafted orthotopically into the prostate (40). In contrast to tumor size, Myc-CaP orthotopic allografts yielded an elevated number of metastases in Sparcl1−/− mice compared to WT mice (Fig. 5C, D). Similar to prior studies in WT mice, most metastases in both WT and Sparcl1−/− mice were to regional and distant lymphatic tissues; however, hematogenous spread to the lungs was observed in one Sparcl1−/− mouse (Fig. 5C). These results suggest that SPARCL1 in the ECM tumor microenvironment restricts metastatic progression.
Figure 5.

SPARCL1 in the tumor microenvironment restricts metastatic progression. A, Photomicrographs of Myc-CaP orthotopic allografts in WT and Sparcl1−/− prostates. B, Average GU weight of Myc-CaP orthotopic allografts in WT (n=7) and Sparcl1−/− prostates (n=7). C, H&E (100× magnification) of metastases from Myc-CaP orthotopic allografts in WT and Sparcl1−/− prostates. D, Average number of metastases from Myc-CaP orthotopic allografts in WT (n=19) and Sparcl1−/− (n=20) prostates. Statistical analyses performed by Student's t test (mean ± SEM; NS=not significant; *P≤0.05).
SPARCL1 modulates collagen-regulated mechanical properties of prostate cancer cells
Collagen has been shown to regulate both biophysical and biochemical dynamics of the tumor microenvironment. How SPARCL1 functions within the framework of a collagen matrix to inhibit cellular migration and invasion and, ultimately tumor progression, is not fully understood (4, 6, 7). To begin to understand the mechanism(s), we used RGD-coated ferrimagnetic microbeads anchored to the cytoskeleton through cell surface integrin receptors (15-18) and probed the material properties of PC3 cells adhered on collagen matrix vs. collagen matrix containing SPARCL1 (collagen+rSPARCL1). Appling forced bead motions with Magnetic Twisting Cytometry (MTC), we first measured cytoskeletal stiffness (g’) and internal friction (g”) of PC3 cells over a wide frequency range (Fig. 6A, B). Throughout the measurement range (oscillatory frequencies from 10−1 to 103 Hz), stiffness of PC3 cells increased with frequency as a weak power law (Fig. 6B). The internal friction also followed a weak power law at low frequencies (below ~10 Hz) but showed stronger frequency dependence at higher frequencies (above ~10 Hz). These classic cellular responses were observed under both conditions (collagen vs. collagen+rSPARCL1). Nevertheless, SPARCL1 in the collagen matrix appreciably decreased PC3 cell stiffness in physiologic range of probing frequencies (Fig. 6B). The internal friction also significantly differed between cells adhered on collagen vs. collagen+rSPARCL1 at both low (1 Hz and below) and high frequencies (100 Hz and above) (Fig. 6B).
Figure 6.

SPARCL1 modulates cytoskeletal stiffness and remodeling dynamics. A, Representative bright field images of RGD-coated ferrimagnetic microbeads incubated with PC3 cells adhered on a collagen or collagen+rSPARCL1 matrix. B, Forced motions using Magnetic Twisting Cytometry were applied to determine mechanical properties including cytoskeletal stiffness (g’) and friction (g”) of cells adhered on a matrix of collagen (geometric mean ± SE; n=219) or collagen+rSPARCL1 (geometric mean ± SE; n=257; *P≤0.05, Student's t-test). The solid lines are the fit of experimental data to the structural damping equation with addition of a Newtonian viscous term as described previously (16). Accordingly, the exponent in the power law, or the slope of the solid lines, describes the material behavior of living cells and is an index along spectrum of solid-like (a Hookean elastic solid) to fluid-like (a Newtonian viscous fluid) states. Fitting was performed by nonlinear regression analysis. C-F, Spontaneous motions of RGD-coated ferrimagnetic beads incubated with PC3 cells adhered on a collagen (n=635) or collagen+rSPARCL1 matrix (n=542). C, The exponent α of cells adhered on Collagen vs. Collagen+rSPARCL1. D-F, SPARCL1 in the collagen matrix decreased the speed of cytoskeletal remodeling as measured by (D) the Diffusion coefficient and computed mean square displacement (MSD) (E) at 10s and (F) at 300s (mean ± SEM; NS=not significant; *P≤0.05).
