

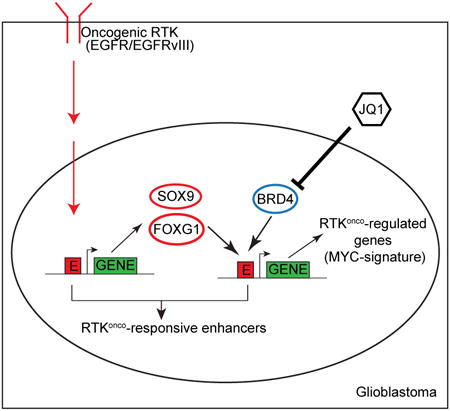
Summary
Epidermal Growth Factor Receptor (EGFR) gene amplification and mutations are the most common oncogenic events in Glioblastoma (GBM), but the mechanisms by which they promote aggressive tumor growth are not well understood. Here, through integrated epigenome and transcriptome analyses of cell lines, genotyped clinical samples and TCGA data, we show that EGFR mutations remodel the activated enhancer landscape of GBM, promoting tumorigenesis through a SOX9 and FOXG1-dependent transcriptional regulatory network in vitro and in vivo. The most common EGFR mutation, EGFRvIII, sensitizes GBM cells to the BET-bromodomain inhibitor JQ1 in a SOX9, FOXG1-dependent manner. These results identify the role of transcriptional/epigenetic remodeling in EGFR-dependent pathogenesis and suggest a mechanistic basis for epigenetic therapy.
Graphical abstract
Introduction
Growth factor receptors are frequently amplified and/or mutated in cancer (Ciriello et al., 2013; Kandoth et al., 2013; Vogelstein et al., 2013). Growth factor receptor mutations activate intracellular signaling cascades that promote growth, at least in part, by regulating transcriptional networks (Lee and Young, 2013). Master transcription factors (TFs) interact with cis-regulatory DNA sequences to control transcriptional repertoires that drive tumor growth and survival (Baylin and Jones, 2011; Suva et al., 2013). However, the mechanisms by which mutated growth factor receptors control the transcriptional machinery and alter the epigenetic landscape of cancer cells to reprogram transcription, are not well understood.
Glioblastoma (GBM) is the most common primary brain cancer of adults and one of the most lethal of all human malignancies (Cloughesy et al., 2014). The epidermal growth factor receptor (EGFR) is amplified and/or mutated in up to 60% of GBMs, promoting tumor growth and survival through persistent activation of signaling networks and metabolic reprogramming (Cloughesy et al., 2014; Furnari et al., 2015). Currently, the impact of EGFR alteration on the transcriptional/epigenetic landscape of tumor cells is not known. Here, using global RNA-seq and ChIP-seq analysis of GBM cell lines, patient-derived tumor cells in neurosphere culture and clinical biopsies genotyped for EGFR mutation status, we identify a SOX9 and FOXG1-dependent transcriptional regulatory network that remodels the enhancer activation landscape to drive EGFRvIII-dependent tumorigenesis in vitro and in vivo and sensitizes GBM cells to the BET-bromodomain inhibitor JQ1. These results link oncogene-induced signaling with a highly specific program of epigenetic remodeling, suggesting that global epigenetic analysis of specific oncogene mutations may provide new therapeutic insights.
Results
Integrative Analyses of Chromatin Landscape Induced by EGFRvIII
We analyzed histone H3 lysine 4 monomethylation (H3K4me1) and histone H3 lysine 27 acetylation (H3K27ac)—two histone modifications associated with poised and active enhancers (Creyghton et al., 2010; Heintzman et al., 2007; Rada-Iglesias et al., 2011)—by ChIP-seq in isogenic U87 GBM cells with, or without, stable expression of the ligand-independent activated EGFR mutation, EGFRvIII. This analysis revealed 2245 putative enhancers that were specifically activated in EGFRvIII-expressing GBM cells (Figure 1A). The activation state of these enhancers was not changed by the addition of serum, which contains growth factors that could activate other cell surface receptors (Figure 1A).
Figure 1. Integrative Analyses of Chromatin and Transcriptional Landscape Induced by Hyperactivated EGFR in GBM.

(A) Heat map of ChIP-seq experiments using antibodies against H3K4me1 and H3K27ac, two histone modifications associated with poised and active candidate enhancers, in U87 and U87EGFRvIII cells. The presence of EGFRvIII results in the activation and silencing of distinct sets of enhancers. Note that these potential enhancers show little response to 10% fetal bovine serum (FBS), which contains various growth factors.
(B) Heat map of RNA-seq experiments shows the transcript levels of TF genes differentially expressed between U87 and U87EGFRvIII cells (see also Figure S1).
(C) EGFRvIII-responsive enhancers are located near EGFRvIII-specific genes.
(D) Motif enrichment analysis indicates that FOX and HMG/SOX binding motifs are enriched in EGFRvIII-specific enhancers.
See also Figure S1 and Table S4.
To gain insight into the regulation of EGFRvIII-specific transcription that might be regulated by these putative enhancers, we performed high-throughput complementary DNA sequencing (RNA-seq) (Figure 1B). The EGFRvIII-activated enhancers identified by H3K4me1 and H3K27ac ChIP-seq were significantly enriched near EGFRvIII-upregulated transcripts, suggesting the presence of a coordinated, EGFRvIII-regulated, transcriptional network (Figure 1C). Analysis of TF recognition motifs within EGFRvIII-activated enhancers showed that binding sites for many of the 41 TFs upregulated in EGFRvIII-expressing GBM cells were highly enriched (Figure S1A). In particular, motifs for the FOX and SOX family of transcription factors were among the most highly enriched motifs (Fig. 1D). SOX9 and FOXG1 transcripts, which have been implicated in brain development and are thought to play a role in cancer (Guo et al., 2012; Rani et al., 2013; Seoane et al., 2004; Swartling et al., 2012; Verginelli et al., 2013; Wang et al., 2012), were both highly elevated by EGFRvIII expression (Figure 1B). Therefore, we set out to determine whether EGFRvIII regulates SOX9 and FOXG1 expression and to study its functional importance.
