

Abstract
In the area of brain injury and neurodegenerative diseases, a plethora of experimental and clinical evidence strongly indicates the promise of therapeutically exploiting the endogenous adaptive system at various levels like triggers, mediators and the end-effectors to stimulate and mobilize intrinsic protective capacities against brain injuries. It is believed that ischemic pre- or post-conditioning are actually the strongest known interventions to stimulate the innate neuroprotective mechanism to prevent or reverse neurodegenerative diseases including stoke and traumatic brain injury. Recently, studies showed the effectiveness of ischemic per-conditioning in some organs. Therefore the term ischemic conditioning, including all interventions applied pre-, per- and post- ischemia, which spans therapeutic windows in 3 time periods, has recently been broadly accepted by scientific communities. In addition, it is extensively acknowledged that ischemia-mediated protection not only affects the neurons but also all the components of the neurovascular network (consisting of neurons, glial cells, vascular endothelial cells, pericytes, smooth muscle cells, and venule/veins). The concept of cerebroprotection has been widely used in place of neuroprotection. Intensive studies on the cellular signaling pathways involved in ischemic conditioning have improved the mechanistic understanding of tolerance to cerebral ischemia. This has added impetus to exploration for potential pharmacologic mimetics, which could possibly induce and maximize inherent protective capacities. However, most of these studies were performed in rodents, and the efficacy of these mimetics remains to be evaluated in human patients. Several classical signaling pathways involving apoptosis, inflammation, or oxidation have been elaborated in the past decades. Newly characterized mechanisms are emerging with the advances in biotechnology and conceptual renewal. In this review we are going to focus on those recently reported methodological and mechanistic discoveries in the realm of ischemic conditioning. Due to the varied time differences of ischemic conditioning in different animal models and clinical trials, it is important to define optimal timing to achieve the best conditioning induced neuroprotection. This brings not only an opportunity in treatment of stroke, but challenges as well, as data is just becoming available and the procedures are not yet optimized. The purpose of this review is to shed light on exploiting these ischemic conditioning modalities to protect the cerebrovascular system against diverse injuries and neurodegenerative disorders.
Keywords: innate cerebroprotection, stroke, ischemia, preconditioning, postconditioning, perconditioning
1 Introduction
Although great endeavor has been devoted to prevention and treatment of stroke, it still remains the second leading cause of death worldwide and the third largest killer in the U.S. (Go et al., 2013) Ischemic Stroke accounts for 85% of all strokes followed by hemorrhagic stroke and transient ischemic attack (Go et al., 2013; Ovbiagele et al., 2013). Patients who survive this unfortunate event, they oftentimes have to live with significant functional limitations that greatly impair their day-to-day life. Stroke is also responsible for enormous expenses in the health care system, accounting for indirect costs of US $ 73.7 billion in 2010 alone (Lloyd-Jones et al., 2010). A number of pharmacological treatments such as mitochondria potassium channel openers, calcium channel blockers, anti-oxidative agents, anti-inflammatory drugs, estrogens, progesterone, iron chelators, stem cells, etc. (Bi and Song, 2011; Hao et al., 2014; Ma et al., 2012; Mangoni et al., 2010; Shao et al., 2012; Sun and Feng, 2013; Tewari et al., 2014; Wali et al., 2014; Zhang et al., 2012) have shown potential in animal stroke models, but most of these failed to function effectively (Bi and Song, 2011; Wali et al., 2014), or caused unwanted side effects in human clinical practice. Considering the unpredictability of the time of a stroke attack, it is of extreme importance to find effective neuroprotective strategies to be delivered (Hahn et al., 2011) either before, during, or after blood flow stoppage to the brain. This could not only decrease costs but also increase the potential for positive health outcomes for patients across the world. With these challenges in mind, new therapeutic and pharmacologic interventions are needed.
Endogenous neuroprotection is the brain’s own ability to mount a response to injury. Naturally, brain cells try to reduce cell death and injury by recruiting its defenses (Huber et al., 1999). Extensive evidence has shown that the brain is able to adapt to adverse events such as cerebral ischemia, thereby improving cell endurance when faced with the possibility of future damage (Huber et al., 1999; Riepe et al., 1997). The response of endogenous neuroprotection pathways to counter cell injury and death depends on the strength of the stimulus. This adaptation requires the institution of a harmonic response at all cell levels. Beyond the limits of ischemic tolerance, cells may be doomed not to function anymore. It is imperative to study and find mechanisms related to improving this response, allowing more explicit opportunities for bridging bench research into bedside clinical trials (Bernaudin et al., 1999).
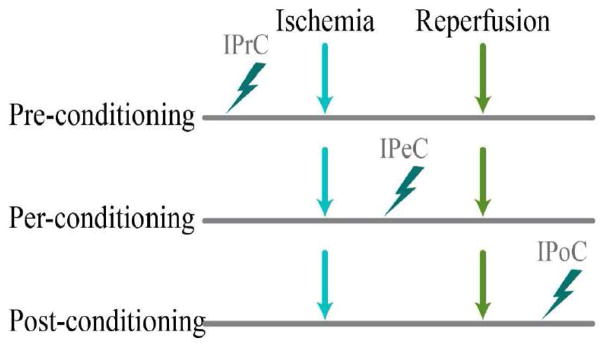
The idea of conditioning is to induce alternating periods of brief ischemia with reperfusion pre, per and post the otherwise prolonged injurious ischemia to trigger endogenous protective mechanisms. The term of ischemic conditioning was proposed to cover the three kinds of ischemic conditioning strategies spanning the three therapeutic windows in ischemic and reperfusion injury (IRI). Interventions during all 3 periods are currently being studied on animal models and some have been applied in clinical trials. Mechanism studies of neuroprotection induced by ischemic conditioning have the potential to be translated into clinical studies. Further, this research could results in the exploration of new drugs that may enhance cerebral auto defense against ischemia, which will ultimately benefit patients at risk for stroke and related neurodegenerative disorders. In the present review, several typical molecules and signaling pathways involved in protecting the brain against ischemic insults will be recapitulated. New avenues discovered in recent years whereby the cerebrovascular system manages to protect itself against ischemia will be highlighted.
2 Ischemic conditioning
2.1 Ischemic pre-conditioning (IPrC)
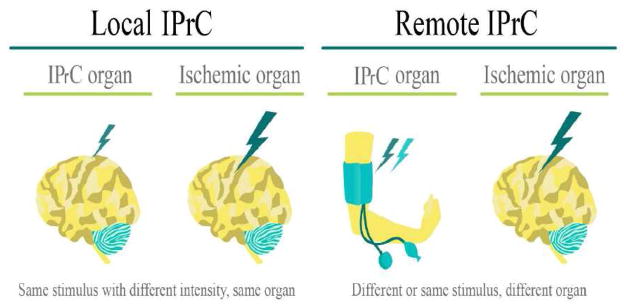
Ischemic pre-conditioning (IPrC) is the application of a brief non-lethal ischemia to cells/tissues/organisms to activate their protective mechanisms (Meller and Simon, 2013) for later injurious ischemia. IPrC is used to induce neuroprotection by delivering brief periods of ischemia prior to major or prolonged ischemia and reperfusion. IPrC has proven thus far to be one of the most effective infarct-limiting strategies available (Lehotsky et al., 2009). Preconditioning is an innovative strategy for treatment of cerebral ischemia and continues to gain interest in the medical community (Koch and Perez-Pinzon, 2013). The idea of IPrC was first described in 1986 when a study done by Murry et al in a model of a rabbit heart, found that induction of mild ischemia prior to a prolonged ischemia event made the organ more tolerant to subsequent lethal ischemic insult (Murry et al., 1986). This has been validated in many experimental disease settings (Bahjat et al., 2013). The type of preconditioning Murry et al. describes is early preconditioning, which lasts anywhere from a few minutes to several hours. There is another phase, the late phase, also known as delayed preconditioning, that starts after 12 hours and lasts up to three days (Ramzy et al., 2006). Distinct neuroprotective responses are elicited at specific time intervals. In the early phase, neuroprotection is achieved by the release of endogenous factors, and is optimized if the second insult is within a 1h period of the first insult (Perez-Pinzon et al., 1996; Perez-Pinzon et al., 1997; Schurr et al., 1986). In the late phase, the activated endogenous pathways will achieve maximal protection if the time interval is prolonged many days after the initial preconditioning stimulus. Furthermore, it will result in a stronger, more longstanding neuroprotection than the early phase (Kato et al., 1992a; Kato et al., 1992b) allowing the cells to fight against ensuing ischemia (Narayanan et al., 2013).
Aside from the local ischemic preconditioning, where periods of brief occlusions to blood flow are made proximal to the brain in order to protect it from longer durations of ischemia, remote ischemic preconditioning (RIPC) is defined as subjecting a remote non-vital and more accessible organ, such as an arm or a leg, to an ischemic stress in order to protect a distant vital organ from more extensive ischemia insults (Ates et al., 2002; Kitagawa et al., 1991). Evidence supporting its safety makes limb transient ischemia the preferred method of inducing remote conditioning (Dezfulian et al., 2013). Evidently, remote activation of protection overcomes some of the limitations of local preconditioning, but it still must be delivered before the onset of ischemia. In turn, the application of local and remote IPrC in the treatment of acute stroke is largely restricted due to the unpredictability of acute stroke onset.
2.2 Ischemic per-conditioning (IPeC)
Ischemic per-conditioning (IPeC) results from application of brief periods of ischemia during a prolonged ischemic insult to render the brain resistant to further otherwise injurious ischemia. Remote ischemic per-conditioning is a type of IPeC, and is similar to remote preconditioning in that it applies short periods of ischemia/reperfusion to a remote organ to protect another vital organ from lethal ischemia. The only difference is that the intermittent ischemia/reperfusion is done during major ischemia. A reduction in infarct size by remote ischemia applied during transport of heart attack victims to the hospital before percutaneous coronary interventions has been demonstrated by human studies on perconditioning (Vinten-Johansen and Shi, 2011). In another study done by Hahn et al. remote ischemic conditioning by transient limb ischemia was delivered in a rat transient middle cerebral artery occlusion model of acute stroke. Compared with control, both pre- and per-conditioning significantly reduced brain infarct size (Hahn et al., 2011). Considering the limitations of IPrC, IPeC was proven to be a more facile, clinically relevant intervention to drive the innate cerebroprotective mechanisms. Recently, the therapeutic potential of remote ischemic perconditioning has also been tested in female rodents. This will strengthen the evidence for safety and effectiveness in future human studies (Hoda et al., 2014). With this in mind, IPeC is a relevant treatment option for stroke intervention, and this is especially true for remote perconditioning during cerebral ischemia.
2.3 Ischemic Post-Conditioning (IPoC)
Ischemic post-conditioning (IPoC) induces neuroprotection by allowing short periods of ischemia to occur at the time of reperfusion (Halkos et al., 2004). IPoC is defined as a novel strategy that induces brief interruptions in blood flow in the early phases of reperfusion, thereby protecting organs from ischemia/reperfusion injury (IRI) (Liu et al., 2007). The effective neuroprotective strategies included improvements in disrupted cerebral blood flow and prevention of cytochrome c translation. Other mechanisms were found in post-conditioned mice, such as protein kinase Akt (Pignataro et al., 2008) and phospoinositide 3 kinase linked pathway activation (Rehni and Singh, 2007). In our group, remote limb ischemic post-conditioning has been found to protect against neonatal hypoxic–ischemic brain injury in rat pups via the opioid receptor/Akt pathway (Zhou et al., 2011). Questions remain about the common pathways involved in all 3 forms of conditioning, and the nuances that separate one form of conditioning from another, which leaves an opportunity for continued study. Translating pre, per, and post-conditioning into clinical settings will require the combination of basic science research and clinical testing (Vinten-Johansen and Shi, 2011).
3 Mechanisms concerning ischemic conditioning
Ischemic conditioning induced neuroprotection has been shown to appear in numerous forms, including but not limited to regulation of neurotrophin expression, strengthening the neurovascular network decreasing inflammation and apoptosis, and also improving cerebral metabolism, etc. (Dirnagl et al., 2003). Although these mechanisms vary greatly, they are intertwined, with significant crosstalk among signaling pathways. Recent literature has elucidated new pathways in neuroprotection, bringing hope to the possibility for translation and for possible pharmacologic treatments.
3.1 Neurovascular network-based ischemic tolerance
Recently the concept of neurovascular unit (neurons, glial cells, vascular endothelial cells, pericytes, smooth muscle cells) has been extended to include veins and venules and for that reason we prefer to use the term neurovascular network. Based on having the structures of the brain, not limited to neurons, as final beneficiaries from IPrC we will use the term cerebroprotection during this review. It is accepted that protection of the Neurovascular network primarily comes from maintaining the function of the blood brain barrier (BBB) as it is the first structure to be injured after ischemic reperfusion injury (IRI) (del Zoppo and Hallenbeck, 2000; del Zoppo and Mabuchi, 2003), and knowing that the injured state that happens following cerebral ischemia represents an important element of BBB disruption, keeping its integrity is one of the desirable end results after IPrC (Deng et al., 2014). Strengthening this system seems to be the solution to not only protect neurons and cerebral integrity, but also to improve functional outcomes after ischemic stroke.
3.1.1 Astrocytes-mediated ischemic tolerance
Astrocytes cover over 90% of the cerebral surface (Igarashi et al., 1999; Janzer and Raff, 1987; Kondo et al., 1996; Willis et al., 2004) and have an important role during the process of ischemia influencing the mechanism of BBB protection. Reactive astrocytes are required for functional recovery after stroke (Hayakawa et al., 2010) and indirectly contribute to post-ischemic angiogenesis, a hypoxic induced event (Liu et al., 2014a), through production of vascular endothelial growth factor (VEGF) (Zhang et al., 2002).
Astrocytes reside in the cerebral side of the BBB increasing its strength (Park et al., 2003; Petty and Lo, 2002) even though their primary function is to restrict permeability of the BBB, accounting for maintenance of the integrity of the NVU (Igarashi et al., 1999; Janzer and Raff, 1987; Kondo et al., 1996; Willis et al., 2004) in the framework of ischemic stroke (del Zoppo and Mabuchi, 2003; Lo et al., 2003). Astrocytes are activated in response to preconditioning, and have an even greater activation during ischemia in preconditioned brains (Bacigaluppi et al., 2010; Biron et al., 1999). This may be due in part to repression of NF-κB-mediated transcription of inflammatory mediators (Biron et al., 1999). In addition, astrocytes assist in ischemic resistance through free radical scavenging, glycogen storage and erythropoietin production (Napoli and Neumann, 2009). Generation by preconditioned astrocytes of anti-inflammatory cytokines such as IL-10, heat shock proteins (for example, HSP27 and HSP32), and trophic factors such as TGFβ, BDNF, GDNF (glial cell-derived neurotrophic factor), and VEGF might also contribute to cerebroprotection (Zhang et al., 2002).
3.1.2 Endothelial cells-mediated ischemic tolerance
Endothelial cells are of paramount importance to the formation of the BBB. They are found connected to astrocytes by integrins to keep the integrity of the of the neurovascular unit (Hayes et al., 2008). Preconditioning increases microvessel formation thus decreasing reductions in blood flow that normally happen to the brain after permanent stroke. In addition, it also reverses the hypoperfusion that occurs early during reperfusion following global ischemia (Nakamura et al., 2006). Chronic cerebral hypoperfusion is beyond the scope of this review and has been shown to be associated with depressive like behaviors in rat model of permanent occlusion of the bilateral common carotid arteries (Lee et al., 2014).