To determine the underlying dynamics of the cytoskeletal network, we then assessed spontaneous motions of the same functionalized microbeads (15). In both conditions (collagen vs. collagen+rSPARCL1), the mean square displacements (MSD) of unforced beads increased with time as a power law with an exponent α greater than unity (Fig. 6C, D). As we have defined elsewhere (15), such anomalous bead motions are not characteristics of Brownian motions that are thermally-driven but are rather characteristics of a discrete molecular-level rearrangements (remodeling) of actin cytoskeleton driven by myosin motors. The exponent α did not differ between cells adhered on collagen vs. cells on collagen+rSPARCL1 (Fig. 6C). Compared with cells adhered on collagen, however, PC3 cells adhered on collagen+rSPARCL1 showed marked decreases in diffusion coefficient D* (a measure of the speed of cytoskeleton remodeling) and computed MSDs (Fig. 6D-F). Similar to PC3 cells grown on collagen+rSPARCL1, Myc-CaP-mSPARCL1 cells had a significantly reduced rate of cytoskeletal remodeling compared to Myc-CaP parental and Myc-CaP-EV controls when adhered to a collagen matrix (Supplementary Fig. S4B). Collectively, these data indicate that SPARCL1 in the collagen matrix acts to slow down the rate at which cells remodel their internal network of cytoskeleton. These biophysical analyses, taken together, suggest that SPARCL1 attenuates collagen-regulated mechanical correlates that may be required for metastatic-invasion of prostate cancer.
SPARCL1 inhibits transmission of cell traction force
The ability of a single living cell or, cell ensemble, to exert traction force upon its surrounding is the necessary prerequisite for diverse biological processes, such as local cellular migrations in development to metastatic-invasion of cancer (17-20). Using Fourier transform traction microscopy (19), we measured the ability of an individual PC3 cell to exert traction force upon its surrounding (collagen vs. collagen+rSPARCL1). On a collagen matrix containing SPARCL1, PC3 cells showed slower adhesion dynamics than on collagen, but exhibit similar dispersion of cell size on both matrices by 24 hours (4). Consistent with this, we found no significant differences in the computed cell projected area between cells on collagen vs. collagen+rSPARCL1 (Fig. 7A, B). Strikingly, however, cells on collagen+rSPARCL1 exhibited a much muted cell traction force than on collagen (Fig. 7C, D). Traction (root mean square) average over the entire cell projected area was significantly reduced by SPARCL1 in the collagen matrix (Fig. 7C). In addition, cells adhered on collagen+rSPARCL1 showed a two-fold reduction in net contractile moment (Fig. 7D), which is a scalar measure of the cell's contractile strength (19). These results suggest that SPARCL1 inhibits transmission of cell traction force, presumably via focal adhesions.
Figure 7.

SPARCL1 restricts traction force and focal adhesion assembly. The contractile stress arising at the interface between each adherent PC3 cell and its substrate (collagen or collagen+rSPARCL1) was measured by Fourier transform traction microscopy. A, Representative phase contrast and traction images of PC3 adhered on collagen or collagen+rSPARCL1. B, Projected cell area of PC3 cells on collagen (n=16) or collagen+rSPARCL1 (n=21). C-D, SPARCL1 in the collagen matrix decreased RMS traction and net contractile moment, a scalar measure of cell's contractile strength (mean ± SEM). E, RMS tractions near RGD-coated beads were significantly increased compared to those near SPARCL1-coated and uncoated. RMS tractions were normalized to number of beads within the defined area. Bars represent median (n=8 randomly chosen localized areas per group). F, Focal adhesion assembly measured by immunofluorescence of Paxillin (focal adhesion), Phallodin (actin fibers), and DAPI (nuclei) of PC3 cells adhered on a matrix of collagen (n=35) or collagen+rSPARCL1 (n=59) (600x magnification). G, Quantification of focal adhesion number per cell. Statistical analyses performed by Student's t test (B, C, D and G) and One-way ANOVA (E) (mean ± SEM; NS= not significant; *P≤0.05, **P≤0.005, ****P≤0.0001).
