

Abstract
As is often the case for rare diseases, the number of published reviews and case reports of Glucose transporter type I deficiency (G1D) approaches or exceeds that of original research. This can indicate medical interest, but also scientific stagnation. In assessing this state of affairs here, we focus not on what is peculiar or disparate about G1D, but on the assumptions that have reigned thus far undisputed, and critique them as a potential impediment to progress. To summarize the most common G1D phenotype, we trace the 25-year story of G1D in parallel with the natural history of one of two index patients, identified in 1990 by one of us (G.M.R.) and brought up to date by the other (J.M.P.) while later examining widely-repeated but little-scrutinized statements. Among them are those that pertain to assumptions about brain fuels; energy-failure; cerebrospinal glucose concentration; the purpose of ketogenic diet; the role of the defective blood brain barrier; genotype-phenotype correlations; a bewildering array of phenotypes; ictogenesis, seizures and the electroencephalogram; the use of mice to model the disorder; and what treatments may and may not be expected to accomplish. We reach the forgone conclusion that the proper study of mankind - and of one of its ailments (G1D) - is man itself (rather than mice, isolated cells or extrapolated inferences), and propose a framework for rigorous investigation that we hope will lead to a better understanding and to better treatments for this and for rare disorders in general. These considerations, together with experience drawn from other disorders, lead, as a logical consequence, to the nullification of the view that therapeutic development (i.e., trials) for rare diseases could or should be accelerated without the most vigorous scientific scrutiny: Trial and error constitute an inseparable couple, such that, at the present time, hastening the former is bound to precipitate the latter.
Keywords: Glut1 deficiency, G1D, natural history, blood brain barrier, astrocytes, synapse, glutamate, GABA, thalamocortical, epilepsy, MRI, ketogenic diet, triheptanoin
INTRODUCTION
The syndrome of glucose transporter type I deficiency (G1D) was first identified and simultaneously inferred to arise from a transporter defect on the basis of clinical analytical evidence (i.e., decreased cerebrospinal glucose concentration) and protein abundance quantification (i.e., diminished glucose transporter abundance in red blood cell membranes) [1–3]. Many patients (often those with the most disabling symptomatology) manifest brain hyperexcitation or deregulated excitability that translates into epilepsy (Table 1). Equally incapacitating can be abnormalities of movement and muscle tone (Table 2). The only genetic locus for G1D known thus far is 1p34.2 [4]. Molecular analysis of SLC2A1 in this locus is productive for diagnostic purposes, but causal mutations remain unidentified in about 15% of patients ([5] and J.M.P., unpublished observations). Nevertheless, to date, diminished brain glucose accumulation has been demonstrated by positron emission tomography in all G1D patients studied with 18F-deoxyglucose used as a surrogate (but not a substitute) of glucose [6] (Figure 1), as autoradiography of the transgenic mouse brain with 14C-2-deoxyglucose (another tracer) also demonstrates [7]. DNA-testing-negative patients can be indistinguishable from DNA-positive patients, who appear to effectively behave as phenocopies (Figure 1). Many –but not all-epileptic, motor and other clinical manifestations respond to a ketogenic diet, which is typically maintained from diagnosis in infancy or childhood until seizures subside in adolescence [8]. Importantly, there is little evidence that the diet has any significant impact on cognition and individual autonomy. Research efforts have concentrated on the identification of further phenotypes or clinical variants and, simultaneously, on understanding the modulation of cellular and synaptic excitability [9, 10] and brain metabolism in G1D mice [11]. More recently, an increased rate of detection of G1D patients with (supposedly) non-epileptic movement disorders [12], which are sometimes considered less-severe disease variants, has kindled interest in the adaptation of the ketogenic diet [13] and nutritional variants, such as the modified Atkins diet, for the treatment of manifestations of –presumably- lesser import than epilepsy, such as dystonia, dyskinesia or ataxia. Indeed, diets are bound to remain the mainstay of therapy until new modifiable targets are identified, as anti-seizure medication and other drugs prove ineffective or insufficient in G1D [5, 14]. In this regard, triheptanoin, a food supplement additive to a regular diet in lieu of a portion of normally-consumed fat, has shown early promise both as a stimulant of brain metabolism in the G1D mouse brain [15] and as a potential therapy in human G1D [16]. Importantly, the generation of an array of animal models of the disease [17–19], among which antisense knockdown mice have proven most informative [7], is expected to shed light on some of these - but probably not on all - standing questions.
