

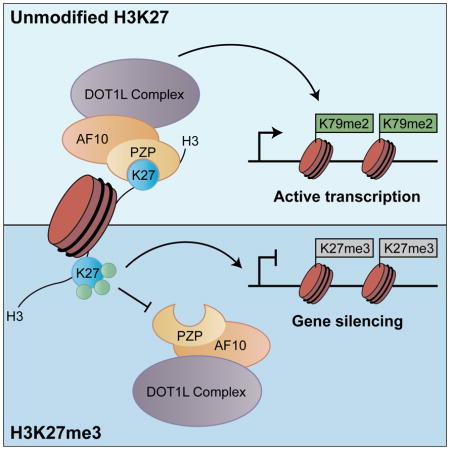
SUMMARY
AF10, a DOT1L cofactor, is required for H3K79 methylation and cooperates with DOT1L in leukemogenesis. However, the molecular mechanism by which AF10 regulates DOT1L-mediated H3K79 methylation is not clear. Here, we report that AF10 contains a “reader” domain that couples unmodified H3K27 recognition to H3K79 methylation. An AF10 region consisting of a PHD finger-Zn knuckle-PHD finger (PZP) folds into a single module that recognizes amino acids 22-27 of H3, and this interaction is abrogated by H3K27 modification. Structural studies reveal that H3-binding triggers rearrangement of the PZP module to form an H3(22-27)-accommodating channel and the unmodified H3K27 side-chain is encased in a compact hydrogen bond acceptor-lined cage. In cells, PZP recognition of H3 is required for H3K79 dimethylation, expression of DOT1L-target genes, and proliferation of DOT1L-addicted leukemic cells. Together, our results uncover a pivotal role for H3K27 – via readout by the AF10 PZP domain – in regulating the cancer-associated enzyme DOT1L.
Graphical abstract
INTRODUCTION
The readout of histone post-translational modifications (PTMs) is crucial for understanding how chromatin dynamics influence the regulation of the genome (Taverna et al., 2007). To date, PHD finger domains represent the largest known class of reader domains that are sensitive to methyl-lysine (Musselman and Kutateladze, 2011). For example, there are dozens of PHD fingers that specifically bind to H3K4me3, (trimethylation of K4 on H3), which is a hallmark of active transcription. These proteins include the second PHD finger of the chromatin remodeler BPTF (Li et al., 2006; Wysocka et al., 2006), PHD fingers from the ING tumor suppressor families (Pena et al., 2006; Shi et al., 2006), the PHD finger of RAG2, an essential component of the RAG1/2 V(D)J recombinase (Matthews et al., 2007) and the PHD finger from the core transcriptional machinery protein TAF3 (Vermeulen et al., 2007). Besides PHD fingers that recognize H3K4me3, a second class of PHD fingers first discovered on the chromatin regulator BHC80, bind to the very N-terminus of H3 tails and are repelled by methylation at H3K4 (Lan et al., 2007).
In the human genome, eleven proteins present in four families (JADE, BRPF, KDM4, and AF10/17) contain the chromatin-associated PHD finger-Zn knuckle-PHD finger (PZP) module (Figure 1A and S1A; (Klein et al., 2014)). The first PHD finger of the PZP domain within the JADE (JADE1/2/3) and BRPF (BRPF1/2/3) families behaves similar to BHC80 to regulate histone acetyltransferase (HAT) complexes (Lalonde et al., 2013; Saksouk et al., 2009). Notably, the second PHD finger cooperates with the first PHD finger to mediate nucleosome binding through a mechanism that is yet to be elucidated (Klein et al., 2014; Lalonde et al., 2013).
Figure 1. The PZP domains of AF10 and AF17 bind to H3 amino acids 15-34 and the interaction is abrogated by H3K27 methylation.

(A) Top: Schematic of full length AF10 with the indicated domains. Bottom: Schematic of motifs that comprise the PZP domain.
(B) The PZP domains of AF10 and AF17 bind to nucleosomes. Western blot of biotinylated recombinant nucleosomes (rNuc) pull-downs incubated with GST, GST-AF10PZP, GST-AF17PZP and GST-JADE1PZP as indicated.
(C) The N-terminal tail of H3 is necessary for AF10PZP and AF17PZP binding to nucleosomes. Pull-down assays as in (B) with nucleosomes containing either wild-type H3 or H3- N (amino acids 27-135) as indicated. Bottom panel: Western blot showing H3 and H3-ΔN used for pull-downs.
(D) Amino acids 15-34 of H3 are sufficient for binding to AF10PZP and AF17PZP. Western blot analysis of histone peptide pull-downs with the indicated proteins and biotinylated H3 peptides.
(E and F) Methylation of H3K27 inhibits binding of AF10PZP and AF17PZP to H3 (aa 15-34) peptides. (E) Peptide pull-down assays as in (D). (F) ITC was used to determine the Kd values for the interaction of AF10PZP with the indicated peptides. Standard deviation was derived from nonlinear fitting. See Figure S3A for curves.
(G) Acetylation of H3K27 inhibits binding of AF10PZP to H3 (aa 15-34) peptides. Pull-down assays as in (D) with GST-AF10PZP and the indicated biotinylated peptides.
(H) Methylation of H3K27 inhibits binding of AF10PZP and AF17PZP to nucleosomes. Nucleosome binding assays as in (B) with the indicated modified rNucs. Bottom three panels: Western of H3K4me3, H3k27me3 and total H3 to control for integrity of reagents.