In order to test this postulate, we coated microbeads (~4.5 μm in diameter) with rSPARCL1 or RGD, allowed the beads to first adhere to the collagen matrix, and then plated PC3 cells. Using both unconstraint and constraint Fourier transform traction cytometry (19), we measured cell traction forces of individual PC3 cells encompassing the beads (rSPARCL1 vs. RGD), as well as the localized tractions near the beads (Supplementary Fig. S5A). Compared with cells adhered directly to collagen (without beads underneath) or through uncoated- beads, PC3 cells adhered to collagen through RGD-coated beads tended to increase cell spreading (Supplementary Fig. S5B). In contrast, cells adhered to collagen through SPARCL1-coated beads tended to decrease cell spreading (Supplementary Fig. S5B). Interestingly, cells adhered to collagen through RGD-coated beads show reinforcement of contractile strength whereas those adhered through SPARCL1-coated beads did not (Supplementary Fig. S5C). Moreover, localized RMS tractions around SPARCL1-coated beads were significantly lower than those around RGD-coated beads (Fig. 7E and Supplementary Fig. S5D, E). Consistent with the notion that SPARCL1 inhibits transmission of cell traction force via focal adhesions, we also observed decreased focal adhesion assembly as measured by immunofluorescent staining of paxillin and vinculin in cells adhered on collagen matrix containing SPARCL1 (Fig. 7F, G and Supplementary Fig. S5F). These results, taken together, support the conclusion that SPARCL1 inhibits collagen-induced formation of focal adhesions and transmission of cell traction forces that are required for metastatic-invasion of prostate cancer (17, 18, 41, 42).
SPARCL1 engages cell-ECM interactions
We previously reported that SPARCL1 loss increases the migratory and invasive properties of prostate cancer cells through Ras homolog gene family, member C (RHOC), a known mediator of metastatic progression (43-47). Hence, the reduction in focal adhesions and cell traction forces by SPARCL1 might be due to steric hindrance of cryptic cell attachment sites on collagen or via a selective and sensitive receptor ligation and subsequent regulation of downstream signaling. To test the latter, we again applied microbeads coated with rSPARCL1 or RGD, and measured their binding kinetics on cell surface. As expected RGD-coated beads, which tether tightly to the underlying cytoskeleton through cell surface integrin receptors, showed super-diffusive motions that tended to grow with incubation time (Supplementary Fig. S6A-C). In contrast, rSPARCL1-coated beads showed slower ligation to cell surface and exhibited largely diffusive motions that tended to decrease with incubation time (Supplementary Fig. S6A-C). Strikingly, at 24 hours, motions of rSPARCL1-coated beads emerged as super-diffusive and did not differ from those of RGD-coated beads (Supplementary Fig. S6A-C), indicating ligation of SPARCL1 to the cell's interior cytoskeletal network. These responses were concentration dependent (Supplementary Fig. S7A-C), and distinct from rSPARCL1-coated beads bound to collagen, suggesting ligation of SPARCL1 to the CSK is not due to time-dependent embedding of the beads but rather specific and selective ligation through cell surface receptors. Collectively, these results suggest that SPARCL1 not only tethers directly to collagen ECM but also binds to cell surface through as yet unidentified receptor class.