EGFRvIII Activates the Transcription of SOX9 and FOXG1
In GBM cells treated with the EGFR tyrosine kinase inhibitor erlotinib, we detected a marked decrease in distinct H3K27ac putative enhancer peaks near SOX9 and FOXG1 (Figure 2A). The specificity of the erlotinib-sensitive H3K27ac peak at a FOXG1 enhancer was confirmed by circularized chromosome confirmation capture (4C-seq) (Figure S2A). Therefore, we examined the effect of EGFRvIII on SOX9 and FOXG1 mRNA and protein levels in multiple GBM cell line contexts. In U87 GBM cells, EGFRvIII expression dramatically increased SOX9 and FOXG1 transcript and protein levels (Figures 1B and 2B), which was inhibited by erlotinib (Figure S2B). The mTOR kinase inhibitor torin1 and the MEK inhibitor U0126 also reduced SOX9 and FOXG1 levels, suggesting that EGFRvIII controls expression of these TFs through downstream signaling (Figures S2B and S2C). To determine whether wild type EGFR and mutant EGFR, EGFRvIII, differentially regulate SOX9 and FOXG1, we generated an siRNA construct targeting exon 9 of EGFR, which knocks down both wild-type EGFR and EGFRvIII, and an siRNA construct targeting exon 2, a region deleted in EGFRvIII, which only knocks down wild type EGFR expression (Figure 2C). As shown in Figure 2D, knock down of EGFRvIII, but not wild type EGFR alone resulted in significantly reduced SOX9 and FOXG1 transcript levels. To test whether EGFRvIII's effect on SOX9 and FOXG1 depended on its kinase activity, we introduced a kinase dead EGFRvIII construct (EGFRvIII-KD; Figure 2E) (Akhavan et al., 2013; Huang et al., 1997). In U87 GBM cells, EGFRvIII, but not kinase dead EGFRvIII, significantly elevated SOX9 and FOXG1 transcript levels in an erlotinib-sensitive fashion (Figure 2F), indicating that: 1) the observed effect of EGFRvIII on SOX9 and FOXG1 transcription was not due to exogenous EGFRvIII expression and 2) the effect of EGFRvIII on SOX9 and FOXG1 expression was dependent on EGFRvIII kinase activity. To confirm the effect of EGFRvIII in different GBM cell line contexts, we expressed EGFRvIII under the control of a doxycycline inducible promoter in LN229 GBM cells and expressed EGFRvIII under the control of a doxycycline repressible promoter in U373 GBM cells. EGFRvIII potently upregulated SOX9 and FOXG1 protein expression in both cell lines (Figure 2G). To determine whether endogenously expressed EGFRvIII similarly regulates SOX9 and FOXG1, we analyzed GBM6 patient-derived cells in neurosphere culture (Nathanson et al., 2014; Sarkaria et al., 2007). Erlotinib treatment greatly reduced SOX9 and FOXG1 protein expression in GBM6 cells, demonstrating that endogenously expressed EGFRvIII also regulates SOX9 and FOXG1 through its kinase activity in a non-engineered cell context (Figures 2H and 2I).
Figure 2. EGFRvIII Activates the Transcription of SOX9 and FOXG1.

(A) Snapshots of UCSC genome browser of ChIP-seq experiments at the loci of SOX9 and FOXG1. Shaded H3K27ac peaks indicated putative EGFRvIII-responsive enhancers.
(B) Western blots of U87 and U87EGFRvIII cells cultured with or without FBS. SOX9 and FOXG1 proteins levels are at much higher levels in U87EGFRvIII cells.
(C) Western blots of U87EGFRvIII cells treated by siRNAs. One siRNA (siEGFR#1 targeting exon 9) knocks down the expression of both wild-type EGFR and EGFRvIII; the other siRNA (siEGFR#2 targeting exon 2) only knocks down wild type EGFR (exons 2-7 are deleted in EGFRvIII).
(D) qRT-PCR experiments show that siEGFR#1, but not siEGFR#2, is able to decrease the transcript levels of SOX9 and FOXG1.
(E) Western blots show the expression of EGFRvIII and kinase dead EGFRvIII (EGFRvIII-KD). Erlotinib treatment (10 μM for 24hr) abrogates the kinase activity of EGFRvIII.
(F) qRT-PCR of cells treated with erlotinib. **: p<0.01; t-test, error bars represent S.D.
(G) EGFRvIII regulates the expression of SOX9 and FOXG1 in the LN229_teton_vIII cell line, in which EGFRvIII is induced by doxycycline (DOX), and in U373_tetoff_vIII cell line, in which EGFRvIII is suppressed by DOX.
(H-I) Erlotinib suppresses the expression of SOX9 and FOG1 in a patient derived neurosphere GBM cell line, GBM6, which endogenously expresses EGFRvIII. **: p<0.01; t-test, error bars represent S.D.
See also Figure S2 and Table S3.
SOX9 and FOXG1 Interact with EGFRvIII-responsive Enhancers
To determine whether SOX9 and FOXG1 bind to EGFRvIII-responsive enhancers we performed ChIP-qPCR on two informative genes, FOXF1 and TFAP2c, using antibodies directed against SOX9 and FOXG1. We focused on these two genes because: 1) they are near EGFRvIII-responsive enhancers (defined by H3K27Ac and H3K27me1, being very high at enhancers near these genes in U87EGFRvIII cells and silent in U87 cells), 2) their expression was greatly elevated by the expression of EGFRvIII and 3) SOX9 or FOXG1 knockdown abrogated their mRNA expression. As shown in Figure 3A, SOX9 and FOX1 both bind to EGFRvIII-responsive enhancers near these two EGFR-regulated genes. Furthermore, we performed ChIP-qPCR on the SOX9 and FOXG1 loci, demonstrating that SOX9 and FOXG1 both bind to enhancers that may regulate them, thus providing compelling evidence for autoregulation (Figure 3B and 3C). Careful inspection of the ChIP-qPCR data suggested that FOXG1 might also cross regulate SOX9. FOXG1 knockdown reduced SOX9 transcript and protein level, and vice versa, raising the possibility of a more complex form of cross regulation (Figures S3A and S3B).
Figure 3. SOX9 and FOXG1 Interact with EGFRvIII-responsive Enhancers.

(A) UCSC genome browser snap shots are shown at the top. ChIP-qPCR was used to detect the binding of SOX9 and FOXG1 with putative EGFRvIII-responsive enhancer elements (H3K27ac high in U87EGFRvIII/low in U87) near FOXF1 and TFAP2c, both of which are induced by EGFRvIII in U87 cells (Figure S1A). PCR targets are shown as filled blocks below corresponding enhancer elements; control PCR target is shown as empty block. Pro: promoter. qPCR experiments are shown at the bottom.
(B) SOX9 and FOXG1 ChIP-qPCR at the SOX9 locus.
(C) SOX9 and FOXG1 ChIP-qPCR at the FOXG1 locus. *: p<0.05; **: p<0.01; ***: p<0.001; t-test. Error bars represent S.D.
See also Figure S3 and Table S3.