See also Figure S1A–C
AF10 and AF17 are both subunits of the multimeric DOT1L complex that mediates H3K79 methylation (Mohan et al., 2010). Besides the PZP domain, both proteins contain an octapeptide motif and leucine zipper (OM-LZ) domain (see schematic, Figure 1A) that directly binds to DOT1L. In addition, the AF10 and AF17 genes are both involved in recurrent chromosomal translocations in human leukemia, suggesting a key role for these proteins in leukemogenesis (Moore et al., 2005; Prasad et al., 1994; Suzukawa et al., 2005). A common theme amongst AF10-translocated leukemias is the aberrant expression of the homebox (HOX) gene cluster, which is implicated in the propagation of the leukemic state (Dik et al., 2005; Okada et al., 2006). Recent studies demonstrated that continued expression of the leukemogenic HOX genes – specifically at the HOXA cluster – is dependent on AF10 (Chen et al., 2013; Deshpande et al., 2014; DiMartino et al., 2002). Importantly, the dependence of HOXA gene expression on AF10 is not limited to AF10-translocated leukemias, suggesting important functions for the intact protein (Deshpande et al., 2014). Thus, AF10 is a potential candidate for therapeutic intervention in leukemia and perhaps other malignancies that are driven by abnormal HOX gene activation.
Here, we characterize the histone-binding activities of the AF10 and AF17 PZP domains. We find that in contrast to other PZP domains or any previously characterized reader domain, the AF10 and AF17 PZP domains recognize a region of H3 that spans amino acids 22 to 27. This interaction with H3 occurs both in the context of peptides and nucleosomes and is inhibited by covalent modification, notably methylation at H3K27. We solved the crystal structure of the AF10 PZP domain bound to H3 peptide, which revealed that all three motifs of the PZP domain cooperate as a unique compact scaffold to mediate H3 recognition. We established a cellular complementation system to functionally investigate the structure and demonstrated that maintenance of H3K79me2 at physiologic levels in cells is dependent upon the ability of AF10 to bind unmodified H3K27. At DOT1L gene targets, the AF10PZP-H3 interaction is needed for AF10 occupancy and enrichment of H3K79me2. The interaction is also required for expression of DOT1L-target genes and for full proliferative capacity of a DOT1L-dependent leukemia cell line. Thus, our study identifies a function for the unmodified state of H3K27 in regulating the evolutionarily conserved and canonical H3K79 methylation mark.
RESULTS
The PZP Domains of AF10 and AF17 Bind Nucleosomes via Amino Acids 15-34 of H3
To test whether the largely identical PZP domains of AF10 and AF17 (AF10PZP and AF17PZP, respectively; 91% identity, see sequence alignment in Figure S1A), like the PZP domains of the JADE and BRPF proteins, interact with nucleosomes, we performed pull-down assays with recombinant PZP domains and recombinant nucleosomes (rNuc) containing biotinylated DNA (Lalonde et al., 2013). As shown in Figure 1B, AF10PZP and AF17PZP as well as the positive control JADE1PZP bound rNuc, whereas the GST control did not. Moreover, like the JADE1 PZP domain, the N-terminal tail of H3 is necessary for the interaction of AF10PZP and AF17PZP with nucleosomes (Figure 1C). To determine if there was a specific region within the H3 N-terminus sufficient for the interaction, pull-down assays were performed using a series of biotinylated peptides spanning the tail region. In contrast to JADE1PZP, which bound as expected to a peptide spanning amino acids 1-21 (H31-21; the very N-terminus of H3) (Saksouk et al., 2009), both AF10PZP and AF17PZP did not bind the H31-21 peptide but rather bound to a peptide spanning amino acids 15-34 (H315-34; Figure 1D). The AF10PZP, in addition to being sufficient, is also necessary for full-length AF10 to bind H315-34 (Figure S1B). Finally, all three domains of the PZP module (see schematic Figure 1A) are necessary for binding to H315-34 as individual domains within the PZP of AF10 and AF17 as well as PHD1-Zn knuckle and Zn knuckle-PHD2 polypeptides did not bind to any of the peptides or to nucleosomes (data not shown). We conclude that in the context of histone binding, AF10PZP and AF17PZP each behave as a single functional unit that binds to a region of H3 that spans amino acids 15-34.
AF10PZP and AF17PZP Binding to H3 is Repelled by Methylation and Acetylation at K27
H3K27 methylation is a canonical and biologically important modification present within the H315-34 peptide (Vizan et al., 2014). As many PHD fingers function as highly sensitive “readers” of the methylation status at H3K4, we asked if the PHD-finger-containing AF10PZP and AF17PZP domains have a similar relationship to the methylation status at H3K27. Notably, in contrast to the H3K27me-binding chromodomain of CBX7 (Yap et al., 2010), the interaction of AF10PZP and AF17PZP to the H315-34 peptide was abolished by mono-, di-, and tri-methylation at K27 (Figure 1E). Quantitation of the interaction by isothermal titration calorimetry (ITC) demonstrated that AF10PZP bound H315-34 peptides with a Kd of ~5 μM and the binding could no longer be detected upon addition of a single methyl moiety, as well as di- and tri-methylation, at K27 (Figure 1F and S1C). Finally, H3K27 is also acetylated in cells, and this modification, like methylation, disrupted recognition of H3 by AF10PZP and AF17PZP (Figure 1G; data not shown).
To address the role of H3K27 modification in the context of nucleosome recognition, we used methyl lysine analog chemistry (Simon et al., 2007) to install tri-methylation at either lysine 4 (H3KC4me3) or lysine 27 (H3KC27me3) on rNucs. Consistent with the results obtained when using peptides, AF10PZP and AF17PZP bound to control and H3KC4me3 rNucs but not to H3KC27me3 nucleosomes (Figure 1H). Thus, we conclude that nucleosome recognition by the PZP domains of AF10 and AF17 is antagonized by methylation at lysine 27.