Discussion
Prostate cancer is exquisitely reliant on AR signaling and elucidation of novel AR-driven programs that mediate metastatic progression hold promise for clinically translatable targets. Our findings presented herein establish insight into a novel AR-regulated pathway. We define an AR-mediated mechanism in the prostate for both physiologic and pathologic suppression of SPARCL1. We show both in vitro and in vivo pharmacological modulation of SPARCL1 expression with anti-androgen therapies including Enzalutamide. This is consistent with previous findings showing decreased Sparcl1 gene expression during phases of androgen-induced physiologic growth in the prostate (4). We mechanistically demonstrate that androgens directly suppress SPARCL1 expression through recruitment of AR to the SPARCL1 locus. In support of this, we demonstrate that SPARCL1 loss concurrently occurs with AR amplification or overexpression in patient-based data. We further show that androgens mediate epigenetic regulation of SPARCL1 and that this can be pharmacologically modulated with HDAC inhibitors, which are currently in clinical trials for metastatic prostate cancer. Thus our data establishes a model whereby androgen-induced AR signaling inhibits SPARCL1 gene expression through chromatin remodeling.
SPARCL1 loss is associated with disease progression in multiple cancer types (4, 6, 48, 49), suggesting that its loss may be a conserved and critical step for metastatic invasion. Alternate mechanisms governing SPARCL1 expression have yet to be fully defined; however, conservation may exist among steroid responsive cancers as Estradiol has been shown to inhibit SPARCL1 gene expression (50). Studies in non-small cell lung cancer support that steroid independent pathways may similarly regulate SPARCL1 at this locus (34). Aberrant epigenetic modulation at this locus via HDACs in androgen independent cells suggests that this mechanism may also occur in castration resistant prostate cancer and in steroid independent cancers. While these studies indicate that aberrant methylation at this locus is not the driving force for SPARCL1 suppression in the prostate, this does not preclude that epigenetic modulation by DNA methylation does not occur in other cancers. In addition to epigenetic regulation of SPARCL1 expression, genomic alterations and loss of heterozygosity (49) may account for a small percentage of SPARCL1 loss observed in advance cancers.
We establish a model, whereby SPARCL1 secreted in the ECM microenvironment restricts the invasive pathways necessary for both local and metastatic disease progression. Temporal loss of SPARCL1 is necessary for epithelial bud invasion during prostate development (4), yet constitutive loss of SPARCL1 in Sparcl1−/− mice was not sufficient to initiate carcinogenesis. Consistent with prior studies (4), our in vivo findings demonstrate that SPARCL1 loss does not lead to aberrant proliferation or activation of oncogenic signaling. Instead, our data suggest that in the context of oncogenic activation, SPARCL1 functions to restrict the invasive steps required for the transition from pre-cancerous lesions (PIN) to cancer. We show that, similar to human prostate cancers, Sparcl1 gene expression was increasingly suppressed during Hi-Myc-induced prostate tumor progression. Genetic deletion of Sparcl1 in Hi-Myc mice led to a significant increase in early prostate cancer incidence with an intermediate phenotype seen in Hi-Myc/Sparcl1+/− mice. Complementary to these studies, overexpression of SPARCL1 in Myc-CaP cells restricted orthotopic allograft size. Interestingly, Myc-CaP orthotopic allografts in WT and Sparcl1−/− mice were similar in size suggesting that SPARCL1 secreted by benign adjacent prostate cells was not sufficient to suppress tumor growth. In contrast to tumor size, SPARCL1 from the host mitigated metastatic progression of Myc-CaP orthotopic allografts. These findings are consistent with prior reports showing that SPARCL1 limits tumor establishment at distant sites (4, 6, 7, 9). These data, taken together, support the conclusion that SPARCL1 functions to suppress adenocarcinoma formation and progression to metastases by attenuating the invasive pathways necessary for local and metastatic cancer invasion.
