

Abstract
Mitochondrial DNA (mtDNA) mutator mice express a mutated form of mtDNA polymerase gamma (PolgA) that results an accelerated accumulation of somatic mtDNA mutations in association with a premature aging phenotype. An exploratory metabolomic analysis of cortical metabolites in sedentary and exercised mtDNA mutator mice and wild-type (WT) littermate controls at 9–10 months of age was performed. Pathway analysis revealed deficits in the neurotransmitters acetylcholine, glutamate and aspartate that were ameliorated by exercise. Nicotinamide adenine dinucleotide (NAD+) depletion and evidence of increased Poly [ADP-ribose] polymerase 1 (PARP-1) activity were apparent in sedentary mtDNA mutator mouse cortex, along with deficits in carnitine metabolites and an upregulated antioxidant response that largely normalized with exercise. These data highlight specific pathways that are altered in the brain in association with an accelerated age-related accumulation of somatic mtDNA mutations. These results may have relevance to age-related neurodegenerative diseases associated with mitochondrial dysfunction, such as Alzheimer’s disease and Parkinson’s disease, and provide insights into potential mechanisms of beneficial effects of exercise on brain function.
Keywords: Aging, exercise, metabolism, NAD, mitochondria, DNA damage
1. Introduction
Somatic point mutations or deletions arise in mitochondrial DNA (mtDNA) through multiple mechanisms, including oxidative damage by reactive oxygen species (Cheng et al., 1992; Kuchino et al., 1987; Yakes and Van Houten, 1997), impaired base excision repair mechanisms (Stuart and Brown, 2006) or mtDNA polymerase γ (POLG) infidelity (Cheng et al., 1992; A. A. Johnson and K. A. Johnson, 2001; Krishnan et al., 2008; Kuchino et al., 1987; Yakes and Van Houten, 1997). Levels of mtDNA mutations increase with age in the brain (Mecocci et al., 1997; Stuart and Brown, 2006) and have been implicated in neurodegenerative diseases such as Alzheimer’s and Parkinson’s disease (Clark et al., 2011; Lin et al., 2012; 2002; Wallace, 2005).
The mtDNA mutator mouse is a well-characterized model of progeroid aging (Kujoth et al., 2005; Trifunovic et al., 2004). These transgenic mice carry a substitution mutation in the PolgA gene that encodes mtDNA polymerase-γ. This PolgAD257A mutation alters the proofreading function of the holoenzyme so that a high number of point mutations are introduced into the mitochondrial genome during replication (Kujoth et al., 2005). mtDNA deletions are also reported in this model (Vermulst et al., 2008), although the absolute level of these deletions has been reported to be quite low (Kraytsberg et al., 2009). The accumulation of high levels of somatic mtDNA mutations as a result of the nuclear PolgAD257A mutation leads to systemic mitochondrial dysfunction and a striking, progressive phenotype from 7–9 months onwards, at which point the mice exhibit weight loss, reduced subcutaneous fat, greying and loss of fur, and pronounced kyphosis. The phenotype also includes osteoporosis, anemia, reduced fertility and enlargement of the heart, as well shortened lifespan (Kujoth et al., 2005; Trifunovic et al., 2004). These abnormalities are dramatically improved by a 5-month endurance exercise regimen beginning at 3 months of age, which attenuates the accumulation of mtDNA mutations in skeletal muscle and reduces brain atrophy (Safdar et al., 2011a). Results are presented here from an untargeted metabolomics screen employed to explore the effect of the same endurance exercise regimen on metabolites in the cortex of sedentary and exercised mtDNA mutator mice.
2 Methods
2.1 PolgAD257A/D257A mouse breeding
The endurance exercise protocol outlined in this study was approved by McMaster University’s Animal Research and Ethics Board under the global Animal Utilization Protocol 12-03-09, and the experimental protocol strictly followed guidelines published by the Canadian Council of Animal Care. Homozygous knock-in mtDNA mutator mice (PolgAD257A/D257A) and WT littermates (PolgA+/+) were bred and maintained at McMaster University’s Central Animal Facility as previously published (Safdar et al., 2011a). The presence of the POLG knock-in mutation was confirmed in this line as previously published (Kujoth et al., 2005). A total of 21 WT (sedentary n = 12, exercised n= 9) and 19 mtDNA mutator mice (sedentary n= 10, exercised n= 9) were bred for this study. Three mtDNA mutator mice from the sedentary group became moribund and were sacrificed two-weeks before the end of the study aged between 8 months and three weeks to nine months of age. Data from these mice were not included in the current study.
2.2 Endurance exercise protocol
At three months of age, male and female mtDNA mutator mice and age-matched WT littermate controls were housed individually in microisolator cages in a temperature and humidity-controlled room with food and water ad libitum (Safdar et al., 2011a). None of the mice had previously been subjected to an exercise regimen. A one-week training pre-period was included to acclimatize the mice to the treadmill (Eco 3/6 treadmill; Columbus Instruments). The endurance exercise protocol was then performed as previously published (Safdar et al., 2011a; 2011b). In brief, mice were subjected to forced treadmill exercise three times a week at 15 meters per minute for 45 minutes for a period of six months. A five-minute warm-up period and a five-minute cool down period at 10 meters per minute were included.
As noted in the prior section, a wild-type exercise group was originally included; however, subsequent analysis of the muscle to ensure that the stimulus/intervention was sufficient to induce mitochondrial biogenesis showed that none of the well-established exercise-responsive transcripts were induced in muscle of the WT exercise group; whereas the well characterized effect of the PolgAD257A/D257A mutation and the response to exercise in mutator mice vs. WT sedentary mice indicated the expected muscle response to the intervention (data not shown). As a consequence of the complete lack of a training effect in the WT exercised mice, metabolomic data for the WT exercised group are not presented here, for it would be scientifically unjustified to attribute any changes (or lack thereof) to exercise training when we have evidence that they did not respond physiologically to the exercise stimulus. Given that we used the same absolute exercise stimulus for the WT and mtDNA mutator mice, and that the mutator mice have lower exercise capacity, the relative exercise intensity for the WT mice was likely not sufficient to elicit a training effect. In the future we will be evaluating the effect of exercise in WT mice using a higher intensity to represent the same relative and not absolute exercise intensity (Friedlander et al., 1998). It is important to note that omitting the data for the exercised WT group does not alter any of the conclusions of this study.
2.3 Sacrifice and tissue processing
After sacrifice by cervical dislocation, each mouse was decapitated and the brain was removed into icecold sterile saline, chilled for five minutes and then dissected on glass placed over ice as described previously (Clark et al., 2012). Briefly, the brain was placed into a chilled brain matrix and sliced into 1 mm thick coronal sections on ice. The sections were then placed into ice-cold saline. Cortex was dissected from the right and left cortical hemispheres of a slice containing the caudal striatum and snapfrozen. This region of cortex roughly corresponds to M1, M2 and S1 cortex.
2.4 Metabolomic analysis
Cortical samples (n=9 per group, except MUT SED where n=7 see explanation in 2.1) were shipped on dry ice to Metabolon (Durham, NC, USA). Unbiased metabolomic analysis was performed by Metabolon according to published methods (Boudonck et al., 2009; Lawton et al., 2008; Sreekumar et al., 2009). Briefly, the protein fraction was removed using a proprietary series of organic and aqueous extractions while maximizing recovery of small molecules. The extracted samples were split for Gas Chromatography (GS)/Mass Spectrometry (MS) and Liquid Chromatography (LC)-MS/MS analysis. Also included were several technical replicate samples created from a homogenous pool containing a small amount of all study samples. Internal standards were added to each sample prior to injection into the mass spectrometers.
2.5 Bioinformatics and statistics
Identification and Grouping of Metabolites
Metabolites were identified by automated comparison of the ion features in the experimental samples to a reference library of chemical standard entries that included retention time, molecular weight (m/z), preferred adducts, and in-source fragments as well as associated MS spectra, and were curated by visual inspection for quality control using Metabolizer software developed at Metabolon (DeHaven et al., 2010). For statistical analyses and data display purposes, any missing values were assumed to be below the limit of detection and these values were imputed with the compound minimum (minimum value imputation). Raw ion intensities were normalized according to the median value observed for a given biochemical. Following median scaling and imputation of missing values, statistical analysis of log-transformed data was performed using “R” (http://cran.r-project.org/), which is a freely available, open-source software package. ANOVA was used to identify biochemicals that differed significantly between the three experimental groups (WT mice (n = 9), sedentary mtDNA mutator mice (n = 7) and exercised mtDNA mutator mice (n = 9)). P-values ≤0.05 were considered statistically significant and p-values <0.10 were reported as trends. Multiple comparisons were accounted for by estimating the false discovery rate (FDR) using q-values (Storey and Tibshirani, 2003). Data in figures are presented as box-and-whisker plots indicating the minimum, the 25th percentile, the median, the 75th percentile, and the maximum values.