Post ischemic endothelium-dependent vasodilation is better preserved in the preconditioned brain (Bastide et al., 2003). This is believed to result from the anti-inflammatory effects of endothelially-derived nitric oxide, due to a preconditioning-induced Akt-dependent phosphorylation of endothelial NOS (Hashiguchi et al., 2004, Furuya et al., 2005) that enhances nitric oxide activity (Puisieux et al., 2000, Furuya et al., 2005). Studies have indicated that post ischemia reductions in selectins and immunoglobulin adhesion molecules expression may decrease infiltration through reduction of leukocyte–endothelial rolling and adherence (Andjelkovic et al., 2003, Stenzel-Poore et al., 2003, Dhodda et al., 2004, Ding et al., 2005).
Prior preconditioning may also activate neovascular responses through the activation of endothelial NF-κB production and responsiveness to VEGF (Wick et al., 2002) and the induction of distinct endothelial cell-specific anti-apoptotic response networks (Alavi et al., 2003), increasing the cerebrovascular endothelium’s ability to cope with post ischemic angiogenesis and microvascular remodeling.
3.1.3 Leukocytes-mediated ischemic tolerance
It is known that stimulation of ICAM-1, P-Selectin and E-Selectin molecules assist in increasing the adherence of leukocytes to the damaged vascular endothelium, thus obstructing microvessels and contributing to the increase in leukocyte diapedesis and worsening infiltration. Ischemic preconditioning reduces the expression of ICAM-1 (Ding et al., 2005), consequently decreasing inflammation and improving cerebroprotection following ischemic reperfusion injury. Ischemic preconditioning affects not only leukocyte-endothelial adhesion, but also circulating leukocytes, as shown by transient suppression of the leukocyte genes encoding adhesion, migration, chemotaxis, and cytokine synthesis after brief ischemia in the human forearm (Konstantinov et al., 2004). In addition, blood monocytes show less activation after ischemia in a preconditioned brain (Rosenzweig et al., 2004). VEGF produced by reactive astrocytes is crucial for post-ischemic angiogenesis (Hong et al., 2004) and its action may require neutrophil matrix metalloproteinases (MMPs), suggesting a link between inflammatory cells and angiogenesis (Douen et al., 2000).
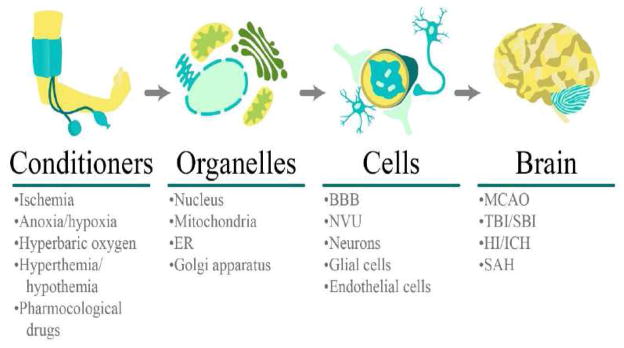
3.2 Subcellular organelles-based ischemic tolerance
Intracellular organelles including mitochondria, Golgi apparatus, and endoplasmic reticulum have not only been shown to have cerebroprotective responses but also to develop ischemic tolerance. All this may set a foundation for future treatment interventions that will aid in stroke recovery and development of novel treatments and preventive mechanisms related to neuronal apoptosis-related diseases.
3.2.1 Mitochondria-mediated cerebroprotection
Mitochondria have a role in metabolic energy production and involvement in apoptosis, thus becoming important regulators of neuronal cell life and death (Yuan and Yankner, 2000). Mitochondrial dysfunction is considered to be one of the key events linking ischemic/recirculation insult with neuronal cell death (Berridge et al., 2003). It is well known that a constant blood flow to the brain is essential for delivering oxygen and glucose to neurons and stoppage of the blood flow even for a short period of time may induce irreversible damage or death (Zemke et al., 2004). There are two ATP-sensitive potassium channels (K+ATP channels): one resides in the inner membrane of mitochondria, while the other is dispersed in the plasma membranes. The mitochondrial K+ATP (mK+ATP) channel has been suggested as “ the key” to ischemic preconditioning. It has been hypothesized that opening the mK+ATP channels may depolarize mitochondrial membrane potentials and promote an increase in the electron transport chain rate and thus increase ATP production (Schultz et al., 1997). To prove that, the use of 5-hydorxydecanoate (5-HD), an mK+ATP blocker, prevented brain ischemic preconditioning induced cerebroprotection. Evidence shows that brain ischemic preconditioning is influenced by the activation of mK+ATP. This has been confirmed by observing a lack of preconditioning-induced protection afforded by adenosine and R-PIA (an adenosine A1 receptor agonist) when K+ATP channels were blocked. Conversely, induction of ischemic tolerance was promoted by K+ATP channel opener (RP-52891, aprikalim) (Auchampach et al., 1992). Another study, done by Perez-Pinzon and Born, using transient infusion of the K+ATP channel antagonist sulfonylurea tolbutamide, demonstrated block of brain ischemic preconditioning induced protection after forebrain ischemia, whereas its promotion in hippocampal slices was achieved with pinacidil, a K+ATP channel agonist (Perez-Pinzon and Born, 1999). The role of p53 translocation and mitochondrial apoptosis will be discussed in the anti-death signaling pathway.
3.2.2 Endoplasmic reticulum-mediated cerebroprotection
In recent years a series of studies have turned attention to the mRNA and protein levels of the ER stress genes after ischemic/reperfusion damage (IRI) in both naive and preconditioned rats (Lehotsky et al., 2009; Pavlikova et al., 2009; Urban et al., 2009). These experiments (Urban et al., 2009; Lehotsky et al. 2009) indicate that time dependent differences in endoplasmic reticular gene expression at both the mRNA and protein levels are initiated by IRI and, furthermore, that pre-ischemic treatment affects endoplasmic gene expression. Added to these experiments, studies suggested not only that attenuation of endoplasmic reticulum (ER) response may be related to IPrC but also that ischemic tolerance has effects on InsP3 receptor mediated Ca2+ signaling an important mediator in cerebroprotection (Bickler et al., 2009). Changes in gene expression of key proteins provide an insight into ER stress pathways, suggesting that enhancing recovery after stroke via these proteins could lead to future therapeutic interventions. Remote ischemic post conditioning also shows protection against ischemia-reperfusion brain injury by attenuating ER stress response-induced apoptosis (Liu et al., 2014b).
3.2.3 Golgi complex-mediated cerebroprotection
A metabolic pathway consisting of endoplasmic reticulum, Golgi complex, secretory vesicles, plasma membrane, and lysosomes is known as the secretory pathway (SP). The SP is involved in many functions such as stress sensing, neuronal aging, transduction of apoptotic signals, etc. by sorting, assembling and functionalizing the newly synthesized proteins (Maag et al., 2003; Sepulveda et al., 2008). The Golgi system, making up part of the SP, has a dynamic store of Ca2+, thereby playing a pivotal role in protein processing and quality control. The secretory pathways Ca2+-ATPase (SPCAs) represent a subfamily of P-type ATPases and function as ion pumps that supply the lumen of the Golgi with a high luminal Ca2+ concentration requisite for the optimal activity of many enzymes and also post-translational processing and trafficking of the newly formed proteins in the Golgi apparatus. There are 2 subtypes of SPCA: SPCA1 and SPCA2. SPCA1 has significant role in SPCA-facilitated transport of Ca2+ for calcium storage within the brain (Wootton et al., 2004).
Lehotsky et al. showed that SPCA activity is damaged by free radicals selectively in vitro (Lehotsky et al., 2002a), and transient ischemia for 15 min induces considerable lipid and protein oxidation in hippocampal membranes. At later stages after the ischemic insult, protein oxidation depressed enzymatic activities of main antioxidant enzymes were (Lehotsky et al., 2002b; Urikova et al., 2006). Thus, IRI-induced oxidative alterations may disturb neuronal ion transport and further inhibit protein synthesis by damaging the SPCA1 activity (Burda et al., 2003; Lehotsky et al., 2002b; Lipton, 1999).
In order to evaluate the effects of severe metabolic stress induced by IRI and/or IPrC on the expression of SPCA1, the mRNA and protein levels of SPCA1 were analyzed (Lehotsky et al., 2002b; Lehotsky et al., 2004). In these experiments, ischemic preconditioning had a partial protective effect on SPCA activity. Ischemic insult after IPrC pretreatment initiates only non-significant inhibition of Ca2+-ATPase activity compared to the preconditioned control. After 1 and 3 h of reperfusion, the Ca2+-ATPase activity exceeded control levels and reached it again after 24 h of reperfusion. However, the changes were not statistically significant at any reperfusion time. The ability of a preconditioned brain to upregulate defense mechanisms against oxidative stress during ischemia helps enable enzymatic activity to return to it normal functioning (Danielisova et al., 2005; Gidday, 2006; Obrenovitch, 2008). IPrC reduced oxidative changes induced by ischemia in the hippocampal membranes in both the ischemic and reperfusion period, possibly due to upregulation of antioxidant enzymes related to preconditioning (Danielisova et al., 2005; Gidday, 2006; Obrenovitch, 2008).
3.3 Synaptic signaling-based ischemic tolerance
In the past several decades, excitotoxicity has been the center stage of stroke research (Lai et al., 2014). It has also been identified as one of the first steps in the pathology of cerebral ischemia (Choi et al., 1987). IPrC was found to ameliorate excitotoxicity by inhibiting glutamate release (Dave et al., 2005; Douen et al., 2000) increasing inhibitory neurotransmitter gamma-aminobutyric acid (GABA) release (Douen et al., 2000) and enhancing GABA presynaptic and postsynaptic activities, thus making neurons more resistant to excitotoxic insult (DeFazio et al., 2009; Sommer et al., 2002; Sommer et al., 1995). These findings suggest that IPrC promotes synaptic modifications that may preserve synaptic function and enhance functional recovery following cerebral ischemia.
3.3.1 Excitatory Glutamate and NMDA receptors
Glutamate is the major excitatory neurotransmitter in the mammalian brain (Yokobori et al., 2013), and is responsible for initiation of postsynaptic excitatory signaling through distinct ionotropic and metabotropic glutamate receptors. The ionotropic glutamate receptors include the N-methyl-D-aspartic acid (NMDA) receptor, the 2-amino-3-(3-hydroxy-5-methylisoxazol-4-yl) proprionate receptor, and the kainate receptor subtypes (Kato et al., 1992b). Ca2+ entry into the postsynaptic neuron happens via NMDA receptor and this allows activation of Ca2+- dependent enzymes and downstream signaling pathways, leading to changes synaptic strength (Dingledine et al., 1999). Though glutamate kills neurons through the NMDA receptor-mediated mechanism, mild NMDA receptor activation is involved in the cerebroprotective mechanism of brain ischemic preconditioning (Gidday, 2006). Exposure of cortical cell cultures to low levels of glutamate or NMDA to induce NMDA receptor activation has a preconditioning effect (Lange-Asschenfeldt et al., 2004). One mechanism underlying NMDA receptor-mediated brain ischemic preconditioning is the rapid adaptation of voltage dependent calcium flux. Since hypoxia/ischemia induced neurodegeneration can be triggered by cytoplasmic calcium overload, the NMDA receptor mediates rapid calcium adaptation in preconditioning that may alleviate the cell damage caused by calcium overload. Glutamate-independent Ca2+ and Na+ overload mechanisms have also been associated with neuronal ischemic cell death due to activation of mitochondrial dysfunction and apoptosis among others. (Pignataro et al., 2014).
The other mechanism involves activation of NMDA receptors leading to the rapid release of brain-derived neurotrophic factor (BDNF). Other key mediators which have been involved in synaptic NMDA receptor-dependent neuroprotection include cyclic AMP responsive element binding protein, phosphatidylinositol 3 (PI3)-kinase, Akt and glycogen synthase kinase 3 beta (GSK3β). These mediators can be induced only by low doses of NMDA via the action potential-dependent route (Kato et al., 1992c; Raval et al., 2003). In addition, glutamate transporter-1 (GLT-1), a predominant subtype of glutamate transporter, has an important role in glutamate regulation. Its upregulation ameliorates brain damage. Ischemic conditioning is responsible for its increase thereby providing further cerebroprotection. Furthermore, exercise preconditioning was believed to be involved in upregulating this transporter by involving different signaling pathways (Gong et al., 2012; Liu et al., 2011; Wang et al., 2014). IPreC also induced cerebroprotection thought the stimulation of GLT-1 mediated by transcriptional activity of the nuclear receptor peroxisome proliferator-activated receptor gamma (PPAR-γ) (Romera et al., 2007). Action potential regulators such as voltage activated potassium channels play a role in cerebroprotection against ischemic injury (Shah and Aizenman, 2014) but their focus is beyond the scope of this review.
3.3.2 Inhibitory GABA and GABA receptors
Gamma-aminobutyric acid (GABA) is a well-known inhibitory neurotransmitter in the brain. GABAergic disinhibition has been attributed to lesion progression because neuronal hyperexcitability associated with sustained downregulation of GABA A receptors was found in peri-infarct regions. Recently, Dave et al found that brain ischemic preconditioning promoted a robust release of GABA in rats after lethal ischemia (Dave et al., 2005; Perez-Pinzon, 2007). They also observed that the activity of glutamate decarboxylase (the rate-limiting enzyme in GABA synthesis in the brain) was higher in the brain ischemic preconditioning group compared with controls and ischemic groups. They further tested the hypothesis that GABA B receptor activation was also cerebroprotective during ischemia or early reperfusion by using an in vitro model (organotypic hippocampal slice culture). They found that administration of the GABA B agonist baclofen during ischemia and the first hour of reperfusion provided significant cerebroprotection. In addition, Sommer’s group has shown that ischemic tolerance in the preconditioned gerbil hippocampus is associated with increased ligand binding to inhibitory GABA-A receptors between 30 min and 48 h of recirculation (Sommer et al., 2003). In summary, preconditioning can cause increased GABA release or increased GABA receptor expression. The changes in both pre- and postsynaptic GABAergic transmission are likely to contribute to a shift of the glutamate/GABA balance toward inhibition in the preconditioning brain (Obrenovitch, 2008). A study conducted by Globus, et al. showed that elevated glutamate levels during ischemia are not enough to incur ischemic damage. Glycine and gamma-aminobutyric acid (GABA), which modulate glutamatergic activity, also played a considerable role. Similar increases in glutamate, GABA, and glycine were observed in the striatum and thalamus after ischemia. For that reason an ‘excitotoxic index’ as: [glutamate] x [glycine]/[GABA] was created to quantitavely describe the composite magnitude of amino acid neurotransmitter changes with ischemia (Globus et al., 1991).
3.3.3. Inhibitory adenosine and adenosine A1 receptor
Adenosine is a neuromodulator and vasomodulator that is normally released when ATP levels decline (Ghiardi et al., 1999). Evidence showed adenosine is released after IPrC and initiates a signaling pathway that promotes ischemic tolerance in brain via activation of A1 receptors (A1AR) (Lange-Asschenfeldt et al., 2004; Perez-Pinzon et al., 1996; Reshef et al., 1996). Inhibition of synaptic activity is observed when adenosine binds to the A1AR (Perez-Pinzon et al., 1996), which is believed to be the key receptor in the induction of ischemic tolerance.