Our data offer new understanding into how SPARCL1 in the ECM microenvironment mechanistically functions as a barrier to tumor initiation and progression. Cellular migration and invasion through the ECM is dependent on the transmission of mechanical and regulatory signals between the matrix and the cell through focal adhesions. Our data demonstrate that SPARCL1 in the ECM limited cellular ability to form focal adhesions on the collagen matrix and thereby attenuated corresponding biophysical dynamics of the cytoskeletal framework which have been shown to potentiate migration. This is consistent with prior reports showing that SPARCL1 mitigates mediators of focal adhesion activation such as RHO family members (4, 51). Integration of ECM-induced biochemical and biophysical signals in the cell ultimately translates into its ability to exert traction force upon the ECM with increasing traction associated with enhanced metastatic potential (41). Our data support that SPARCL1 inhibits the transmission of cell traction force on the matrix. Similar to other matricellular proteins (52), we determined that SPARCL1 in the ECM readily tethers to the collagen ECM (53) while also binding with slower kinetics to the cell cytoskeleton. This suggests that SPARCL1 may restrict cellular invasion by binding to collagen as well as through cell receptor interactions. From the data presented herein, we proposed a model in which cells secrete SPARCL1 in the microenvironment to limit cellular traction and consequent migration and invasion, and thus cancers may optimize their metastatic potential by suppressing SPARCL1 expression.
Supplementary Material
ACKNOWLEDGMENTS
We thank Ben Park and John Isaacs for helpful discussions and critical review of this manuscript. We thank Sai Vangala and Nikhil Adapa for technical support. We thank Cagla Eroglu at Duke University for the Sparcl1 knock-out mice. We thank Jennifer Meyers at the JHU Next Generation Sequencing Facility and Lillian Dasko-Vincent at the JHU Cell Imaging Core Facility. Supported by The Prostate Cancer Foundation Hagen Challenge Award (EMS, PJH); The Patrick C. Walsh Prostate Cancer Fund (PJH, EMS); The Flight Attendant Medical Research Institute Young Clinical Scientist Award (PJH); The Hinman Urologic Endowed Fund Scholarship (PJH); and the National Heart, Lung, and Blood Institute grant RO1HL107361 (SSA).
REFERENCES
- 1.Augello MA, Den RB, Knudsen KE. AR function in promoting metastatic prostate cancer. Cancer metastasis reviews. 2014;33:399–411. doi: 10.1007/s10555-013-9471-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schaeffer EM, Marchionni L, Huang Z, Simons B, Blackman A, Yu W, et al. Androgen-induced programs for prostate epithelial growth and invasion arise in embryogenesis and are reactivated in cancer. Oncogene. 2008;27:7180–91. doi: 10.1038/onc.2008.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pritchard C, Mecham B, Dumpit R, Coleman I, Bhattacharjee M, Chen Q, et al. Conserved gene expression programs integrate mammalian prostate development and tumorigenesis. Cancer Res. 2009;69:1739–47. doi: 10.1158/0008-5472.CAN-07-6817. [DOI] [PubMed] [Google Scholar]
- 4.Hurley PJ, Marchionni L, Simons BW, Ross AE, Peskoe SB, Miller RM, et al. Secreted protein, acidic and rich in cysteine-like 1 (SPARCL1) is down regulated in aggressive prostate cancers and is prognostic for poor clinical outcome. Proc Natl Acad Sci U S A. 2012;109:14977–14982. doi: 10.1073/pnas.1203525109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nelson PS, Plymate SR, Wang K, True LD, Ware JL, Gan L, et al. Hevin, an antiadhesive extracellular matrix protein, is down-regulated in metastatic prostate adenocarcinoma. Cancer Res. 1998;58:232–6. [PubMed] [Google Scholar]
