

Abstract
The therapeutic promise of microRNA in cancer has yet to be realized. In this study, we identified and therapeutically exploited a new role for miR-10b at the metastatic site, which links its overexpression to tumor cell viability and proliferation. In the protocol developed, we combined a miR-10b-inhibitory nanodrug with low-dose anthracycline to achieve complete durable regressions of metastatic disease in a murine model of metastatic breast cancer. Mechanistic investigations suggested a potent anti-proliferative, pro-apoptotic effect of the nanodrug in the metastatic cells, potentiated by a cell-cycle arrest produced by administration of the low-dose anthracycline. miR-10b was overexpressed specifically in cells with high metastatic potential, suggesting a role for this miRNA as a metastasis-specific therapeutic target. Taken together, our results implied the existence of pathways that regulate the viability and proliferation of tumor cells only after they have acquired the ability to grow at distant metastatic sites. As illustrated by miR-10b targeting, such metastasis-dependent apoptotic pathways would offer attractive targets for further therapeutic exploration.
Keywords: cancer, nanotherapy, miRNA, metastasis, breast cancer
INTRODUCTION
Oncogenic microRNAs (oncomiRs) have been shown to promote the migration, proliferation, and survival of tumor cells (1,2). Therefore, by targeting microRNA species it should be possible to achieve an effective and persistent therapeutic response in cancer patients. Our hypothesis is based on the rationale that the tumor cell phenotype is critically dependent on fundamental molecular pathways of oncogenesis and that altering these pathways can result in very specific and robust therapeutic effects. microRNAs, in particular, are a promising target because they are uniquely altered in tumor cells and represent a “hub” of carcinogenesis, since a single microRNA can coordinately affect the expression of multiple genes resulting in a comprehensive therapeutic response. In addition, because of the fundamental role played by microRNAs in defining tumor cell phenotypes, evasion of this therapeutic intervention by mutation is less likely.
This rationale is experimentally supported by the phenomenon of oncomiR addiction, defined as the incapacity of the tumor cell to survive in the absence of certain oncogenic microRNAs. An example is presented by microRNA-21. In a model of lymphoma, inactivation of microRNA-21 led to a complete tumor regression in a matter of days (3). These prior studies and our results described here support the feasibility of curing human cancers through pharmacological inactivation of miRNAs.
The paucity of options for patients with advanced and recurring metastatic breast cancer prompted us to further investigate the potential for targeting miR-10b implicated in the formation of metastasis (4,5). To inhibit miR-10b, we used a nanodrug (MN-anti-miR10b) composed of magnetic nanoparticles conjugated to locked-nucleic-acid antisense oligonucleotides (6). Our choice of magnetic nanoparticles (20-nm MN) extends from our earlier work, which demonstrated that these nanoparticles represent very efficient oligonucleotide delivery vehicles for RNAi-targeted therapy and imaging (6–12). These nanoparticles have favorable biodistribution and circulation half-life (~6 hrs in rodents and 24 hrs in humans) (11). Following intravenous injection, the nanoparticles distribute through the circulation and progressively accumulate in the tumor interstitium via the well-known enhanced permeability and retention (EPR) effect (13,14). Once inside the tumoral interstitium, the nanoparticles are endocytosed by the tumor cells through macropinocytosis and are ultimately released into the cytosol, where they become available to the RISC, as described in detail in (9).
In the present study, we showed that the combination of MN-anti-miR10b with low-dose doxorubicin led to a complete and persistent regression and elimination of metastatic cancer with no evidence of systemic toxicity. We also found that the mechanism behind this response involved a dramatic effect of the nanodrug on tumor cell viability, akin to oncomir addiction, potentiated by a cell-cycle inhibitory effect of low-dose doxorubicin. These studies present a new possibility for curing human metastatic cancers through pharmacological disruption of metastamirs. The clinical translation of this knowledge is particularly urgent in patients diagnosed with advanced metastatic breast cancer or in patients with disseminated recurrent disease.
MATERIALS AND METHODS
Locked Nucleic Acids
The short locked nucleic acid (LNA) oligonucleotide sequences used in this study were 5′-ThioMC6-D/GTGTAACACGTCTATACGCCCA-3′ (against miRNA-10b) and 5′-ThioMC6-D/CACAAATTCGGTTCTACAGGGTA-3′ (scrambled oligo). Both oligos were designed and synthesized by Exiqon Inc., (Woburn, MA). The 5′-Thiol-Modifier C6 disulfide (5′-ThioMC6) was inserted into both sequences for conjugation to magnetic nanoparticles (MN). The disulfide on the oligonucleotide was activated by 3% Tris (2-carboxyethyl) phosphine hydrochloride (TCEP, Thermoscientific Co., Rockford, IL), followed by purification with ammonium acetate/ethanol precipitation treatment prior to conjugation to the nanoparticles, as described previously (6,7).
MN-anti-miR10b Synthesis and Characterization
Aminated magnetic nanoparticles (MN) were synthesized following a protocol published previously (6). To afford near-infrared imaging, nanoparticles were labeled with Cy5.5 near-infrared optical dye a described previously (7). Nanoparticles with 74 amino groups per MN and a size of 20.3±0.6 nm were used for conjugation to the LNA. MN was conjugated the to the heterobifunctional linker N-Succinimidyl 3-[2-pyridyldithio]-propionate (SPDP, Thermoscientific Co., Rockford, IL) and activated LNA oligos sequentially. Briefly, SPDP was dissolved in anhydrous DMSO and incubated with MN, which has a maleimide group for thioether linkage with the LNA oligos. The 5′-ThioMC6 of the LNA oligo was activated to release the thiol via 3% TCEP treatment in nuclease free PBS. The LNA oligos were purified using an ammonium acetate/ethanol precipitation method. After TCEP-activation and purification, the oligos were dissolved in water and incubated with the SPDP modified MN overnight. MN-anti-miR10b was freshly prepared each week and the number of LNA per MN was determined as 8.0±0.7 following the electrophoresis analysis method described previously (6,7). Schematic representation of MN-anti-miR10b is shown in Fig. 1A.
Figure 1.