Principle Component Analysis
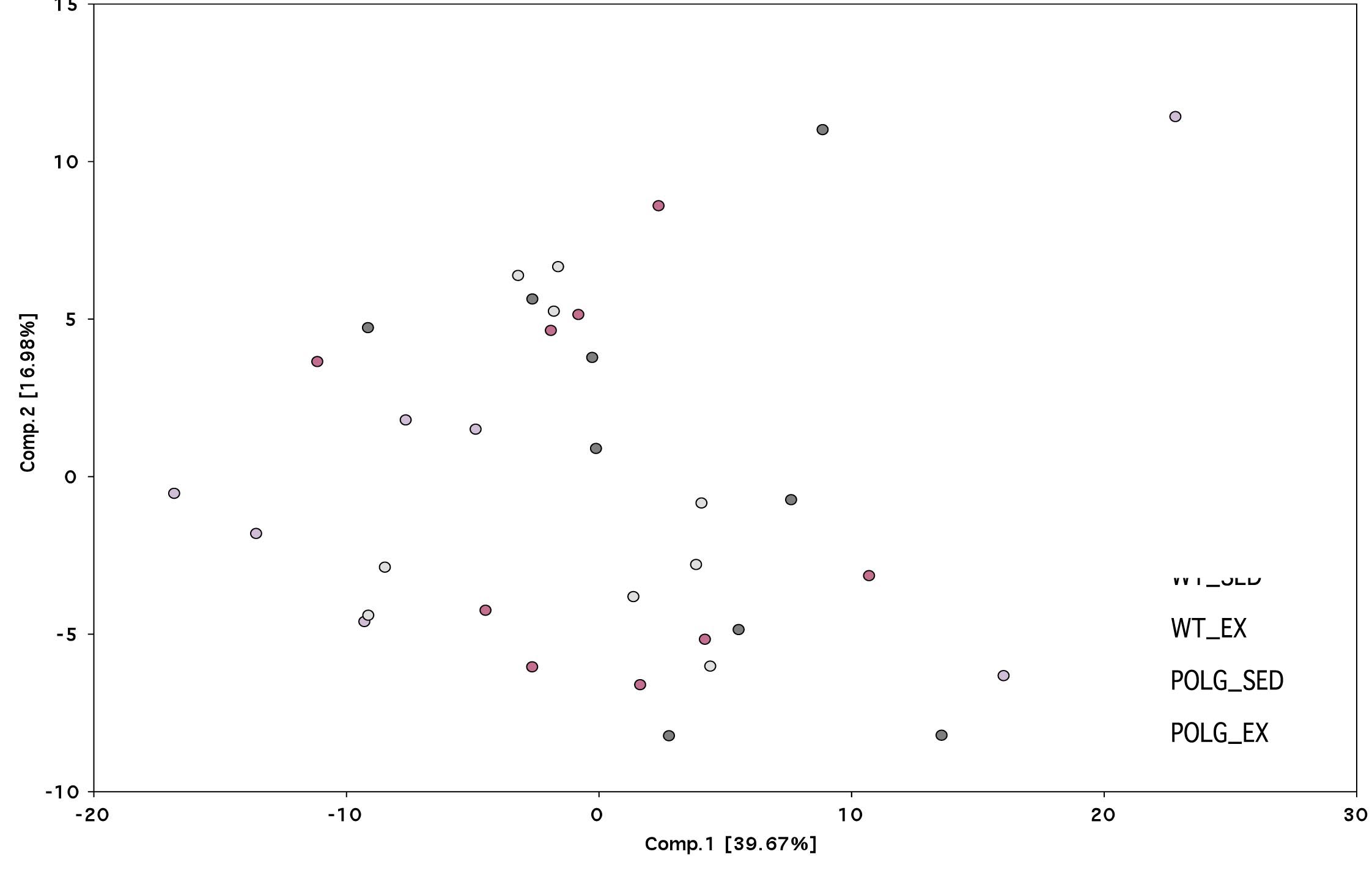
Principle component analysis (PCA) to determine separation of the three groups as a function of the brain metabolome demonstrated a substantial amount of overlap among groups. In this analysis, a large number of metabolic variables were transformed into a smaller number of orthogonal variables in order to analyze variation between the groups and in order to group differing populations separately. Results from the PCA corroborate the modest number of significant biochemical changes that were observed when comparing the groups (Supplementary Figure 1).
Metabolic Set Enrichment Analysis (MSEA)
Hotelling’s T2 test, a multivariate generalization of the t-test, was performed to test whether the mean vectors (consisting of multiple metabolites grouped according to sub-pathway) are different or not. The comparisons performed were WT SED vs. POLG SED, POLG SED vs. POLG EX and WT SED vs. POLG EX. In order to compute the p-value the sub pathway must contain fewer compounds than the total number of samples from both groups being compared, therefore this introduced the following constraints: WT_SED vs POLG_SED (9 vs. 7 – no more than 15 compounds), POLG_SED vs POLG_EX (7 vs. 9 – no more than 15 compounds), WT_SED vs POLG_EX (9 vs. 9 – no more than 17 compounds). The lysolipid group was broken up into the 1 and 2 forms because of the high number of metabolites that fall into this classification. However, the 1-form was still too large for these comparisons. Additionally, while sub-pathways with a single compound (e.g. the neurotransmitter sub-pathway that consists only of acetylcholine) can be calculated, the p-value is identical to that calculated using a regular two sample t-test.
2.6 HPLC measurements of CoA and acetyl-CoA
Pieces of adjacent cortex were used for HPLC analysis of CoA and acetyl-CoA (n = 6 per group). 200µl of ice-cold 5% PCA containing 50 µmol DTT was added to each piece of cortex (2–10mg). After vortexing, the sample was briefly sonicated for 6 – 8 seconds. The homogenate was set on ice for 10–15 min for better extraction, vortexed and centrifuged twice at 14,000rpm for 20 min at 4°C. After the second centrifugation, 100–150µl of clear supernatant was transferred into the HPLC vial for direct HPLC analysis. The concentration of CoA and acetyl-CoA in each sample was calculated based on a calibration curve equation and per mg of wet tissue.
The Waters HPLC system consisted of the following: A model 2489 UV/VIS detector, 2707 autosampler, and 1525 binary pump. Chromatographic data collection and data analysis was controlled by Breeze2 software installed on a Dell computer. Wavelength for UV detection was 259nm. The chromatographic method involved the use of 100 mmol/L NaH2PO4 and 75 mmol/L CH3COONa: acetonitrile (96:6, v/v) mobile phase, pH 4.6 adjusted by adding of concentrated H3PO4. Coenzyme A and acetyl coenzyme A separation was performed on ESA, RP-C18, 150×3mm, 3µm, 120A (PN# 70-0636) analytical column equipped with Phenomenex Security Guard column (cartridge C18, 4×2mm, PN# AJ0-4286). The columns were maintained at room temperature, injection volume was 30µl, and flow rate was 0.5ml/min. Under these conditions coenzyme A and acetyl coenzyme A eluted at 3.75min and 8.15min respectively. A set of acetyl CoA and CoA standards was prepared by serial dilutions in deionized water in a range of 0.08–5µM.
3 Results
3.1 Mutator mice exhibit deficiencies in cortical acetylcholine
Metabolomic analysis revealed that acetylcholine levels were significantly lower in the cortex of sedentary mtDNA mutator mice relative to WT sedentary controls (Fig. 1A). Exercise in mtDNA mutator mice tended to normalize acetylcholine levels, and levels in the Polg-exercise group were not significantly different from that of WT (Fig. 1A).
Fig. 1. Deficits in metabolites relating to acetylcholine.

A. Acetylcholine levels in sedentary and exercised WT and mutator mouse cortex as detected by the metabolomic screen. B. Pantothenic Acid levels in sedentary and exercised WT and mutator mouse cortex as detected by the metabolomic screen. C. Acetyl-CoA levels as measured by HPLC.