Structure of AF10PZP in the Free State
In order to understand the molecular basis of AF10PZP-H3K27 interaction, we first determined the structure of AF10PZP in the apo-form. We focused on AF10, as this protein but not AF17 has been shown to regulate H3K79 methylation (Mohan et al., 2010). The structure of free AF10PZP was solved at 1.6 Å resolution (x-ray statistics listed in Table 1). The protein adopts a unique fold composed of PHD finger 1 (in blue) coordinated by Zn1 and Zn2, a Zn-knuckle (in green) coordinated by Zn3, and PHD finger 2 (in magenta) coordinated by Zn4 and Zn5 (Figure S2A). The first PHD finger adopts a mixed α/β fold characteristic of canonical PHD fingers, while PHD finger 2 is atypical and is composed solely of six α-helical segments with a disordered segment spanning residues 179–188 (Figure S2A). The Zn knuckle plays a bridging role and forms contacts with both PHD fingers 1 (Figure S2B) and 2 (Figure S2C), resulting in an overall compact fold for the PZP motif (Figure S2A).
Table 1.
Data collection and refinement statistics.
| AF10 (1-208) | AF10 (18-208)-(GS)6-H3(1-36) | |
|---|---|---|
| Data collection | ||
| Space group | P 41 21 2 | P 21 |
| Cell dimensions | ||
| a, b, c (Å) | 51.9, 51.9, 147.9 | 42.9, 80, 59.3 |
| a, b, g () | 90, 90, 90 | 90, 102.1, 90 |
| Resolution (Å) | 50 - 1.6 (1.63 - 1.60) | 50 - 2.62 (2.76 -2.62) |
| Wavelength (Å) | 1.283 | 0.979 |
| Rsym or Rmerge | 0.075 (0.683) | 0.148 (0.545) |
| I/sI | 55.0 (2.56) | 10.7 (2.56) |
| Completeness (%) | 99.8 (99.3) | 98.1 (99.2) |
| Redundancy | 7.9 (4.6) | 3.1 (3.1) |
| Refinement | ||
| Resolution (Å) | 50 - 1.6 (1.63 - 1.60) | 50 - 2.62 (2.76 - 2.62) |
| No. reflections | 50458 (3652) | 11659 (1167) |
| Rwork/Rfree | 0.19/0.21 | 0.18/0.24 |
| No. atoms | 1432 | 2857 |
| Protein | 1319 | 2784 |
| Zn | 5 | 10 |
| Water | 108 | 63 |
| Wilson B | 21.97 | 24.93 |
| B-factors | 29.68 | 29.33 |
| Protein | 29.33 | 29.26 |
| Zn | 23.56 | 22.15 |
| Water | 34.25 | 24.11 |
| Ramachandran outliers | 0 | 0 |
| Ramachandran allowed | 1.2 % | 3.3 % |
| Ramachandran favored | 98.8 % | 96.7 % |
| R.m.s deviations | ||
| Bond lengths (Å) | 0.007 | 0.004 |
| Bond angles (°) | 1.001 | 0.805 |
Highest resolution shell is shown in parenthesis.
Structure of AF10PZP Reveals Dynamic Conformational Transition upon H3 Binding
Next, the crystal structure of AF10PZP bound to an H3 peptide was solved. Our attempts at crystallization of H3(1-36) peptide bound to AF10PZP were unsuccessful. We therefore fused the C-terminus of the AF10 PZP domain (aa 18-208) to the N-terminus of H3(1-36) using a (Gly-Ser)6 linker to obtain diffraction quality crystals of the hybrid construct. We solved the 2.62 Å crystal structure of the covalently-linked AF10PZP-H3(1-36) peptide construct (x-ray statistics in Table 1). The structure of the complex is shown in Figure 2A with the protein in a color-coded ribbon representation and the bound peptide in a yellow stick representation. A segment of the H3(1-36) peptide spanning residues A21 to K27 is bound with directionality (see 2Fo-Fc composite simulated annealing omit map contoured at 1.2σ in Figure 2B) within a surface channel of the PZP domain (Figure 2C); this minimal peptide was sufficient for binding, though with weaker affinity than the longer peptide (Figure S1D). Within this channel, intermolecular contacts involving H3 residues T22 (Figure 2D and S2E), K23 (Figure 2E and S2F), A24 (Figure 2F and S2G), R26 (Figure 2G and S2H) and K27 (Figure 2H and S2I) span all sub-domains (PHD finger 1, Zn knuckle and PHD finger 2) of the PZP domain. The ε-NH3+ group of unmodified K23 is anchored through hydrogen bond formation with the carboxylate group of E179 of the PZP domain (Figure 2E). By contrast, the ε-NH3+ group of unmodified K27 is anchored through hydrogen bond formation with three backbone carbonyl groups of the PZP domain, leaving no room for even mono-methylation at K27 (Figure 2H and S2I).
Figure 2. Structural basis of H3 recognition by AF10PZP.

(A) 2.6 Å crystal structure of AF10PZP (18-208) complexed with H3(1-36) peptide.
(B) Fo-Fc omit map (2.5σ level) of the bound H3 peptide (side chain density can be traced from A21 to K27) in the structure of AF10PZP-H3 complex.
(C) Positioning of the bound H3 peptide (A21 to K27 in stick representation) within the AF10PZP binding site (electrostatic surface representation).
(D–H) Details of intermolecular contacts involving residues T22 (panel D), K23 (panel E), A24 (panel F), R26 (panel G) and K27 (panel H) of the bound H3 peptide with the AF10PZP in the structure of the complex.
(I) Formation of an anti-parallel β-sheet within the PHD2 finger of AF10PZP upon complex formation with H3(1-36) peptide.
See also Figures S1D and S2
A segment within PHD finger 2 of AF10PZP, that spans amino acids 179-188, is disordered in the 1.60 Å structure in the free state (Figure S2A). Upon complex formation with H3(1-36) peptide, this region becomes ordered to form an anti-parallel β-sheet (Figure 2I, marked by arrow; free and bound structures superimposition shown in Figure S2D). This dynamic β-sheet forms part of the binding channel wall within the PZP domain and contributes to the alignment of the bound H3 peptide with residue E179, which emanates from this β-sheet platform, directly involved in recognition of the ε-NH3+ group of K23 (Figure 2E).