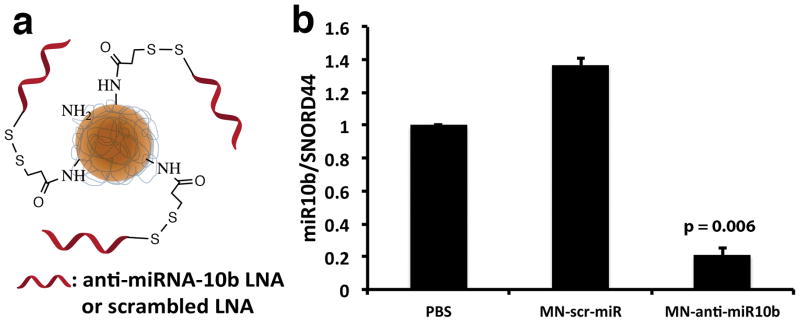
Synthesis and testing of MN-anti-miR10b. a. MN-anti-miR10b consisted of dextran-coated magnetic nanoparticles conjugated to an antisense LNA oligonucleotide targeting miRNA-10b. b. qRT-PCR showed significant inhibition of miRNA-10b, following a 48-hr incubation of human metastatic breast cancer cells with MN-anti-miR10b (Data represent average±s.d.; two-tailed t-test; n = 3).
Cell culture
The metastatic human breast cancer cell line, MDA-MB-231-luc-D3H2LN, was obtained from Perkin Elmer, Hopkinton, MA. The line was authenticated by the supplier using the IMPACT Profile I (PCR). The cells were cultured in Dulbecco’s modified Eagle’s medium (Sigma, St. Louis, MO), supplemented with 10% FBS (Thermoscientific, Waltham, MA), 1% antibiotics (Invitrogen, Carlsbad, CA), and 2 mM L-glutamine, per the supplier’s instructions. The invasive murine breast cancer cell line, 4T1, was obtained from American Type Culture Collection, Manassas VA. The line was authenticated by the supplier based on viability, recovery, growth, morphology, and isoenzymology. The cells were cultured in high glucose DMEM supplemented 5% FBS, and antibiotics (100 units/ml penicillin and 100 μg/ml streptomycin), as recommended by the supplier. The luciferase-transformed SUM149PT and SUM159PT human breast cancer cell lines were provided by Dr. Kornelia Polyak (Dana-Farber Cancer Institute). The lines were authenticated using STR and SNP6 arrays, as well as whole exome sequencing. The cells were cultured in a mixture of 50% Dulbecco’s modified Eagle’s medium/F-12 50/50, 1x (Corning, Cellgro, Manassas, VA) supplemented with 10% FBS and antibiotics (100 units/ml penicillin and 100 μg/ml streptomycin), and 50% Mammary Epithelial Cell Growth Medium (Cell Applications, Inc., San Diego, CA).
Generation of knockout cell line
A pair of TALEN (transcription-activator-like proteins fused to fok nickase) expressing clones designed to generate a double stranded cut in the genomic region of miR-10b were purchased from GeneCopoeia (Rockville, MD). MDA-MB-231-luc-D3H2LN cells were transfected following standard protocols and selected over hygromycin. Individual clones originating from a single cell were obtained by serial dilution. PCR was performed on genomic DNA to determine indel formation.
In vitro migration and invasion assays
Invasion assay was performed using 8 μm pore inserts coated with extracellular matrix proteins (Cell Biolabs, INC, San Diego, CA). Uncoated inserts served as migration controls. MDA-MB-231-luc-D3H2LN cells were serum starved overnight, in MEM medium. Following, 1.12 × 104 cells were plated inside the inserts. Drugs (MN-anti-miR10b or MN-scr-miR, 5 μM or 21.7 μg of ASO) with or without 0.1 μM doxorubicin were added directly to the cells. Outer chamber contained media supplemented with 10% FBS. Inserts were incubated for 24 h for migration assay and 48 h for invasion assay, respectively. Cells that crossed the pore were stained as described by the manufacturer and counted under a light microscope.
In vitro Apoptosis and Cell Proliferation
For apoptosis assay, 1 × 104 cells were plated in 8-chambered slides (Lab-Tek, ThermoFisher, Waltham, MA) along with MN-anti-miR10b or MN-scr-miR (5 μM of ASO) in combination with 0.1 μM of doxorubicin. Slides were incubated for 60 h and stained for apoptosis using TUNEL assay kit (Chemicon International, Temecula, CA) and mounted in anti-fade medium containing DAPI (Vector Laboratories, Inc. Burlingame, CA). Apoptotic and total cells were counted in 6 different fields of view for each treatment and averaged. Cell proliferation was determined by incubating cells for 48 h with drug combinations as above and assayed by MTT assay. In addition, cell viability in luciferase-expressing SUM149PT and SUM159PT cells was assessed by bioluminescence optical imaging, following addition of D-Luciferin potassium salt in DPBS (200 μl of 15mg ml−1; Perkin Elmer, Waltham, MA). Images were acquired using the IVIS Spectrum Imaging System (Perkin Elmer, Waltham, MA). Identical imaging acquisition settings (time, 0.5 ~ 60 sec; F-stop, 2; binning, medium) and the same area of regions of interest (ROIs) was used to obtain total radiance (photons sec−1 cm−2 sr−1). Images were processed using the Living Image Software (Ivis Spectrum, Perkin Elmer, Waltham, MA). The total radiance from the bioluminescence readings was used for signal quantification.
Cell Cycle Analysis
For cell cycle analysis, 37,500 cells/ml were plated in a 6-well culture plate. Cells were incubated with MN-anti-miR10b or MN-scr-miR (5 μM of ASO) alone or in combination with doxorubicin (0.1 μM) for 48 hrs. Next, cells were washed with PBS, collected by trypsinization and fixed in 70% ethanol pre-cooled to −20 °C. The cells were washed again with PBS and treated with RNase A (Worthington-Biochemical, Lakewood, NJ) for 30 min at RT before adding propidium iodide. Cells were analyzed on FACSCalibur (BD Biosciences, San Jose, CA).