SOX9 and FOXG1 Correlate with EGFR Amplification/mutation in Clinical GBM Samples
Having demonstrated that EGFRvIII controls SOX9 and FOXG1 expression in relatively simplified isogenic GBM cell culture systems, we next examined the correlation between EGFR/EGFRvIII and SOX9/FOXG1 expression in a large cohort of clinical GBM samples in The Cancer Genome Atlas (TCGA) consortium (http://cancergenome.nih.gov/), consisting of gene expression microarray data from 598 GBMs (Brennan et al., 2013). Among the relatively large HMG/SOX and FOX transcription factor families, the expression of SOX9 and FOXG1 were the most highly correlated with that of EGFR (pFOXG1 = 1.27E-18; pSOX9 = 1.10E-41, Matlab corr. Function; Figures 4A, S4A and S4B). In a separate TCGA dataset using RNA-seq profiles from 169 GBM tumor samples (Brennan et al., 2013), SOX9 and FOXG1 expression were significantly elevated in those GBMs bearing EGFR amplification and mutations, including EGFRvIII (pFOXG1 < 0.0038 ; pSOX9 < 0.0017, Wilcoxon, Figures 4B and S4D).
Figure 4. SOX9 and FOXG1 Correlate with EGFR Amplification/mutation in Clinical GBM Samples.

(A) Correlation of the transcript levels of SOX and FOX families with EGFR in 598 tumors in the TCGA GBM microarray database. SOX9 and FOXG1 show the highest correlation with EGFR among these two families.
(B) Expression levels of EGFR, SOX9, and FOXG1 in TCGA GBM RNA-seq database. SOX9 and FOXG1 are expressed at higher levels in tumors with EGFR amplification and mutation.
(C) The levels of commonly amplified RTKs (table S1) can accurately predict SOX9 and FOXG1 levels in GBM using a random forest classifier.
(D) Heat map of ChIP-seq using tissue samples, including 6 GBMs (4 expressing high levels of EGFR and 2 expressing high levels of FGFR3, Figure S4J), 1 low grade glioma (which carries IDH1 R132H mutation), and 6 normal brain tissues (Table S2). Comparison of H3K27ac peaks in these tissue samples revealed two clusters of enhancers that are active specifically in GBM with high levels of EGFR or FGFR3. Ant.caudate: anterior caudate. Cing.gyrus: cingulate gyrus. DLFC: dorsolateral prefrontal cortex. hipp: hippocampus. Inf.temp lobe: inferior temporal lobe. Mid frontal lobe: middle frontal lobe.
(E) Putative enhancers active in high grade GBM are enriched with binding motifs HMG/SOX and FOX proteins.
(F) GBM-specific enhancers are enriched near genes showing differential levels between EGFRvIII+ and EGFR euploid tumors in the TCGA GBM RNA-seq database.
(G-H) Immunohistochemistry of tissue microarray containing normal and GBM tissue sections. Representative anti-SOX9 and anti-FOXG1 stains are shown. N: normal. T: tumor. Scale bar: 30 μm.
(I) Strong SOX9 and FOXG1 stains are significantly associated with high levels of phosphor-EGFR stain (marker of activated EGFR), while showing little correlation with phospho-PDGFR and phospho-MET. Statistics was performed using Fisher's exact test; one-tail p-values are shown in the table. Asterisk indicates p<0.05.
See also Figure S4 and Tables S2 and S4.
A random forest classifier demonstrated that genetic alteration of growth factor receptors accurately predicted the levels of SOX9 (AUC = 0.94, p = 2.3e-15, Z test) and FOXG1 (AUC = 0.83, p < 1e-15, Z test), in GBM clinical samples driven largely by genetic alterations of EGFR (Figures 4C, S4E and S4F). Furthermore, FGFR3 overexpression, which is associated with gene amplification and/or mutation (Parker et al., 2013; Singh et al., 2012), was associated with elevated SOX9 levels in the TCGA clinical GBM samples and ligand-induced FGFR3 activation increases SOX9 levels in vitro (Figures S4E and S4G). Taken together with the mechanistic data showing that EGFRvIII promotes SOX9 and FOXG1 (Figure 2), these results indicate a strong association between EGFR genetic alterations and SOX9 and FOXG1 expression, as well as an association between FGFR3 and SOX9, in GBM. This association was also confirmed at the protein level. An immunohistochemical analysis of GBM tissue microarrays demonstrated significant correlations between phospho-EGFR, SOX9 and FOXG1 proteins, all of which were correlated with each other and with tumor cell proliferation rate, as measured by Ki67 staining (Figures 4G, 4H and 4I). Analysis of TCGA data from other tumor types also demonstrated elevated SOX9 and FOXG1 in association with amplification of various growth factor receptors in a variety of cancers (Figures S4H and S4I; Table S1), suggesting that SOX9 and FOXG1 may be common transcriptional effectors downstream of growth factor alterations in multiple cancer types.
To further verify if growth factor induced transcriptional program and remodeling of epigenome identified in the above cell models is present in GBM tumors, we performed ChIP-seq to profile the H3K4me1 and H3K27ac status of regulatory DNA sequences in a set of GBM clinical samples. Fourteen frozen GBM tissue samples were screened for quality and six were selected based upon the presence of greater than 70% tumor cells in the tissue sample (Table S2). This included four GBMs with EGFR amplification and EGFRvIII mutation, and two GBMs with elevated FGFR3 transcript expression (Figure S4J). We also performed H3K4me1 and H3K27ac ChIP-seq on one low grade glioma bearing no receptor tyrosine kinase (RTK) mutations, but containing an IDH1 R132H mutation (Table S2). As a basis of comparison, we obtained H3K27ac ChIP-seq profiles from six different normal brain regions. Two distinct clusters of growth factor-associated enhancers were detected in the GBMs, which were not found in either the IDH1-mutant low grade glioma or in the normal brain samples (Figure 4D). These putative EGFR/FGFR3-activated enhancers were significantly enriched for binding sequence motifs of the SOX and FOX family of transcription factors (Figure 4E), and are located near genes differentially expressed between EGFRvIII+ and EGFReuploid GBMs (Figure 4F). Taken together, these results indicate the presence of a coordinated transcriptional program with epigenetic remodeling downstream of RTK genetic alterations in clinical GBM samples.