Adenosine inhibits synaptic transmission, decreases K+- stimulated glutamate release, and inhibits presynaptic calcium fluxes via adenosine A1AR. The response to calcium influx is also important for the development of protection against ischemia, because calcium influx has been linked to the production of reactive oxygen species and the initiation of a number of signaling cascades leading to cell damage (Kristian and Siesjo, 1998). Many reports have shown that preconditioning-induced cerebroprotection is dependent on adenosine A1 receptors. In rats, ischemic preconditioning increased adenosine A1 receptor immunoreactivity in the hippocampal CA1 region at days 1, 3, and 7 after preconditioning induction, within the window of ischemic tolerance (Zhou et al., 2004). Some unspecific and specific adenosine A1 receptor antagonists abolished ischemic tolerance. Moreover, Liu et al demonstrated that isoflurane-induced tolerance against focal cerebral ischemia in the rat middle cerebral artery occlusion (MCAO) model was attenuated by adenosine A1receptor antagonists (Liu et al., 2006).
3.4 Protein degradation systems-based ischemic tolerance
The mechanisms involved in this section collaborate with either cell survival or cell death. Three pathways show how these mechanisms relate to autophagy. Autophagy in neurons is likely a fundamental step in brain reparation as it removes of toxic protein aggregates and damaged organelles during the post ischemic phase (Luo et al., 2013) and its activation has been indicated to contribute to tolerance towards an ischemic insult (Chen et al., 2014b). The first two pathways are the ubiquitone-proteasome pathway and the lysosomal pathway; both are related to protein degradation systems in mammalian cells. The final pathway reviewed is the AMPK mediated pathway (5′ adenosine monophosphate-activated protein kinase).
3.4.1 The ubiquitin-proteasomal pathway
Proteasomes consist of multiple protein subunits and degraded cytosolic proteins along with misfolded proteins that failed quality assurance in the ER (Wu et al., 2006). These proteins are identified by ER-specific E3 ligases that mediate polyubiquitination of the misfolded proteins on the cytosolic side of ER and are lastly degraded by proteasomes (Chen et al., 2008). Degradation of Ubi-proteins is ATP-dependent and these proteins act as signals to induce heat shock transcription factors to stimulate expression of molecular chaperones (Voellmy, 2004) that will inhibit protein accumulation. ATPases are the main cellular chaperones and facilitate protein folding through several series of binding and release of unfolded protein substrates by hydrolysis of ATP. Along with ischemia preconditioning, ubiquitin–proteasome pathway and molecular chaperons can prevent protein aggregation in lethal ischemia (Liu et al., 2005).
The ubiquitin-proteasomal pathway has been studied in rat forebrain ischemia models and transient cerebral ischemia followed by reperfusion. Hippocampal CA1 pyramidal neurons that underwent ischemia followed by delayed neuronal death progressively accumulated protein aggregates (Liu et al., 2009). The accumulation of unfolded proteins causes, through signaling pathways, reaction from mitochondria, endoplasmic reticulum (ER) and cytoplasm, thereby contributing to neuronal damage. At the same time, ubiquitin-proteasome and chaperones work to counter protein agglomeration and facilitate protein folding (Ghaemmaghami et al., 2003; Hebert and Molinari, 2007). Additionally, Liu et al. demonstrated that IPrC greatly reduced protein aggregation in CA1 neurons after ischemia. After IPrC, Ubiquitin-conjugated proteins were decreased and a reduction in free ubiquitin depletion after brain ischemia was observed. Decreased aggregate -containing fractions of HSP 70 and HSP 40 were reduced after IPrC (Liu et al., 2005).
3.4.2 Autophagy/Lysosomal Pathway
Autophagy and Lysosomal pathways have been under the spotlight for several years. There are ongoing debates regarding autophagy and neuronal death/survival despite studies reporting activation of autophagy after ischemia (Adhami et al., 2006; Balduini et al., 2009; Carloni et al., 2008; Koike et al., 2008; Rami et al., 2008) and after permanent middle cerebral artery occlusion (pMCAO)(Wen et al., 2008). Autophagy is a conserved step participating in cellular energy controlled by degrading and recycling its components, and has been proposed as a downstream target of AMPK (Jiang et al., 2014).
Participation of autophagy in the pathogenesis of cerebral ischemia is debatable and uncertain (Wei et al., 2012), though previous studies have pointed out AMPK’s ability to provide cerebroprotection against ischemia in peripheral organs (Matsui et al., 2007; Nepal and Park, 2013; Pauly et al., 2012; Wang et al., 2013a; Xie et al., 2011; Zaouali et al., 2013). For instance, studies have shown an AMPK mediated pathway is associated with induction of autophagy, thereby promoting reduction in infarct volume, cell apoptosis, and neurological deficits caused by pMCAO (Jiang et al., 2014). This brings light to the hypothesis that pre activation of autophagy through the AMPK pathway may provide therapeutic cerebroprotection and prevent stroke. With this in mind, Jiang et al. found AMPK mediated autophagy contributes to cerebroprotection after IPC. IPrC upregulated HSP 70 is decreased by autophagy inhibition improving cerebroprotection. These studies support the idea of strong relationships between autophagy and IPrC in cerebroprotection against lethal ischemia (Sheng et al., 2010).
Jiang et al. demonstrated increased activation of p-AMKP/AMPK in rat brain led to intensification of autophagy, and other studies showed activation of AMPK via metformin, giving rise to enhanced autophagy in rat brain (Kariko et al., 2004). Furthermore, mTOR inhibition by AMPK activation enhanced autophagy (Zheng and Zuo, 2003). ULK1 phosphorylation directed by AMPK could also augment autophagy (Zheng and Zuo, 2004). No increment of autophagy was seen when AMPK was inhibited (Kariko et al., 2004). These studies show activation of autophagy with AMPK through several signaling pathways. Intracellular ATP and amino acids were renewed after IPrC induced autophagy, degradation of proteins, lipids, and glycogen granules, thereby augmenting tolerance of neurons to deadly ischemic exposure and providing cerebroprotection (Nakajima et al., 2004). In addition, autophagy efficiently facilitates cellular energy production via removal of dysfunctional mitochondria allowing efficient use of these substrates (Wick et al., 2002). These findings further support the hypothesis that pre-activation of brain autophagy could protect against subsequent permanent cerebral ischemia (Schultz et al., 1997; Zhang et al., 2001).
Some studies show autophagy activation after strokes worsens neuronal injury (Wei et al., 2013; Wen et al., 2008). Preconditioning poses a challenge in that massive stimulation of autophagy may lead to excessive self-digestion while its activation can also lead to clearing injured organelles and unfolded proteins. Others show autophagy pre activation increases tolerance against ischemic insults (Sheng et al., 2012; Sheng et al., 2010). Activation of autophagy by IPrC can decrease cell apoptosis by inhibiting overwhelming stress on the ER and inflammatory responses following ischemia (Gao et al., 2013; Sheng et al., 2012). Some studies suggest that deterioration of neurons occurs during the reperfusion phase after induction of autophagy (Bu et al., 2014; Gao et al., 2012).
3.5 Classical anti-death signaling pathways
Although there are multiple key signaling pathways that mediate preconditioning, it is plausible that significant cross-talk occurs among pro-survival kinases following IPrC.
3.5.1 Anti-inflammation pathways
It is well known that inflammation leads to secondary damage and further tissue injury in acute ischemia. Preconditioning heavily influences cytokine and chemokine expression, suggesting they have an important role in acute stroke. Downregulation of inflammation and reduction of the activation of neutrophils and leucocyte endothelial interactions are included in the protective effects of ischemic preconditioning (Castillo et al., 2003; Della-Morte et al., 2012). Pro-inflammatory cytokines IL-1 and IL-6, modulators of the immune response, are downregulated and anti-inflammatory factor IL-10 is upregulated (Petcu et al., 2008; Warzecha et al., 2007). Ohtsuki et al. showed increased levels of IL-1 and 1 β after ischemic preconditioning in Mongolian gerbils (Ohtsuki et al., 1996) and that possible mechanisms include the release of arachidonic acid, enhancement of NMDA activation, and stimulation of nitric oxide synthase. These findings are further supported by data showing the IL-1 receptor antagonist blocked ischemic tolerance after preconditioning (Huang et al., 2006).
Another important cytokine in conferring ischemic tolerance is TNF-α (Castillo et al., 2003), which has anti-inflammatory and cytoprotective effects through activation of NF-κB and is expressed in the ischemic brain (Ginis et al., 2002). Nawashiro et al., studied the effects of pretreatment with TNF-α administered intracisternally in mice, finding a significant reduction in infarct size in mice pretreated with TNF-α after being subjected to MCAO 48 hours later (Nawashiro et al., 1997). Liu et al., demonstrated that TNF-α preconditioning induced tolerance to hypoxia, similar to the effects of hypoxia preconditioning of rat cortical neurons (Liu et al., 2000). Administering TNF-α-neutralizing antibody attenuated hypoxia preconditioning. Pre-treatment with TNF-α induces further tolerance to TNF-α exposure thereby improving tolerance to ischemia. Inhibition of IFN-γ have also been shown to possibly provide neuroprotection, due to a decrease in the recruitment of pro-inflammatory cells, preventing activation of microglia/macrophges in the brain and improving neural cell survival (Seifert and Pennypacker, 2014). In addition, it has been reported that microglial galectin 3 plays a potentially toxic role by promoting pro-inflammatory cascades (Rabinovich and Toscano, 2009). Microglial activation in ischemic injury has been correlated with increased galectin-3 level (Lalancette-Hebert et al., 2012; Rotshenker, 2003) leading to the demise of hippocampal neurons (Satoh et al., 2011). However, preconditioning strategies or hypothermia both result in the downregulation of microglial galectin-3 and attenuate these deleterious events (Lalancette-Hebert et al., 2012; Satoh et al., 2011). The main mechanism by which the brain benefits by protection from ischemia is not well known, but astrocytes, microglia and leukocytes proliferation may play a role according to recent studies and their investigation following cerebral ischemia is warranted to improve understanding of the inflammatory response that follows brain injury, thus benefiting the development of therapeutic targets for devastating conditions such as stroke (Leng et al., 2014).
When discussing stimuli that promote brain ischemic tolerance it is worth mentioning “ischemic cross-tolerance” which describes the tolerance to ischemia provided by non-ischemic noxious insults through inducing genetic reprogramming or post-translational changes. Many examples of such cross-tolerance have now been recognized (Dirnagl et al., 2003; Gidday, 2006), among which, LPS, (Stenzel-Poore et al., 2007) NMDA, and TNF-α are well-documented. In addition, epileptic seizures can decrease subsequent seizure induced neuronal damage (epileptic tolerance) and serve as another example of cross-tolerance (Plamondon et al., 1999). These studies suggest that different stressful stimuli other than ischemia itself can furnish ischemic tolerance. While the specific genes and proteins involved in such processes may differ, the resulting effect appears to be the reduction in cell death resulting from the exposure to a normally harmful stimulus.
Endogenous neuroprotection by preconditioning with a sublethal ischemic stimulus largely depends on de novo expression of neuroprotective genes mediated by transcription factors like HIF-1(hypoxia-inducible factor), and CREB (cAMP-response element binding protein) (Mergenthaler and Dirnagl, 2011).
Ischemic conditioning acts on various targets to protect the brain. The release of locally or remotely acting metabolites (e.g. adenosine) and activation of genetic and epigenetic responses either directly through sensors (HIF-1) in complex signaling pathways, or via neuronal pathways (e.g. activating the sympathetic nervous system or the hypothalamic–pituitary axis) are stimulated by ischemic conditioning, leading to a complex cascade. (Mergenthaler and Dirnagl, 2011)
HIF-1 is not the only transcription factor that contributes to IPC induced cerebroprotection. Non-HIF transcription factors such as CREB and NFkB, among others, directly or indirectly regulate hypoxia-responsive gene expression. CREB is a transcription factor activated in response to intracelular calcium elevation, a process regulated by NMDA receptor activation (Hu et al., 1999). The penumbral region of preconditioned rats have high amounts of phosphorylated CREB (Nakajima et al., 2002), thus being associated with the NMDA receptor preconditioning process.
3.5.2 Anti-apoptosis pathways
Besides the role of mK+ATP channels, mitochondria have also been linked to apoptosis. The level of pro-apoptotic proteins of bax, p53 and anti-apoptotic protein of bcl-2, bcl-xl are involved in apoptotic processes induced by ischemic reperfusion injury. These proteins were analyzed by Western blot analysis in both cortical and hippocampal mitochondria regions (Racay et al., 2009; Racay et al., 2007). In that study, these proteins led to increase of p53 level in hippocampal mitochondria, with significant differences after 3 h, 24 h, and 72 h of reperfusion. Also, ischemia-induced translocation of p53 to mitochondria was completely abolished by IPrC since no significant changes in mitochondrial p53 level were observed after preconditioned ischemia. Similar to naive ischemia, the levels of both bax and bcl-xl were not affected by IPrC. In addition, the study showed that IPrC was protective not only to ischemia-induced DNA fragmentation and also to a number of positive Fluoro-Jade C staining cells. This indicates that IPrC abolished almost completely both initiation and execution of mitochondrial apoptosis induced by global brain ischemia in the vulnerable CA1 layer of rat hippocampus (Racay et al., 2009; Racay et al., 2007). Thus, by acting at both levels, IPrC will protect from ischemia-associated changes and preserve the integrity of mitochondrial membranes. In the face of ischemic brain injury, IPrC may then provide neuronal survival by inhibiting mitochondrial p53 pathway and also activating inhibition of p53 translocation to mitochondria (Otani, 2008).
3.5.3 Anti-oxidation pathways
Deadly hypoxic situations are restrained with the use of transcription factors. The factor nuclear factor erythroid 2 related factor (Nrf2) senses oxidative stress in the cells, and its targeted gene’s expression, which are involved in maintaining the redox state of the cell, are increased in both rat and human astrocytes following ischemia. They are glutathione and glutathione related enzyme (Bell et al., 2011). In contrast, thioredoxin, an Nrf2 regulated protein involved in reducing oxidized thiol groups in the cell is increased following hypoxic preconditioning and has been observed in rat neocortex (Stroev et al., 2004) and hippocampus (Stroev et al., 2009). In addition, transcription factor family of signal transducers and activators of transcription (STATs), are also stimulated and confer cerebroprotection following IPrC through increments in antioxidants (Kim et al., 2008; Lin et al., 2011; Perez-Pinzon et al., 2012). Therefore, a strong cerebroprotective mechanism of IPrC may be seen through reduction of oxidative stress through antioxidant expression.
Oxidative stress also affects oxidation of DNA, and depending on the extent of the damage, stress will incite pro-apoptotic pathways. Recruitment of DNA repair enzyme poly (ADP-ribose) polymerase-1 (PARP-1) consumes NAD+ and ATP cellular stores (Lavignon et al., 1992; Li et al., 2006). β-polymerase-mediated and apurinic/apyrimidinic endonuclease-mediated base excision repair (BER) is also increased following IPrC in rats and improves ischemic tolerance (Li et al., 2006). Therefore, finding possible therapy aimed at increasing DNA repair ability after oxidative stress could provide cerebroprotection (Narayanan et al., 2013).