SDS-PAGE and Western Blotting
Cells treated with various drug combinations were washed with cold PBS and lysed in RIPA buffer containing 1mg ml−1 DNAse (Roche Life Science, Indianapolis, IN) and supplemented with protease inhibitor cocktail (Roche Life Science, Indianapolis, IN). Cell lysates, stored frozen at −80 °C were resolved on 4–20% SDS-PAGE gels and transferred onto a nitrocellulose membrane. The membranes were first stained with anti-BIM (Cell Signaling Technology, Danvers, MA), and anti-HOXD10 (Santa Cruz Biotechnology, Dallas, TX) antibody cocktail followed by peroxidase conjugated-anti-rabbit antibody. ECL (Roche Life Science, Indianapolis, IN) was used to develop blots. Signal intensity was recorded on an IVIS Spectrum imaging station (Perkin Elmer, Hopkinton, MA). After documenting signal, the blot was subjected to a second round of immunostaining using an anti-actin-HRP antibody (Santa Cruz Biotechnology, Dallas, TX).
Fluorescence Microscopy and Nuclear Size Distribution
Cells grown in chambered slides were treated with drug combinations and stained for Vimentin using Alexa Fuor-647-labeled anti-vimentin antibody as instructed by the manufacturer (Santa Cruz Biotechnology, Dallas, TX). Slides were mounted in DAPI containing antifade mounting medium (Prolong Gold, Life Technologies, Grand Island, NY) and observed under fluorescence microscope. For nuclear size distribution studies, DAPI stained images were imported in ImageJ (ver. 1.48v) image processing software. Calibration was applied, nuclei were counted, and their area was determined. 4–7 images were analyzed to get data for at least 1000 cells. Nuclei whose area fell within 10 and 350 μm2 were selected and their distribution was plotted.
Animal model
Six-week-old female nude mice (NIH III nude) were implanted orthotopically under the top third mammary fat pad with the MDA-MB-231-luc-D3H2LN cell line (2 × 106 cells). These animals formed primary tumors by 2 weeks that metastasized to lymph nodes within 4 to 5 weeks. These cells expressed luciferase and could be detected by noninvasive bioluminescence imaging (BLI) for corresponding analysis of tumor burden. Akin to clinical treatment of metastatic breast cancer, the primary tumor was surgically removed once lymph node metastases were confirmed by BLI. Therapeutic treatment started from the day after primary tumor removal and was repeated once a week as described below. All animal experiments were performed in compliance with institutional guidelines and approved by the Institutional Animal Care and Use Committee at Massachusetts General Hospital.
3-Dimensional Fluorescence Imaging Tomography
Cy5.5 labeled MN was administered by i.v. injection into a mouse with bilateral secondary tumors in axillary lymph nodes confirmed by bioluminescence imaging. A set of fluorescence images was collected using an IVIS Spectrum imaging station (Perkin Elmer, Waltham, MA) for the reconstruction of 3D fluorescence imaging tomography (3D-FLIT) using Living Image software (ver. 4.4, Perkin Elmer, Waltham, MA). The excitation wavelength was fixed at 675nm and the emission wavelength was 720nm. Overall, 18 fluorescence images were processed to reconstruct 3D-FLIT for the illustration of fluorescence sources in the body. From these, 2-D images were extracted in the coronal and transaxial planes.
Bioluminescence Optical Imaging
To evaluate metastatic burden we used bioluminescence imaging. Anesthetized mice were injected in the lower left abdominal quadrant with D-Luciferin potassium salt in DPBS (200 μl of 15mg ml−1; Perkin Elmer, Waltham, MA) twelve minutes before image acquisition. Identical imaging acquisition settings (time, 0.5 ~ 60 sec; F-stop, 2; binning, medium) and the same area of regions of interest (ROIs) was used to obtain total radiance (photons sec−1 cm−2 sr−1) of the metastatic tumor signals. Bioluminescence imaging (IVIS Spectrum, Perkin Elmer, Waltham, MA) was performed for 6 ~ 12 min to obtain the maximum radiance from the metastatic tumors and all images were processed using the Living Image Software (IVIS Spectrum, Perkin Elmer, Waltham, MA). The total radiance from the bioluminescence readings was used for signal quantification.
Therapy
Therapy was delivered to animals after the surgical removal of primary tumors, once lymph node metastases were confirmed by BLI. Treatment was initiated on the week of tumor removal. The therapeutic protocol consisted of concurrent injections of MN-anti-miR10b or MN-scr-miR intravenously, 15 mg kg−1 as iron, 10 mg kg−1 as LNA) and low-dose doxorubicin (i.p. 4 mg kg−1, Sigma, St. Louis, MO) for the following treatment groups were used: Group 1 - PBS only (n = 2), Group 2 - MN-scr-miR without doxorubicin (n = 6), Group 3 -MN-scr-miR with doxorubicin (n = 10), Group 4 -MN-anti-miR10b only (n = 7), and Group 5 -MN-scr-miR10b with doxorubicin (n = 10). Therapy was administered weekly until the disappearance of BLI-visible metastasis (4 weeks to all mice in Group 5, 12 weeks to all the mice in Groups 1 to 4). All mice were monitored weekly by bioluminescence imaging to assess metastatic burden for a maximum of 20 weeks after the first treatment or until animals become moribund. Mouse body weight was monitored before each imaging session.
Real-time quantitative reverse transcription-PCR
To measure the extent of miR-10b inhibition by MN-anti-miR10b, MDA-MB-231-luc-D3H2LN cells were incubated with MN-anti-miR10b or MN-scr-miR (45 μg of iron, 4nmols LNA) for 48 h at 37 °C. From the total extracted RNA, the miRNA-enriched fraction was harvested using a miRNeasy mini kit, according to the manufacturer’s protocol (Qiagen Inc., Hilden, Germany). Following the same protocol, metastatic lymph nodes harvested after two weekly intravenous injections were homogenized to extract the miRNA-enriched fraction. Relative levels of miR-10b were determined by real-time quantitative reverse transcription-PCR (qRT-PCR; Taqman protocol) and compared to the internal housekeeping gene, SNORD44. Taqman analysis was carried out using an ABI Prism 7700 sequence detection system (Applied Biosystems, Foster City, CA). The primers (Hs-miR-10b-3 miScript Primer, Hs-SNORD44-11 miScript Primer) and assay kit (miScript PCR Starter Kit) were purchased from Qiagen (Hilden, Germany).