3.2 Mutator mice are deficient in pantothenic acid, a precursor of co-enzyme A
Acetylcholine is synthesized from choline and acetyl-CoA. Acetyl-CoA, considered the hub of metabolism as it feeds Krebs cycle, is a thioester of acetic acid or pyruvic acid and coenzyme A (CoA): CoA itself is synthesized from cysteamine and pantothenic acid (Kyoto Enyclopedia of Genes and Genomes (KEGG) map00770). Metabolomic analysis revealed low pantothenic acid (vitamin B5) levels in sedentary mutator mouse cortex compared to sedentary WT mice (Fig.1B). Exercise increased cortical levels of pantothenic acid in mtDNA mutator mouse cortex relative to the sedentary condition (Fig. 1B), and levels in the Polg-exercise group were not significantly different from levels in sedentary WT mice.
3.3 Exercise lowers acetyl-CoA levels in mtDNA mutator mice
Given the lower levels of pantothenic acid and acetylcholine observed in sedentary mtDNA mutator mice, it was hypothesized that CoA or acetyl-CoA would be lower in sedentary mtDNA mutator mice and would increase with exercise. In contrast, CoA levels did not vary between any of the groups, and acetyl-CoA was lower in exercised mtDNA mutator mice (Figure 1C). This may be indicative of increased utilization of acetyl-CoA after exercise and/or deficits in acetyl-CoA production in the mtDNA mutator mice.
3.4 Mutator mice display alterations in membrane phospholipid precursors and degradation products
Pathway analysis identified significant baseline alterations in three metabolites associated with membrane remodeling in sedentary mtDNA mutator mice. These metabolites; choline phosphate, glycerophosphorylcholine (GPC), phosphoethanolamine are closely linked to acetylcholine formation (KEGG map00564). Levels of the precursors glycerol and ethanolamine were not altered in mutator mice compared to WT littermates.
Significant reductions in cortical levels of choline phosphate (Fig. 2A) and GPC (Fig.2B), the two main cellular stores of choline, in sedentary mtDNA mutator mice relative to WT suggest a broad dysregulation of choline-containing metabolites in the mutator mouse brain. GPC is a required intermediate in the synthesis of the phospholipid phosphatidylcholine via the cytidine pathway (Infante, 1985). Cytidine itself was reduced relative to WT with borderline significance in the mutator sedentary condition (data not shown; sed mut/sed WT = 0.83, p=0.064) but the phosphatidylcholine intermediate cytidine 5'-diphosphocholine was not affected (sed mut/sed WT = 0.85, p=0.151).
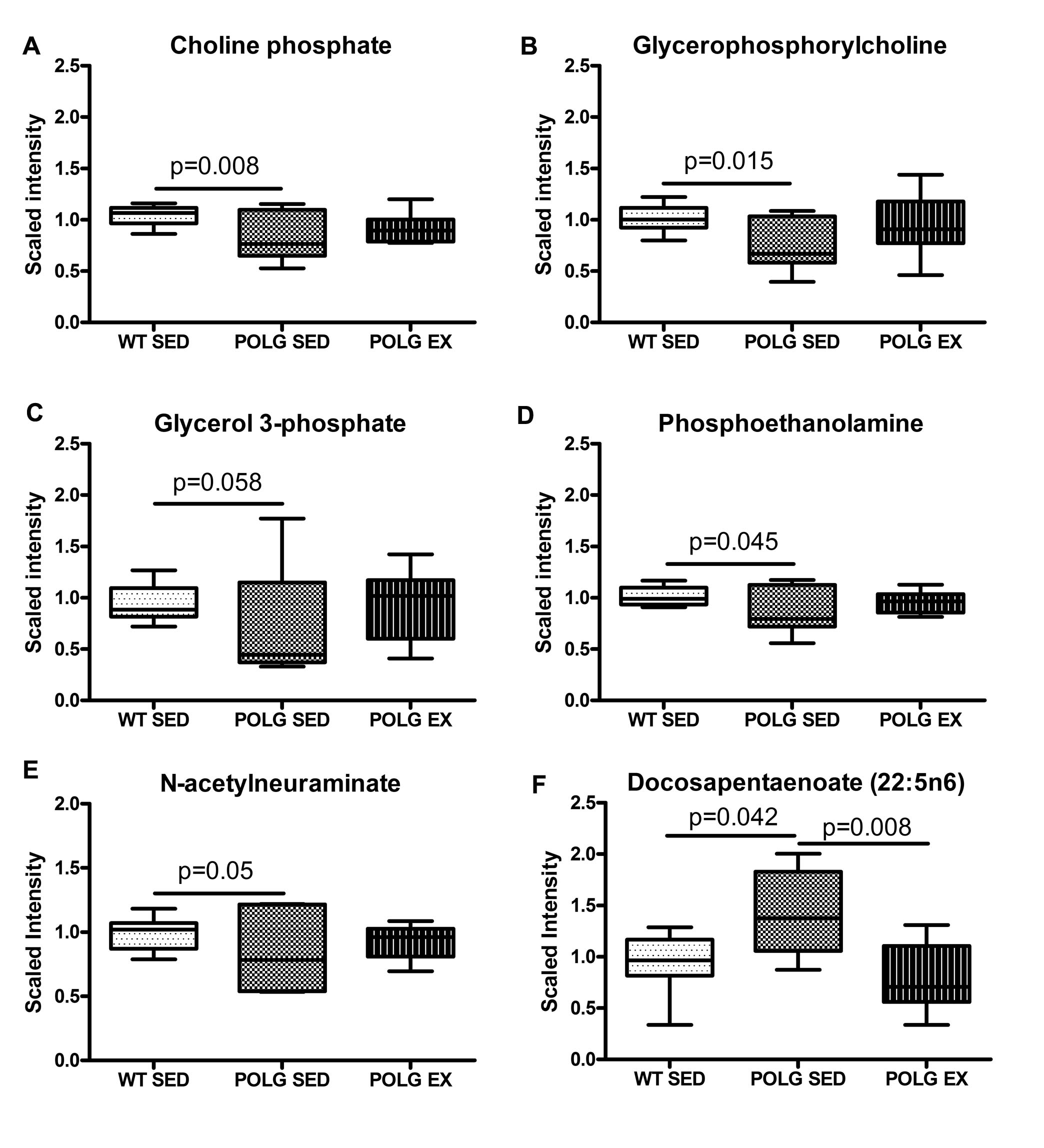
Fig. 2. Alterations in membrane phospholipid and acetylcholine precursors.

A. Choline phosphate B. Glycerphosphocholine C. Glycerol-3-phosphate D. Phosphoethanolamine E. N-acetylneuraminate and F. Docosapentaenoate levels in sedentary and exercised WT and mutator mouse cortex as detected by the metabolomic screen.
GPC is a precursor to glycerol-3-phosphate (G3P). Consistent with the decrease in GPC, metabolomics analysis revealed lower levels of G3P in the cortex of the sedentary mutator mice of borderline significance (Fig. 2C; p=0.058). Deficits in phosphoethanolamine were also observed in sedentary mutator mice compared to WT (Fig. 2D). Phosphoethanolamine is a precursor to choline phosphate therefore the reduction in this metabolite is upstream of the deficit in choline phosphate.
Exercised mtDNA mutator mice exhibited choline phosphate, GPC and phosphoethanolamine levels that were not significantly higher than that of sedentary mutator mouse cortex (Fig. 2A & 2B).
Levels of N-acetylneuraminate (Neu5Ac), a sialic acid involved in membrane remodeling, were decreased in sedentary mutator mice relative to cortex (Fig. 2E). Neu5Ac is a critical component of gangliosides that form the plasma membrane in the brain and may have important roles in development and neuronal regeneration (McClay et al., 2012; Schnaar et al., 2014). Neu5Ac is an important nutrient that can also be synthesized by the brain with the early steps of synthesis dependent on acetyl-CoA availability (KEGG ec00520). The lower level of Neu5Ac in mutator cortex likely contributed to the significant p value obtained by comparison to WT for the aminosugars metabolism sub-pathway by MSEA. The same analysis also identified a significant difference between sedentary WT and mutator mice for glycerolipid metabolism (supplementary figure 2).
Cortical levels of fatty acids, which make up the tails of phospholipids, were largely unaffected by the presence of the PolgD257A mutation or exercise, with the exception of the omega-6 essential fatty acid docosapentaenoate (n6 DPA; 22:5n6), which was significantly increased in sedentary mutator mice compared to sedentary WT mice (Fig.2F). This is likely the reason why MSEA identified the essential fatty acid sub-pathway as being significantly different between sedentary and exercised mutator mice (supplementary table 2). Docosapentaenoate primarily functions as a minor component of phospholipids in cell membranes and accumulates in neuronal membranes during n-3 fatty acid deficiency (Kim et al., 2003), possibly indicating a fatty acid deficiency in mutator mice that is corrected by our endurance exercise protocol.