- 6.Hu H, Zhang H, Ge W, Liu X, Loera S, Chu P, et al. Secreted Protein Acidic and Rich in Cysteines-Like 1 Suppresses Aggressiveness and Predicts Better Survival in Colorectal Cancers. Clin Cancer Res. doi: 10.1158/1078-0432.CCR-12-0124. [DOI] [PubMed] [Google Scholar]
- 7.Xiang Y, Qiu Q, Jiang M, Jin R, Lehmann BD, Strand DW, et al. SPARCL1 suppresses metastasis in prostate cancer. Molecular oncology. 2013;7:1019–30. doi: 10.1016/j.molonc.2013.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Claeskens A, Ongenae N, Neefs JM, Cheyns P, Kaijen P, Cools M, et al. Hevin is down-regulated in many cancers and is a negative regulator of cell growth and proliferation. Br J Cancer. 2000;82:1123–30. doi: 10.1054/bjoc.1999.1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Esposito I, Kayed H, Keleg S, Giese T, Sage EH, Schirmacher P, et al. Tumor-suppressor function of SPARC-like protein 1/Hevin in pancreatic cancer. Neoplasia. 2007;9:8–17. doi: 10.1593/neo.06646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brekken RA, Sullivan MM, Workman G, Bradshaw AD, Carbon J, Siadak A, et al. Expression and characterization of murine hevin (SC1), a member of the SPARC family of matricellular proteins. The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society. 2004;52:735–48. doi: 10.1369/jhc.3A6245.2004. [DOI] [PubMed] [Google Scholar]
- 11.Kleeberger W, Bova GS, Nielsen ME, Herawi M, Chuang AY, Epstein JI, et al. Roles for the stem cell associated intermediate filament Nestin in prostate cancer migration and metastasis. Cancer Res. 2007;67:9199–206. doi: 10.1158/0008-5472.CAN-07-0806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haffner MC, Aryee MJ, Toubaji A, Esopi DM, Albadine R, Gurel B, et al. Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nat Genet. 42:668–75. doi: 10.1038/ng.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yegnasubramanian S, Lin X, Haffner MC, DeMarzo AM, Nelson WG. Combination of methylated-DNA precipitation and methylation-sensitive restriction enzymes (COMPARE-MS) for the rapid, sensitive and quantitative detection of DNA methylation. Nucleic acids research. 2006;34:e19. doi: 10.1093/nar/gnj022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aryee MJ, Liu W, Engelmann JC, Nuhn P, Gurel M, Haffner MC, et al. DNA methylation alterations exhibit intraindividual stability and interindividual heterogeneity in prostate cancer metastases. Science translational medicine. 2013;5:169ra10. doi: 10.1126/scitranslmed.3005211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bursac P, Fabry B, Trepat X, Lenormand G, Butler JP, Wang N, et al. Cytoskeleton dynamics: fluctuations within the network. Biochem Biophys Res Commun. 2007;355:324–30. doi: 10.1016/j.bbrc.2007.01.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fabry B, Maksym GN, Butler JP, Glogauer M, Navajas D, Fredberg JJ. Scaling the microrheology of living cells. Phys Rev Lett. 2001;87:148102. doi: 10.1103/PhysRevLett.87.148102. [DOI] [PubMed] [Google Scholar]
- 17.Kim JJ, Yin B, Christudass CS, Terada N, Rajagopalan K, Fabry B, et al. Acquisition of paclitaxel resistance is associated with a more aggressive and invasive phenotype in prostate cancer. Journal of cellular biochemistry. 2013;114:1286–93. doi: 10.1002/jcb.24464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gajula RP, Chettiar ST, Williams RD, Thiyagarajan S, Kato Y, Aziz K, et al. The twist box domain is required for Twist1-induced prostate cancer metastasis. Molecular cancer research : MCR. 2013;11:1387–400. doi: 10.1158/1541-7786.MCR-13-0218-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Butler JP, Tolic-Norrelykke IM, Fabry B, Fredberg JJ. Traction fields, moments, and strain energy that cells exert on their surroundings. Am J Physiol Cell Physiol. 2002;282:C595–605. doi: 10.1152/ajpcell.00270.2001. [DOI] [PubMed] [Google Scholar]