Histology and fluorescence microscopy of tissue sections
To analyze the metastatic lesions ex vivo and evaluate histopathology, excised tissues (lymph nodes, lungs, liver, spleen, intestine, brain, kidney) were embedded in Tissue-Tek OCT compound (Sakura Finetek, Torrance, CA) and snap frozen in liquid nitrogen. The tissues were cut into 7 μm sections. To determine MN-anti-miR10b accumulation in tissue, the sections were stained using incubation with an anti-firefly luciferase antibody (1:50 dilution; Abcam, Cambridge, MA) at 4°C overnight, followed by a FITC-conjugated anti-dextran monoclonal antibody (1:50 dilution; Stemcell Technologies, Vancouver, BC, Canada) and a Texas red-conjugated goat anti-rabbit secondary antibody (1:50 dilution, Santa Cruz Biotechnology, Santa Cruz, CA) at RT for 1h. Apoptosis in tumor tissues was evaluated by performing a terminal deoxynucleotidyl transferase–mediated dUTP nick end labeling (TUNEL) assay (ApopTag Fluorescein In situ Apoptosis Detection kit, Chemicon International, Temecula, CA) according to the manufacturer’s protocol. For determining proliferation, frozen tumor and lymph node sections were incubated with an anti-Ki67 antibody (1:100 dilution; Abcam, Cambridge, MA) followed by a DyLight-488-labeled secondary antibody at RT for 1 h. Afterwards, the slides were counterstained and mounted with Vectashield mounting medium with DAPI (Vector Laboratories, Inc. Burlingame, CA). The sections were analyzed by fluorescence microscopy using a Nikon Eclipse 50i fluorescence microscope (Nikon, Tokyo, Japan), equipped with the necessary filter sets (Chroma Technology Corporation, Bellows Falls, VT). Images were acquired using a charge coupled device camera with near-IR sensitivity (SPOT 7.4 Slider RTKE; Diagnostic Instruments, Sterling Heights, MI). The images were analyzed using SPOT 4.0 Advance version software (Diagnostic Instruments). For histopathology, the tissues were stained with hematoxylin and eosin (H&E).
Systemic toxicity
Blood was collected at necropsy via cardiac puncture and serum was separated by centrifugation at 1600 × g for 15 min. Serum chemistries from each mouse recipient were analyzed by using Catalyst Dx® Chemisty Analyzer, by IDEXX Laboratories (Westbrook, ME).
Statistical Analysis
Data were expressed as mean ± s.d. or s.e.m., where indicated. Statistical comparisons were drawn using a two-tailed t-test (SigmaStat 3.0; Systat Software, Richmond, CA). A value of P < 0.05 was considered statistically significant.
RESULTS
MN-anti-miR10b can target miRNA-10b in breast tumor cells and has a profound effect on metastatic tumor cell phenotype.
To demonstrate that MN-anti-miR10b synthesized as designed (Fig. 1a) was functional in the human lymph-node metastatic MDA-MB-231-luc-D3H2LN cell line, we incubated these cells with MN-anti-miR10b. This resulted in significant 86.3 ± 5.8% inhibition of the target miRNA-10b, compared to the inactive control MN-scr-miR functionalized with an irrelevant oligonucleotide (Fig. 1b). These results indicated that the MN-anti-miR10b design could be used to very efficiently inhibit miRNA-10b in tumor cells.
In order to assess the therapeutic potential of this methodology, we investigated the effect of miR-10b inhibition by MN-anti-miR10b on tumor cell apoptosis, proliferation, invasion and migration. We found that miR-10b inhibition by MN-anti-miR10b led to a significant induction of apoptosis, inhibition of proliferation, and reduction in invasion and migration (Fig. 2). We also hypothesized that by co-administering a low dose of a cytotoxic chemotherapeutic (e.g., doxorubicin), we could amplify the effectiveness of MN-anti-miR10b. Namely, we posited that low-dose doxorubicin would serve to inhibit the cell cycle and slow down the rate of cell division, as shown by us previously (8). Since the loss of these nanoparticles from tumor cells is governed by cell division and not degradation or excretion (15), the addition of low-dose doxorubicin is needed to achieve higher, therapeutic doses of MN-anti-miR10b in tumor cells. We observed that upon the combination treatment with MN-anti-miR10b (0.5 μM) and a low-dose doxorubicin (0.05 μM, <IC10), the tumor cells acquired a spindle-like shape, detached, and died (Fig. 2a). This corresponded to a significant induction of apoptosis (Fig. 2b) and decrease in proliferation (Fig. 2c). Importantly, increased apoptosis was not seen in the absence of MN-anti-miR10b nor was it seen in cells treated with doxorubicin alone. The observed loss of cell viability had a broad relevance to invasive disease, because it was also seen in murine 4T1, as well as human SUM149PT and SUM159PT cells (Supplementary Fig. S1). Importantly, the induction of apoptosis by MN-anti-miR10b was highest in the more mesenchymal 4T1 and SUM159PT cells and less pronounced in the more epithelial SUM149PT cells (Supplementary Fig. S1b).
Figure 2.

Phenotypic effects of treatment with MN-anti-miR10b and low-dose doxorubicin in human metastatic breast cancer cells. a. Cell morphology. By 48-hr of incubation with MN-anti-miR10b and dox, cells assumed a spindle shape, detached, and died. b. Apoptosis. There was a significant increase in apoptosis in cells treated with MN-anti-miR10b and dox. c. Proliferation. There was a significant decrease in proliferation in cells treated with MN-anti-miR10b and dox. d. Cell cycle analysis. Doxorubicin-induced G2/M cell cycle arrest. The combination treatment with MN-anti-miR10b and doxorubicin led to cell cycle arrest and aneuploidy. e. Invasion and migration. Invasion and migration of tumor cells were both inhibited by the combination treatment. f. Western blot analysis of the expression of the known miR-10b target, HOXD10 and the predicted target BIM, which is a member of the Bcl-2 family of proapoptotic proteins (Data represent average±s.d.; two-tailed t-test; n = 3).