4 Clinical considerations
4.1 Naturally occurring ischemic preconditioning
Naturally occurring ischemic preconditioning is ischemic tolerance achieved through the natural course of some diseases. Data has shown a decrease in severity of stroke or myocardial infarction when preceded by transient ischemic attack or angina. This was supported with measurement and analysis of creatine kinase, infarct size on imaging, development of malignant arrhythmia, mortality and neurologic disability (Abete et al., 1997; Castillo et al., 2003; Lorgis et al., 2012; Moncayo et al., 2000; Wegener et al., 2004). Also, patients with peripheral vascular disease (PVD) and sleep apnea (through repetitive hypoxic crises) may have experienced naturally occurring ischemic preconditioning (Connolly et al., 2013; Koch et al., 2007). PVD patients had better functional outcomes and smaller stroke volumes according to an observational study. Cerebroprotection was felt to be the result of chronic limb ischemia (Connolly et al., 2013). The mechanisms involved in ischemic conditioning have been derived primarily from IPrC research. IPeC and IPoC have less studies available in the literature and for that reason are not as well known as IPrC.
4.2 Application limitations
4.2.1 Limitations from narrow safety margin of preconditioning
The central principle of preconditioning when faced with ischemic injury is balance: to have an inducible dose high enough to produce an effect, but at a subthreshold level that will not cause damage. Among the possible limitations that may interfere with preconditioning using RIPC are changes in intensity, duration, and frequency of specific stimulus, as these are variables that will determine the outcomes from the intervention. According to Dirnagl et al., there are no clear limits between the acquisition of ischemic tolerance and cellular apoptosis/necrosis (Dirnagl et al., 2009). For this reason, the therapeutic advantage of IPrC in clinical practice may be limited by narrow safety margins (Durukan and Tatlisumak, 2010).
4.2.2 Limitations from animal models
The translation of rodent models to the clinical setting has limitations. Studies show that rodents lack similar brain architecture and that their white matter and grey matter ratios are different from humans (Oberheim et al., 2006). Not only that, but the typical age of rodents used in IPrC studies does not parallel the age of typical patients in need of IPrC therapy (Quinn, 2005). This creates a problem as studies show that aging decreases the capacity to induce IPrC (He et al., 2005). Some evidence suggests that ischemic tolerance is less likely achieved in the aged heart and brain. In animal studies, transient ischemia leads to a different gene expression in older animals when compared with younger animals (Della-Morte et al., 2012). Rodent models also used a consistent, controlled ischemic insult; however, stroke oftentimes happens oftentimes without immediate notice in humans (Koch et al., 2012). Faced with these challenges, there is a need not only to better investigate IPrC related mechanisms to increase comprehension of a specific method or emulation of IPrC to facilitate clinical translation (Narayanan et al., 2013), but also to decode IPrC related cerebroprotection that converts animal models closer to real clinical scenarios as implied by the Stroke Therapy Academic Industry Roundtable criteria (STAIR) (Fisher et al., 2009).
4.3 Clinical trials and indications of preconditioning
With thousands of entries into MEDLINE 1948 to present, the combination of “preconditioning” and “brain” have preclinical studies as the bulk of reports (Koch, 2013). In order for the rodent preclinical model to have clinical applicability, it first has to be translated to fit into a clinical context. With that in mind, remote ischemic preconditioning (RIPC) could give hope to the idea of successful clinical translation. RIPC involves cycles of temporary occlusion and restoration of blood flow in a forelimb far removed from the desired site of protection (Narayanan et al., 2013). Due to its relative simplicity and perceived safety, clinical trials have previously applied this concept by using blood pressure cuffs to cause temporary occlusion and reestablishment of blood flow in arms or thighs of patients, translating the RIPC model (Kharbanda et al., 2002) and allowing the pursuit of efficacy using this approach in clinical medicine (Keep et al., 2014). This model was also translated by promoting inflation of inter-arterial balloons with deflation just prior to coronary angioplasty or coronary artery bypass procedures, simulating IPrC cytoprotection. RIPC has caused no harm in critically ill patients with subarachnoid hemorrhage (Koch et al., 2011), thereby further supporting this model as a safe and practicable preventive strategy (Narayanan et al., 2013).
Patients subjected to lengthy or invasive surgeries (with possible ischemia as an outcome), might not only have improved outcomes after ischemic preconditioning treatment, but IPrC also serves as ischemic and inflammatory shields in those with chronic conditions such as metabolic syndrome, cardiovascular and cerebrovascular disease, or those at risk for recurrent ischemic attacks (Narayanan et al., 2013). In addition, pharmacological preconditioning with nitroglycerine (Jneid et al., 2005), and anesthetic-PC (Frassdorf et al., 2009) have also been tested in clinical trials to protect the heart from cardio vascular interventions with high risk of cardiac ischemic event. Results are promising and give hope that clinical trials of PC can be applied to cerebroprotection in high risk situations once this avenue of PC is proven safe (Durukan and Tatlisumak, 2010).
With RIPC being the most likely therapeutic intervention in acute stroke, its timing is a fundamental determinant in acquiring the desired protections. Various timing regiments including cycle times, duration, and PC starting time (Lin et al., 2011; Murry et al., 1986; Narayanan et al., 2013; Ramzy et al., 2006) have been extensively studied in animal models, but are still far from translational applicability. This is due in part to the differences between human and animal models. For this reason, more research is necessary to better understand optimal approaches for future interventions. In addition, the various PC interventions differ in specific timing and duration, and this is further complicated by the course of the stroke and differences in gender, age, and comorbidities. In spite of all these challenges, different timing of preconditioning can still be used for cerebroprotection for elective brain surgeries.
4.4 Future directions
4.4.1 From ischemic conditioning toward pharmacological conditioning
Mechanical and pharmacologic preconditioning are two currently available methods of preconditioning. Translating mechanical preconditioning into a clinical context is challenging due to the difficulty in foreseeing the onset of an ischemic event. Even with its predictability, applying short term mechanical preconditioning is improbable before cerebral ischemia. For that reason, there are limits to utilization of mechanical preconditioning (Narayanan et al., 2013). This emphasizes the need to discover pharmacologic agents to induce preconditioning and better elucidation of its underlying mechanisms, allowing better translation from mechanical to pharmacologic preconditioning in the clinical settings (Keep et al., 2014; Tang et al., 2011).
Numerous triggers have been identified as being able to elicit and reproduce the effects of ischemic preconditioning. It is a challenge to decide the optimal approach to induce pathways critical for preconditioning and the best choice of medications and their administration. Perhaps the use of a pharmacologic agent that can activate pathways critical for IPrC induced cerebroprotection would be more clinically applicable. A study has shown the contribution of Sirtuin 1(SIRT1, a class III NAD+-dependent histone deacetylase) to mediate delayed IPrC-induced neuroprotection (Della-Morte et al., 2009). Therefore, a potent activator of SIRT1such as the polyphenol resveratrol could represent a potential therapy for cerebral ischemia, since resveratrol is safe in humans (Johnson et al., 2011). However, mechanistic clarification is lacking for the use of resveratrol preceding IPrC. In addition, SIRT1 deserves more studies regarding its role in the promotion of neuronal survival and its activities on either lowering energy demand or increasing energy supply (Petegnief and Planas, 2013).
Another example is the use of isoflurane, a fast induction anesthetic, that mimics IPrC by activation of mK+ATP channels (Tanaka et al., 2003). Thus, brief patient exposures to isoflurane prior to invasive surgery could serve as another clinical use of IPrC.
4.4.2 Development of Biomarkers
Development of a biomarker will assure that a particular treatment induces the desired biologic activity and the final clinical effect. In addition, it is also thought to be paramount in translation of basic IPrC from the laboratory to the clinical setting (Koch, 2010; Koch and Perez-Pinzon, 2013). Development of specific biomarkers would help specify the magnitude of response and the best preconditioning stimulus in patients which would help in dose selection in the layout of phase II clinical trials (Koch et al., 2014). So developing easily measurable end points of biologic activity to facilitate the translational success of novel neuroprotective therapies has been a recommendation of the 2010 National Heart Blood and Lung Institute (NHBL) Workshop on cardioprotection and the STAIR (Stroke Therapy Academic Industry Roundtable) on cerebroprotection (Fisher et al., 2009; Schwartz Longacre et al., 2011). In 2013, Wang reviewed the applicability of these consensuses and proposed paying special attention to safety and dosages of PC treatments, meticulously testing for a match of pre-clinical models to the human condition, and lastly, attention to the timing of both the initiation and discontinuation of the PC stimulus relative to the injury ictus (Wang et al., 2013b).
4.4.2.1 Serum biomarkers
The characterization of a systemic state of ischemic tolerance is the objective of biomarker development for preconditioning. Serum biomarkers are of particular interest given their historical use in clinical medicine. Animal studies have also shown widespread effects of preconditioning on the expression of platelet aggregates, tissue plasminogen activator(tPA), tPA inhibitor, and D-dimer involved in hemostasis and fibrinolysis, supporting that proteins involved in the coagulation cascade can be potential biomarkers of the preconditioning response (Koch et al., 2014). Even more, they are readily available and can be serially separated. A recent study of remote ischemic preconditioning in healthy volunteers undergoing repetitive cycles of arm preconditioning found that a global proteomic response was stimulated in the serum, in that proteins involved in hemostasis were altered, further supporting the study of biomarkers (Hepponstall et al., 2012).
Most of the clinical work in assessing a human preconditioning response has involved the immune and inflammatory systems. Of interest is that a positive TNF-a/IL-6 ratio has been proposed as a marker for ischemic tolerance in humans. A study of 283 patients found that those who had a transient ischemic attack prior to their first ischemic stroke had a reduced infarct size on CT scan and improved functional outcomes at 3 months. They showed immunomodulatory effects of ischemic preconditioning, leading to the hypothesis that the state of cerebroprotection is remarkably associated with elevated levels of TNF-a/IL-6 ratio (Castillo et al., 2003). Lin et al. also added to the clinical scenario by showing the postoperative rise in malondialdehyde, IL-6 and IL-8 within 24 hours after orthopedic surgery in patients after leg preconditioning.
4.4.2.1 Imaging biomarkers
Aside from serum analysis, the preconditioning response has also been evaluated through blood flow, perfusion imaging, and vasomotor testing. Improved blood flow in the target organ during and after ischemia is among the physiologic effects of IPrC. Zhao et al. demonstrated in animal models of focal cerebral ischemia that preconditioning improves blood flow in the ischemic penumbra when compared to animals not preconditioned (Zhao and Nowak, 2006). In addition to that, improvements in blood flow/cerebral perfusion have also been shown by single photon emission computed tomography and transcranial Doppler with repetitive bilateral arm preconditioning for secondary prevention of stroke inpatients with intracranial stenosis (Meng et al., 2012). Also a study of remote ischemic limb preconditioning in patients with subarachnoid hemorrhage using transcranial Doppler and cerebral microdialysis, showed the correlation of cell membrane preservation and the reduction in the lactate/pyruvate ratio and glycerol to transcranial Doppler measured cerebral hemodynamics (Gonzalez et al., 2013). This opens an avenue to monitor metabolic responses to preconditioning in some critically ill patients requiring invasive monitoring for clinical purposes (Koch et al., 2014).
5. Conclusion
As reviewed above, the challenges of translating basic science research to the bed side will require intensive study and cooperation among researchers, in order to discover more mechanisms associated with cerebroprotection. As mentioned in the abstract, ischemic pre-conditioning stimulates the innate neuroprotective mechanism to prevent or reverse neurodegenerative diseases including stoke and traumatic brain injury. In TBI, pre-conditioning is linked to reduction in primary and secondary brain damage and reduction of necrosis-mediated damage associated with the glutaminergic system. For those reasons, the challenges and intensive work to study, research, and translate the topic will pay off in the future (Yokobori et al., 2013).
Collaboration will also be necessary to achieve these goals and for the development of serum and imaging biomarkers that will enable monitoring the body’s response to a conditioning stimulus and also identify differences among individuals, animal models and rat strains, as these differences may help understand genetic contributions to injury (Kunze et al., 2014), thus decreasing the factors limiting possibilities of translating per-conditioning ischemia to clinical practice. In addition, research must continue in order to develop more pharmacological agents mimicking ischemic preconditioning and to further delineate the human genetic and proteomic responses to preconditioning. With all the above combined, we hope to answer questions on how to design a better conditioning strategy to demonstrate the influence of such a strategy on the prevention of neurodegenerative disorders, such as the functions of ion channels in regulation of neuronal regenerative activities (Chen et al., 2014a), improvements in outcomes of stroke patients, and to show a way to address other questions that may come up as new evidences arise in the literature. It is certain that a great amount of work is needed to clarify questions existing up to this date. Despite that, with all that has been done so far we can say that there is a promising light at the end of the tunnel to improve the life of millions of people around the world.
Figure 1.
Three types of ischemic conditioning
Figure 2.
Local IPrC and Remote IPrC
Figure 3.

mk+ATP channel mediated ischemic tolerance
Figure 4.

Synaptic signaling based ischemic tolerance
Figure 5.
Conditioner induced cerebroprotection at different levels
Table 1.
Effects of IPrC on cerebroprotection at cellular and subcellular levels
| Neurovascular network | Suborganelles | Synaptic Signaling | Anti - Death Signaling Pathways | Protein Degradation Systems | |
|---|---|---|---|---|---|
|
|
|
|
|
|
|
|
|
|
|
|
|
↑ Activity or quantity incrasses. ↓ Activity or quantity decreases.
VEGF: Vascular Endothelial Growth Factors; HSP27: Heat Shock Protein 27; NF-κB: Nuclear Fator κB; A1AR: Adenosine Receptor A1; Nrf2: Nuclear Factor Erythroid 2 Related Factor; PARP-1: Polymerase-1; STATs: Signal Transducers and Activators of Transcription; AMPK: 5′ Adenosine Monophosphate-Activated Protein Kinase; IL: Interleukin; GABA: Gamma-Aminobutyric Acid; BDNF: Brain-Derived Neurotrophic Fator; NMDA: N-methyl-D-aspartic acid; ICAM: Intercellular Adhesion Molecule.
Highlights.
Ischemic conditioning induces endogenous brain protection pre, per, and post stroke.
Ischemic conditioning mediated protection affects neurovascular network.