SOX9 and FOXG1 Promote GBM in vitro and in vivo
To determine the functional role of SOX9 and FOXG1 in EGFRvIII-dependent tumor growth, we generated GBM cells stably expressing short hairpin RNAs targeting SOX9 and FOXG1 and performed ChIP-seq analysis to determine the effect of these knockdowns on activation state of EGFRvIII-responsive enhancers. Knockdown of either TF significantly decreased H3K27 acetylation and H3K4 monomethylation of EGFRvIII-responsive enhancers (Figure 5A and 5B). Further, knockdown of either SOX9 or FOXG1 severely impaired the growth of U87EGFRvIII cells in culture and almost completely blocked colony formation in soft agar (Figure 5C; Figures S5A, S5B, and S5C). In vivo, SOX9 or FOXG1 knockdown resulted in a marked delay in tumor development along with significant reductions in tumor sizes in mice bearing SOX9 or FOXG1-knockdown tumors, and this was associated with significantly longer survival (Figures 5D, 5E and 5F; Figures S5D-S5L).
Figure 5. SOX9 and FOXG1 are required for the Growth of EGFRvIII-expressing GBM.

(A-B) CHIP-seq experiments indicate that SOX9 and FOXG1 knockdowns significantly decrease H3K27 acetylation and H3K4 monomethylation in the chromatin of EGFRvIII-responsive enhancers.
(C) Knockdown of SOX9 or FOXG1 reduces the proliferation of U87EGFRvIII cells grown in petri dish. **: p<0.01; t-test. Error bars represent S.D.
(D) Fluorescence scan of an intracranial xenograft model of U87EGFRvIII cells transplanted into the mouse brain. The cells were engineered to constitutively express the near-infrared fluorescent protein iRFP720 so that tumor mass can be monitored by 3D fluorescence tomography. White arrows indicate the sites where cancer cells were injected. Shown are representative images of tumor scan at the 20th day post-injection.
(E) SOX9 or FOXG1 knockdown slowed down the formation of tumors by U87EGFRvIII cells in vivo. At the 20th day post-injection, the fluorescence intensities of tumors formed by U87EGFRvIIIshSOX9 or U87EGFRvIIIshFOXG1 cells were significantly lower than those formed by U87EGFRvIIIshScr cells (pshSOX9≤ 0.0001, pshFOXG1≤ 0.0001, t-test, n=6 per group, Error bars represent S.D.).
(F) Kaplan-Meier survival curves of host mice for the xenograft experiments. The mice containing SOX9 or FOXG1 knockdown cells lived longer than the control mice, pshSOX9≤ 0.0001, pshFOXG1≤ 0.0001, log-rank (Mantel-Cox) test.
(G) Venn diagram of the comparison of RNA-seq experiments using U87EGFRvIII, U87EGFRvIIIshSOX9, U87EGFRvIIIshFOXG1 cells. SOX9 and FOXG1 are both required for the expression of 397 genes in U87EGFRvIII.
(H) Gene ontology (GO) analysis indicates that SOX9 and FOXG1 co-regulated genes are associated with a variety of RTK-driven cancers.
(I) Gene set enrichment analysis (GSEA) indicates that SOX9 and FOXG1 knockdowns led to the loss of gene signatures associated with the EGFR pathway and the proto-oncogenic c-MYC.
See also Figure S5.
To identify the global transcriptional repertoires controlled by SOX9 and FOXG1, we performed RNA-seq analysis of U87EGFRvIII cells with or without shRNA knockdown of SOX9 or FOXG1. This analysis identified 993 and 1920 genes whose expression was markedly reduced by SOX9 and FOXG1 knockdown, respectively, including 376 genes that are regulated by both TFs (Figure 5G). These 376 genes were highly overrepresented in gene ontologies that are associated with glioma and other cancer types, indicating a potentially important oncogenic function (Figure 5H). Taken together, these results indicate that SOX9 and FOXG1 collaborate to control a subset of EGFRvIII-regulated genes in GBM.
To identify actionable transcriptional programs regulated by SOX9 or FOXG1, we performed gene set enrichment analysis (GSEA) on the transcriptomes of SOX9/FOXG1 knockdown cells. This analysis indicated significant enrichment for previously identified c-MYC target genes and EGFR-regulated genes (Figure 5I), suggesting that SOX9 and FOXG1-coregulated genes play a role in promoting EGFR-dependent tumor cell growth.
EGFRvIII Sensitizes GBM cells to JQ1-induced Cell Death through SOX9 and FOXG1
c-MYC expression, and transcription of its target genes, is regulated by BRD4, a bromodomain and extra-terminal (BET) protein family member, which acts as a transcriptional cofactor (Shi and Vakoc, 2014). Notably, among the BET family members, BRD4 transcript level was significantly correlated with that of SOX9 and FOXG1 in the TCGA database of GBMs (Figures 6A and 6B). BRD4 transcription was also significantly increased in the classical GBM subtype that is enriched for EGFR-genetic alterations (Figure 6C). Consistent with earlier studies (Delmore et al., 2011; Filippakopoulos et al., 2010), BRD4 knockdown or treatment with pan-BET bromodomain small molecule inhibitor JQ1 dramatically lowered c-MYC levels (Figures 6D, 6E and S6A). Interestingly, SOX9 or FOXG1 knockdown markedly diminished both BRD4 and c-MYC protein levels in U87EGFRvIII GBM cells (Figures 6F and 6G). Taken together, these results demonstrate that EGFRvIII controls c-MYC levels through SOX9 and FOXG1 mediated regulation of BRD4, consistent with recent evidence that c-MYC is critical for EGFRvIII-dependent tumorigenesis (Babic et al., 2013; Masui et al., 2013).
Figure 6. EGFRvIII Sensitizes GBMs to the BET-bromodomain inhibitor JQ1.

(A-C) Correlation of BRD4 with SOX9, and FOXG1 in TCGA GBM gene expression data.
(D-E) Western blots indicate that BRD4 knockdown decreases c-MYC in U87EGFRvIII cells. JQ1 treatment (1 μM) for 48 hrs decreases c-MYC while increasing the level of BRD4, suggesting that BRD4 histone-binding activity is essential for the expression of c-MYC.
(F-G) Western blots show that BRD4 and c-MYC are depleted by SOX9 or FOXG1 knockdown in U87EGFRvIII cells. (H) Annexin V/PI FACS analysis of apoptotic cells in a series of stable cell lines following 5-day treatment with 1 μM JQ1. U87EGFRvIII cells experienced significantly higher apoptosis levels compared to all the other cell lines. **: p <0.01, ***: p <0.001, ANOVA test with Dunnett's multiple comparison test. Error bars represent S.D.
(I) Heightened sensitivity to JQ1 relies on EGFRvIII's kinase activity. **: p<0.01, ***: p<0.001, N.S.: not significant. t-test. Error bars represent SD.