Analysis of the effect of the combination treatment on cell cycle confirmed our hypothesis regarding the mechanism underlying the observed synergy between doxorubicin and MN-anti-miR10b. Namely, doxorubicin induced polyploidy and G2/M arrest (Fig. 2d). This indicated that in the described scenario, doxorubicin, by inhibiting the cell cycle, acted as a sensitizer to a pro-apoptotic trigger provided by MN-anti-miR10b. In line with this hypothesis, when low-dose doxorubicin and MN-anti-miR10b were combined, there was an increase of both the pre-G1 apoptotic and G2/M arrested cell populations. Interestingly, the inhibition of the cell cycle appeared to extend solely from the doxorubicin component of the combination treatment. MN-anti-miR10b did not demonstrate an appreciable effect on the cell cycle, suggesting a cell-cycle independent apoptotic effect (Fig. 2d).
A further look into the phenotypic effects of the combination treatment revealed that, consistent with the literature (4) and our prior studies (6), the combination treatment inhibited tumor cell migration and invasion. The effect extended from MN-anti-miR10b alone and was not influenced by the presence of doxorubicin (Fig. 2e).
As noted above, treatment with MN-anti-miR10b caused a drastic change in morphology (Fig. 2a) that was further augmented by co-treatment with low dose doxorubicin. This change was reflected in profound reorganization of the cytoskeleton (Supplementary Fig. S2). The shape and size of the nucleus were also affected. We have previously described that low dose doxorubicin results in cell cycle arrest and nuclear swelling (8). Interestingly, MN-anti-miR10b treatment reduced the size of the nucleus and this effect was not reversed by the addition of low-dose doxorubicin (Supplementary Fig. S2). These results indicated that the pro-apoptotic effects of the combination treatment manifested as disruption in cytoskeletal and nuclear structure.
To further investigate the basis for the observed decrease in tumor cell viability, we analyzed the expression of the pro-apoptotic member of the BCL-2 family member, BIM, which has previously been implicated as a direct target of miR-10b in glioblastoma (16). We focused on BIM, in addition to the well-established miR-10b target HOXD10 implicated in tumor cell invasion and metastasis (4). Consistent with our phenotypic analysis, miR-10b inhibition in the presence or absence of doxorubicin led to upregulation of both HOXD10 and BIM (Fig. 2f). The induction of BIM by MN-anti-miR10b pointed to a molecular mechanism behind the observed link between miR-10b and apoptosis in metastatic breast cancer cells, which is a novel finding.
These studies indicate that by combining a low dose cytotoxic, such as doxorubicin and miR-10b inhibition using MN-anti-miR10b, it is possible to mediate a profound effect on tumor cell phenotype manifested at its endpoint by tumor cell death and loss of invasive properties. These results also suggested that miR-10b is a metastamir essential for tumor cell survival, akin to the mechanism behind oncomir addiction. Further proof for this hypothesis came from experiments in which we attempted to generate a miR-10b knock-out subline derived from the MDA-MB-231-luc-D3H2LN cell line (Supplementary Fig. S3a). We observed that within 24 h of transfection with the knock-out vector set (TALENs L+R), the cells acquired a spindle-shaped conformation and began to detach. By 72 h of transfection, there were no viable cells (Supplementary Fig. S3b). This latter result suggested that a robust therapeutic effect could also be achieved by treatment with a higher dose of MN-anti-miR10b alone, in the absence of doxorubicin. However, at higher doses of MN-anti-miR10b, issues of oligotoxicity begin to emerge, making monotherapy less optimal.
Combination therapy with MN-anti-miR10b and low-dose chemotherapy can regress breast cancer metastasis.
With an outlook towards clinical translation of our therapeutic approach, we performed therapeutic studies using a combination of MN-anti-miR10b and low-dose doxorubicin in a murine model of lymph-node metastatic breast cancer. To validate miR-10b as a metastasis-specific target in this model, we first compared the expression of miR-10b in the primary tumors and lymph node metastases. miR-10b is upregulated in metastatic breast cancer cell lines and in the primary tumors of patients with breast cancer metastasis relative to non-metastatic patients (4). It is also upregulated in metastatic samples relative to matched primary tumors (17). Consistent with these findings, we also observed highly significant overexpression of miR-10b in the lymph node metastases relative to the primary tumors of the animal model used here (Supplementary Fig. S4).
Following a clinical scenario we surgically removed the primary tumors after confirmation of lymph node metastasis. Since inhibition of miR-10b does not affect primary tumor growth (6), removal of the tumor was necessary to permit long-term studies without mortality. Accumulation of MN-anti-miR10b in the lymph nodes 24 hrs after intravenous injection was confirmed by in vivo near infrared (NIRF) optical imaging (Fig. 3a) and verified by fluorescence microscopy (Fig. 3b). Effective miR-10b inhibition in metastatic tumor cells was confirmed by RT-PCR (Fig. 3c).
Figure 3.
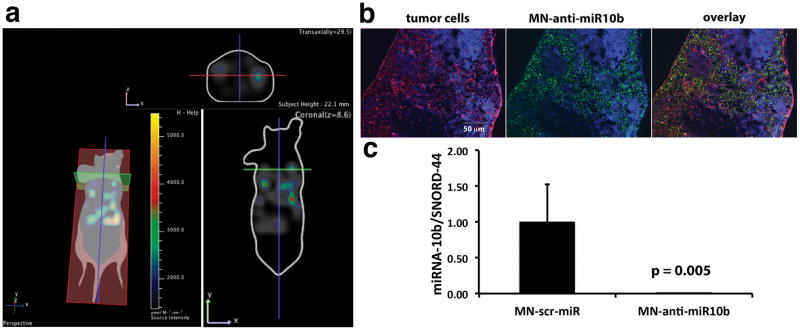
MN-anti-miR10b accumulation and miR-10b inhibition 24 hrs after intravenous injection into mice bearing lymph node metastases. a. 3D-FLIT was reconstructed to identify sources of fluorescence within the mouse body (left). 2D-images were then extracted in the coronal (bottom right) and transaxial (top right) planes to confirm the accumulation of MN-anti-miR10b. This mouse had bilateral metastasis in the axillary lymph nodes. Fluorescence was visible in the metastatic lymph nodes, indicating MN-anti-miR10b accumulation. b. Fluorescence microscopy of MN-anti-miR10b accumulation in lymph-node metastatic tumor cells (Red- tumor cells stained for luciferase, green- MN-anti-miR10b stained for dextran, blue- nuclei). c. miR-10b inhibition by MN-anti-miR10b. The intravenously injected MN-anti-miR10b significantly inhibited miR-10b in the lymph node of metastasis bearing mice (Data represent average±s.d.; two-tailed t-test; n = 3).