Ischemic conditioning in terms of mechanisms and translational use.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abete P, Ferrara N, Cacciatore F, Madrid A, Bianco S, Calabrese C, Napoli C, Scognamiglio P, Bollella O, Cioppa A, Longobardi G, Rengo F. Angina-induced protection against myocardial infarction in adult and elderly patients: a loss of preconditioning mechanism in the aging heart? Journal of the American College of Cardiology. 1997;30:947–954. doi: 10.1016/s0735-1097(97)00256-8. [DOI] [PubMed] [Google Scholar]
- Adhami F, Liao G, Morozov YM, Schloemer A, Schmithorst VJ, Lorenz JN, Dunn RS, Vorhees CV, Wills-Karp M, Degen JL, Davis RJ, Mizushima N, Rakic P, Dardzinski BJ, Holland SK, Sharp FR, Kuan CY. Cerebral ischemia-hypoxia induces intravascular coagulation and autophagy. The American journal of pathology. 2006;169:566–583. doi: 10.2353/ajpath.2006.051066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ates E, Genc E, Erkasap N, Erkasap S, Akman S, Firat P, Emre S, Kiper H. Renal protection by brief liver ischemia in rats. Transplantation. 2002;74:1247–1251. doi: 10.1097/00007890-200211150-00009. [DOI] [PubMed] [Google Scholar]
- Auchampach JA, Grover GJ, Gross GJ. Blockade of ischaemic preconditioning in dogs by the novel ATP dependent potassium channel antagonist sodium 5-hydroxydecanoate. Cardiovascular research. 1992;26:1054–1062. doi: 10.1093/cvr/26.11.1054. [DOI] [PubMed] [Google Scholar]
- Bacigaluppi M, Comi G, Hermann DM. Animal models of ischemic stroke. Part two: modeling cerebral ischemia. The open neurology journal. 2010;4:34–38. doi: 10.2174/1874205X01004020034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahjat FR, Gesuete R, Stenzel-Poore MP. Steps to translate preconditioning from basic research to the clinic. Translational stroke research. 2013;4:89–103. doi: 10.1007/s12975-012-0223-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balduini W, Carloni S, Buonocore G. Autophagy in hypoxia-ischemia induced brain injury: evidence and speculations. Autophagy. 2009;5:221–223. doi: 10.4161/auto.5.2.7363. [DOI] [PubMed] [Google Scholar]
- Bell KF, Fowler JH, Al-Mubarak B, Horsburgh K, Hardingham GE. Activation of Nrf2-regulated glutathione pathway genes by ischemic preconditioning. Oxidative medicine and cellular longevity. 2011;2011:689524. doi: 10.1155/2011/689524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernaudin M, Marti HH, Roussel S, Divoux D, Nouvelot A, MacKenzie ET, Petit E. A potential role for erythropoietin in focal permanent cerebral ischemia in mice. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 1999;19:643–651. doi: 10.1097/00004647-199906000-00007. [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nature reviews Molecular cell biology. 2003;4:517–529. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- Bi Q, Song Z. The effects of combination anti-inflammatory and anti-oxidation drugs in acute stroke. Zhonghua nei ke za zhi. 2011;50:140–143. [PubMed] [Google Scholar]
- Bickler PE, Fahlman CS, Gray J, McKleroy W. Inositol 1,4,5-triphosphate receptors and NAD(P)H mediate Ca2+ signaling required for hypoxic preconditioning of hippocampal neurons. Neuroscience. 2009;160:51–60. doi: 10.1016/j.neuroscience.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biron CA, Nguyen KB, Pien GC, Cousens LP, Salazar-Mather TP. Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annual review of immunology. 1999;17:189–220. doi: 10.1146/annurev.immunol.17.1.189. [DOI] [PubMed] [Google Scholar]
- Bu Q, Liu X, Zhu Y, Liu Y, Wang Y. w007B protects brain against ischemia-reperfusion injury in rats through inhibiting inflammation, apoptosis and autophagy. Brain research. 2014;1558:100–108. doi: 10.1016/j.brainres.2014.02.034. [DOI] [PubMed] [Google Scholar]
- Burda J, Hrehorovska M, Bonilla LG, Danielisova V, Cizkova D, Burda R, Nemethova M, Fando JL, Salinas M. Role of protein synthesis in the ischemic tolerance acquisition induced by transient forebrain ischemia in the rat. Neurochemical research. 2003;28:1213–1219. doi: 10.1023/a:1024232513106. [DOI] [PubMed] [Google Scholar]
- Carloni S, Buonocore G, Balduini W. Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury. Neurobiology of disease. 2008;32:329–339. doi: 10.1016/j.nbd.2008.07.022. [DOI] [PubMed] [Google Scholar]
- Castillo J, Moro MA, Blanco M, Leira R, Serena J, Lizasoain I, Davalos A. The release of tumor necrosis factor-alpha is associated with ischemic tolerance in human stroke. Annals of neurology. 2003;54:811–819. doi: 10.1002/ana.10765. [DOI] [PubMed] [Google Scholar]
- Chen D, Yu SP, Wei L. Ion channels in regulation of neuronal regenerative activities. Translational stroke research. 2014a;5:156–162. doi: 10.1007/s12975-013-0320-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Qi Z, Luo Y, Hinchliffe T, Ding G, Xia Y, Ji X. Non-pharmaceutical therapies for stroke: mechanisms and clinical implications. Progress in neurobiology. 2014b;115:246–269. doi: 10.1016/j.pneurobio.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JJ, Lin F, Qin ZH. The roles of the proteasome pathway in signal transduction and neurodegenerative diseases. Neuroscience bulletin. 2008;24:183–194. doi: 10.1007/s12264-008-0183-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW, Maulucci-Gedde M, Kriegstein AR. Glutamate neurotoxicity in cortical cell culture. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1987;7:357–368. doi: 10.1523/JNEUROSCI.07-02-00357.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly M, Bilgin-Freiert A, Ellingson B, Dusick JR, Liebeskind D, Saver J, Gonzalez NR. Peripheral vascular disease as remote ischemic preconditioning, for acute stroke. Clinical neurology and neurosurgery. 2013;115:2124–2129. doi: 10.1016/j.clineuro.2013.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielisova V, Nemethova M, Gottlieb M, Burda J. Changes of endogenous antioxidant enzymes during ischemic tolerance acquisition. Neurochemical research. 2005;30:559–565. doi: 10.1007/s11064-005-2690-4. [DOI] [PubMed] [Google Scholar]
- Dave KR, Lange-Asschenfeldt C, Raval AP, Prado R, Busto R, Saul I, Perez-Pinzon MA. Ischemic preconditioning ameliorates excitotoxicity by shifting glutamate/gamma-aminobutyric acid release and biosynthesis. Journal of neuroscience research. 2005;82:665–673. doi: 10.1002/jnr.20674. [DOI] [PubMed] [Google Scholar]
- DeFazio RA, Raval AP, Lin HW, Dave KR, Della-Morte D, Perez-Pinzon MA. GABA synapses mediate neuroprotection after ischemic and epsilonPKC preconditioning in rat hippocampal slice cultures. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2009;29:375–384. doi: 10.1038/jcbfm.2008.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Zoppo GJ, Hallenbeck JM. Advances in the vascular pathophysiology of ischemic stroke. Thrombosis research. 2000;98:73–81. doi: 10.1016/s0049-3848(00)00218-8. [DOI] [PubMed] [Google Scholar]
- del Zoppo GJ, Mabuchi T. Cerebral microvessel responses to focal ischemia. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2003;23:879–894. doi: 10.1097/01.WCB.0000078322.96027.78. [DOI] [PubMed] [Google Scholar]
- Della-Morte D, Dave KR, DeFazio RA, Bao YC, Raval AP, Perez-Pinzon MA. Resveratrol pretreatment protects rat brain from cerebral ischemic damage via a sirtuin 1-uncoupling protein 2 pathway. Neuroscience. 2009;159:993–1002. doi: 10.1016/j.neuroscience.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Della-Morte D, Guadagni F, Palmirotta R, Ferroni P, Testa G, Cacciatore F, Abete P, Rengo F, Perez-Pinzon MA, Sacco RL, Rundek T. Genetics and genomics of ischemic tolerance: focus on cardiac and cerebral ischemic preconditioning. Pharmacogenomics. 2012;13:1741–1757. doi: 10.2217/pgs.12.157. [DOI] [PubMed] [Google Scholar]
- Deng J, Lei C, Chen Y, Fang Z, Yang Q, Zhang H, Cai M, Shi L, Dong H, Xiong L. Neuroprotective gases--fantasy or reality for clinical use? Progress in neurobiology. 2014;115:210–245. doi: 10.1016/j.pneurobio.2014.01.001. [DOI] [PubMed] [Google Scholar]
- Dezfulian C, Garrett M, Gonzalez NR. Clinical application of preconditioning and postconditioning to achieve neuroprotection. Translational stroke research. 2013;4:19–24. doi: 10.1007/s12975-012-0224-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacological reviews. 1999;51:7–61. [PubMed] [Google Scholar]
- Dirnagl U, Becker K, Meisel A. Preconditioning and tolerance against cerebral ischaemia: from experimental strategies to clinical use. The Lancet Neurology. 2009;8:398–412. doi: 10.1016/S1474-4422(09)70054-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirnagl U, Simon RP, Hallenbeck JM. Ischemic tolerance and endogenous neuroprotection. Trends in neurosciences. 2003;26:248–254. doi: 10.1016/S0166-2236(03)00071-7. [DOI] [PubMed] [Google Scholar]
- Douen AG, Akiyama K, Hogan MJ, Wang F, Dong L, Chow AK, Hakim A. Preconditioning with cortical spreading depression decreases intraischemic cerebral glutamate levels and down-regulates excitatory amino acid transporters EAAT1 and EAAT2 from rat cerebal cortex plasma membranes. Journal of neurochemistry. 2000;75:812–818. doi: 10.1046/j.1471-4159.2000.0750812.x. [DOI] [PubMed] [Google Scholar]
- Durukan A, Tatlisumak T. Preconditioning-induced ischemic tolerance: a window into endogenous gearing for cerebroprotection. Experimental & translational stroke medicine. 2010;2:2. doi: 10.1186/2040-7378-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher M, Feuerstein G, Howells DW, Hurn PD, Kent TA, Savitz SI, Lo EH, Group S. Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke; a journal of cerebral circulation. 2009;40:2244–2250. doi: 10.1161/STROKEAHA.108.541128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frassdorf J, Borowski A, Ebel D, Feindt P, Hermes M, Meemann T, Weber R, Mullenheim J, Weber NC, Preckel B, Schlack W. Impact of preconditioning protocol on anesthetic-induced cardioprotection in patients having coronary artery bypass surgery. The Journal of thoracic and cardiovascular surgery. 2009;137:1436–1442. 1442 e1431–1432. doi: 10.1016/j.jtcvs.2008.04.034. [DOI] [PubMed] [Google Scholar]
- Gao B, Zhang XY, Han R, Zhang TT, Chen C, Qin ZH, Sheng R. The endoplasmic reticulum stress inhibitor salubrinal inhibits the activation of autophagy and neuroprotection induced by brain ischemic preconditioning. Acta pharmacologica Sinica. 2013;34:657–666. doi: 10.1038/aps.2013.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Jiang T, Guo J, Liu Y, Cui G, Gu L, Su L, Zhang Y. Inhibition of autophagy contributes to ischemic postconditioning-induced neuroprotection against focal cerebral ischemia in rats. PloS one. 2012;7:e46092. doi: 10.1371/journal.pone.0046092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O’Shea EK, Weissman JS. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- Ghiardi GJ, Gidday JM, Roth S. The purine nucleoside adenosine in retinal ischemia-reperfusion injury. Vision research. 1999;39:2519–2535. doi: 10.1016/s0042-6989(99)00038-3. [DOI] [PubMed] [Google Scholar]
- Gidday JM. Cerebral preconditioning and ischaemic tolerance. Nature reviews Neuroscience. 2006;7:437–448. doi: 10.1038/nrn1927. [DOI] [PubMed] [Google Scholar]
- Ginis I, Jaiswal R, Klimanis D, Liu J, Greenspon J, Hallenbeck JM. TNF-alpha-induced tolerance to ischemic injury involves differential control of NF-kappaB transactivation: the role of NF-kappaB association with p300 adaptor. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2002;22:142–152. doi: 10.1097/00004647-200202000-00002. [DOI] [PubMed] [Google Scholar]
- Globus MY, Ginsberg MD, Busto R. Excitotoxic index--a biochemical marker of selective vulnerability. Neuroscience letters. 1991;127:39–42. doi: 10.1016/0304-3940(91)90889-2. [DOI] [PubMed] [Google Scholar]
- Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Magid D, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, Moy CS, Mussolino ME, Nichol G, Paynter NP, Schreiner PJ, Sorlie PD, Stein J, Turan TN, Virani SS, Wong ND, Woo D, Turner MB American Heart Association Statistics C, Stroke Statistics S. Executive summary: heart disease and stroke statistics--2013 update: a report from the American Heart Association. Circulation. 2013;127:143–152. doi: 10.1161/CIR.0b013e318282ab8f. [DOI] [PubMed] [Google Scholar]
- Gong SJ, Chen LY, Zhang M, Gong JX, Ma YX, Zhang JM, Wang YJ, Hu YY, Sun XC, Li WB, Zhang Y. Intermittent hypobaric hypoxia preconditioning induced brain ischemic tolerance by up-regulating glial glutamate transporter-1 in rats. Neurochemical research. 2012;37:527–537. doi: 10.1007/s11064-011-0639-3. [DOI] [PubMed] [Google Scholar]
- Gonzalez NR, Hamilton R, Bilgin-Freiert A, Dusick J, Vespa P, Hu X, Asgari S. Cerebral hemodynamic and metabolic effects of remote ischemic preconditioning in patients with subarachnoid hemorrhage. Acta neurochirurgica Supplement. 2013;115:193–198. doi: 10.1007/978-3-7091-1192-5_36. [DOI] [PubMed] [Google Scholar]
- Hahn CD, Manlhiot C, Schmidt MR, Nielsen TT, Redington AN. Remote ischemic per-conditioning: a novel therapy for acute stroke? Stroke; a journal of cerebral circulation. 2011;42:2960–2962. doi: 10.1161/STROKEAHA.111.622340. [DOI] [PubMed] [Google Scholar]
- Halkos ME, Kerendi F, Corvera JS, Wang NP, Kin H, Payne CS, Sun HY, Guyton RA, Vinten-Johansen J, Zhao ZQ. Myocardial protection with postconditioning is not enhanced by ischemic preconditioning. The Annals of thoracic surgery. 2004;78:961–969. doi: 10.1016/j.athoracsur.2004.03.033. discussion 969. [DOI] [PubMed] [Google Scholar]
- Hao L, Zou Z, Tian H, Zhang Y, Zhou H, Liu L. Stem cell-based therapies for ischemic stroke. BioMed research international. 2014;2014:468748. doi: 10.1155/2014/468748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa K, Nakano T, Irie K, Higuchi S, Fujioka M, Orito K, Iwasaki K, Jin G, Lo EH, Mishima K, Fujiwara M. Inhibition of reactive astrocytes with fluorocitrate retards neurovascular remodeling and recovery after focal cerebral ischemia in mice. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2010;30:871–882. doi: 10.1038/jcbfm.2009.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes K, Sprague S, Guo M, Davis W, Friedman A, Kumar A, Jimenez DF, Ding Y. Forced, not voluntary, exercise effectively induces neuroprotection in stroke. Acta neuropathologica. 2008;115:289–296. doi: 10.1007/s00401-008-0340-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Z, Crook JE, Meschia JF, Brott TG, Dickson DW, McKinney M. Aging blunts ischemic-preconditioning-induced neuroprotection following transient global ischemia in rats. Current neurovascular research. 2005;2:365–374. doi: 10.2174/156720205774962674. [DOI] [PubMed] [Google Scholar]