(J-K) Inducible EGFRvIII-expressing GBM cell lines are sensitive to JQ1-induced apoptosis. **: p<0.01, N.S.: not significant. t-test. Error bars represent SD.
See also Figure S6.
Recent studies have shown that JQ1 is effective in suppressing the growth of certain cancers that rely on amplified transcription for maintaining their oncogenic state (Lin et al., 2012; Loven et al., 2013). Therefore, we asked whether EGFRvIII sensitizes GBMs to JQ1 and whether this is mediated through the SOX9 and FOXG1 transcriptional network. In U87 GBM cells, the presence of EGFRvIII significantly increased the amount of apoptosis in response to JQ1 (Figures 6H and S6B). Short hairpins targeting SOX9, FOXG1 or BRD4 all reversed the EGFRvIII-dependent apoptotic sensitivity to JQ1 (Figure 6H), demonstrating that EGFRvIII-expressing GBM cells have heightened apoptotic responsiveness to JQ1, which is mediated by SOX9 and FOXG1 and is BRD4-dependent. This heightened response to JQ1 is attributable to the kinase activity of EGFRvIII, because no difference was observed in the level of JQ1-induced cell death in U87 GBM cells expressing kinase dead EGFRvIII (Figure 6I). The effect of EGFRvIII on sensitizing GBM cells to JQ1 was also observed in LN229 and U373 GBM cells in which EGFRvIII was under the control of doxycycline-regulatable promoters (LN229 tet-on; U373 tet-off; Figures 6J and 6K).
To further confirm these findings, we applied a live-cell activated caspase-3/7 imaging assay to assess whether this JQ1-mediated apoptosis was correlated with increased caspase activation and/or EGFRvIII kinase activity. By this method, JQ1 had only minor effects on U87 cells. In contrast, JQ1 treatment caused dose-dependent increases in caspase activity levels in both U87 EGFRvIII and kinase dead EGFRvIII cells. However, JQ1 exerted its effects on the U87 EGFRvIII cells at significantly lower concentrations relative to those expressing the catalytically inactive mutant (Figures 7A, 7B and S6C). Hence, the effects of JQ1 on cell death are likely caspase-dependent and dramatically enhanced by EGFRvIII kinase activity. Furthermore, the patient-derived neurosphere cell line GBM6, which carries endogenous EGFR amplification and EGFRvIII mutation, is sensitive to JQ1-induced cell death and its viability is dependent on the expression of SOX9 and FOXG1 (Figure 7C).
Figure 7. JQ1 suppresses EGFRvIII-dependent tumor growth.

(A) Representative images from the live-cell imaging assay for activated caspase-3/7 (green) in U87 cell lines treated with 0.3 μM JQ1. Nuclei are stained with Hoechst (blue). At this inhibitor concentration, high levels of caspase activity are only evident in the EGFRvIII-expressing cells.
(B) JQ1 dose response in caspase activity assay. JQ1 has minor effects on U87 cells and more potently increases caspase activity in U87 cells expressing catalytically active EGFRvIII relative to those expressing the kinase dead receptor [EC50(U87 EGFRvIII) = 0.20 μM; EC50(U87 EGFRvIII kinase dead) ∼ 10 μM; EC50(U87) ≫ 10 μM]. **: p<0.0001, extra sum of squares F test. Error bars represent S.D.
(C) GBM6, a patient-derived GBM neurosphere cell line that endogenously expresses EGFRvIII, is sensitive to JQ1-induced apoptosis. Knockdowns of SOX9 or FOXG1 decrease the viability of GBM6 in vitro. *: p<0.05, **: p<0.01, t-test. Error bars represent S.D.
(D) FMT fluorescence scan of intracranially transplanted U87EGFRvIII_iRFP720 cells in mice treated with JQ1 or vehicle at 18th day post-injection and quantitation of fluorescence intensity of tumors formed by U87EGFRvIII_iRFP720 cells in mice. Vehicle or JQ1 (50 mg/kg of body weight, twice per day) treatment started at the 6th day post-injection (n=12 per group). **: p<0.01, t-test. Error bars represent S.D.
See also Figure S7.
Lastly, we examined the effect of JQ1, which was highly brain-penetrant (Figure S7), on U87 EGFRvIII GBM growth in the brain in a mouse xenograft model. JQ1 administered twice daily at 50 mg/kg by oral gavage significantly decreased intracranial tumor growth (Figure 7D).
Discussion
Cancer arises from the intertwined processes of spontaneous somatic mutation and sequential selection for aggressive subclones (Stratton, 2011). Cancer is also an epigenetic disease. Mutations in transcription factors, chromatin regulators and even non-coding intergenic sequences including putative super-enhancer sequences, contribute to tumor formation and progression (Lee and Young, 2013; Mansour et al., 2014), consistent with the critical role for epigenetic alterations in tumorigenesis. There is also a complex interplay between genetic and epigenetic mechanisms in cancer, as oncogenes remodel cis-regulatory and transcription factor networks to promote tumor formation and progression (Baylin and Jones, 2011; Hanahan and Weinberg, 2011; Lee and Young, 2013; Rivera and Ren, 2013; Shen and Laird, 2013; Suva et al., 2013). Currently, the mechanisms by which oncogenic mutations remodel the epigenome are incompletely understood.
Here, we set out to determine the impact of EGFR mutations, one of the signature molecular lesions in GBM, on epigenetic reprogramming. GBM is a particularly compelling tumor for this type of integrated analysis. GBM is one of the most deeply genomically characterized forms of cancer (Lawrence et al., 2014), revealing a remarkably high prevalence of EGFR amplification and mutations (Brennan et al., 2013), even down the single cell level (Patel et al., 2014). Extensive research has begun to identify the signaling pathways and metabolic events by which EGFR mutations promote GBM pathogenesis (Cloughesy et al., 2014; Furnari et al., 2015). However, the global impact of EGFR mutations on epigenetic remodeling in GBM is not understood. Therefore, we applied global RNA-seq and ChIP-seq approaches to GBM cell lines, patient-derived tumor cells in neurosphere culture, and clinical biopsies genotyped for EGFR mutation status to interrogate the global impact of EGFRvIII on epigenetic remodeling, revealing a SOX9 and FOXG1-dependent transcriptional regulatory network that remodels the enhancer activation landscape to drive EGFRvIII-dependent tumorigenesis.
There are likely to be many other epigenetic states and transcription factors that are critical for GBM pathogenesis, including the ones recently described that control the stem-like state and in vivo tumor propagation capacity (Rheinbay et al., 2013; Suva et al., 2014). We have previously shown that endogenously expressed EGFRvIII does not appear to alter the stem-like state of GBM cells (Nathanson et al., 2014). Thus, it is not surprising that our analyses might not pick up those previously detected critical transcription factor networks involved in stem cell fate reprogramming. Instead, our study enabled us to detect a critical transcription network and epigenetic state by which EGFRvIII promotes tumor formation and progression.