Validation of Intermolecular Contacts in Covalently-linked Complex from Site-specific Mutation Studies
Alanine substitutions of key amino acids in AF10PZP that interact with H3 largely abolished or decreased binding to H3 peptides as determined in pull-down and ITC assays (Figure 3A, 3B and S1E). These AF10 residues include those that interact with H3T22, H3K23, H3A24, H3R26, and H3K27 (Figure 2D–H). In contrast, mutation of residues located on the opposite side of the H3-binding surface had no impact on the AF10PZP-H315-34 interaction (Figure S1F). In complementary experiments, alanine substitution of two H3 residues contacted by AF10PZP, H3R26A and H3K27A, disrupt AF10PZP and AF17PZP binding to rNucs (Figure 3C). The structure revealed that the bound H3 tail adopts an α-helical conformation, with the side chain of H3A25 not making intermolecular contacts with AF10PZP (see Figure 2A). H3A25P substitution interrupted α-helix formation and prevented nucleosome binding by AF10PZP and AF17PZP (Figure 3C). Finally, the H3K27M substitution associated with pediatric gliomas (Lewis et al., 2013) also disrupted binding (data not shown). Taken together, the biochemical, biophysical, and structural data point to a specific interaction between AF10PZP and H3 that is exquisitely sensitive to chemical alteration of K27.
Figure 3. Structure-guided mutations disrupt AF10PZP binding to H3 in vitro in cells.

(A and B) Structure-guided mutations in AF10PZP disrupt binding to H3(15-34) peptides. (A) Peptide pull-down assays with the indicated mutants and peptides. (B) ITC as in Figure 1F with the AF10PZP derivatives as in (A) binding to H3(15-34) peptide. Standard deviation was derived from nonlinear fitting. See Figure S3C for curves.
(C) Mutations of key residues on H3 disrupt AF10PZP-binding to nucleosomes. Pull-down assays as in Figure 1B with nucleosomes containing either wild-type H3 or H3 with the indicated point mutations. Bottom panel: Western blot showing H3 as a loading control.
(D–F) The AF10PZP-H3 interaction stabilizes AF10 at chromatin. (D) Western blot analysis with indicated antibodies of lysates biochemically separated into soluble and chromatin-enriched (Chrom) fractions from U2OS cells expressing wild-type or mutant GFP-AF10. WCE: whole cell extract is shown to control for total AF10 expression. Tubulin and H3 levels are shown as control for the integrity of fractionation. (E) In situ fractionation of HeLa cells expressing GFP-AF10 or GFP-AF10L107A. Representative image of GFP localization determined in the indicated cell lines ± detergent treatment prior to fixation and visualization by fluorescence microscopy. DAPI is shown to indicate nuclei of cells. (F) Quantification of in situ fractionation experiments as in (E). Normalized fluorescence refers to the average intensity of signal in the absence of detergent treatment (see methods). Error bars represent the standard error of the mean (SEM) based on counting 30–60 cells from three independent fields and two biological replica.
See also Figures S1E–F and S2J
It is important to note that the key intermolecular contacts involving H3(22-27) and AF10PZP in the crystal structure of the covalently-linked AF10PZP-(Gly-Ser)6-H3(1-36) complex have been validated using site-specific mutations of both H3 and AF10PZP residues (Figures 3A–C, and S1C-F), ruling out impact on intermolecular recognition resulting from the covalent linkage between components of the complex.
The PZP-H3 Interaction Stabilizes AF10 at Chromatin
We next investigated the role of H3-binding by the PZP domain on AF10 function in cells. AF10 – as a direct binding partner of DOT1L and regulator of H3K79 methylation – is likely chromatin-associated under physiologic conditions. We postulated that the interaction between the PZP domain and H3 might therefore regulate AF10 stability at chromatin. To test this idea, we established a tetracycline-inducible system in U2OS cells to express GFP-AF10 and GFP-AF10L107A, a mutant that abolishes H3 binding (Figure 3A, B) and is involved in both K23 and K27 side-chain recognition (Figure S2J). Analysis of lysates biochemically separated into chromatin-enriched and soluble fractions demonstrated that AF10L107A enrichment at chromatin was reduced in comparison to wild-type AF10, with more mutant being detected in the soluble fraction (Figure 3D). Decreased chromatin-association for AF10L107A versus wild-type AF10 was also observed by in situ fractionation as an independent method (Figure 3E; see 3F for quantification). Thus, we conclude that PZP binding to H3 plays a role in stabilizing AF10 at chromatin in cells.
The AF10-H3 Interaction is Essential for Global H3K79 Dimethylation
We next knocked out AF10 in U2OS cells using the CRISPR/Cas9 system and two independent small guide RNAs (sgRNAs) targeting splice sites on the AF10 gene, which resulted in loss of AF10 expression and no change in DOT1L expression (Figure S3A and S3B). Relative to wild-type cells, knockout of AF10 led to depletion of H3K79me2 levels as determined by both Western analysis (Figure 4A) and by mass spectrometry (Figure S3C), consistent with previous reports investigating AF10 function and its connection to DOT1L catalytic activity (Deshpande et al., 2014; Mohan et al., 2010). Of the three possible methyl states at H3K79, the loss of dimethyl was most pronounced, and no changes were detected in the methylation levels at several other histone residues (Figures 4A, S3C, and Table S1). Similar results were observed when we depleted AF10 in HT1080 cells as a second independent cell line (Figure S3D and S3E), whereas depletion of AF17 had no affect on H3K79 methylation (Figure S3F), consistent with previous reports (Deshpande et al., 2014; Mohan et al., 2010). We note that peptide arrays were employed to test the epitope and methyl-state specificity of antibodies used in our studies (data not shown; (Bua et al., 2009; Fuchs et al., 2011)). Finally, the levels of monoubiquitination of H2B (H2Bubi), an upstream signal that promotes H3K79 methylation (Lee et al., 2007), were unaltered upon AF10 depletion (Figure S3G). Taken together, our results are consistent with previous reports that AF10 regulates DOT1L activity and is preferentially required for the generation of the dimethyl state at H3K79.