- Hebert DN, Molinari M. In and out of the ER: protein folding, quality control, degradation, and related human diseases. Physiological reviews. 2007;87:1377–1408. doi: 10.1152/physrev.00050.2006. [DOI] [PubMed] [Google Scholar]
- Hepponstall M, Ignjatovic V, Binos S, Monagle P, Jones B, Cheung MH, d’Udekem Y, Konstantinov IE. Remote ischemic preconditioning (RIPC) modifies plasma proteome in humans. PloS one. 2012;7:e48284. doi: 10.1371/journal.pone.0048284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoda MN, Bhatia K, Hafez SS, Johnson MH, Siddiqui S, Ergul A, Zaidi SK, Fagan SC, Hess DC. Remote ischemic perconditioning is effective after embolic stroke in ovariectomized female mice. Translational stroke research. 2014;5:484–490. doi: 10.1007/s12975-013-0318-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu BR, Fux CM, Martone ME, Zivin JA, Ellisman MH. Persistent phosphorylation of cyclic AMP responsive element-binding protein and activating transcription factor-2 transcription factors following transient cerebral ischemia in rat brain. Neuroscience. 1999;89:437–452. doi: 10.1016/s0306-4522(98)00352-2. [DOI] [PubMed] [Google Scholar]
- Huang J, Upadhyay UM, Tamargo RJ. Inflammation in stroke and focal cerebral ischemia. Surgical neurology. 2006;66:232–245. doi: 10.1016/j.surneu.2005.12.028. [DOI] [PubMed] [Google Scholar]
- Huber R, Kasischke K, Ludolph AC, Riepe MW. Increase of cellular hypoxic tolerance by erythromycin and other antibiotics. Neuroreport. 1999;10:1543–1546. doi: 10.1097/00001756-199905140-00027. [DOI] [PubMed] [Google Scholar]
- Igarashi Y, Utsumi H, Chiba H, Yamada-Sasamori Y, Tobioka H, Kamimura Y, Furuuchi K, Kokai Y, Nakagawa T, Mori M, Sawada N. Glial cell line-derived neurotrophic factor induces barrier function of endothelial cells forming the blood-brain barrier. Biochemical and biophysical research communications. 1999;261:108–112. doi: 10.1006/bbrc.1999.0992. [DOI] [PubMed] [Google Scholar]
- Janzer RC, Raff MC. Astrocytes induce blood-brain barrier properties in endothelial cells. Nature. 1987;325:253–257. doi: 10.1038/325253a0. [DOI] [PubMed] [Google Scholar]
- Jiang T, Yu JT, Zhu XC, Zhang QQ, Tan MS, Cao L, Wang HF, Shi JQ, Gao L, Qin H, Zhang YD, Tan L. Ischemic Preconditioning Provides Neuroprotection by Induction of AMP-Activated Protein Kinase-Dependent Autophagy in a Rat Model of Ischemic Stroke. Molecular neurobiology. 2014 doi: 10.1007/s12035-014-8725-6. [DOI] [PubMed] [Google Scholar]
- Jneid H, Leessar M, Bolli R. Cardiac preconditioning during percutaneous coronary interventions. Cardiovascular drugs and therapy/sponsored by the International Society of Cardiovascular Pharmacotherapy. 2005;19:211–217. doi: 10.1007/s10557-005-2466-8. [DOI] [PubMed] [Google Scholar]
- Johnson WD, Morrissey RL, Usborne AL, Kapetanovic I, Crowell JA, Muzzio M, McCormick DL. Subchronic oral toxicity and cardiovascular safety pharmacology studies of resveratrol, a naturally occurring polyphenol with cancer preventive activity. Food and chemical toxicology: an international journal published for the British Industrial Biological Research Association. 2011;49:3319–3327. doi: 10.1016/j.fct.2011.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kariko K, Weissman D, Welsh FA. Inhibition of toll-like receptor and cytokine signaling--a unifying theme in ischemic tolerance. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2004;24:1288–1304. doi: 10.1097/01.WCB.0000145666.68576.71. [DOI] [PubMed] [Google Scholar]
- Kato H, Araki T, Kogure K. Preserved neurotransmitter receptor binding following ischemia in preconditioned gerbil brain. Brain research bulletin. 1992a;29:395–400. doi: 10.1016/0361-9230(92)90074-8. [DOI] [PubMed] [Google Scholar]
- Kato H, Araki T, Murase K, Kogure K. Induction of tolerance to ischemia: alterations in second-messenger systems in the gerbil hippocampus. Brain research bulletin. 1992b;29:559–565. doi: 10.1016/0361-9230(92)90123-f. [DOI] [PubMed] [Google Scholar]
- Kato H, Liu Y, Araki T, Kogure K. MK-801, but not anisomycin, inhibits the induction of tolerance to ischemia in the gerbil hippocampus. Neuroscience letters. 1992c;139:118–121. doi: 10.1016/0304-3940(92)90871-4. [DOI] [PubMed] [Google Scholar]
- Keep RF, Wang MM, Xiang J, Hua Y, Xi G. Full steam ahead with remote ischemic conditioning for stroke. Translational stroke research. 2014;5:535–537. doi: 10.1007/s12975-014-0363-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharbanda RK, Mortensen UM, White PA, Kristiansen SB, Schmidt MR, Hoschtitzky JA, Vogel M, Sorensen K, Redington AN, MacAllister R. Transient limb ischemia induces remote ischemic preconditioning in vivo. Circulation. 2002;106:2881–2883. doi: 10.1161/01.cir.0000043806.51912.9b. [DOI] [PubMed] [Google Scholar]
- Kim EJ, Raval AP, Perez-Pinzon MA. Preconditioning mediated by sublethal oxygen-glucose deprivation-induced cyclooxygenase-2 expression via the signal transducers and activators of transcription 3 phosphorylation. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2008;28:1329–1340. doi: 10.1038/jcbfm.2008.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa K, Matsumoto M, Kuwabara K, Tagaya M, Ohtsuki T, Hata R, Ueda H, Handa N, Kimura K, Kamada T. ‘Ischemic tolerance’ phenomenon detected in various brain regions. Brain research. 1991;561:203–211. doi: 10.1016/0006-8993(91)91596-s. [DOI] [PubMed] [Google Scholar]
- Koch S. Preconditioning the human brain: practical considerations for proving cerebral protection. Translational stroke research. 2010;1:161–169. doi: 10.1007/s12975-010-0025-5. [DOI] [PubMed] [Google Scholar]
- Koch S. Moving towards preconditioning for neurological disorders: are we ready for clinical trials? Translational stroke research. 2013;4:15–18. doi: 10.1007/s12975-012-0220-7. [DOI] [PubMed] [Google Scholar]
- Koch S, Della-Morte D, Dave KR, Sacco RL, Perez-Pinzon MA. Biomarkers for ischemic preconditioning: finding the responders. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2014;34:933–941. doi: 10.1038/jcbfm.2014.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch S, Katsnelson M, Dong C, Perez-Pinzon M. Remote ischemic limb preconditioning after subarachnoid hemorrhage: a phase Ib study of safety and feasibility. Stroke; a journal of cerebral circulation. 2011;42:1387–1391. doi: 10.1161/STROKEAHA.110.605840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch S, Perez-Pinzon MA. Proceedings of the 2nd translational preconditioning meeting Miami. Translational stroke research. 2013;4:1–2. doi: 10.1007/s12975-012-0252-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch S, Sacco RL, Perez-Pinzon MA. Preconditioning the brain: moving on to the next frontier of neurotherapeutics. Stroke; a journal of cerebral circulation. 2012;43:1455–1457. doi: 10.1161/STROKEAHA.111.646919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch S, Zuniga S, Rabinstein AA, Romano JG, Nolan B, Chirinos J, Forteza A. Signs and symptoms of sleep apnea and acute stroke severity: is sleep apnea neuroprotective? Journal of stroke and cerebrovascular diseases: the official journal of National Stroke Association. 2007;16:114–118. doi: 10.1016/j.jstrokecerebrovasdis.2007.01.002. [DOI] [PubMed] [Google Scholar]
- Koike M, Shibata M, Tadakoshi M, Gotoh K, Komatsu M, Waguri S, Kawahara N, Kuida K, Nagata S, Kominami E, Tanaka K, Uchiyama Y. Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. The American journal of pathology. 2008;172:454–469. doi: 10.2353/ajpath.2008.070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo T, Kinouchi H, Kawase M, Yoshimoto T. Astroglial cells inhibit the increasing permeability of brain endothelial cell monolayer following hypoxia/reoxygenation. Neuroscience letters. 1996;208:101–104. doi: 10.1016/0304-3940(96)12555-6. [DOI] [PubMed] [Google Scholar]
- Kristian T, Siesjo BK. Calcium in ischemic cell death. Stroke; a journal of cerebral circulation. 1998;29:705–718. doi: 10.1161/01.str.29.3.705. [DOI] [PubMed] [Google Scholar]
- Kunze A, Zierath D, Drogomiretskiy O, Becker K. Variation in behavioral deficits and patterns of recovery after stroke among different rat strains. Translational stroke research. 2014;5:569–576. doi: 10.1007/s12975-014-0337-y. [DOI] [PubMed] [Google Scholar]
- Lai TW, Zhang S, Wang YT. Excitotoxicity and stroke: identifying novel targets for neuroprotection. Progress in neurobiology. 2014;115:157–188. doi: 10.1016/j.pneurobio.2013.11.006. [DOI] [PubMed] [Google Scholar]
- Lalancette-Hebert M, Swarup V, Beaulieu JM, Bohacek I, Abdelhamid E, Weng YC, Sato S, Kriz J. Galectin-3 is required for resident microglia activation and proliferation in response to ischemic injury. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2012;32:10383–10395. doi: 10.1523/JNEUROSCI.1498-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange-Asschenfeldt C, Raval AP, Dave KR, Mochly-Rosen D, Sick TJ, Perez-Pinzon MA. Epsilon protein kinase C mediated ischemic tolerance requires activation of the extracellular regulated kinase pathway in the organotypic hippocampal slice. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2004;24:636–645. doi: 10.1097/01.WCB.0000121235.42748.BF. [DOI] [PubMed] [Google Scholar]
- Lavignon M, Tounekti N, Rayner B, Imbach JL, Keith G, Paoletti J, Malvy C. Inhibition of murine leukemia viruses by nuclease-resistant alpha-oligonucleotides. Antisense research and development. 1992;2:315–324. doi: 10.1089/ard.1992.2.315. [DOI] [PubMed] [Google Scholar]
- Lee SR, Choi B, Paul S, Seo JH, Back DB, Han JS, Choi DH, Kwon KJ, Shin CY, Lee J, Han SH, Kim HY. Depressive-Like Behaviors in a Rat Model of Chronic Cerebral Hypoperfusion. Translational stroke research. 2014 doi: 10.1007/s12975-014-0385-3. [DOI] [PubMed] [Google Scholar]
- Lehotsky J, Burda J, Danielisova V, Gottlieb M, Kaplan P, Saniova B. Ischemic tolerance: the mechanisms of neuroprotective strategy. Anatomical record. 2009;292:2002–2012. doi: 10.1002/ar.20970. [DOI] [PubMed] [Google Scholar]
- Lehotsky J, Kaplan P, Matejovicova M, Murin R, Racay P, Raeymaekers L. Ion transport systems as targets of free radicals during ischemia reperfusion injury. General physiology and biophysics. 2002a;21:31–37. [PubMed] [Google Scholar]
- Lehotsky J, Kaplan P, Murin R, Raeymaekers L. The role of plasma membrane Ca2+ pumps (PMCAs) in pathologies of mammalian cells. Frontiers in bioscience: a journal and virtual library. 2002b;7:d53–84. doi: 10.2741/A769. [DOI] [PubMed] [Google Scholar]
- Lehotsky J, Murin R, Strapkova A, Urikova A, Tatarkova Z, Kaplan P. Time course of ischemia/reperfusion-induced oxidative modification of neural proteins in rat forebrain. General physiology and biophysics. 2004;23:401–415. [PubMed] [Google Scholar]
- Leng T, Shi Y, Xiong ZG, Sun D. Proton-sensitive cation channels and ion exchangers in ischemic brain injury: new therapeutic targets for stroke? Progress in neurobiology. 2014;115:189–209. doi: 10.1016/j.pneurobio.2013.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Luo Y, Zhang F, Signore AP, Gobbel GT, Simon RP, Chen J. Ischemic preconditioning in the rat brain enhances the repair of endogenous oxidative DNA damage by activating the base-excision repair pathway. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2006;26:181–198. doi: 10.1038/sj.jcbfm.9600180. [DOI] [PubMed] [Google Scholar]
- Lin HW, Thompson JW, Morris KC, Perez-Pinzon MA. Signal transducers and activators of transcription: STATs-mediated mitochondrial neuroprotection. Antioxidants & redox signaling. 2011;14:1853–1861. doi: 10.1089/ars.2010.3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton P. Ischemic cell death in brain neurons. Physiological reviews. 1999;79:1431–1568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- Liu AJ, Hu YY, Li WB, Xu J, Zhang M. Cerebral ischemic pre-conditioning enhances the binding characteristics and glutamate uptake of glial glutamate transporter-1 in hippocampal CA1 subfield of rats. Journal of neurochemistry. 2011;119:202–209. doi: 10.1111/j.1471-4159.2011.07396.x. [DOI] [PubMed] [Google Scholar]
- Liu C, Chen S, Kamme F, Hu BR. Ischemic preconditioning prevents protein aggregation after transient cerebral ischemia. Neuroscience. 2005;134:69–80. doi: 10.1016/j.neuroscience.2005.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Ginis I, Spatz M, Hallenbeck JM. Hypoxic preconditioning protects cultured neurons against hypoxic stress via TNF-alpha and ceramide. American journal of physiology Cell physiology. 2000;278:C144–153. doi: 10.1152/ajpcell.2000.278.1.C144. [DOI] [PubMed] [Google Scholar]
- Liu J, Wang Y, Akamatsu Y, Lee CC, Stetler RA, Lawton MT, Yang GY. Vascular remodeling after ischemic stroke: mechanisms and therapeutic potentials. Progress in neurobiology. 2014a;115:138–156. doi: 10.1016/j.pneurobio.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Chen H, Zhan B, Xing B, Zhou J, Zhu H, Chen Z. Attenuation of reperfusion injury by renal ischemic postconditioning: the role of NO. Biochemical and biophysical research communications. 2007;359:628–634. doi: 10.1016/j.bbrc.2007.05.129. [DOI] [PubMed] [Google Scholar]
- Liu X, Zhao S, Liu F, Kang J, Xiao A, Li F, Zhang C, Yan F, Zhao H, Luo M, Luo Y, Ji X. Remote ischemic postconditioning alleviates cerebral ischemic injury by attenuating endoplasmic reticulum stress-mediated apoptosis. Translational stroke research. 2014b;5:692–700. doi: 10.1007/s12975-014-0359-5. [DOI] [PubMed] [Google Scholar]