In therapeutic time course studies, there was complete regression of lymph node metastasis in the experimental mice treated with MN-anti-miR10b and low-dose doxorubicin after 4 weeks of therapy (Fig. 4a, b). By contrast, in the control groups, there was metastatic progression. In the experimental mice, treatment was discontinued once complete metastatic regression was observed at week 4. Still, by the endpoint of the study at week 12, no recurrence was observed (Fig. 4a and b). Ex vivo analysis revealed the presence of lymph node metastasis and metastatic dissemination to the lungs in control animals treated with PBS or MN-scr-miR with or without doxorubicin (Fig. 4c). In the mice treated with MN-anti-miR10b only, there was evidence of metastasis to the lymph node but not to the lungs. In contrast, in mice treated with MN-anti-miR10b and doxorubicin no gross lymph node or lung metastases could be detected (Fig. 4c).
Figure 4.

Metastatic burden and survival of mice treated with MN-anti-miR10b and low-dose dox. a. Representative bioluminescence images of metastatic burden showing complete regression of metastases in animals treated with MN-anti-miR10b and dox. b. Quantitative analysis of metastatic burden from all treatment groups indicating complete regression of metastatic burden in the lymph nodes of experimental animals treated with MN-anti-miR10b and dox after just 4 weekly treatments. Background counts are derived from non-tumor bearing animals. c. Ex vivo BLI showing the absence of detectable lymph node or lung metastases in mice treated with MN-anti-miR10b and dox. In animals treated with MN-anti-miR10b alone, there were lymph node but not lung metastases. In all other groups, there were both lymph node and lung metastases. d. Animal weight. The groups treated with MN-anti-miR10b with or without dox continued to gain weight throughout the time course of the study. e. Mortality. Only in the group of animals treated with MN-anti-miR10b and dox there was no mortality from carcinoma. (Data represent average±s.e.m.; Within-Subjects ANOVA: p < 0.05. PBS, n = 2; MN-scr-miR, n = 6; MN-scr-miR+dox, n = 10; MN-anti-miR10b, n = 7; MN-anti-miR10b+dox, n = 10).
Histopathological analysis indicated that in the control groups treated with PBS or MN-scr-miR in the presence or absence of doxorubicin, there was lymph node and lung infiltration by tumor cells. In mice treated with MN-anti-miR10b alone, there were metastases in the lymph nodes with no evidence of lung involvement. By contrast, in mice treated with MN-anti-miR10b and doxorubicin, the lymph nodes and lungs appeared clear of metastasis (Supplementary Fig. S5).
The effect of therapy with MN-anti-miR10b and low-dose doxorubicin also translated into significant improvement in body weight (Fig. 4d) and survival (Fig. 4e). Unlike control groups, by week 14 after the beginning of therapy, none of the experimental animals (MN-anti-miR10b/doxorubicin) had succumbed to cancer. Furthermore, combination therapy with doxorubicin and MN-anti-miR10b was superior to monotherapy with MN-anti-miR10b (Fig. 4a, b, c, and e). We need to note that the MN-scr-miR control nanodrug also had an effect on the overall survival. This could be assigned to the presence of a CpG motif in the scr oligo (but not on the anti-miR10b oligo) that is known to stimulate signaling through Toll-like receptors (18).
By the 15th week of therapy, the differences between experimental and control animals were apparent by simple anatomical observation. The control animals treated with MN-scr-miR and doxorubicin displayed gross lymphadenopathy (Supplementary Fig. S6a and Supplementary Video S8) and cachexia (Supplementary Fig. S6b and Supplementary Video S8). The experimental animals treated with MN-anti-miR10b and doxorubicin appeared healthy (Supplementary Fig. S6 and Supplementary Video S8). Long-term observation indicated that the remission of metastatic disease was permanent and that the treatment resulted in an effective cure (Supplementary Fig. S7).
Ex vivo examination of the lymph nodes revealed both induction of apoptosis and reduction of proliferation in the MN-anti–miR10b groups compared with the MN-scr-miR groups either with or without chemotherapy (Fig. 5). This finding is expected based on our in vitro results.
Figure 5.
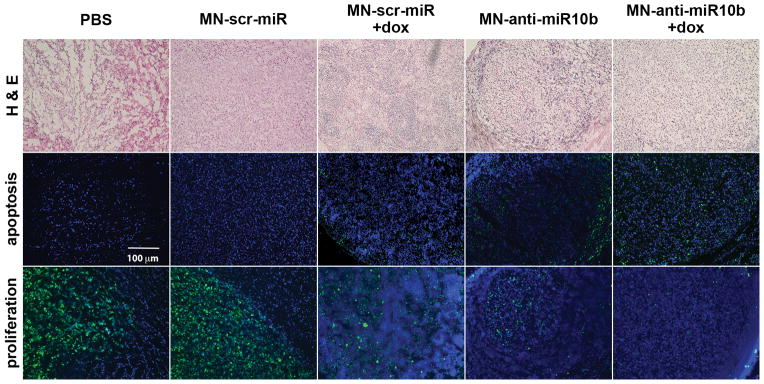
Levels of apoptosis and proliferation in lymph nodes of mice treated with MN-anti-miR10b and low-dose doxorubicin. H&E staining (top), TUNEL assay (middle) and immunostaining for Ki67 (bottom) in lymph node sections from animals injected with PBS, MN-scr-miR, MN-scr-miR + dox, MN-anti-miR10b, and MN-anti-miR10b + dox. There was evidence of increased apoptosis and reduced proliferation in the lymph nodes of mice treated with MN-anti-miR10b + dox relative to the control groups. In these studies, tissues representing the MN-anti-miR10b + dox group were derived from an animal sacrificed after two weeks of treatment when lymph node metastases had not completely regressed. Still, metastatic burden in that animal was lower that in the control groups. TUNEL (green), Ki67 (green), DAPI (blue).