Figure 4. An intact AF10 PZP domain is required for DOT1L enzymatic and cellular functions in leukemic cells.

(A) Depletion of AF10 using the CRISPR/Cas9 system specifically decreases global H3K79me2 level in U2OS cells. Western analysis with the indicated antibodies of whole cell extract (WCE) stably expressing control sgRNA or two independent sgRNAs targeting AF10. H3 is shown as a loading control.
(B) AF10PZP-binding to H3 is required for global H3K79me2. Western analysis as in (A) with cell lines expressing either control or AF10-targeting sgRNA. Dox: cells were treated with doxycycline to induce expression of GFP, GFP-AF10, and GFP-AF10L107A as shown.
(C) AF10PZP-binding to H3 regulates expression of DOT1L-target gene PABPC1. Quantitative real-time PCR (qPCR) analysis of PABPC1 mRNA expression level in the indicated cell lines. Error bars indicate SEM from three experiments.
(D) An intact AF10PZP domain promotes H3K79me2 enrichment at the PABPC1 gene. Top panel: schematic of PABPC1 gene with the position of the two primers used for direct chromatin immunoprecipitation (ChIP) assays indicated. Middle panel: Key showing the cell lines used for the ChIP assays. Left bottom panel: H3K79me2 enrichment at PABPC1 with the H3K79me2 signal normalized to total H3 signal. Right bottom panel: GFP enrichment at PABPC1 shown as % GFP signal/input. Error bars indicate the SEM from three experiments.
(E) The AF10-H3 interaction regulates H3K79me2 in ML2 leukemia cells. Western analysis as in (A) in ML2 cells treated as indicated.
(F) AF10 binding to H3 required for full expression of DOT1L-target genes in ML2 leukemia cells. Analysis of HOXA10 and RUNX1 mRNA expression as in (C) in ML2 cells treated as indicated in the key. Error bars represent the SEM from three experiments.
(G) AF10PZP-binding to H3 is required for ML2 leukemia cells to maintain normal proliferative rate. Growth curve of cell lines treated as indicated in the key. Error bars represent the SEM from three independent experiments.
See also Figures S3 and S4
Exogenous AF10 can be expressed in the presence of the AF10-sgRNAs, as the target sequences (splice site junctions) are absent from the cDNA. Therefore, to test the functional consequences of the AF10-H3 interaction in a cellular context, we established a complementation system by knocking out endogenous AF10 in the inducible GFP-AF10 wild-type and mutant U2OS cell lines. As shown in Figure 4B and S3C, in an AF10 deletion background, induction of wild-type GFP-AF10, but not the L107A H3-binding mutant, reconstituted H3K79me2 levels to those observed in control cells. In mammals, H3K79 methylation (both me2 and me3) correlates with gene activity and is thought to promote expression, potentially through positive regulation of transcription elongation (McLean et al., 2014; Nguyen and Zhang, 2011; Steger et al., 2008). In this context, loss of AF10 led to a modest reduction in expression of the DOT1L-target gene PABPC1 (Figure 4C) (Li et al., 2014; Steger et al., 2008). PABPC1 expression was reconstituted close to control levels upon induction of GFP-AF10 but not with GFP-AF10L107A (Figures 4C). At the level of chromatin, H3K79me2 is enriched proximal to the transcription start site (TSS) and gene body of the PABPC1 gene (Figure 4D). This signal, which is lost in AF10 deleted cells, is reconstituted upon induction of GFP-AF10, but not upon induction of GFP-AF10L107A (Figure 4D). Consistent with these results, GFP-AF10L107A occupancy at the PABPC1 gene was low relative to wild-type GFP-AF10 (Figure 4D), suggesting a model in which the inability of the H3-binding mutant to reconstitute H3K79me2 is due to reduced affinity for chromatin.
AF10-Mediated H3K79me2 Regulates Expression of Key DOT1L-Target Genes
For most cell types, including U2OS cells, loss of H3K79 methylation due to inhibition of DOT1L is well tolerated ((Chen et al., 2013; Deshpande et al., 2013; McLean et al., 2014; Nguyen and Zhang, 2011); unpublished observations). In contrast, cell lines derived from MLL-fusion leukemias are frequently addicted to DOT1L and inhibitors of DOT1L are being developed as chemotherapeutics to treat these difficult cancers (for review see (Daigle et al., 2013)). We postulated that the AF10PZP-H3 interaction might be important for a subset of MLL-fusion leukemic cells in which the fusion partner lacks chromatin-binding activity (see discussion). To test this idea, we established an AF10 complementation system in ML2 leukemia cells, a line in which H3K79me2 and H3K27me3 are largely mutually exclusive at the protein level as determined by Western analysis and quantitative mass spectrometry (Figure S4A and S4B). ML2 cells contain an MLL-AF6 fusion (AF6: ALL1-Fused gene from chromosome 6, a protein normally involved in cell-cell junctions) and are sensitive to DOT1L inhibition (Deshpande et al., 2013)). Like with U2OS and HT1080 cells, maintenance of H3K79me2 levels in ML2 cells requires an intact AF10 PZP domain (Figure 4E, S4C, and S4D). AF10 depletion with loss of H3K79me2 resulted in decreased expression in several key DOT1L-target genes, such as HOXA10, and did not affect DOT1L protein levels (Figure 4F, S4E, and S4F). The expression of these genes was restored in cells complemented with wild-type AF10 but not with AF10L107A (Figure 4F and S4E). Further, AF10 depletion in ML2 cells decreased the cellular proliferation rates relative to control depleted cells, a phenotype that was reversed upon reconstitution of AF10-depleted ML2 cells with wild-type AF10, but not with mutant AF10L107A (Figure 4G). These data argue that the PZP-mediated interaction between AF10 and H3 plays a critical role in regulating both the expression of DOT1L-target genes and the proliferation of leukemic cells.