- Liu XQ, Sheng R, Qin ZH. The neuroprotective mechanism of brain ischemic preconditioning. Acta pharmacologica Sinica. 2009;30:1071–1080. doi: 10.1038/aps.2009.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Xiong L, Chen S, Wang Q. Isoflurane tolerance against focal cerebral ischemia is attenuated by adenosine A1 receptor antagonists. Canadian journal of anaesthesia = Journal canadien d’anesthesie. 2006;53:194–201. doi: 10.1007/BF03021827. [DOI] [PubMed] [Google Scholar]
- Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G, Ferguson TB, Ford E, Furie K, Gillespie C, Go A, Greenlund K, Haase N, Hailpern S, Ho PM, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, McDermott MM, Meigs J, Mozaffarian D, Mussolino M, Nichol G, Roger VL, Rosamond W, Sacco R, Sorlie P, Stafford R, Thom T, Wasserthiel-Smoller S, Wong ND, Wylie-Rosett J American Heart Association Statistics C, Stroke Statistics S. Executive summary: heart disease and stroke statistics--2010 update: a report from the American Heart Association. Circulation. 2010;121:948–954. doi: 10.1161/CIRCULATIONAHA.109.192666. [DOI] [PubMed] [Google Scholar]
- Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nature reviews Neuroscience. 2003;4:399–415. doi: 10.1038/nrn1106. [DOI] [PubMed] [Google Scholar]
- Lorgis L, Gudjoncik A, Richard C, Mock L, Buffet P, Brunel P, Janin-Manificat L, Beer JC, Brunet D, Touzery C, Rochette L, Cottin Y, Zeller M. Pre-infarction angina and outcomes in non-ST-segment elevation myocardial infarction: data from the RICO survey. PloS one. 2012;7:e48513. doi: 10.1371/journal.pone.0048513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo T, Park Y, Sun X, Liu C, Hu B. Protein misfolding, aggregation, and autophagy after brain ischemia. Translational stroke research. 2013;4:581–588. doi: 10.1007/s12975-013-0299-5. [DOI] [PubMed] [Google Scholar]
- Ma J, You C, Hao L. Iron chelators for acute stroke. The Cochrane database of systematic reviews. 2012;9:CD009280. doi: 10.1002/14651858.CD009280.pub2. [DOI] [PubMed] [Google Scholar]
- Maag RS, Hicks SW, Machamer CE. Death from within: apoptosis and the secretory pathway. Current opinion in cell biology. 2003;15:456–461. doi: 10.1016/s0955-0674(03)00075-9. [DOI] [PubMed] [Google Scholar]
- Mangoni AA, Woodman RJ, Gilbert AL, Knights KM. Use of non-steroidal anti-inflammatory drugs and risk of ischemic and hemorrhagic stroke in the Australian veteran community. Pharmacoepidemiology and drug safety. 2010;19:490–498. doi: 10.1002/pds.1945. [DOI] [PubMed] [Google Scholar]
- Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circulation research. 2007;100:914–922. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- Meller R, Simon RP. Tolerance to ischemia - an increasingly complex biology. Translational stroke research. 2013;4:40–50. doi: 10.1007/s12975-012-0246-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng R, Asmaro K, Meng L, Liu Y, Ma C, Xi C, Li G, Ren C, Luo Y, Ling F, Jia J, Hua Y, Wang X, Ding Y, Lo EH, Ji X. Upper limb ischemic preconditioning prevents recurrent stroke in intracranial arterial stenosis. Neurology. 2012;79:1853–1861. doi: 10.1212/WNL.0b013e318271f76a. [DOI] [PubMed] [Google Scholar]
- Mergenthaler P, Dirnagl U. Protective conditioning of the brain: expressway or roadblock? The Journal of physiology. 2011;589:4147–4155. doi: 10.1113/jphysiol.2011.209718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moncayo J, de Freitas GR, Bogousslavsky J, Altieri M, van Melle G. Do transient ischemic attacks have a neuroprotective effect? Neurology. 2000;54:2089–2094. doi: 10.1212/wnl.54.11.2089. [DOI] [PubMed] [Google Scholar]
- Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- Nakajima T, Iwabuchi S, Miyazaki H, Okuma Y, Inanami O, Kuwabara M, Nomura Y, Kawahara K. Relationship between the activation of cyclic AMP responsive element binding protein and ischemic tolerance in the penumbra region of rat cerebral cortex. Neuroscience letters. 2002;331:13–16. doi: 10.1016/s0304-3940(02)00752-8. [DOI] [PubMed] [Google Scholar]
- Nakajima T, Iwabuchi S, Miyazaki H, Okuma Y, Kuwabara M, Nomura Y, Kawahara K. Preconditioning prevents ischemia-induced neuronal death through persistent Akt activation in the penumbra region of the rat brain. Journal of Veterinary Medical Science. 2004;66:521–527. doi: 10.1292/jvms.66.521. [DOI] [PubMed] [Google Scholar]
- Napoli I, Neumann H. Microglial clearance function in health and disease. Neuroscience. 2009;158:1030–1038. doi: 10.1016/j.neuroscience.2008.06.046. [DOI] [PubMed] [Google Scholar]
- Narayanan SV, Dave KR, Perez-Pinzon MA. Ischemic preconditioning and clinical scenarios. Current opinion in neurology. 2013;26:1–7. doi: 10.1097/WCO.0b013e32835bf200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawashiro H, Tasaki K, Ruetzler CA, Hallenbeck JM. TNF-alpha pretreatment induces protective effects against focal cerebral ischemia in mice. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 1997;17:483–490. doi: 10.1097/00004647-199705000-00001. [DOI] [PubMed] [Google Scholar]
- Nepal S, Park PH. Activation of autophagy by globular adiponectin attenuates ethanol-induced apoptosis in HepG2 cells: involvement of AMPK/FoxO3A axis. Biochimica et biophysica acta. 2013;1833:2111–2125. doi: 10.1016/j.bbamcr.2013.05.013. [DOI] [PubMed] [Google Scholar]
- Oberheim NA, Wang X, Goldman S, Nedergaard M. Astrocytic complexity distinguishes the human brain. Trends in neurosciences. 2006;29:547–553. doi: 10.1016/j.tins.2006.08.004. [DOI] [PubMed] [Google Scholar]
- Obrenovitch TP. Molecular physiology of preconditioning-induced brain tolerance to ischemia. Physiological reviews. 2008;88:211–247. doi: 10.1152/physrev.00039.2006. [DOI] [PubMed] [Google Scholar]
- Ohtsuki T, Ruetzler CA, Tasaki K, Hallenbeck JM. Interleukin-1 mediates induction of tolerance to global ischemia in gerbil hippocampal CA1 neurons. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 1996;16:1137–1142. doi: 10.1097/00004647-199611000-00007. [DOI] [PubMed] [Google Scholar]
- Otani H. Ischemic preconditioning: from molecular mechanisms to therapeutic opportunities. Antioxidants & redox signaling. 2008;10:207–247. doi: 10.1089/ars.2007.1679. [DOI] [PubMed] [Google Scholar]
- Ovbiagele B, Goldstein LB, Higashida RT, Howard VJ, Johnston SC, Khavjou OA, Lackland DT, Lichtman JH, Mohl S, Sacco RL, Saver JL, Trogdon JG American Heart Association Advocacy Coordinating C, Stroke C. Forecasting the future of stroke in the United States: a policy statement from the American Heart Association and American Stroke Association. Stroke; a journal of cerebral circulation. 2013;44:2361–2375. doi: 10.1161/STR.0b013e31829734f2. [DOI] [PubMed] [Google Scholar]
- Park JA, Choi KS, Kim SY, Kim KW. Coordinated interaction of the vascular and nervous systems: from molecule- to cell-based approaches. Biochemical and biophysical research communications. 2003;311:247–253. doi: 10.1016/j.bbrc.2003.09.129. [DOI] [PubMed] [Google Scholar]
- Pauly M, Daussin F, Burelle Y, Li T, Godin R, Fauconnier J, Koechlin-Ramonatxo C, Hugon G, Lacampagne A, Coisy-Quivy M, Liang F, Hussain S, Matecki S, Petrof BJ. AMPK activation stimulates autophagy and ameliorates muscular dystrophy in the mdx mouse diaphragm. The American journal of pathology. 2012;181:583–592. doi: 10.1016/j.ajpath.2012.04.004. [DOI] [PubMed] [Google Scholar]
- Pavlikova M, Tatarkova Z, Sivonova M, Kaplan P, Krizanova O, Lehotsky J. Alterations induced by ischemic preconditioning on secretory pathways Ca2+-ATPase (SPCA) gene expression and oxidative damage after global cerebral ischemia/reperfusion in rats. Cellular and molecular neurobiology. 2009;29:909–916. doi: 10.1007/s10571-009-9374-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Pinzon MA. Mechanisms of neuroprotection during ischemic preconditioning: lessons from anoxic tolerance. Comparative biochemistry and physiology Part A, Molecular & integrative physiology. 2007;147:291–299. doi: 10.1016/j.cbpa.2006.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Pinzon MA, Born JG. Rapid preconditioning neuroprotection following anoxia in hippocampal slices: role of the K+ ATP channel and protein kinase C. Neuroscience. 1999;89:453–459. doi: 10.1016/s0306-4522(98)00560-0. [DOI] [PubMed] [Google Scholar]
- Perez-Pinzon MA, Mumford PL, Rosenthal M, Sick TJ. Anoxic preconditioning in hippocampal slices: role of adenosine. Neuroscience. 1996;75:687–694. doi: 10.1016/0306-4522(96)00311-9. [DOI] [PubMed] [Google Scholar]
- Perez-Pinzon MA, Stetler RA, Fiskum G. Novel mitochondrial targets for neuroprotection. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2012;32:1362–1376. doi: 10.1038/jcbfm.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Pinzon MA, Xu GP, Dietrich WD, Rosenthal M, Sick TJ. Rapid preconditioning protects rats against ischemic neuronal damage after 3 but not 7 days of reperfusion following global cerebral ischemia. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 1997;17:175–182. doi: 10.1097/00004647-199702000-00007. [DOI] [PubMed] [Google Scholar]
- Petcu EB, Kocher T, Kuhr A, Buga AM, Kloting I, Herndon JG, Kessler C, Popa-Wagner A. Mild systemic inflammation has a neuroprotective effect after stroke in rats. Current neurovascular research. 2008;5:214–223. doi: 10.2174/156720208786413424. [DOI] [PubMed] [Google Scholar]
- Petegnief V, Planas AM. SIRT1 regulation modulates stroke outcome. Translational stroke research. 2013;4:663–671. doi: 10.1007/s12975-013-0277-y. [DOI] [PubMed] [Google Scholar]
- Petty MA, Lo EH. Junctional complexes of the blood-brain barrier: permeability changes in neuroinflammation. Progress in neurobiology. 2002;68:311–323. doi: 10.1016/s0301-0082(02)00128-4. [DOI] [PubMed] [Google Scholar]
- Pignataro G, Meller R, Inoue K, Ordonez AN, Ashley MD, Xiong Z, Gala R, Simon RP. In vivo and in vitro characterization of a novel neuroprotective strategy for stroke: ischemic postconditioning. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2008;28:232–241. doi: 10.1038/sj.jcbfm.9600559. [DOI] [PubMed] [Google Scholar]
- Pignataro G, Sirabella R, Anzilotti S, Di Renzo G, Annunziato L. Does Na(+)/Ca(2)(+) exchanger, NCX, represent a new druggable target in stroke intervention? Translational stroke research. 2014;5:145–155. doi: 10.1007/s12975-013-0308-8. [DOI] [PubMed] [Google Scholar]
- Plamondon H, Blondeau N, Heurteaux C, Lazdunski M. Mutually protective actions of kainic acid epileptic preconditioning and sublethal global ischemia on hippocampal neuronal death: involvement of adenosine A1 receptors and K(ATP) channels. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 1999;19:1296–1308. doi: 10.1097/00004647-199912000-00002. [DOI] [PubMed] [Google Scholar]
- Quinn R. Comparing rat’s to human’s age: how old is my rat in people years? Nutrition. 2005;21:775–777. doi: 10.1016/j.nut.2005.04.002. [DOI] [PubMed] [Google Scholar]
- Rabinovich GA, Toscano MA. Turning ‘sweet’ on immunity: galectin-glycan interactions in immune tolerance and inflammation. Nature reviews Immunology. 2009;9:338–352. doi: 10.1038/nri2536. [DOI] [PubMed] [Google Scholar]
- Racay P, Chomova M, Tatarkova Z, Kaplan P, Hatok J, Dobrota D. Ischemia-induced mitochondrial apoptosis is significantly attenuated by ischemic preconditioning. Cellular and molecular neurobiology. 2009;29:901–908. doi: 10.1007/s10571-009-9373-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racay P, Tatarkova Z, Drgova A, Kaplan P, Dobrota D. Effect of ischemic preconditioning on mitochondrial dysfunction and mitochondrial p53 translocation after transient global cerebral ischemia in rats. Neurochemical research. 2007;32:1823–1832. doi: 10.1007/s11064-007-9437-3. [DOI] [PubMed] [Google Scholar]
- Rami A, Langhagen A, Steiger S. Focal cerebral ischemia induces upregulation of Beclin 1 and autophagy-like cell death. Neurobiology of disease. 2008;29:132–141. doi: 10.1016/j.nbd.2007.08.005. [DOI] [PubMed] [Google Scholar]
- Ramzy D, Rao V, Weisel RD. Clinical applicability of preconditioning and postconditioning: the cardiothoracic surgeons’s view. Cardiovascular research. 2006;70:174–180. doi: 10.1016/j.cardiores.2006.01.020. [DOI] [PubMed] [Google Scholar]
- Raval AP, Dave KR, Mochly-Rosen D, Sick TJ, Perez-Pinzon MA. Epsilon PKC is required for the induction of tolerance by ischemic and NMDA-mediated preconditioning in the organotypic hippocampal slice. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2003;23:384–391. doi: 10.1523/JNEUROSCI.23-02-00384.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehni AK, Singh N. Role of phosphoinositide 3-kinase in ischemic postconditioning-induced attenuation of cerebral ischemia-evoked behavioral deficits in mice. Pharmacological reports: PR. 2007;59:192–198. [PubMed] [Google Scholar]
- Reshef A, Sperling O, Zoref-Shani E. Preconditioning of primary rat neuronal cultures against ischemic injury: characterization of the “time window of protection’. Brain research. 1996;741:252–257. doi: 10.1016/s0006-8993(96)00939-0. [DOI] [PubMed] [Google Scholar]
- Riepe MW, Kasischke K, Raupach A. Acetylsalicylic acid increases tolerance against hypoxic and chemical hypoxia. Stroke; a journal of cerebral circulation. 1997;28:2006–2011. doi: 10.1161/01.str.28.10.2006. [DOI] [PubMed] [Google Scholar]
- Romera C, Hurtado O, Mallolas J, Pereira MP, Morales JR, Romera A, Serena J, Vivancos J, Nombela F, Lorenzo P, Lizasoain I, Moro MA. Ischemic preconditioning reveals that GLT1/EAAT2 glutamate transporter is a novel PPARgamma target gene involved in neuroprotection. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2007;27:1327–1338. doi: 10.1038/sj.jcbfm.9600438. [DOI] [PubMed] [Google Scholar]
- Rotshenker S. Microglia and macrophage activation and the regulation of complement-receptor-3 (CR3/MAC-1)-mediated myelin phagocytosis in injury and disease. Journal of molecular neuroscience: MN. 2003;21:65–72. doi: 10.1385/JMN:21:1:65. [DOI] [PubMed] [Google Scholar]
- Satoh K, Niwa M, Goda W, Binh NH, Nakashima M, Takamatsu M, Hara A. Galectin-3 expression in delayed neuronal death of hippocampal CA1 following transient forebrain ischemia, and its inhibition by hypothermia. Brain research. 2011;1382:266–274. doi: 10.1016/j.brainres.2011.01.049. [DOI] [PubMed] [Google Scholar]