3.5 High levels of mitochondrial mutations lower the NAD+ pool in mtDNA mutator mouse cortex
MSEA identified nicotinate and nicotinamide metabolism as a sub-pathway that significantly differed between sedentary WT and mutator mice (supplementary table 2). Levels of the coenzyme NAD+ were observed to be significantly lower in sedentary mtDNA mutator mice relative to WT in the primary analysis (Fig.3A). This NAD+ deficit in mtDNA mutator mice may be linked to increased activity of the Poly [ADP-ribose] polymerase (PARP) family of enzymes as highly elevated adenosine 5'diphosphoribose (ADPR) is observed in Polg mutator mouse cortex compared to WT (Fig.3C). Elevation of poly (ADP) ribose (PAR) and depletion of NAD+ are hallmarks of PARP activation (Shah et al., 2011). PARP enzymes synthesize ADPR by cleaving the ADP-ribose moiety from NAD+ with subsequent depletion of the cytosolic NAD+ pool. ADPR can then go on to form chains of PAR (Alano et al., 2010; Berger, 1985).
Fig. 3. NAD+ and related metabolites.

A. NAD+ B. NADH C. ADPR and D. Nicotinamide levels in sedentary and exercised WT and mutator mouse cortex as detected by the metabolomic screen.
Levels of adenosine diphosphate (ADP) and adenosine monophosphate (AMP) were similar in all groups studied (data not shown). ATP was not detected in any samples using this method.
3.6 Excitatory amino acids (EAAs) are low in Polg mutator mouse cortex and increase with exercise
The metabolomic screen also exposed a deficit in the excitatory amino acids (EAAs) glutamate (Fig. 4A) and aspartate (Fig. 4B) in sedentary mtDNA mutator mouse cortex compared to WT controls. No such deficit was apparent in the exercised mtDNA mutator mice compared to WT. MSEA also identified alanine and aspartate metabolism and glutamate metabolism sub-pathways as being significantly altered between sedentary WT and mutator mice, but by this measure the alanine and aspartate subpathway was still significantly different between sedentary WT and exercised mutator mice (supplementary table 2).
Fig. 4. Amino acid and amino acid metabolite alterations.

A. Glutamate B. Aspartate C. Pyrogulatamine D. Gamma-glutamyl-glutamate E. Lysine F. Tyrosine and G. Urea levels in sedentary and exercised WT and mutator mouse cortex as detected by the metabolomic screen.
Glutamate can be produced from transamination of α-ketoglutarate with the amino group being transferred from either alanine or aspartate, or via glutaminase where the amino group of glutamine is hydrolyzed to produce glutamate and ammonium. Brain alanine and glutamine levels were unaffected by the presence of the PolgAD257A mutation or the exercise paradigm. However, pyroglutamine (a cyclic derivative of glutamine) was significantly lower in sedentary and exercised mutator cortex relative to WT (Fig. 3C).
Glutamate and aspartate were not the only amino acids altered in mutator mice. Levels of lysine (Fig. 4E) and tyrosine (Fig. 4F) were also lowered in sedentary mutator mouse cortex relative to WT, with lysine increased in exercised mutator mice relative to sedentary mutator mice (Fig. 4E). The lysine subpathway was identified as significantly altered between sedentary WT and mutator mice and also between sedentary WT mice and exercised mutator mice by MSEA (supplementary table 2). These ketogenic amino acids are also involved in the formation of acetyl-CoA and acetoacetate and reductions in these amino acids in sedentary mtDNA mutator mouse cortex could represent increased utilization to synthesize the raw materials required for Krebs cycle.
Urea is the main nitrogenous end product of protein metabolism and was decreased relative to WT in sedentary mutator mice (Fig. 6A). Other products of the urea cycle such as N-acetyl-glutamate, arginine and ornithine were unaffected by the PolgAD257A mutation or exercise (data not shown). Lower levels of urea in sedentary mtDNA mutator mice may be related to depleted muscle mass and lower amino acid flux to the liver in these animals.
Fig. 6. Alterations in the antioxidant profile.

A. Reduced glutathione (GSH) and B. Oxidized glutathione levels in sedentary and exercised WT and mutator mouse cortex as detected by the metabolomic screen. C. GSH:GSSG ratio calculated from scaled imputed values D. Cystathionine E. Carnosine F. Homocarnosine G. Ergothionine levels in sedentary and exercised WT and mutator mouse cortex as detected by the metabolomic screen.
3.7 Carnitine metabolism is altered in mtDNA mutator mice
Metabolomic analysis detected lower levels of carnitine in sedentary mutator mouse cortex compared to sedentary WT (Fig. 5A). In contrast, exercised mutator mouse brain exhibited levels of carnitine that were indistinguishable from sedentary WT mice. This pattern was also seen by MSEA (supplementary table 2). Depletion of carnitine in the sedentary mutator mice may reflect a general reduction in the number of mitochondria as mtDNA is depleted in these mice (Dai et al., 2013; Safdar et al., 2011a), or a defect in carnitine synthesis. Levels of carnitine in the exercised mutants were not significantly different from sedentary WT, but did not significantly differ from levels observed in sedentary mutator mice. Further studies are required to determine if this reflects experimental variation, or if this is indicative of enhanced carnitine synthesis after exercise.
Fig. 5. Changes in carnitine metabolism.

A. Carnitine B. Deoxycarnitine C. Acetylcarnitine D. Propionylcarnitine levels in sedentary and exercised WT and mutator mouse cortex as detected by the metabolomic screen.
Deoxycarnitine a carnitine precursor, and acetylcarnitine an acetylated form of carnitine, followed the same pattern as carnitine (Fig. 5B & 5C). Other carnitine derivatives were unaffected by either the presence of the PolgD257A mutation or endurance exercise.
3.8 Sedentary mtDNA mutator mice exhibit alterations in levels of antioxidants
In addition to its role in fatty acid transport into mitochondria, carnitine can also act as an antioxidant (Mingorance et al., 2011; Ribas et al., 2014). Levels of ergothioneine, another antioxidant, also are lower in sedentary mutator mouse cortex (Fig. 6G) relative to sedentary WT cortex. Ergothioneine is a dietary antioxidant that has been reported to protect against excitotoxicity (McClay et al., 2012). Collectively, these findings indicate that certain components of the brain’s anti-oxidant defenses are lower in the mutator mice.
In contrast, other components of the antioxidant defense system were higher in mtDNA mutator mouse cortex. The mean reduced glutathione to oxidized glutathione ratio (GSH:GSSG) for sedentary mutator mice was significantly higher than for sedentary WT mice (1.65 vs. 1.19, respectively; Fig. 6C, p < 0.05), although the level of reduced glutathione did not significantly differ from WT, MSEA did identify glutathione metabolism as a sub-pathway that significantly differed between sedentary and exercised mutator groups (supplementary table 2). This may indicate greater capacity for antioxidant activity via the GSH system in mutator mouse brain. Similarly, Kolesar et al. have recently reported an increase in non-enzymatic antioxidant capacity in brain of sedentary mutator mice vs. sedentary WT mice (Kolesar et al., 2014). Glutathione is synthesized from cysteine, glutamate and glycine. Cystathionine, a precursor to cysteine that is not effectively converted in neurons (Enokido, 2005) was increased in sedentary mutator mouse brain relative to sedentary WT (Fig. 5D). Reduced levels of glutamate in sedentary mutator mouse cortex may be related to increased use of glutamate in the synthesis of glutathione.
Carnosine (Fig. 6E) and homocarnosine (Fig. 6F) levels are both increased in cortex from sedentary mtDNA mutator mice relative to sedentary WT. Carnosine levels typically decrease with age in human muscle (Bellia et al., 2009; Everaert et al., 2010), along with glutathione (Currais and Maher, 2013; Emir et al., 2011) so these findings are unexpected in a mouse model of progeriod aging. Carnosine is an important pH buffer and the higher levels may be a compensatory attempt to deal with the increased proton burden secondary to the much higher glycolytic flux in the mutator mice (Saleem et al., 2015).
3.9 Other metabolic alterations in mtDNA mutator mouse brain
1,5-anhydroglucitol (1,5-AG) is a monosaccharide derived from ingestion of food and has a slightly different structure to glucose. Levels of 1,5-AG were nearly doubled in sedentary mutator mice vs. sedentary WT mice (Fig. 7). 1,5-AG is used as a marker for type 2 diabetes and is lower in individuals with hyperglycemia and glucosuria because high levels of glucose block reabsorption of 1,5-AG in the proximal tubule of the kidney (Kilpatrick et al., 1999). The significance of high levels of 1,5-AG in brain is unclear. It may be related to lower levels of glucose in the brain causing increased reabsorption of 1,5- AG, but we are unable to assess this possibility as glucose was not detected by the metabolomic screen.