DISCUSSION
Here, we identify the PZP domains of AF10 and AF17 as reader domains that have the hereto-unknown specificity for H3 in a region spanning K27. In contrast to any previously described readers, the AF10 and AF17 PZP-mediated H3-recognition events are specific for the unmodified state of H3K27 and obstructed by chemical modification at K27. This residue is known to be a highly methylated in embryonic stem cells (Ferrari et al., 2014). In the cell lines used in our study, we also we observed a high degree of H3K27 methylation, whereas the majority of H3K79 is unmodified, consistent with the notion that H3K79 modification is repelled by H3K27 methylation (Figure S4G and S4H). Functionally, the interaction between the AF10 PZP domain and H3 suggests a molecular mechanism where AF10 regulates the stability of DOT1L at chromatin targets, and by extension, the generation of H3K79 methylation. Our structural and biochemical analyses have defined the key recognition elements that contribute to complex formation. These include the dynamic formation of a β-sheet within PHD finger 2 on complex formation that contributes to completion of an H3 peptide-binding channel formation (Figure 2 and S2D). In addition, the side-chain of unmodified H3K27 is accommodated within a compact, hydrogen bond acceptor-lined binding pocket in which even mono-methylation of K27 cannot be tolerated due to steric constraints (Figure 1F and S1C).
The exquisite sensitivity of H3-recognition by AF10 to chemical modification at K27 elucidates a molecular connection between K27 and K79. Indeed, in recent reports focusing on either leukemic or somatic cell reprogramming (Bernt et al., 2011; Onder et al., 2012), H3K79me2 and H3K27me3 showed a genome-wide anti-correlation in their enrichment patterns, consistent with their antagonistic relationship in regulating transcription and our proteomic analysis indicating mutual exclusivity of H3K79me2 and H3K27me3 at the protein level (Figure S4A and S4B). We propose that H3-recognition by AF10PZP serves to ensure the mutual exclusivity of H3K79me2 and H3K27me3 at co-regulated genes by directing DOT1L to regions devoid of H3K27 methylation. The crosstalk between these two residues also highlights the diversity of functional states and signaling potential associated with dynamic modification of H3K27, which includes mono-, di-, tri-methylation, acetylation, and the unmodified state. Moreover, our findings suggest a model in which H3K27 methylation, in addition to its well-characterized and important role in actively directing epigenetic silencing (Vizan et al., 2014), also implicitly promotes silencing by repelling AF10 and DOT1L activity from target sites.
In MLL-fusion leukemias, aberrant recruitment of DOT1L to target genes is thought to activate a pathologic transcriptional program that promotes leukemogenesis (Deshpande et al., 2012; Krivtsov and Armstrong, 2007; McLean et al., 2014; Nguyen and Zhang, 2011; Stein and Tallman, 2015). In the majority of cases, the MLL-fusion partner is a DOT1L-associated protein and directly recruits DOT1L to chromatin. However, for a significant number of AML patients with MLL translocations, MLL is not fused to a core DOT1L complex component. In these cases, the mechanism by which DOT1L is targeted to chromatin is unclear, yet these cell lines require DOT1L-mediated activation of oncogenic genes. We focused on one such fusion (MLL-AF6) and provided evidence that the AF10-H3 interaction is important for DOT1L functions at chromatin. We postulate that developing compounds targeting the binding pocket of AF10PZP for H3 will disrupt H3K79 methylation and provide therapeutic value for patients suffering from atypical MLL fusion AMLs, potentially in combination with DOT1L inhibitors to mitigate the risk of cancers developing resistance. Together, our findings have identified the molecular basis of AF10-dependent crosstalk between H3K27 and H3K79, two crucial H3 residues with roles in chromatin regulation, epigenetics, and human disease.
EXPERIMENTAL PROCEDURES
AF10PZP, AF17PZP, JADE1PZP, and CBX7 domains were cloned into pGEX-6P-1 for in vitro binding experiments. Histone H3 and mutants were cloned into pET3 or pET21a for recombinant nucleosome reconstitution. Full length AF10 (NM_004641.3) was originally cloned into pENTR3C and then recombined into pCDNA5-Hygro-FRT-TO-GFP-N DEST and pLenti CMV Hygro DEST (W117-1) (Addgene). See Supplementary information for detailed methods.
Supplementary Material
Highlights.
AF10 PZP domain recognizes unmodified H3K27 to regulate H3K79 dimethylation
The PZP domain functions as a single unit to read the modification state at H3K27
PZP-dependent chromatin binding by AF10 regulates DOT1L target genes
AF10 binding to H3K27 regulates proliferation of MLL-AF6 rearranged leukemic cells
Acknowledgments
We thank S. Blacklow for the T-REx U2OS cell. This work was supported in part by grants from the Leukemia and Lymphoma Society and STARR Foundation to D.J.P. and from the NIH to O.G. (R01 GM079641), S.A. (CA66996 and CA176745), B.A.G. (GM110174 and AI118891), and B.D.S. (GM110058). A.W. was supported by NIH training grant (T32 GM007276). S.A. is a consultant for Epizyme, Inc. O.G. and B.D.S. are co-founders of EpiCypher, Inc.
Footnotes
ACCESSION NUMBERS
The coordinates and structure factors for the AF10-apo structure and for the AF10-H3(1-36) hybrid construct have been deposited in the Protein Data Bank under accession numbers PDB_ID 5DAG and PDB_ID 5DAH, respectively.