- 20.Garzon-Muvdi T, Schiapparelli P, ap Rhys C, Guerrero-Cazares H, Smith C, Kim DH, et al. Regulation of brain tumor dispersal by NKCC1 through a novel role in focal adhesion regulation. PLoS biology. 2012;10:e1001320. doi: 10.1371/journal.pbio.1001320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kucukdereli H, Allen NJ, Lee AT, Feng A, Ozlu MI, Conatser LM, et al. Control of excitatory CNS synaptogenesis by astrocyte-secreted proteins Hevin and SPARC. Proc Natl Acad Sci U S A. 108:E440–9. doi: 10.1073/pnas.1104977108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2:401–4. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karnes RJ, Bergstralh EJ, Davicioni E, Ghadessi M, Buerki C, Mitra AP, et al. Validation of a Genomic Classifier that Predicts Metastasis Following Radical Prostatectomy in an At Risk Patient Population. J Urol. 2013;190:2047–53. doi: 10.1016/j.juro.2013.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Erho N, Crisan A, Vergara IA, Mitra AP, Ghadessi M, Buerki C, et al. Discovery and validation of a prostate cancer genomic classifier that predicts early metastasis following radical prostatectomy. PloS one. 2013;8:e66855. doi: 10.1371/journal.pone.0066855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Best CJ, Gillespie JW, Yi Y, Chandramouli GV, Perlmutter MA, Gathright Y, et al. Molecular alterations in primary prostate cancer after androgen ablation therapy. Clin Cancer Res. 2005;11:6823–34. doi: 10.1158/1078-0432.CCR-05-0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nelson PS, Clegg N, Arnold H, Ferguson C, Bonham M, White J, et al. The program of androgen-responsive genes in neoplastic prostate epithelium. Proc Natl Acad Sci U S A. 2002;99:11890–5. doi: 10.1073/pnas.182376299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bolton EC, So AY, Chaivorapol C, Haqq CM, Li H, Yamamoto KR. Cell- and gene-specific regulation of primary target genes by the androgen receptor. Genes Dev. 2007;21:2005–17. doi: 10.1101/gad.1564207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu J, Mani RS, Cao Q, Brenner CJ, Cao X, Wang X, et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell. 17:443–54. doi: 10.1016/j.ccr.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Esteller M. Epigenetics in cancer. The New England journal of medicine. 2008;358:1148–59. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 32.Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nature reviews Cancer. 2011;11:726–34. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nelson WG, De Marzo AM, Yegnasubramanian S. Epigenetic alterations in human prostate cancers. Endocrinology. 2009;150:3991–4002. doi: 10.1210/en.2009-0573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Isler SG, Ludwig CU, Chiquet-Ehrismann R, Schenk S. Evidence for transcriptional repression of SPARC-like 1, a gene downregulated in human lung tumors. International journal of oncology. 2004;25:1073–9. [PubMed] [Google Scholar]
- 35.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, et al. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karlic R, Chung HR, Lasserre J, Vlahovicek K, Vingron M. Histone modification levels are predictive for gene expression. Proc Natl Acad Sci U S A. 107:2926–31. doi: 10.1073/pnas.0909344107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ellwood-Yen K, Graeber TG, Wongvipat J, Iruela-Arispe ML, Zhang J, Matusik R, et al. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell. 2003;4:223–38. doi: 10.1016/s1535-6108(03)00197-1. [DOI] [PubMed] [Google Scholar]
- 38.Watson PA, Ellwood-Yen K, King JC, Wongvipat J, Lebeau MM, Sawyers CL. Context-dependent hormone-refractory progression revealed through characterization of a novel murine prostate cancer cell line. Cancer Res. 2005;65:11565–71. doi: 10.1158/0008-5472.CAN-05-3441. [DOI] [PubMed] [Google Scholar]