Fig. 7.

1,5-anhydroglucitol levels in sedentary and exercised WT and mutator mouse cortex as detected by the metabolomic screen.
4 Discussion
This work identifies neurotransmitter deficits in the cortex of mtDNA mutator mice through an unbiased metabolomics screen. Acetylcholine, glutamate and aspartate are all affected. Acetylcholine is associated with regulation of the sleep-wake cycle as well as modulation of cognitive function and learning and memory (Saper et al., 2005; Schliebs and Arendt, 2011; Woolf and Butcher, 2011). Metabolomic analysis of mtDNA mutator mouse cortex revealed lower levels of multiple metabolites that converge onto acetylcholine synthesis (Fig. 8), although levels of two acetylcholine precursors, CoA and acetyl-CoA, were not lower in sedentary mtDNA mutator mice. Instead, acetyl-CoA was only decreased in exercised mutator mice, paradoxically a condition under which pantothenic acid was increased above sedentary levels.
Fig. 8. Convergence of metabolite deficiencies onto acetylcholine.

Shaded boxes and ovals indicate a reduction in this metabolite in the cortex of sedentary mtDNA mutator mice relative to sedentary WT mice.
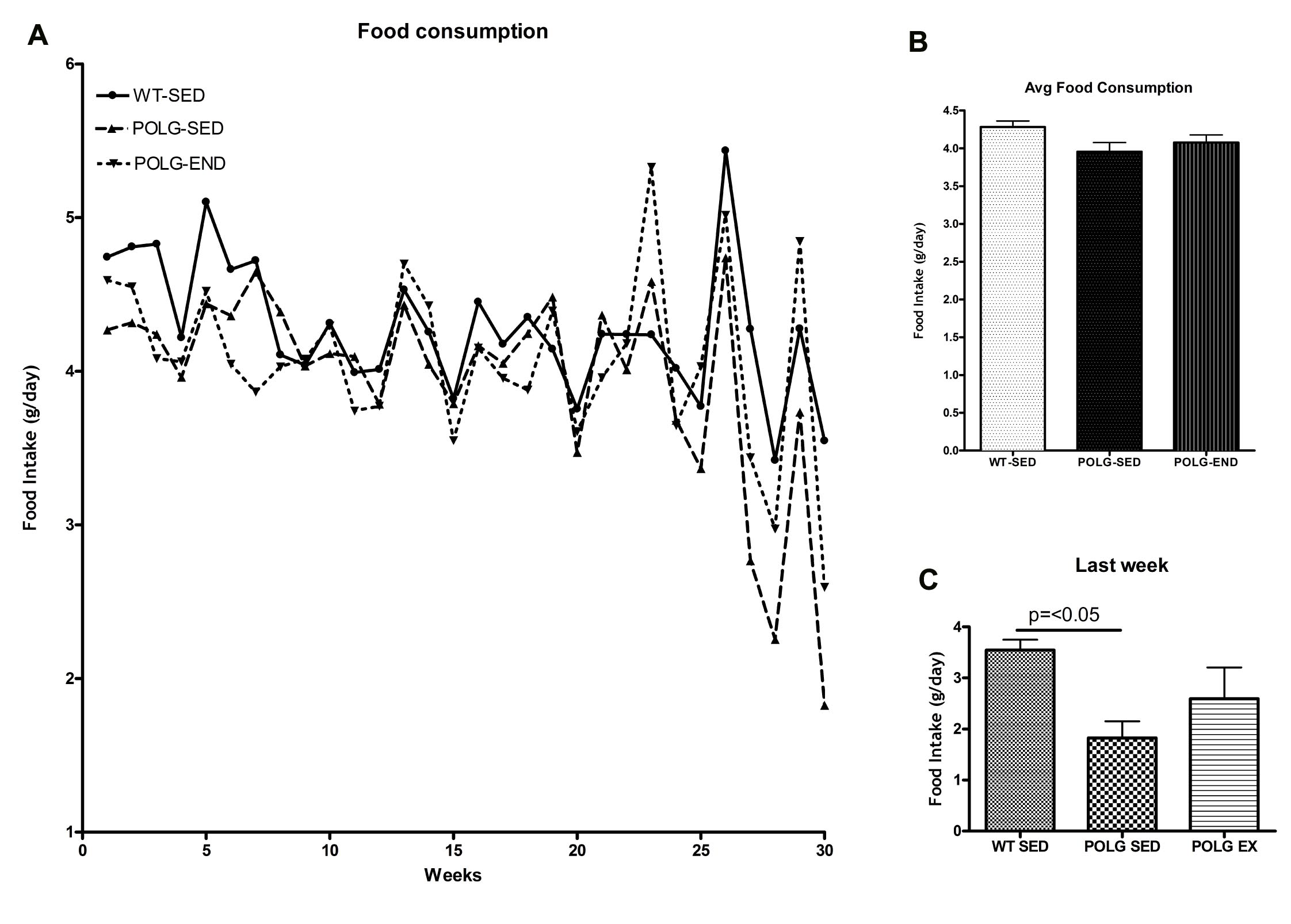
Pantothenic acid, a precursor to CoA synthesis that was lowered in the sedentary mutator mouse cortex and increased with exercise, is ubiquitous in the diet and deficiency in rare in humans for this reason. Pantothenic acid was present in the diet of all the mice at 23 mg/kg (Teklad 22/5 Rodent Diet). Food intake data did not show a statistically significant difference in food intake between sedentary mtDNA mutator mice and exercised mtDNA mutator mice (Supplementary Figure 2), suggesting that decreased food consumption is not an explanation for the pantothenic acid deficit in the Polg mutator mice. Recently, Fox et al. reported malabsorption of nutrients in mtDNA mutator mice secondary to increased apoptosis and perturbed stem-progenitor cell cycling as a result of mitochondrial dysfunction (Fox et al., 2012). In 2011 Safdar et al. reported improved mitochondrial function and decreased apoptosis in the duodenum of mtDNA mutator mice with exercise (Safdar et al., 2011a); therefore it is possible that improved absorption of micronutrients is the reason for increased pantothenic acid in the exercised mtDNA mutator mice. Interestingly, deficiency in pantothenic acid has been shown to cause anemia and greying of the fur in experimental animals (Daft and Kornberg, 1945; Kuo et al., 2007), a prominent phenotype of the mtDNA mutator mice.
Reduced levels of a subset of acetylcholine precursors suggest that a decrease in synthesis may contribute to the acetylcholine deficit in this mouse aging model. However, in future studies it will be important to examine other potential contributors, such as degeneration of the cholinergic neurons as seen with aging and in Alzheimer’s disease (Schliebs and Arendt, 2011).
Glutamate and aspartate are excitatory amino acid neurotransmitters that frequently coexist in neurons (Hill et al., 2000) with glutamate being the predominant excitatory neurotransmitter in cortex. These neurotransmitters (along with acetylcholine) are important for learning, memory and cognition (Riederer and Hoyer, 2006), and are decreased in regions of the AD brain compared to normal controls (Gueli and Taibi, 2013).
As well as being a neurotransmitter, glutamate is an important metabolite in brain; it is a precursor not only to aspartate (which is also lower in mtDNA mutator mouse cortex) but also to GABA and glutamine (unaffected in mtDNA mutator mouse cortex), and can stimulate glycolysis (García-Espinosa et al., 2004; Hill et al., 2000). Glutamate metabolism is closely linked to the Krebs cycle: glutamate and the coenzyme NAD+ are converted to the important Krebs cycle intermediate α-ketoglutarate (Krebs and Veech, 1969). Decreased glutamate in mtDNA mutator mouse brain could be a result of increased metabolism to α-ketoglutarate to provide Krebs cycle intermediates. α-ketoglutarate was not detected on the metabolomics screen. However, MSEA identified significant differences in the glycolysis, gluconeogenesis and pyruvate metabolism, and Krebs cycle sub-pathways between WT and mutator sedentary mice. The difference in glycolysis, gluconeogenesis and pyruvate metabolism between WT and mutator mice remained even after exercise (supplementary table 2). This is perhaps unsurprising as mutator mice have an increased dependence on glycolysis for energy (Saleem et al., 2015).
Alternatively, as glutamate is synthesized from glutamine provided by astrocytes (Hawkins, 2009) and glutamine levels in cortex were unaffected by the PolgAD257A mutation, the mutator mouse glutamatergic deficit may be indicative of specific loss of cortical neurons.