- Schultz JE, Qian YZ, Gross GJ, Kukreja RC. The ischemia-selective KATP channel antagonist, 5-hydroxydecanoate, blocks ischemic preconditioning in the rat heart. Journal of molecular and cellular cardiology. 1997;29:1055–1060. doi: 10.1006/jmcc.1996.0358. [DOI] [PubMed] [Google Scholar]
- Schurr A, Reid KH, Tseng MT, West C, Rigor BM. Adaptation of adult brain tissue to anoxia and hypoxia in vitro. Brain research. 1986;374:244–248. doi: 10.1016/0006-8993(86)90418-x. [DOI] [PubMed] [Google Scholar]
- Schwartz Longacre L, Kloner RA, Arai AE, Baines CP, Bolli R, Braunwald E, Downey J, Gibbons RJ, Gottlieb RA, Heusch G, Jennings RB, Lefer DJ, Mentzer RM, Murphy E, Ovize M, Ping P, Przyklenk K, Sack MN, Vander Heide RS, Vinten-Johansen J, Yellon DM National Heart L, Blood Institute N.I.o.H. New horizons in cardioprotection: recommendations from the 2010 National Heart, Lung, and Blood Institute Workshop. Circulation. 2011;124:1172–1179. doi: 10.1161/CIRCULATIONAHA.111.032698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert HA, Pennypacker KR. Molecular and cellular immune responses to ischemic brain injury. Translational stroke research. 2014;5:543–553. doi: 10.1007/s12975-014-0349-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepulveda MR, Marcos D, Berrocal M, Raeymaekers L, Mata AM, Wuytack F. Activity and localization of the secretory pathway Ca2+-ATPase isoform 1 (SPCA1) in different areas of the mouse brain during postnatal development. Molecular and cellular neurosciences. 2008;38:461–473. doi: 10.1016/j.mcn.2008.02.012. [DOI] [PubMed] [Google Scholar]
- Shah NH, Aizenman E. Voltage-gated potassium channels at the crossroads of neuronal function, ischemic tolerance, and neurodegeneration. Translational stroke research. 2014;5:38–58. doi: 10.1007/s12975-013-0297-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao B, Cheng Y, Jin K. Estrogen, neuroprotection and neurogenesis after ischemic stroke. Current drug targets. 2012;13:188–198. doi: 10.2174/138945012799201702. [DOI] [PubMed] [Google Scholar]
- Sheng R, Liu XQ, Zhang LS, Gao B, Han R, Wu YQ, Zhang XY, Qin ZH. Autophagy regulates endoplasmic reticulum stress in ischemic preconditioning. Autophagy. 2012;8:310–325. doi: 10.4161/auto.18673. [DOI] [PubMed] [Google Scholar]
- Sheng R, Zhang LS, Han R, Liu XQ, Gao B, Qin ZH. Autophagy activation is associated with neuroprotection in a rat model of focal cerebral ischemic preconditioning. Autophagy. 2010;6:482–494. doi: 10.4161/auto.6.4.11737. [DOI] [PubMed] [Google Scholar]
- Sommer C, Fahrner A, Kiessling M. [3H]muscimol binding to gamma-aminobutyric acid(A) receptors is upregulated in CA1 neurons of the gerbil hippocampus in the ischemia-tolerant state. Stroke; a journal of cerebral circulation. 2002;33:1698–1705. doi: 10.1161/01.str.0000016404.14407.77. [DOI] [PubMed] [Google Scholar]
- Sommer C, Fahrner A, Kiessling M. Postischemic neuroprotection in the ischemia-tolerant state gerbil hippocampus is associated with increased ligand binding to inhibitory GABA(A) receptors. Acta neuropathologica. 2003;105:197–202. doi: 10.1007/s00401-002-0632-7. [DOI] [PubMed] [Google Scholar]
- Sommer C, Gass P, Kiessling M. Selective c-JUN expression in CA1 neurons of the gerbil hippocampus during and after acquisition of an ischemia-tolerant state. Brain pathology. 1995;5:135–144. doi: 10.1111/j.1750-3639.1995.tb00587.x. [DOI] [PubMed] [Google Scholar]
- Stenzel-Poore MP, Stevens SL, King JS, Simon RP. Preconditioning reprograms the response to ischemic injury and primes the emergence of unique endogenous neuroprotective phenotypes: a speculative synthesis. Stroke; a journal of cerebral circulation. 2007;38:680–685. doi: 10.1161/01.STR.0000251444.56487.4c. [DOI] [PubMed] [Google Scholar]
- Stroev SA, Gluschenko TS, Tjulkova EI, Spyrou G, Rybnikova EA, Samoilov MO, Pelto-Huikko M. Preconditioning enhances the expression of mitochondrial antioxidant thioredoxin-2 in the forebrain of rats exposed to severe hypobaric hypoxia. Journal of neuroscience research. 2004;78:563–569. doi: 10.1002/jnr.20282. [DOI] [PubMed] [Google Scholar]
- Stroev SA, Tyul’kova EI, Glushchenko TS, Tugoi IA, Samoilov MO, Pelto-Huikko M. Thioredoxin-1 expression levels in rat hippocampal neurons in moderate hypobaric hypoxia. Neuroscience and behavioral physiology. 2009;39:1–5. doi: 10.1007/s11055-008-9091-5. [DOI] [PubMed] [Google Scholar]
- Sun HS, Feng ZP. Neuroprotective role of ATP-sensitive potassium channels in cerebral ischemia. Acta pharmacologica Sinica. 2013;34:24–32. doi: 10.1038/aps.2012.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Weihrauch D, Ludwig LM, Kersten JR, Pagel PS, Warltier DC. Mitochondrial adenosine triphosphate-regulated potassium channel opening acts as a trigger for isoflurane-induced preconditioning by generating reactive oxygen species. Anesthesiology. 2003;98:935–943. doi: 10.1097/00000542-200304000-00021. [DOI] [PubMed] [Google Scholar]
- Tang LH, Xia ZY, Zhao B, Wei XD, Luo T, Meng QT. Phosphocreatine preconditioning attenuates apoptosis in ischemia-reperfusion injury of rat brain. Journal of biomedicine & biotechnology. 2011;2011:107091. doi: 10.1155/2011/107091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewari A, Mahendru V, Sinha A, Bilotta F. Antioxidants: The new frontier for translational research in cerebroprotection. Journal of anaesthesiology, clinical pharmacology. 2014;30:160–171. doi: 10.4103/0970-9185.130001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban P, Pavlikova M, Sivonova M, Kaplan P, Tatarkova Z, Kaminska B, Lehotsky J. Molecular analysis of endoplasmic reticulum stress response after global forebrain ischemia/reperfusion in rats: effect of neuroprotectant simvastatin. Cellular and molecular neurobiology. 2009;29:181–192. doi: 10.1007/s10571-008-9309-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urikova A, Babusikova E, Dobrota D, Drgova A, Kaplan P, Tatarkova Z, Lehotsky J. Impact of Ginkgo Biloba Extract EGb 761 on ischemia/reperfusion - induced oxidative stress products formation in rat forebrain. Cellular and molecular neurobiology. 2006;26:1343–1353. doi: 10.1007/s10571-006-9030-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinten-Johansen J, Shi W. Perconditioning and postconditioning: current knowledge, knowledge gaps, barriers to adoption, and future directions. Journal of cardiovascular pharmacology and therapeutics. 2011;16:260–266. doi: 10.1177/1074248411415270. [DOI] [PubMed] [Google Scholar]
- Voellmy R. Transcriptional regulation of the metazoan stress protein response. Progress in nucleic acid research and molecular biology. 2004;78:143–185. doi: 10.1016/S0079-6603(04)78004-6. [DOI] [PubMed] [Google Scholar]
- Wali B, Ishrat T, Won S, Stein DG, Sayeed I. Progesterone in experimental permanent stroke: a dose-response and therapeutic time-window study. Brain: a journal of neurology. 2014;137:486–502. doi: 10.1093/brain/awt319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LT, Chen BL, Wu CT, Huang KH, Chiang CK, Hwa Liu S. Protective role of AMP-activated protein kinase-evoked autophagy on an in vitro model of ischemia/reperfusion-induced renal tubular cell injury. PloS one. 2013a;8:e79814. doi: 10.1371/journal.pone.0079814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang MM, Xi G, Keep RF. Should the STAIR criteria be modified for preconditioning studies? Translational stroke research. 2013b;4:3–14. doi: 10.1007/s12975-012-0219-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhang M, Yang SD, Li WB, Ren SQ, Zhang J, Zhang F. Pre-ischemic treadmill training alleviates brain damage via GLT-1-mediated signal pathway after ischemic stroke in rats. Neuroscience. 2014;274:393–402. doi: 10.1016/j.neuroscience.2014.05.053. [DOI] [PubMed] [Google Scholar]
- Warzecha Z, Dembinski A, Ceranowicz P, Dembinski M, Cieszkowski J, Kusnierz-Cabala B, Naskalski JW, Jaworek J, Konturek SJ, Pawlik WW, Tomaszewska R. Influence of ischemic preconditioning on blood coagulation, fibrinolytic activity and pancreatic repair in the course of caerulein-induced acute pancreatitis in rats. Journal of physiology and pharmacology: an official journal of the Polish Physiological Society. 2007;58:303–319. [PubMed] [Google Scholar]
- Wegener S, Gottschalk B, Jovanovic V, Knab R, Fiebach JB, Schellinger PD, Kucinski T, Jungehulsing GJ, Brunecker P, Muller B, Banasik A, Amberger N, Wernecke KD, Siebler M, Rother J, Villringer A, Weih M, Stroke MRIiASSGotGCN. Transient ischemic attacks before ischemic stroke: preconditioning the human brain? A multicenter magnetic resonance imaging study. Stroke; a journal of cerebral circulation. 2004;35:616–621. doi: 10.1161/01.STR.0000115767.17923.6A. [DOI] [PubMed] [Google Scholar]
- Wei K, Wang P, Miao CY. A double-edged sword with therapeutic potential: an updated role of autophagy in ischemic cerebral injury. CNS neuroscience & therapeutics. 2012;18:879–886. doi: 10.1111/cns.12005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei N, Yu SP, Gu XH, Chen DD, Whalin MK, Xu GL, Liu XF, Wei L. The involvement of autophagy pathway in exaggerated ischemic brain damage in diabetic mice. CNS neuroscience & therapeutics. 2013;19:753–763. doi: 10.1111/cns.12123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen YD, Sheng R, Zhang LS, Han R, Zhang X, Zhang XD, Han F, Fukunaga K, Qin ZH. Neuronal injury in rat model of permanent focal cerebral ischemia is associated with activation of autophagic and lysosomal pathways. Autophagy. 2008;4:762–769. doi: 10.4161/auto.6412. [DOI] [PubMed] [Google Scholar]
- Wick A, Wick W, Waltenberger J, Weller M, Dichgans J, Schulz JB. Neuroprotection by hypoxic preconditioning requires sequential activation of vascular endothelial growth factor receptor and Akt. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2002;22:6401–6407. doi: 10.1523/JNEUROSCI.22-15-06401.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis CL, Nolan CC, Reith SN, Lister T, Prior MJ, Guerin CJ, Mavroudis G, Ray DE. Focal astrocyte loss is followed by microvascular damage, with subsequent repair of the blood-brain barrier in the apparent absence of direct astrocytic contact. Glia. 2004;45:325–337. doi: 10.1002/glia.10333. [DOI] [PubMed] [Google Scholar]
- Wootton LL, Argent CC, Wheatley M, Michelangeli F. The expression, activity and localisation of the secretory pathway Ca2+-ATPase (SPCA1) in different mammalian tissues. Biochimica et biophysica acta. 2004;1664:189–197. doi: 10.1016/j.bbamem.2004.05.009. [DOI] [PubMed] [Google Scholar]
- Wu JC, Liang ZQ, Qin ZH. Quality control system of the endoplasmic reticulum and related diseases. Acta biochimica et biophysica Sinica. 2006;38:219–226. doi: 10.1111/j.1745-7270.2006.00156.x. [DOI] [PubMed] [Google Scholar]
- Xie Z, Lau K, Eby B, Lozano P, He C, Pennington B, Li H, Rathi S, Dong Y, Tian R, Kem D, Zou MH. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes. 2011;60:1770–1778. doi: 10.2337/db10-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokobori S, Mazzeo AT, Hosein K, Gajavelli S, Dietrich WD, Bullock MR. Preconditioning for traumatic brain injury. Translational stroke research. 2013;4:25–39. doi: 10.1007/s12975-012-0226-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Yankner BA. Apoptosis in the nervous system. Nature. 2000;407:802–809. doi: 10.1038/35037739. [DOI] [PubMed] [Google Scholar]
- Zaouali MA, Boncompagni E, Reiter RJ, Bejaoui M, Freitas I, Pantazi E, Folch-Puy E, Abdennebi HB, Garcia-Gil FA, Rosello-Catafau J. AMPK involvement in endoplasmic reticulum stress and autophagy modulation after fatty liver graft preservation: a role for melatonin and trimetazidine cocktail. Journal of pineal research. 2013;55:65–78. doi: 10.1111/jpi.12051. [DOI] [PubMed] [Google Scholar]
- Zemke D, Smith JL, Reeves MJ, Majid A. Ischemia and ischemic tolerance in the brain: an overview. Neurotoxicology. 2004;25:895–904. doi: 10.1016/j.neuro.2004.03.009. [DOI] [PubMed] [Google Scholar]
- Zhang J, Yang J, Zhang C, Jiang X, Zhou H, Liu M. Calcium antagonists for acute ischemic stroke. The Cochrane database of systematic reviews. 2012;5:CD001928. doi: 10.1002/14651858.CD001928.pub2. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Wu YX, Hao YB, Dun Y, Yang SP. Role of endogenous opioid peptides in protection of ischemic preconditioning in rat small intestine. Life sciences. 2001;68:1013–1019. doi: 10.1016/s0024-3205(00)01004-3. [DOI] [PubMed] [Google Scholar]
- Zhang ZG, Zhang L, Jiang Q, Chopp M. Bone marrow-derived endothelial progenitor cells participate in cerebral neovascularization after focal cerebral ischemia in the adult mouse. Circulation research. 2002;90:284–288. doi: 10.1161/hh0302.104460. [DOI] [PubMed] [Google Scholar]
- Zhao L, Nowak TS., Jr CBF changes associated with focal ischemic preconditioning in the spontaneously hypertensive rat. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2006;26:1128–1140. doi: 10.1038/sj.jcbfm.9600269. [DOI] [PubMed] [Google Scholar]
- Zheng S, Zuo Z. Isoflurane preconditioning reduces purkinje cell death in an in vitro model of rat cerebellar ischemia. Neuroscience. 2003;118:99–106. doi: 10.1016/s0306-4522(02)00767-4. [DOI] [PubMed] [Google Scholar]
- Zheng S, Zuo Z. Isoflurane preconditioning induces neuroprotection against ischemia via activation of P38 mitogen-activated protein kinases. Molecular pharmacology. 2004;65:1172–1180. doi: 10.1124/mol.65.5.1172. [DOI] [PubMed] [Google Scholar]
- Zhou AM, Li WB, Li QJ, Liu HQ, Feng RF, Zhao HG. A short cerebral ischemic preconditioning up-regulates adenosine receptors in the hippocampal CA1 region of rats. Neuroscience research. 2004;48:397–404. doi: 10.1016/j.neures.2003.12.010. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Fathali N, Lekic T, Ostrowski RP, Chen C, Martin RD, Tang J, Zhang JH. Remote limb ischemic postconditioning protects against neonatal hypoxic-ischemic brain injury in rat pups by the opioid receptor/Akt pathway. Stroke; a journal of cerebral circulation. 2011;42:439–444. doi: 10.1161/STROKEAHA.110.592162. [DOI] [PMC free article] [PubMed] [Google Scholar]