EGFR mutations, including EGFRvIII, are compelling drug targets in GBM. However, EGFR tyrosine kinase inhibitors have failed to show durable benefit for GBM patients in part because it has not been possible to achieve sufficient intratumoral drug levels to adequately inhibit EGFR phosphorylation (Vivanco et al., 2012). This failure of target inhibition results in feedback activation of other receptor tyrosine kinases such as PDGFRβ to maintain downstream signal flux and/or reversible loss of EGFRvIII from extrachromosomal DNA to promote drug resistance (Akhavan et al., 2013; Furnari et al., 2015; Nathanson et al., 2014). Until new drug formulations or better dosing approaches are developed that safely achieve higher dose levels and more effectively target inhibition within the tumor in the brain, alternative therapeutic strategies for EGFR activated GBMs need to be considered.
Our finding that EGFRvIII sensitizes GBM cells to the BET-bromodomain inhibitor, suggests a potentially clinically actionable “precision medicine” strategy. We find that EGFRvIII enhances JQ1-dependent apoptosis via its effect on SOX9 and FOXG1, which converge to control BRD4 and c-MYC protein levels. JQ1 treatment results in a near total loss of c-MYC protein on Western blot (Figure 6D), whereas the decrement in c-MYC transcript is more modest (Figure S6A). Similarly, SOX9 and FOXG1 knockdowns precipitously lower c-MYC protein levels (Figures 6F and 6G), despite relatively modest effects on expression of c-MYC transcript. These observations, although limited in scope, raise the possibility that SOX9 and FOXG1 may regulate c-MYC protein translation and/or stability in a BRD4 (or other BET bromodomain protein) dependent fashion. Future studies will be needed to test this possibility; to determine if it is detected in other cell contexts, and to identify a potential underlying mechanism.
Many cancer types depend on c-MYC for growth and proliferation, and c-MYC is a common target for amplification in many of these tumors (Lee and Young, 2013; Lin et al., 2012; Nie et al., 2012). In contrast in GBM, c-MYC is rarely amplified or mutated (Brennan et al., 2013). We have previously demonstrated that EGFRvIII signaling through mTORC1 enhances c-MYC activity through the alternative splicing of the c-MYC heterodimerization partner Max to generate Delta Max, a gain of function variant (Babic et al., 2013), and we have found that EGFRvIII controls c-MYC protein level through a FOXO 1, 3- acetylation signaling cascade (Masui et al., 2013). Thus, our finding that EGFRvIII regulates c-MYC levels through the SOX9 and FOXG1 transcriptional activity further demonstrates the critical nature of c-MYC in EGFRvIII-dependent GBM pathogenesis, expands our understanding of its regulation, and potentially explains the enhanced apoptotic sensitivity to JQ1. It is also interesting to speculate that BRD4 and elevated c-MYC activity downstream of growth factor receptor mutations in adult GBMs may be a common pathogenic mechanism shared with pediatric high grade gliomas that have H3K27M mutations, but lack growth factor receptor alterations (Herz et al., 2014; Lewis et al., 2013; Sturm et al., 2014).
Experimental Procedures
Cell Lines and Tissue Samples
The human glioma cell line U87 (a.k.a. U-87MG) was purchased from ATCC. U87EGFRvIII, U87EGFRvIII-KD, LN229_teton_vIII, U373_tetoff_vIII, and GBM6 cell lines were described before (Akhavan et al., 2013; Nathanson et al., 2014; Wang et al., 2006). Tumor samples were obtained from UCLA Brain Tumor Translational Resource. Normal brain tissues were obtained from the National Institute of Child Health and Human Development (NICHD) Brain Bank for Developmental Disorders. Ethics approval was obtained from the University of California Los Angeles for use of the GBM tumor samples, and from the University Health Network and the Hospital for Sick Children for use of the normal brain tissues.
RNA-seq
Total RNA was extracted from 1-2 million cells cultured in petri dish or 50-100 mg of tissue using the RNeasy mini kit (Qiagen). 5∼10 μg of total RNA was used to prepare RNA-seq libraries according to Illumina protocol. 4-6 libraries were mixed for multiplexed pair-end sequencing using Illumina HiSeq 2000 (Illumina).
ChIP-seq and ChIP-qPCR
Chromatin immunoprecipitation (ChIP) was performed as previously described (Heintzman et al., 2007). immunoprecipitated DNA was purified after phenol extraction and was used for quantitative PCR (KAPA Biosystems) or for preparing barcoded high throughput sequencing libraries according to Illumina protocols with minor modifications. 4-12 library preps were mixed for multiplexed single-read sequencing using Illumina Hi-seq 2000 (Illumina). Antibodies used for ChIP were polyclonal rabbit anti-H3K27ac (Active Motif, Cat#39133), polyclonal rabbit anti-H3K4me3 (Active Motif, Cat#39159), polyclonal rabbit anti-H3K4me1 (Abcam, Cat#8895), monoclonal rabbit IgG (Abcam, Cat#ab172730), polyclonal rabbit anti-SOX9 (Millipore, Cat#AB5535), and polyclonal rabbit anti-FOXG1 (Pierce, Cat# PA5-26794).
ChIP-seq Analysis
For a given locus, histone modification enrichment is quantified as E = log2 (ChIP RPKM / input RPKM), where RPKM is defined as the number of reads per kilobase of locus per million mapped reads, in either ChIP or input samples. To avoid division by zero, a pseudocount of 0.05 is added to both numerator and denominator. Given a set of differentially expressed genes, we created a set of bins +/-500kb from the transcription start sites of these genes. We also repeated this process to create 5000 random sets of bins corresponding to the same number of random genes. Given a set of enhancers, we assessed enrichment by overlapping with bins of differentially expressed genes and comparing with the overlap for random bin sets. Motif finding was performed using Homer v4.2 (Heinz et al., 2010).
RNA-seq Analysis
For pairwise comparisons between two sets of samples, the number of sense exonic reads were quantified and input to edgeR (Robinson et al., 2010) to normalize and call differentially expressed genes at a p-value cutoff of 0.05. To perform Gene Set Enrichment Analysis (GSEA) of RNA-seq data on U87EGFRvIIIshSox9 or U87EGFRvIIIshFoxg1 compared to U87EGFRvIII, we represented expression of genes as the mean of RPKM values from biological replicates, calculated the log ratio of knockdown to U87EGFRvIII cells, and ran GSEA v2.1.0 (Subramanian et al., 2005).