- 39.Magnon C, Hall SJ, Lin J, Xue X, Gerber L, Freedland SJ, et al. Autonomic nerve development contributes to prostate cancer progression. Science. 2013;341:1236361. doi: 10.1126/science.1236361. [DOI] [PubMed] [Google Scholar]
- 40.Ellis L, Lehet K, Ramakrishnan S, Adelaiye R, Pili R. Development of a castrate resistant transplant tumor model of prostate cancer. Prostate. 2012;72:587–91. doi: 10.1002/pros.21465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kraning-Rush CM, Califano JP, Reinhart-King CA. Cellular traction stresses increase with increasing metastatic potential. PloS one. 2012;7:e32572. doi: 10.1371/journal.pone.0032572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mierke CT, Rosel D, Fabry B, Brabek J. Contractile forces in tumor cell migration. European journal of cell biology. 2008;87:669–76. doi: 10.1016/j.ejcb.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arpaia E, Blaser H, Quintela-Fandino M, Duncan G, Leong HS, Ablack A, et al. The interaction between caveolin-1 and Rho-GTPases promotes metastasis by controlling the expression of alpha5-integrin and the activation of Src, Ras and Erk. Oncogene. 31:884–96. doi: 10.1038/onc.2011.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hakem A, Sanchez-Sweatman O, You-Ten A, Duncan G, Wakeham A, Khokha R, et al. RhoC is dispensable for embryogenesis and tumor initiation but essential for metastasis. Genes Dev. 2005;19:1974–9. doi: 10.1101/gad.1310805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hall CL, Dubyk CW, Riesenberger TA, Shein D, Keller ET, van Golen KL. Type I collagen receptor (alpha2beta1) signaling promotes prostate cancer invasion through RhoC GTPase. Neoplasia. 2008;10:797–803. doi: 10.1593/neo.08380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vega FM, Fruhwirth G, Ng T, Ridley AJ. RhoA and RhoC have distinct roles in migration and invasion by acting through different targets. J Cell Biol. 193:655–65. doi: 10.1083/jcb.201011038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu M, Wu ZF, Rosenthal DT, Rhee EM, Merajver SD. Characterization of the roles of RHOC and RHOA GTPases in invasion, motility, and matrix adhesion in inflammatory and aggressive breast cancers. Cancer. 116:2768–82. doi: 10.1002/cncr.25181. [DOI] [PubMed] [Google Scholar]
- 48.Cao F, Wang K, Zhu R, Hu YW, Fang WZ, Ding HZ. Clinicopathological significance of reduced SPARCL1 expression in human breast cancer. Asian Pacific journal of cancer prevention : APJCP. 2013;14:195–200. doi: 10.7314/apjcp.2013.14.1.195. [DOI] [PubMed] [Google Scholar]
- 49.Li P, Qian J, Yu G, Chen Y, Liu K, Li J, et al. Down-regulated SPARCL1 is associated with clinical significance in human gastric cancer. J Surg Oncol. 105:31–7. doi: 10.1002/jso.22025. [DOI] [PubMed] [Google Scholar]
- 50.Okamoto OK, Carvalho AC, Marti LC, Vencio RZ, Moreira-Filho CA. Common molecular pathways involved in human CD133+/CD34+ progenitor cell expansion and cancer. Cancer cell international. 2007;7:11. doi: 10.1186/1475-2867-7-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sullivan MM, Puolakkainen PA, Barker TH, Funk SE, Sage EH. Altered tissue repair in hevin-null mice: inhibition of fibroblast migration by a matricellular SPARC homolog. Wound Repair Regen. 2008;16:310–9. doi: 10.1111/j.1524-475X.2008.00370.x. [DOI] [PubMed] [Google Scholar]
- 52.Murphy-Ullrich JE, Sage EH. Revisiting the matricellular concept. Matrix Biol. 2014;37:1–14. doi: 10.1016/j.matbio.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hambrock HO, Nitsche DP, Hansen U, Bruckner P, Paulsson M, Maurer P, et al. SC1/hevin. An extracellular calcium-modulated protein that binds collagen I. J Biol Chem. 2003;278:11351–8. doi: 10.1074/jbc.M212291200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.