Having demonstrated that the combination treatment is efficacious, we needed to address the important issue of systemic toxicity. We analyzed a panel of blood chemistry markers including indicators of renal, liver toxicity, etc. and found no significant deviations that could be assigned to components of MN-anti-miR10b (Fig. 6a). In addition, histopathology of major organs demonstrated the absence of any gross tissue abnormalities (Fig. 6b), suggesting that there was no chronic toxicity as a result of treatment.
Figure 6.
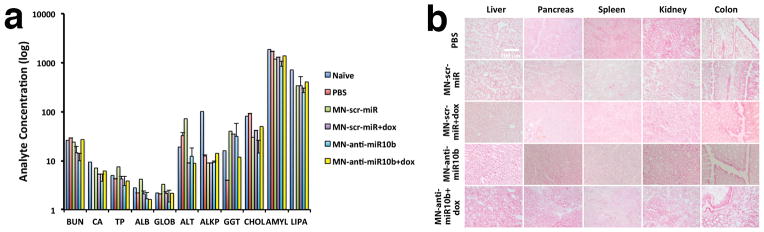
Systemic toxicity of treatment with MN-anti-miR10b and low-dose doxorubicin. a. Blood chemistries. b. Histopathology of major organs. There was no evidence of impaired organ function or abnormal tissue structure as a result of the treatment. (Data represent average±s.d.; two-tailed t-test; n = 3).
Taken together, these results provide compelling evidence of the potential of miR-10b as a target for the therapy of metastatic breast cancer. Importantly, efficient therapeutic delivery vehicles, such as the one described here, hold one of the keys to the effectiveness of therapeutic intervention.
DISCUSSION
Here we describe a new therapeutic strategy for the treatment of metastatic breast cancer that combines a molecularly targeted therapeutic agent that inhibits the pro-metastatic miRNA-10b with a low dose of doxorubicin. The choice of miR-10b as a target is based on the fact that it is involved in tumor cell invasion and migration in metastatic cancer (4,17,19). Ma et al (4,5) were the first to establish that microRNA-10b (miR-10b) initiates breast cancer invasion and metastasis. The clinical importance of miR-10b is underscored by the finding that although it is expressed at low levels in samples from metastasis-free breast cancer patients, it is overexpressed in the patients with metastatic disease (4).
The choice of miR-10b is further motivated by its potentially broad relevance to cancer. Recent studies have demonstrated that the influence of this microRNA extends beyond breast cancer (5,6,20–22) and also includes lung, colorectal, gastric, bladder, pancreatic, ovarian, hepatocellular and brain cancer, suggesting that the proposed therapeutic approach may have a more global relevance to metastatic disease (16,17).
The key advancement presented by the current investigation extends from the discovery of a previously unknown effect of miR-10b on the viability of metastatic cancer cells. Specifically, miR-10b inhibition in these cells resulted in a complete loss of viability through apoptosis. Given the specific overexpression of miR-10b in metastatic cells and their inability to survive once miR-10b is efficiently inhibited, we have termed this phenomenon “metastamir addiction”, akin to “oncomir addiction”. miR-10b metastamir addiction is at the root of the robust therapeutic response engendered by the combination of MN-anti-miR10b and a low-dose cytostatic, such as doxorubicin. The mechanism behind this response relies most profoundly on the effect of the miR-10b inhibitor on metastatic cell viability. In our studies, whereas low-dose doxorubicin strictly effected cell-cycle arrest, the miR-10b-inhibitory MN-anti-miR10b limited migration and invasion through its target HOXD10 (4), and induced apoptosis partially by upregulating the pro-apoptotic Bcl-2 protein, BIM. While known for its relevance to brain cancer (16), BIM has not been described as a miR-10b target in metastatic breast cancer cells and represents a novel finding.
Given the more pronounced pro-apoptotic effect of the nanodrug in cells with a more mesenchymal phenotype and in line with our earlier studies, which suggested that miR-10b plays a context-dependent role in tumorigenesis (6), the observed effects are likely relevant to highly invasive, metastatic tumor cells. These findings suggest the possibility that miR-10b could influence the proliferation and/or viability of a distinct subpopulation of highly invasive cells, as the ones used in this study. Indeed, miR-10b is upregulated in metastatic breast cancer cell lines and in the primary tumors of patients with breast cancer metastasis relative to non-metastatic patients (4). It is also upregulated in metastatic samples relative to matched primary tumors (17).
On a broader scale, these results support the existence of pathways that regulate the viability of tumor cells only after these cells have acquired the ability to migrate. The identification and targeting of these metastasis-dependent apoptotic pathways would represent an attractive area of therapeutic development for cancer.
The significance of our work derives from the fact that, by presenting the possibility of regression and elimination of metastases, the described approach can have a transformative impact on cancer. If successful, this therapeutic strategy is poised for immediate clinical translation, considering that MN-anti-miR10b is based on clinically approved components. Specifically, with an outlook towards clinical translation, which requires FDA approval, we employed a simplified MN-anti-miR10b design, in line with our previous studies that elucidated essential MN-anti-miR10b components (7). This high translational potential underscores the impact of the work, as a stepping-stone toward the introduction of a novel therapeutic modality for metastatic breast cancer.
Supplementary Material
PRECIS.
Striking results suggest the existence of pathways that regulate the viability and proliferation of tumor cells only after they have acquired the ability to grow at distant metastatic sites, with important implications for selective targeting of advanced cancers.
Acknowledgments
Financial Support: The study was supported under grants R00CA129070 and R01CA16346101A1 from the National Cancer Institute to ZM, the Young Investigator Award by the Breast Cancer Alliance to ZM, and 5T32CA009502 to AM.