TCGA Data Analysis
Processed TCGA data were downloaded through the TCGA data portal, and mapped RNA-seq BAM files were acquired through the Cancer Genomics Hub (https://cghub.ucsc.edu/). The EGFR status of TCGA samples are designated as euploid, regionally amplified, focally amplified, or EGFRvIII based on annotations previously published (Brennan et al., 2013). Specifically, EGFRvIII samples are samples with non-zero Δ2-7 values, and euploid / regionally amplified / focally amplified samples are non-EGFRvIII samples labeled as “Euploid” / “Regional gain” / “Focal Amplification”. Random forest analysis was performed using the TreeBagger function defined in Matlab. Given the expression of RTKs known to be amplified in a given cancer type as described in the TCGA Copy Number Portal (Mermel et al., 2011), the random forest was first trained and then used to predict whether each sample is a high (top 100) or low (bottom 100) expresser of SOX9/FOXG1.
Intracranial Xenograft
Athymic nu/nu mice 5 weeks of age were purchased from Harlan Sprague Dawley Inc. 1×105 U87EGFRvIII_iRFP720 cells in 5 μl of phosphate-buffered saline (PBS) were intracranially injected to the mouse brain (Ozawa and James, 2010). Tumor growth was monitored using an FMT 2500 Fluorescence Tomography System (PerkinElmer). For drug treatment studies, vehicle (10% of hydroxypropyl beta cyclodextrin, Sigma-Aldrich Cat#: C0926-10G) or JQ1 (5 mg/ml) were administered to mice (10 μl per gram of body weight, or 50 mg/kg) via gavage twice daily starting from the 6th day post-injection. All procedures have been reviewed and approved by the Institutional Animal Use and Care Committee at University of California San Diego.
Supplementary Material
Acknowledgments
The infra-red Fluorescent Protein 720 (IRFP720) expression construct was kindly given by Dr. V. V. Verkhusha. This work is supported by the National Institute for Neurological Diseases and Stroke grant NS73831 to (P.S.M), the Defeat GBM Program of the National Brain Tumor Society and generous donations from the Ziering Family Foundation in memory of Sigi Ziering (to P.S.M, T.F.C), the Ben and Catherine Ivy Foundation (to P.S.M, T.F.C, W.H.Y); the Ludwig Institute for Cancer Research (to B.R, A.K.S, T.C.G); UCSD Core Facility Stimulus Funding (to B.R); the National Institute of Health grant R01-NS080939 (to F.B.F); the National Cancer Institute grant F31CA186668 (to G.R.V); the Hospital for Sick Children Psychiatric Endowment Fund (to C. L. B); Art of the Brain (to T.F.C, W.H.Y); the Uncle Kory Foundation (to T.F.C). W.K.C is a Fellow of the National Foundation for Cancer Research.
Footnotes
Accession Numbers: The ChIP-seq and RNA-seq data reported in this study have been deposited with the Gene Expression Omnibus under the accession ID: GSE72468.
Supplemental Information: Supplemental Information includes detailed Extended Experimental Procedures, seven figures, three tables, and one data file.
Author Contributions: F.L., G.C.H., B.R. and P.S.M. conceived the project and designed the research. F.L., G.R.V., K.M.T., S.I., H.Y., Z.Y., A.Y.L., C.Z., and D.J. performed experiments. G.C.H. and B.L. conducted high-throughput sequencing data analysis. C.L.B., B.W., G.L., T.F.C., W.H.Y. provided tissue samples, clinical information and helped with interpretation of data. F.B.F., T.C.G., W.Z., A.K.S. and W.K.C. provided new reagents and analytic tools and provided intellectual contributions to design of experiments and interpretation of data. F.L., G.C.H., A.K.S., W.K.C., B.R. and P.S.M. wrote the paper. All authors discussed the results and commented on the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akhavan D, Pourzia AL, Nourian AA, Williams KJ, Nathanson D, Babic I, Villa GR, Tanaka K, Nael A, Yang H, et al. De-repression of PDGFRbeta transcription promotes acquired resistance to EGFR tyrosine kinase inhibitors in glioblastoma patients. Cancer discovery. 2013;3:534–547. doi: 10.1158/2159-8290.CD-12-0502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babic I, Anderson ES, Tanaka K, Guo D, Masui K, Li B, Zhu S, Gu Y, Villa GR, Akhavan D, et al. EGFR mutation-induced alternative splicing of Max contributes to growth of glycolytic tumors in brain cancer. Cell metabolism. 2013;17:1000–1008. doi: 10.1016/j.cmet.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nature reviews. Cancer. 2011;11:726–734. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C. Emerging landscape of oncogenic signatures across human cancers. Nature genetics. 2013;45:1127–1133. doi: 10.1038/ng.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloughesy TF, Cavenee WK, Mischel PS. Glioblastoma: from molecular pathology to targeted treatment. Annual review of pathology. 2014;9:1–25. doi: 10.1146/annurev-pathol-011110-130324. [DOI] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furnari FB, Cloughesy TF, Cavenee WK, Mischel PS. Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nature reviews. Cancer. 2015;15:302–310. doi: 10.1038/nrc3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Keckesova Z, Donaher JL, Shibue T, Tischler V, Reinhardt F, Itzkovitz S, Noske A, Zurrer-Hardi U, Bell G, et al. Slug and Sox9 cooperatively determine the mammary stem cell state. Cell. 2012;148:1015–1028. doi: 10.1016/j.cell.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nature genetics. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Molecular cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz HM, Morgan M, Gao X, Jackson J, Rickels R, Swanson SK, Florens L, Washburn MP, Eissenberg JC, Shilatifard A. Histone H3 lysine-to-methionine mutants as a paradigm to study chromatin signaling. Science. 2014;345:1065–1070. doi: 10.1126/science.1255104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HS, Nagane M, Klingbeil CK, Lin H, Nishikawa R, Ji XD, Huang CM, Gill GN, Wiley HS, Cavenee WK. The enhanced tumorigenic activity of a mutant epidermal growth factor receptor common in human cancers is mediated by threshold levels of constitutive tyrosine phosphorylation and unattenuated signaling. The Journal of biological chemistry. 1997;272:2927–2935. doi: 10.1074/jbc.272.5.2927. [DOI] [PubMed] [Google Scholar]
- Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TI, Young RA. Transcriptional regulation and its misregulation in disease. Cell. 2013;152:1237–1251. doi: 10.1016/j.cell.2013.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis PW, Muller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, Garcia BA, Muir TW, Becher OJ, Allis CD. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science. 2013;340:857–861. doi: 10.1126/science.1232245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Loven J, Rahl PB, Paranal RM, Burge CB, Bradner JE, Lee TI, Young RA. Transcriptional amplification in tumor cells with elevated c-Myc. Cell. 2012;151:56–67. doi: 10.1016/j.cell.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, Young RA. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–334. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour MR, Abraham BJ, Anders L, Berezovskaya A, Gutierrez A, Durbin AD, Etchin J, Lawton L, Sallan SE, Silverman LB, et al. Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science. 2014;346:1373–1377. doi: 10.1126/science.1259037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masui K, Tanaka K, Akhavan D, Babic I, Gini B, Matsutani T, Iwanami A, Liu F, Villa GR, Gu Y, et al. mTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and upregulation of c-Myc. Cell metabolism. 2013;18:726–739. doi: 10.1016/j.cmet.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome biology. 2011;12:R41. doi: 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathanson DA, Gini B, Mottahedeh J, Visnyei K, Koga T, Gomez G, Eskin A, Hwang K, Wang J, Masui K, et al. Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science. 2014;343:72–76. doi: 10.1126/science.1241328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, Wang R, Green DR, Tessarollo L, Casellas R, et al. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell. 2012;151:68–79. doi: 10.1016/j.cell.2012.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa T, James CD. Establishing intracranial brain tumor xenografts with subsequent analysis of tumor growth and response to therapy using bioluminescence imaging. Journal of visualized experiments : JoVE. 2010 doi: 10.3791/1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker BC, Annala MJ, Cogdell DE, Granberg KJ, Sun Y, Ji P, Li X, Gumin J, Zheng H, Hu L, et al. The tumorigenic FGFR3-TACC3 gene fusion escapes miR-99a regulation in glioblastoma. The Journal of clinical investigation. 2013;123:855–865. doi: 10.1172/JCI67144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470:279–283. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rani SB, Rathod SS, Karthik S, Kaur N, Muzumdar D, Shiras AS. MiR-145 functions as a tumor-suppressive RNA by targeting Sox9 and adducin 3 in human glioma cells. Neuro-oncology. 2013;15:1302–1316. doi: 10.1093/neuonc/not090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rheinbay E, Suva ML, Gillespie SM, Wakimoto H, Patel AP, Shahid M, Oksuz O, Rabkin SD, Martuza RL, Rivera MN, et al. An aberrant transcription factor network essential for Wnt signaling and stem cell maintenance in glioblastoma. Cell reports. 2013;3:1567–1579. doi: 10.1016/j.celrep.2013.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera CM, Ren B. Mapping human epigenomes. Cell. 2013;155:39–55. doi: 10.1016/j.cell.2013.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkaria JN, Yang L, Grogan PT, Kitange GJ, Carlson BL, Schroeder MA, Galanis E, Giannini C, Wu W, Dinca EB, et al. Identification of molecular characteristics correlated with glioblastoma sensitivity to EGFR kinase inhibition through use of an intracranial xenograft test panel. Molecular cancer therapeutics. 2007;6:1167–1174. doi: 10.1158/1535-7163.MCT-06-0691. [DOI] [PubMed] [Google Scholar]
- Seoane J, Le HV, Shen L, Anderson SA, Massague J. Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell. 2004;117:211–223. doi: 10.1016/s0092-8674(04)00298-3. [DOI] [PubMed] [Google Scholar]
- Shen H, Laird PW. Interplay between the cancer genome and epigenome. Cell. 2013;153:38–55. doi: 10.1016/j.cell.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Vakoc CR. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Molecular cell. 2014;54:728–736. doi: 10.1016/j.molcel.2014.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh D, Chan JM, Zoppoli P, Niola F, Sullivan R, Castano A, Liu EM, Reichel J, Porrati P, Pellegatta S, et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science. 2012;337:1231–1235. doi: 10.1126/science.1220834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratton MR. Exploring the genomes of cancer cells: progress and promise. Science. 2011;331:1553–1558. doi: 10.1126/science.1204040. [DOI] [PubMed] [Google Scholar]
- Sturm D, Bender S, Jones DT, Lichter P, Grill J, Becher O, Hawkins C, Majewski J, Jones C, Costello JF, et al. Paediatric and adult glioblastoma: multiform (epi)genomic culprits emerge. Nature reviews. Cancer. 2014;14:92–107. doi: 10.1038/nrc3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suva ML, Rheinbay E, Gillespie SM, Patel AP, Wakimoto H, Rabkin SD, Riggi N, Chi AS, Cahill DP, Nahed BV, et al. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell. 2014;157:580–594. doi: 10.1016/j.cell.2014.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suva ML, Riggi N, Bernstein BE. Epigenetic reprogramming in cancer. Science. 2013;339:1567–1570. doi: 10.1126/science.1230184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartling FJ, Savov V, Persson AI, Chen J, Hackett CS, Northcott PA, Grimmer MR, Lau J, Chesler L, Perry A, et al. Distinct neural stem cell populations give rise to disparate brain tumors in response to N-MYC. Cancer cell. 2012;21:601–613. doi: 10.1016/j.ccr.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verginelli F, Perin A, Dali R, Fung KH, Lo R, Longatti P, Guiot MC, Del Maestro RF, Rossi S, di Porzio U, et al. Transcription factors FOXG1 and Groucho/TLE promote glioblastoma growth. Nature communications. 2013;4:2956. doi: 10.1038/ncomms3956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivanco I, Robins HI, Rohle D, Campos C, Grommes C, Nghiemphu PL, Kubek S, Oldrini B, Chheda MG, Yannuzzi N, et al. Differential sensitivity of glioma- versus lung cancer-specific EGFR mutations to EGFR kinase inhibitors. Cancer discovery. 2012;2:458–471. doi: 10.1158/2159-8290.CD-11-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, He S, Yuan J, Mao X, Cao Y, Zong J, Tu Y, Zhang Y. Oncogenic role of SOX9 expression in human malignant glioma. Medical oncology. 2012;29:3484–3490. doi: 10.1007/s12032-012-0267-z. [DOI] [PubMed] [Google Scholar]
- Wang MY, Lu KV, Zhu S, Dia EQ, Vivanco I, Shackleford GM, Cavenee WK, Mellinghoff IK, Cloughesy TF, Sawyers CL, et al. Mammalian target of rapamycin inhibition promotes response to epidermal growth factor receptor kinase inhibitors in PTEN-deficient and PTEN-intact glioblastoma cells. Cancer research. 2006;66:7864–7869. doi: 10.1158/0008-5472.CAN-04-4392. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.