Levels of NAD+ were lower in sedentary mtDNA mutator mice relative to WT. This likely has important consequences as NAD+ is essential to many energy transduction redox reactions. Although NAD+ was low in the cortex of sedentary mtDNA mice, NADH did not increase and the NADH/NAD ratio did not significantly differ between groups (data not shown), which may be due to failure to detect NADH for some samples, requiring use of imputed data points in those cases. Lower levels of NAD+ are likely due to an increase in reactions that consume NAD+, such as ADP-ribosylation by the DNA repair enzyme PARP-1, rather than increased oxidation of NAD+. ADP-ribosylation is apparent in the cortex of mtDNA mutator mice as our unbiased metabolomic screen detected high levels of ADPR, a hallmark of PARP-1 activity, which may reflect PARP activation in both sedentary and exercised groups of mutator mice relative to WT. This PARP activation may be a direct response to mitochondrial DNA damage (Rossi et al., 2009), or could be due to secondary damage to nuclear DNA. However, nuclear DNA damage has not been assessed in this model. In either case, our results suggest that inhibition of poly(ADP-ribose) polymerases could be a powerful tool to ameliorate the aging phenotype in this mouse model of aging.
Levels of NAD+ decrease with age (Gomes et al., 2013) and recent work has shown that low NAD+ in forebrain cortical neurons leads to cortical atrophy and cognitive dysfunction in mice (Stein et al., 2014). Also, increased PARP activity has been associated with AD (Love, 1999). The cognitive status of the mtDNA mutation mice has not been carefully studied, but the cholinergic, glutamatergic and NAD+ deficits in the cortex lead us to hypothesize that this model may have cognitive deficits, a hypothesis that should be tested in future studies.
In prior work, aerobic exercise has been shown to upregulate mitochondrial biogenesis and activity which may help maintain NAD+ availability (Hipkiss, 2010), although this effect has not yet been shown in brain, and we did not detect a significant difference in NAD levels in the cortex of exercised mtDNA mutator mice relative to WT.
Mutator mice have deficits in oxidative phosphorylation (Hiona et al., 2010). Defective oxidative phosphorylation leads to increased production of reactive oxygen species (ROS). Some studies have reported that the oxidative phosphorylation deficits exhibited by mtDNA mutator mice do not result in oxidative damage (Hiona et al., 2010; Kujoth et al., 2005), but recent studies have shown oxidative damage in muscle (Kolesar et al., 2014), and heart that is alleviated by mitochondrial-targeted catalase (Dai et al., 2010), and increased mitochondrial hydrogen peroxide in older mtDNA mutator mice (Logan et al., 2014). Our observation of changes in antioxidants in the brain of sedentary mutator mice is consistent with altered oxidative stress in mtDNA mutator mice. Similarly, Kolesar et al., 2014 have reported an adaptive increase in non-enzymatic antioxidant capacity (modulated in part by cellular glutathione levels) in brain from sedentary mutator mice vs. sedentary WT mice (Kolesar et al., 2014). Therefore the observed up-regulation of glutathione production may be a compensatory response to increased oxidative stress. We also found that exercise normalized many of the antioxidants to levels indistinguishable from WT. Previously, Safdar et al. (2011) have also shown prevention of brain atrophy in mutator mice subjected to exercise. These data may suggest that exercise reduces oxidative stress in the brain and so reduces the need for up-regulating glutathione production.
Reductions in carnitine may be observed as a secondary deficit of impaired oxidative phosphorylation (Scholte et al., 1987). Both carnitine and acetylcarnitine were lower in mtDNA mutator mouse cortex compared to WT, but were not significantly different from WT in the exercised cohort. Carnitine deficiency often manifests with decreases in mitochondrially-produced docosahexaenoate (22:6n-3) and a compensatory increase in microsome -produced docosapentaenoate (n6 DPA; 22:5n6) (Infante and Huszagh, 2000), although we observed an increase in docosapentaenoate there was no depletion of docosahexaenoate (data not shown). Data on docosapentanoate accumulation islimited, but it may be associated with impaired function of peroxisomes (Llanos et al., 2005). Interestingly, elevated serum docosapentaenoate was recently identified as a marker of dementia (Mousavi et al., 2014).
In this paper we describe metabolic alterations in the cortex of mtDNA mutator mice and identified for the first time several cortical metabolic abnormalities in this model, such as impaired cholinergic and glutamatergic neurotransmission, lower NAD+ levels and ADPR generation, dysregulation of carnitine metabolism, and an altered antioxidant profile. This study also demonstrates that physical exercise is a powerful tool for ameliorating some of these deficits.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Average food intake for in grams per day for each week of the experiment.
{kind=link}
Heat map showing statistically significant changes in biochemicals profiled in this study.
Table showing results from the metabolic set enrichment analysis with p-value <0.05.
Highlights.
Polg mtDNA mutator mice exhibit alterations in the neurotransmitters acetylcholine and glutamate compared to age-matched WT littermate controls.
Evidence of increased poly-ADP-ribose polymerase (PARP) activity is apparent due to lower levels of nicotinamide adenine dinucleotide (NAD) and increased ADP-ribose in the cortex of mutator mice at baseline.
Carnitine metabolism is disrupted in mutator mice at baseline.
The antioxidant profile of mutator mice is disrupted at baseline.
Endurance exercise normalizes the majority of these alterations to WT levels.
Acknowledgements
This study was funded by the National Institute of Neurological Disorders and Stroke grant NINDS 1R21NS079324-01A1 to David K. Simon.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- Alano CC, Garnier P, Ying W, Higashi Y, Kauppinen TM, Swanson RA. NAD+ Depletion Is Necessary and Sufficient forPoly(ADP-Ribose) Polymerase-1-Mediated Neuronal Death. Journal of Neuroscience. 2010;30:2967–2978. doi: 10.1523/JNEUROSCI.5552-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellia F, Calabrese V, Guarino F, Cavallaro M, Cornelius C, De Pinto V, Rizzarelli E. Carnosinase levels in aging brain: redox state induction and cellular stress response. Antioxid. Redox Signal. 2009;11:2759–2775. doi: 10.1089/ars.2009.2738. [DOI] [PubMed] [Google Scholar]
- Berger NA. Poly(ADP-ribose) in the cellular response to DNA damage. Radiat. Res. 1985;101:4–15. [PubMed] [Google Scholar]
- Boudonck KJ, Mitchell MW, Nemet L, Keresztes L, Nyska A, Shinar D, Rosenstock M. Discovery of Metabolomics Biomarkers for Early Detection of Nephrotoxicity. Toxicologic Pathology. 2009;37:280–292. doi: 10.1177/0192623309332992. [DOI] [PubMed] [Google Scholar]
- Cheng KC, Cahill DS, Kasai H, Nishimura S, Loeb LA. 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G----T and A----C substitutions. Journal of Biological Chemistry. 1992;267:166–172. [PubMed] [Google Scholar]
- Clark J, Dai Y, Simon DK. Do somatic mitochondrial DNA mutations contribute to Parkinson's disease? Parkinsons Dis. 2011;2011:659694. doi: 10.4061/2011/659694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark J, Silvaggi JM, Kiselak T, Zheng K, Clore EL, Dai Y, Bass CE, Simon DK. Pgc-1α Overexpression Downregulates Pitx3 and Increases Susceptibility to MPTP Toxicity Associated with Decreased Bdnf. PLoS ONE. 2012;7:e48925. doi: 10.1371/journal.pone.0048925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currais A, Maher P. Functional Consequences of Age-Dependent Changes in Glutathione Status in the Brain. Antioxid. Redox Signal. 2013;19:813–822. doi: 10.1089/ars.2012.4996. [DOI] [PubMed] [Google Scholar]