AUTHOR CONTRIBUTIONS
S.C. performed crystallographic structure determination and ITC measurements under the supervision of D.J.P., Y.Z. and A.W.W. performed the molecular and cellular studies under the supervision of O.G. A.J.D. and S.A. provided reagents and contributed to experimental design. S.S. and B.A.G. performed mass spectrometry experiments. K.K. and B.D.S. generated peptides. O.G. and D.J.P. wrote the manuscript with contributions by all the authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bernt KM, Zhu N, Sinha AU, Vempati S, Faber J, Krivtsov AV, Feng Z, Punt N, Daigle A, Bullinger L, et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer cell. 2011;20:66–78. doi: 10.1016/j.ccr.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bua DJ, Kuo AJ, Cheung P, Liu CL, Migliori V, Espejo A, Casadio F, Bassi C, Amati B, Bedford MT, et al. Epigenome microarray platform for proteome-wide dissection of chromatin-signaling networks. PLoS One. 2009;4:e6789. doi: 10.1371/journal.pone.0006789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Deshpande AJ, Banka D, Bernt KM, Dias S, Buske C, Olhava EJ, Daigle SR, Richon VM, Pollock RM, et al. Abrogation of MLL-AF10 and CALM-AF10-mediated transformation through genetic inactivation or pharmacological inhibition of the H3K79 methyltransferase Dot1l. Leukemia. 2013;27:813–822. doi: 10.1038/leu.2012.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle SR, Olhava EJ, Therkelsen CA, Basavapathruni A, Jin L, Boriack-Sjodin PA, Allain CJ, Klaus CR, Raimondi A, Scott MP, et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood. 2013;122:1017–1025. doi: 10.1182/blood-2013-04-497644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande AJ, Bradner J, Armstrong SA. Chromatin modifications as therapeutic targets in MLL-rearranged leukemia. Trends in immunology. 2012;33:563–570. doi: 10.1016/j.it.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande AJ, Chen L, Fazio M, Sinha AU, Bernt KM, Banka D, Dias S, Chang J, Olhava EJ, Daigle SR, et al. Leukemic transformation by the MLL-AF6 fusion oncogene requires the H3K79 methyltransferase Dot1l. Blood. 2013;121:2533–2541. doi: 10.1182/blood-2012-11-465120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande AJ, Deshpande A, Sinha AU, Chen L, Chang J, Cihan A, Fazio M, Chen CW, Zhu N, Koche R, et al. AF10 regulates progressive H3K79 methylation and HOX gene expression in diverse AML subtypes. Cancer cell. 2014;26:896–908. doi: 10.1016/j.ccell.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dik WA, Brahim W, Braun C, Asnafi V, Dastugue N, Bernard OA, van Dongen JJ, Langerak AW, Macintyre EA, Delabesse E. CALM-AF10+ T-ALL expression profiles are characterized by overexpression of HOXA and BMI1 oncogenes. Leukemia. 2005;19:1948–1957. doi: 10.1038/sj.leu.2403891. [DOI] [PubMed] [Google Scholar]
- DiMartino JF, Ayton PM, Chen EH, Naftzger CC, Young BD, Cleary ML. The AF10 leucine zipper is required for leukemic transformation of myeloid progenitors by MLL-AF10. Blood. 2002;99:3780–3785. doi: 10.1182/blood.v99.10.3780. [DOI] [PubMed] [Google Scholar]
- Ferrari KJ, Scelfo A, Jammula S, Cuomo A, Barozzi I, Stutzer A, Fischle W, Bonaldi T, Pasini D. Polycomb-dependent H3K27me1 and H3K27me2 regulate active transcription and enhancer fidelity. Molecular cell. 2014;53:49–62. doi: 10.1016/j.molcel.2013.10.030. [DOI] [PubMed] [Google Scholar]
- Fuchs SM, Krajewski K, Baker RW, Miller VL, Strahl BD. Influence of combinatorial histone modifications on antibody and effector protein recognition. Curr Biol. 2011;21:53–58. doi: 10.1016/j.cub.2010.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein BJ, Lalonde ME, Cote J, Yang XJ, Kutateladze TG. Crosstalk between epigenetic readers regulates the MOZ/MORF HAT complexes. Epigenetics: official journal of the DNA Methylation Society. 2014;9:186–193. doi: 10.4161/epi.26792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nature reviews Cancer. 2007;7:823–833. doi: 10.1038/nrc2253. [DOI] [PubMed] [Google Scholar]
- Lalonde ME, Avvakumov N, Glass KC, Joncas FH, Saksouk N, Holliday M, Paquet E, Yan K, Tong Q, Klein BJ, et al. Exchange of associated factors directs a switch in HBO1 acetyltransferase histone tail specificity. Genes & development. 2013;27:2009–2024. doi: 10.1101/gad.223396.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan F, Collins RE, De Cegli R, Alpatov R, Horton JR, Shi X, Gozani O, Cheng X, Shi Y. Recognition of unmethylated histone H3 lysine 4 links BHC80 to LSD1-mediated gene repression. Nature. 2007;448:718–722. doi: 10.1038/nature06034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Shukla A, Schneider J, Swanson SK, Washburn MP, Florens L, Bhaumik SR, Shilatifard A. Histone crosstalk between H2B monoubiquitination and H3 methylation mediated by COMPASS. Cell. 2007;131:1084–1096. doi: 10.1016/j.cell.2007.09.046. [DOI] [PubMed] [Google Scholar]