Footnotes
Conflict of Interest: The authors have no conflicts to disclose.
References
- 1.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6(11):857–66. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 2.Esquela-Kerscher A, Slack FJ. Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer. 2006;6(4):259–69. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- 3.Medina PP, Nolde M, Slack FJ. OncomiR addiction in an in vivo model of microRNA-21-induced pre-B-cell lymphoma. Nature. 2010;467(7311):86–90. doi: 10.1038/nature09284. [DOI] [PubMed] [Google Scholar]
- 4.Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007;449(7163):682–88. doi: 10.1038/nature06174. [DOI] [PubMed] [Google Scholar]
- 5.Ma L, Reinhardt F, Pan E, Soutschek, Bhat B, Marcusson EG, et al. Therapeutic silencing of miR-10b inhibits metastasis in a mouse mammary tumor model. Nat Biotechnol. 2010;28(4):341–47. doi: 10.1038/nbt.1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yigit MV, Ghosh SK, Kumar M, Petkova V, Kavishwar A, Moore A, et al. Context-dependent differences in miR-10b breast oncogenesis can be targeted for the prevention and arrest of lymph node metastasis. Oncogene. 2013;32(12):1530–8. doi: 10.1038/onc.2012.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoo B, Ghosh SK, Kumar M, Moore A, Yigit MV, Medarova Z. Design of Nanodrugs for miRNA Targeting in Tumor Cells. J Biomed Nanotechnol. 2014;10(6):1114–22. doi: 10.1166/jbn.2014.1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ghosh SK, Yigit MV, Uchida M, Ross AW, Barteneva N, Moore A, et al. Sequence-dependent combination therapy with doxorubicin and a survivin-specific small interfering RNA nanodrug demonstrates efficacy in models of adenocarcinoma. Int J Cancer. 2014;134(7):1758–66. doi: 10.1002/ijc.28499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yoo B, Kavishwar A, Ross A, Pantazopoulos P, Moore A, Medarova Z. In Vivo Detection of miRNA Expression in Tumors Using an Activatable Nanosensor. Mol Imaging Biol. 2015 doi: 10.1007/s11307-015-0863-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoo B, Ifediba MA, Ghosh S, Medarova Z, Moore A. Combination treatment with theranostic nanoparticles for glioblastoma sensitization to TMZ. Mol Imaging Biol. 2014;16(5):680–9. doi: 10.1007/s11307-014-0734-3. [DOI] [PubMed] [Google Scholar]
- 11.Medarova Z, Pham W, Farrar C, Petkova V, Moore A. In vivo imaging of siRNA delivery and silencing in tumors. Nat Med. 2007;13(3):372–7. doi: 10.1038/nm1486. [DOI] [PubMed] [Google Scholar]
- 12.Kumar M, Yigit M, Dai G, Moore A, Medarova Z. Image-guided breast tumor therapy using a small interfering RNA nanodrug. Cancer Res. 2010;70(19):7553–61. doi: 10.1158/0008-5472.CAN-10-2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Branca RT, Cleveland ZI, Fubara B, Kumar CS, Maronpot RR, Leuschner C, et al. Molecular MRI for sensitive and specific detection of lung metastases. Proc Natl Acad Sci U S A. 2010;107(8):3693–7. doi: 10.1073/pnas.1000386107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leuschner C, Kumar CS, Hansel W, Soboyejo W, Zhou J, Hormes J. LHRH-conjugated magnetic iron oxide nanoparticles for detection of breast cancer metastases. Breast Cancer Res Treat. 2006;99(2):163–76. doi: 10.1007/s10549-006-9199-7. [DOI] [PubMed] [Google Scholar]
- 15.Arbab AS, Bashaw LA, Miller BR, Jordan EK, Lewis BK, Kalish H, et al. Characterization of biophysical and metabolic properties of cells labeled with superparamagnetic iron oxide nanoparticles and transfection agent for cellular MR imaging. Radiology. 2003;229(3):838–46. doi: 10.1148/radiol.2293021215. [DOI] [PubMed] [Google Scholar]
- 16.Gabriely G, Yi M, Narayan RS, Niers JM, Wurdinger T, Imitola J, et al. Human glioma growth is controlled by microRNA-10b. Cancer Res. 2011;71(10):3563–72. doi: 10.1158/0008-5472.CAN-10-3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baffa R, Fassan M, Volinia S, O’Hara B, Liu CG, Palazzo JP, et al. MicroRNA expression profiling of human metastatic cancers identifies cancer gene targets. J Pathol. 2009;219(2):214–21. doi: 10.1002/path.2586. [DOI] [PubMed] [Google Scholar]
- 18.Krepler C, Wacheck V, Strommer S, Hartmann G, Polterauer P, Wolff K, et al. CpG oligonucleotides elicit antitumor responses in a human melanoma NOD/SCID xenotransplantation model. J Invest Dermatol. 2004;122(2):387–91. doi: 10.1046/j.0022-202X.2004.22202.x. [DOI] [PubMed] [Google Scholar]
- 19.Ma L, Weinberg RA. MicroRNAs in malignant progression. Cell Cycle. 2008;7(5):570. doi: 10.4161/cc.7.5.5547. [DOI] [PubMed] [Google Scholar]
- 20.Moriarty CH, Pursell B, Mercurio AM. miR-10b Targets Tiam1 Implications for Rac Activation and Carcinoma Migration. J Biol Chem. 2010;285(27):20541–46. doi: 10.1074/jbc.M110.121012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tian Y, Luo A, Cai Y, Su Q, Ding F, Chen H, et al. MicroRNA-10b promotes migration and invasion through KLF4 in human esophageal cancer cell lines. J Biol Chem. 2010;285(11):7986–94. doi: 10.1074/jbc.M109.062877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guessous F, Alvarado-Velez M, Marcinkiewicz L, Zhang Y, Kim J, Heister S, et al. Oncogenic effects of miR-10b in glioblastoma stem cells. J Neuro-oncol. 2013:1–11. doi: 10.1007/s11060-013-1047-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.