- Daft FS, Kornberg A. Anemia and Granulocytopenia in Rats Fed a Diet Low in Pantothenic Acid. Public Health Reports. 1945;60:1201–1215. [PubMed] [Google Scholar]
- Dai D-F, Chen T, Wanagat J, Laflamme M, Marcinek DJ, Emond MJ, Ngo CP, Prolla TA, Rabinovitch PS. Age-dependent cardiomyopathy in mitochondrial mutator mice is attenuated by overexpression of catalase targeted to mitochondria. Aging Cell. 2010;9:536–544. doi: 10.1111/j.1474-9726.2010.00581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Y, Kiselak T, Clark J, Clore E, Zheng K, Cheng A, Kujoth GC, Prolla TA, Maratos-Flier E, Simon DK. Behavioral and metabolic characterization of heterozygous and homozygous POLG mutator mice. Mitochondrion. 2013;13:1–10. doi: 10.1016/j.mito.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeHaven CD, Evans AM, Dai H, Lawton KA. Journal of Cheminformatics. 2010;2:9. doi: 10.1186/1758-2946-2-9. 1758-2946-2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emir UE, Raatz S, McPherson S, Hodges JS, Torkelson C, Tawfik P, White T, Terpstra M. Noninvasive quantification of ascorbate and glutathione concentration in the elderly human brain. NMR Biomed. 2011;24:888–894. doi: 10.1002/nbm.1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enokido Y. Cystathionine -synthase, a key enzyme for homocysteine metabolism, is preferentially expressed in the radial glia/astrocyte lineage of developing mouse CNS. The FASEB Journal. 2005 doi: 10.1096/fj.05-3724fje. [DOI] [PubMed] [Google Scholar]
- Everaert I, Mooyaart A, Baguet A, Zutinic A, Baelde H, Achten E, Taes Y, De Heer E, Derave W. Vegetarianism, female gender and increasing age, but not CNDP1 genotype, are associated with reduced muscle carnosine levels in humans. Amino Acids. 2010;40:1221–1229. doi: 10.1007/s00726-010-0749-2. [DOI] [PubMed] [Google Scholar]
- Fox RG, Magness S, Kujoth GC, Prolla TA, Maeda N. Mitochondrial DNA polymerase editing mutation, PolgD257A, disturbs stem-progenitor cell cycling in the small intestine and restricts excess fat absorption. AJP: Gastrointestinal and Liver Physiology. 2012;302:G914–G924. doi: 10.1152/ajpgi.00402.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedlander AL, Casazza GA, Horning MA, Buddinger TF, Brooks GA. Effects of exercise intensity and training on lipid metabolism in young women. Am J Physiol. 1998;275:E853–E863. doi: 10.1152/ajpendo.1998.275.5.E853. [DOI] [PubMed] [Google Scholar]
- García-Espinosa MA, Rodrigues T, Sierra A, Benito M, Fonseca C, Gray HL, Bartnik BL, García-Martín ML, Ballesteros P, Cerdan S. Cerebral glucose metabolism and the glutamine cycle as detected by in vivo and in vitro 13C NMR spectroscopy. Neurochemistry International. 2004;45:297–303. doi: 10.1016/j.neuint.2003.08.014. [DOI] [PubMed] [Google Scholar]
- Gomes AP, Price NL, Ling AJY, Moslehi JJ, Montgomery MK, Rajman L, White JP, Teodoro JS, Wrann CD, Hubbard BP, Mercken EM, Palmeira CM, de Cabo R, Rolo AP, Turner N, Bell EL, Sinclair DA. Declining NAD(+) induces a pseudohypoxic state disrupting nuclearmitochondrial communication during aging. Cell. 2013;155:1624–1638. doi: 10.1016/j.cell.2013.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gueli MC, Taibi G. Alzheimer’s disease: amino acid levels and brain metabolic status. Neurol Sci. 2013;34:1575–1579. doi: 10.1007/s10072-013-1289-9. [DOI] [PubMed] [Google Scholar]
- Hawkins RA. The blood-brain barrier and glutamate. American Journal of Clinical Nutrition. 2009;90:867S–874S. doi: 10.3945/ajcn.2009.27462BB. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill E, Kalloniatis M, Tan SS. Glutamate, GABA and precursor amino acids in adult mouse neocortex: cellular diversity revealed by quantitative immunocytochemistry. Cereb. Cortex. 2000;10:1132–1142. doi: 10.1093/cercor/10.11.1132. [DOI] [PubMed] [Google Scholar]
- Hiona A, Sanz A, Kujoth GC, Pamplona R, Seo AY, Hofer T, Someya S, Miyakawa T, Nakayama C, Samhan-Arias AK, Servais S, Barger JL, Portero-Otín M, Tanokura M, Prolla TA, Leeuwenburgh C. Mitochondrial DNA Mutations Induce Mitochondrial Dysfunction, Apoptosis and Sarcopenia in Skeletal Muscle of Mitochondrial DNA Mutator Mice. PLoS ONE. 2010;5:e11468. doi: 10.1371/journal.pone.0011468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hipkiss AR. Aging, proteotoxicity, mitochondria, glycation, NAD+ and carnosine: possible interrelationships and resolution of the oxygen paradox. Frontiers in aging neuroscience. 2010 doi: 10.3389/fnagi.2010.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Infante JP. Impaired glycerophosphorylcholine synthesis in murine muscular dystrophy. Medical Biology. 1985;2:81–87. [PubMed] [Google Scholar]
- Infante JP, Huszagh VA. Secondary carnitine deficiency and impaired docosahexaenoic (22: 6 n-3) acid synthesis: a common denominator in the pathophysiology of diseases of oxidative phosphorylation and β-oxidation. FEBS Letters. 2000;468:1–5. doi: 10.1016/s0014-5793(00)01083-8. [DOI] [PubMed] [Google Scholar]
- Johnson AA, Johnson KA. Exonuclease proofreading by human mitochondrial DNA polymerase. Journal of Biological Chemistry. 2001;276:38097–38107. doi: 10.1074/jbc.M106046200. [DOI] [PubMed] [Google Scholar]
- Kilpatrick ESE, Keevilt BGB, Richmond KLK, Newland PP, Addison GMG. Plasma 1,5-anhydroglucitol concentrations are influenced by variations in the renal threshold for glucose. Diabet Med. 1999;16:496–499. doi: 10.1046/j.1464-5491.1999.00093.x. [DOI] [PubMed] [Google Scholar]
- Kim H-Y, Akbar M, Lau A. Effects of docosapentaenoic acid on neuronal apoptosis. Lipids. 2003;38:453–457. doi: 10.1007/s11745-003-1083-z. [DOI] [PubMed] [Google Scholar]
- Kolesar JE, Safdar A, Abadi A, Mac Neil LG, Crane JD, Tarnopolsky MA, Kaufman BA. Defects in mitochondrial DNA replication and oxidative damage in muscle of mtDNA mutator mice. Free Radical Biology and Medicine. 2014:1–11. doi: 10.1016/j.freeradbiomed.2014.07.038. [DOI] [PubMed] [Google Scholar]
- Kraytsberg Y, Simon DK, Turnbull DM, Khrapko K. Do mtDNA deletions drive premature aging in mtDNA mutator mice? Aging Cell. 2009;8:502–506. doi: 10.1111/j.1474-9726.2009.00484.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs HA, Veech RL. Equilibrium relations between pyridine nucleotides and adenine nucleotides and their roles in the regulation of metabolic processes. Advances in enzyme regulation. 1969;7:397–413. doi: 10.1016/0065-2571(69)90030-2. [DOI] [PubMed] [Google Scholar]
- Krishnan KJ, Reeve AK, Samuels DC, Chinnery PF, Blackwood JK, Taylor RW, Wanrooij S, Spelbrink JN, Lightowlers RN, Turnbull DM. What causes mitochondrial DNA deletions in human cells? Nat Genet. 2008;40:275–279. doi: 10.1038/ng.f.94. [DOI] [PubMed] [Google Scholar]
- Kuchino Y, Mori F, Kasai H, Inoue H, Iwai S, Miura K, Ohtsuka E, Nishimura S. Misreading of DNA templates containing 8-hydroxydeoxyguanosine at the modified base and at adjacent residues. Nature. 1987;327:77–79. doi: 10.1038/327077a0. [DOI] [PubMed] [Google Scholar]
- Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, Morrow JD, Van Remmen H, Sedivy JM, Yamasoba T, Tanokura M, Weindruch R, Leeuwenburgh C, Prolla T. Mitochondrial DNA Mutations, Oxidative Stress, and Apoptosis in Mammalian Aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- Kuo YM, Hayflick SJ, Gitschier J. Deprivation of pantothenic acid elicits a movement disorder and azoospermia in a mouse model of pantothenate kinase-associated neurodegeneration. J Inherit Metab Dis. 2007;30:310–317. doi: 10.1007/s10545-007-0560-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawton KA, Berger A, Mitchell M, Milgram KE, Evans AM, Guo L, Hanson RW, Kalhan SC, Ryals JA, Milburn MV. Analysis of the adult human plasma metabolome. Pharmacogenomics. 2008;9:383–397. doi: 10.2217/14622416.9.4.383. [DOI] [PubMed] [Google Scholar]