- Lewis PW, Muller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, Garcia BA, Muir TW, Becher OJ, Allis CD. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science. 2013;340:857–861. doi: 10.1126/science.1232245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Ilin S, Wang W, Duncan EM, Wysocka J, Allis CD, Patel DJ. Molecular basis for site-specific read-out of histone H3K4me3 by the BPTF PHD finger of NURF. Nature. 2006;442:91–95. doi: 10.1038/nature04802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wen H, Xi Y, Tanaka K, Wang H, Peng D, Ren Y, Jin Q, Dent SY, Li W, et al. AF9 YEATS domain links histone acetylation to DOT1L-mediated H3K79 methylation. Cell. 2014;159:558–571. doi: 10.1016/j.cell.2014.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews AG, Kuo AJ, Ramon-Maiques S, Han S, Champagne KS, Ivanov D, Gallardo M, Carney D, Cheung P, Ciccone DN, et al. RAG2 PHD finger couples histone H3 lysine 4 trimethylation with V(D)J recombination. Nature. 2007;450:1106–1110. doi: 10.1038/nature06431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CM, Karemaker ID, van Leeuwen F. The emerging roles of DOT1L in leukemia and normal development. Leukemia. 2014;28:2131–2138. doi: 10.1038/leu.2014.169. [DOI] [PubMed] [Google Scholar]
- Mohan M, Herz HM, Takahashi YH, Lin C, Lai KC, Zhang Y, Washburn MP, Florens L, Shilatifard A. Linking H3K79 trimethylation to Wnt signaling through a novel Dot1-containing complex (DotCom) Genes & development. 2010;24:574–589. doi: 10.1101/gad.1898410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore SD, Strehl S, Dal Cin P. Acute myelocytic leukemia with t(11;17)(q23;q12-q21) involves a fusion of MLL and AF17. Cancer genetics and cytogenetics. 2005;157:87–89. doi: 10.1016/j.cancergencyto.2004.06.015. [DOI] [PubMed] [Google Scholar]
- Musselman CA, Kutateladze TG. Handpicking epigenetic marks with PHD fingers. Nucleic Acids Res. 2011;39:9061–9071. doi: 10.1093/nar/gkr613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen AT, Zhang Y. The diverse functions of Dot1 and H3K79 methylation. Genes & development. 2011;25:1345–1358. doi: 10.1101/gad.2057811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y, Jiang Q, Lemieux M, Jeannotte L, Su L, Zhang Y. Leukaemic transformation by CALM-AF10 involves upregulation of Hoxa5 by hDOT1L. Nature cell biology. 2006;8:1017–1024. doi: 10.1038/ncb1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onder TT, Kara N, Cherry A, Sinha AU, Zhu N, Bernt KM, Cahan P, Marcarci BO, Unternaehrer J, Gupta PB, et al. Chromatin-modifying enzymes as modulators of reprogramming. Nature. 2012;483:598–602. doi: 10.1038/nature10953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pena PV, Davrazou F, Shi X, Walter KL, Verkhusha VV, Gozani O, Zhao R, Kutateladze TG. Molecular mechanism of histone H3K4me3 recognition by plant homeodomain of ING2. Nature. 2006;442:100–103. doi: 10.1038/nature04814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad R, Leshkowitz D, Gu Y, Alder H, Nakamura T, Saito H, Huebner K, Berger R, Croce CM, Canaani E. Leucine-zipper dimerization motif encoded by the AF17 gene fused to ALL-1 (MLL) in acute leukemia. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:8107–8111. doi: 10.1073/pnas.91.17.8107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saksouk N, Avvakumov N, Champagne KS, Hung T, Doyon Y, Cayrou C, Paquet E, Ullah M, Landry AJ, Cote V, et al. HBO1 HAT complexes target chromatin throughout gene coding regions via multiple PHD finger interactions with histone H3 tail. Molecular cell. 2009;33:257–265. doi: 10.1016/j.molcel.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Hong T, Walter KL, Ewalt M, Michishita E, Hung T, Carney D, Pena P, Lan F, Kaadige MR, et al. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature. 2006;442:96–99. doi: 10.1038/nature04835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon MD, Chu F, Racki LR, de la Cruz CC, Burlingame AL, Panning B, Narlikar GJ, Shokat KM. The site-specific installation of methyl-lysine analogs into recombinant histones. Cell. 2007;128:1003–1012. doi: 10.1016/j.cell.2006.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steger DJ, Lefterova MI, Ying L, Stonestrom AJ, Schupp M, Zhuo D, Vakoc AL, Kim JE, Chen J, Lazar MA, et al. DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells. Mol Cell Biol. 2008;28:2825–2839. doi: 10.1128/MCB.02076-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein EM, Tallman MS. Mixed lineage rearranged leukaemia: pathogenesis and targeting DOT1L. Current opinion in hematology. 2015;22:92–96. doi: 10.1097/MOH.0000000000000123. [DOI] [PubMed] [Google Scholar]
- Suzukawa K, Shimizu S, Nemoto N, Takei N, Taki T, Nagasawa T. Identification of a chromosomal breakpoint and detection of a novel form of an MLL-AF17 fusion transcript in acute monocytic leukemia with t(11;17)(q23;q21) International journal of hematology. 2005;82:38–41. doi: 10.1532/IJH97.05025. [DOI] [PubMed] [Google Scholar]
- Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol. 2007;14:1025–1040. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeulen M, Mulder KW, Denissov S, Pijnappel WW, van Schaik FM, Varier RA, Baltissen MP, Stunnenberg HG, Mann M, Timmers HT. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell. 2007;131:58–69. doi: 10.1016/j.cell.2007.08.016. [DOI] [PubMed] [Google Scholar]
- Vizan P, Beringer M, Ballare C, Di Croce L. Role of PRC2-associated factors in stem cells and disease. The FEBS journal. 2014 doi: 10.1111/febs.13083. [DOI] [PubMed] [Google Scholar]
- Wysocka J, Swigut T, Xiao H, Milne TA, Kwon SY, Landry J, Kauer M, Tackett AJ, Chait BT, Badenhorst P, et al. A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature. 2006;442:86–90. doi: 10.1038/nature04815. [DOI] [PubMed] [Google Scholar]
- Yap KL, Li S, Munoz-Cabello AM, Raguz S, Zeng L, Mujtaba S, Gil J, Walsh MJ, Zhou MM. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Molecular cell. 2010;38:662–674. doi: 10.1016/j.molcel.2010.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.