- Lin MT, Castelvetri IC, Zheng KK, et al. Somatic mitochondrial DNA mutations in early Parkinson and incidental Lewy body disease. Ann Neurol. 2012;71:850–854. doi: 10.1002/ana.23568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MT, Simon DK, Ahn CH, Kim LM, Beal MF. High aggregate burden of somatic mtDNA point mutations in aging and Alzheimer's disease brain. Human Molecular Genetics. 2002;11:133–145. doi: 10.1093/hmg/11.2.133. [DOI] [PubMed] [Google Scholar]
- Llanos A, Li Y, Mena P, Salem N, Uauy R. Infants with Intrauterine Growth Restriction Have Impaired Formation of Docosahexaenoic Acid in Early Neonatal Life: A Stable Isotope Study. Pediatr Res. 2005;58:735–740. doi: 10.1203/01.PDR.0000180542.68526.A2. [DOI] [PubMed] [Google Scholar]
- Logan A, Shabalina IG, Prime TA, Rogatti S, Kalinovich AV, Hartley RC, Budd RC, Cannon B, Murphy MP. In vivo levels of mitochondrial hydrogen peroxide increase with age in mtDNA mutator mice. Aging Cell n/a–n/a. 2014 doi: 10.1111/acel.12212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love S. Increased poly(ADP-ribosyl)ation of nuclear proteins in Alzheimer's disease. Brain. 1999;122:247–253. doi: 10.1093/brain/122.2.247. [DOI] [PubMed] [Google Scholar]
- McClay JL, Adkins DE, Vunck SA, Batman AM, Vann RE, Clark SL, Beardsley PM, Oord EJCG. Large-scale neurochemical metabolomics analysis identifies multiple compounds associated with methamphetamine exposure. Metabolomics. 2012;9:392–402. doi: 10.1007/s11306-012-0456-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecocci P, Beal MF, Cecchetti R, Polidori M, Cherubini A, Chionne F, Avelini L, Romano G, Senin U. Mitochondrial membrane fluidity and oxidative damage to mitochondrial DNA in aged and AD human brain. Molecular and Chemical Neuropathology. 1997;31:63–74. doi: 10.1007/BF02815160. [DOI] [PubMed] [Google Scholar]
- Mingorance C, Rodriguez-Rodriguez R, Justo ML, Herrera MD, de Sotomayor MA. Pharmacological effects and clinical applications of propionyl-L-carnitine. Nutrition Reviews. 2011;69:279–290. doi: 10.1111/j.1753-4887.2011.00387.x. [DOI] [PubMed] [Google Scholar]
- Mousavi M, Jonsson P, Antti H, Adolfsson R, Nordin A, Bergdahl J, Eriksson K, Moritz T, Nilsson L-G, Nyberg L. Serum Metabolomic Biomarkers of Dementia. Dement Geriatr Cogn Disord Extra. 2014;4:252–262. doi: 10.1159/000364816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas GS, Vargas CR, Wajner M. l-carnitine supplementation as a potential antioxidant therapy for inherited neurometabolic disorders. Gene. 2014;533:469–476. doi: 10.1016/j.gene.2013.10.017. [DOI] [PubMed] [Google Scholar]
- Riederer P, Hoyer S. From benefit to damage. Glutamate and advanced glycation end products in Alzheimer brain. J Neural Transm. 2006;113:1671–1677. doi: 10.1007/s00702-006-0591-6. [DOI] [PubMed] [Google Scholar]
- Ross JM, Öberg J, Brené S, Coppotelli G, Terzioglu M, Pernold K, Goiny M, Sitnikov R, Kehr J, Trifunovic A, Larsson N-G, Hoffer BJ, Olson L. High brain lactate is a hallmark of aging and caused by a shift in the lactate dehydrogenase A/B ratio. Proc. Natl. Acad. Sci. U.S.A. 2010;107:20087–20092. doi: 10.1073/pnas.1008189107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi MN, Carbone M, Mostocotto C, Mancone C, Tripodi M, Maione R, Amati P. Mitochondrial Localization of PARP-1 Requires Interaction with Mitofilin and Is Involved in the Maintenance of Mitochondrial DNA Integrity. J. Biol. Chem. 2009;284:31616–31624. doi: 10.1074/jbc.M109.025882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safdar A, Bourgeois JM, Ogborn DI, Little JP, Hettinga BP, Akhtar M, Thompson JE, Melov S, Mocellin NJ, Kujoth GC, Prolla TA, Tarnopolsky MA. Endurance exercise rescues progeroid aging and induces systemic mitochondrial rejuvenation in mtDNA mutator mice. Proc. Natl. Acad. Sci. U.S.A. 2011a;108:4135–4140. doi: 10.1073/pnas.1019581108. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Safdar A, Little JP, Stokl AJ, Hettinga BP, Akhtar M, Tarnopolsky MA. Exercise Increases Mitochondrial PGC-1 Content and Promotes Nuclear-Mitochondrial Cross-talk to Coordinate Mitochondrial Biogenesis. Journal of Biological Chemistry. 2011b;286:10605–10617. doi: 10.1074/jbc.M110.211466. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Saleem A, Safdar A, Kitaoka Y, Ma X, Marquez OS, Akhtar M, Nazli A, Suri R, Turnbull J, Tarnopolsky MA. Polymerase gamma mutator mice rely on increased glycolytic flux for energy production. MITOCH. 2015:1–8. doi: 10.1016/j.mito.2014.12.001. [DOI] [PubMed] [Google Scholar]
- Saper CB, Scammell TE, Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature. 2005;437:1257–1263. doi: 10.1038/nature04284. [DOI] [PubMed] [Google Scholar]
- Schliebs R, Arendt T. The cholinergic system in aging and neuronal degeneration. Behavioural Brain Research. 2011;221:555–563. doi: 10.1016/j.bbr.2010.11.058. [DOI] [PubMed] [Google Scholar]
- Schnaar RL, Gerardy-Schahn R, Hildebrandt H. Sialic Acids in the Brain: Gangliosides and Polysialic Acid in Nervous System Development, Stability, Disease, and Regeneration. Physiol. Rev. 2014;94:461–518. doi: 10.1152/physrev.00033.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholte HR, Busch HF, Luyt-Houwen IE, Vaandrager-Verduin MH, Przyrembel H, Arts WF. Defects in oxidative phosphorylation. Biochemical investigations in skeletal muscle and expression of the lesion in other cells. J Inherit Metab Dis. 1987;10(Suppl 1):81–97. doi: 10.1007/BF01812849. [DOI] [PubMed] [Google Scholar]
- Shah GM, Kandan-Kulangara F, Montoni A, Shah RG, Brind’Amour J, Vodenicharov MD, Affar EB. Methods in Molecular Biology, Methods in Molecular Biology. Totowa, NJ: Humana Press; 2011. Approaches to Detect PARP-1 Activation In Vivo, In Situ, and In Vitro; pp. 3–34. [DOI] [PubMed] [Google Scholar]
- Sreekumar A, Poisson LM, Rajendiran TM, Khan AP, Cao Q, Yu J, Laxman B, Mehra R, Lonigro RJ, Li Y, Nyati MK, Ahsan A, Kalyana-Sundaram S, Han B, Cao X, Byun J, Omenn GS, Ghosh D, Pennathur S, Alexander DC, Berger A, Shuster JR, Wei JT, Varambally S, Beecher C, Chinnaiyan AM. Nature. 2009;457:910–914. doi: 10.1038/nature07762. nature07762. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- Stein LR, Wozniak DF, Dearborn JT, Kubota S, Apte RS, Izumi Y, Zorumski CF, Imai SI. Expression of Nampt in Hippocampal and Cortical Excitatory Neurons Is Critical for Cognitive Function. Journal of Neuroscience. 2014;34:5800–5815. doi: 10.1523/JNEUROSCI.4730-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. U.S.A. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart JA, Brown MF. Mitochondrial DNA maintenance and bioenergetics. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 2006;1757:79–89. doi: 10.1016/j.bbabio.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly-Y M, Gidlof S, Oldfors A, Wiborn R, Tornell J, Jacobs HT, Larsson N-G. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature Publishing Group. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- Vermulst M, Wanagat J, Kujoth GC, Bielas JH, Rabinovitch PS, Prolla TA, Loeb LA. DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nat Genet. 2008;40:392–394. doi: 10.1038/ng.95. [DOI] [PubMed] [Google Scholar]
- Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Genetics. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf NJ, Butcher LL. Cholinergic systems mediate action from movement to higher consciousness. Behavioural Brain Research. 2011 doi: 10.1016/j.bbr.2009.12.046. [DOI] [PubMed] [Google Scholar]
- Yakes FM, Van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. U.S.A. 1997;94:514–519. doi: 10.1073/pnas.94.2.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Average food intake for in grams per day for each week of the experiment.
Heat map showing statistically significant changes in biochemicals profiled in this study.
Table showing results from the metabolic set enrichment analysis with p-value <0.05.