TABLE 1.
G1D syndromes
TABLE 2.
Movement disorders in G1D
| Abnormal movements | References |
|---|---|
| Spastic ataxia | [3] |
| Paroxysmal exercise-induced dystonia (DYT18) | [124] |
| Paroxysmal choreoathetosis/spasticity (DYT9) | [125] |
| Paroxysmal non-exertion dyskinesia | John H. Menkes and J.M.P., unpublished |
| Episodic ataxia | J.M.P., unpublished |
| Chorea | [128] |
| Dystonic tremor | [129] |
| Dysarthria | [3] and any other peer-reviewed publications |
Figure 1.
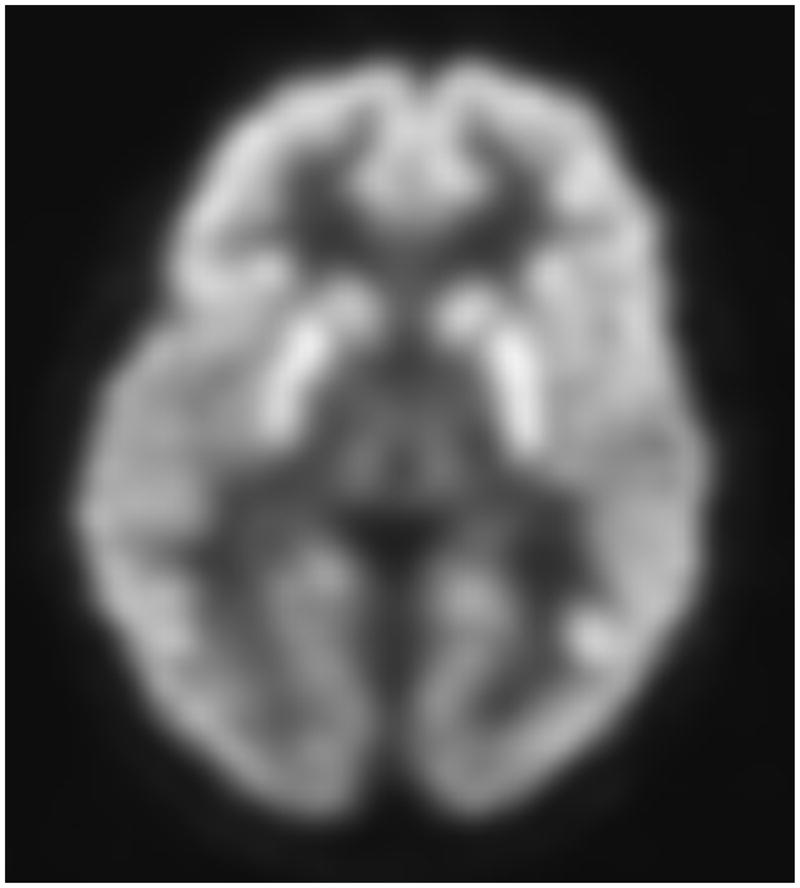
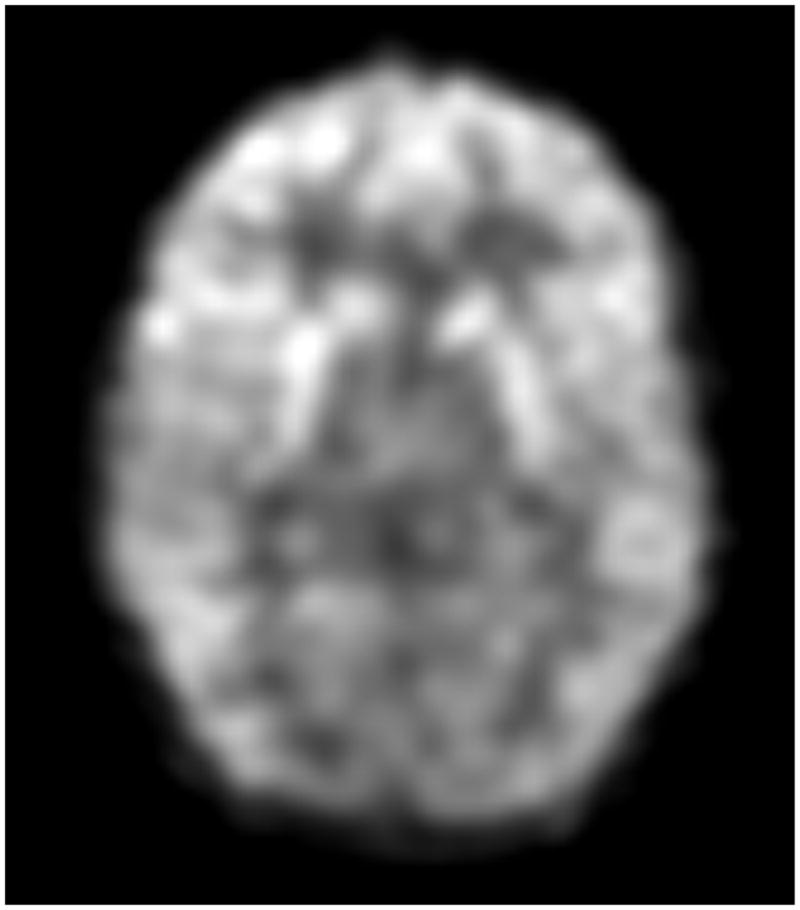
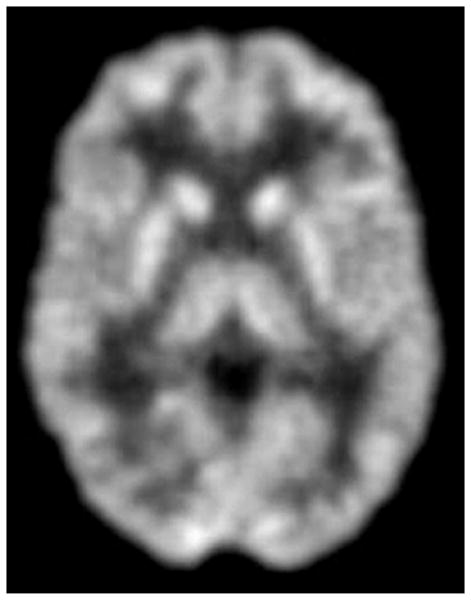
Transaxial FDG-PET images of a G1D patient of 9 years of age harboring a missense mutation (A) and of a 8 year old G1D patient with no mutation in the coding region of SLC2A1 (B). (C) illustrates a 20-year old normal scan for comparison. The images in A and B are virtually indistinguishable, illustrating the fact that DNA-mutation negative patients are phenocopies (in this and all other respects) of patients genetically diagnosed.
As summarized in Table 3, we review assumptions, established facts, and prospects for G1D, emphasizing how the various statements that have prevailed, whether new or old, have influenced progress for patients. At the outset, we adopt the view that many seemingly unsolvable scientific problems are not solved, but simply dissolved once they are placed in proper context and their premises accurately critiqued. The diagnosis and natural history of one of the index patients followed for 25 years serves as our guide in this endeavor. This case report part of the review illustrates the most common form of disease presentation while prefacing a discussion of clinically-relevant mechanisms that can or should inform therapeutic decisions.
TABLE 3.
Developmental milestones and neurological signs of the G1D index patient
| Age in months | Clinical observations and examination |
|---|---|
| 1–2 | Smiles, raises head |
| 2 | Fails to follow completely with eyes |
| 5 | Both hand fisted, no purposeful hand movements |
| 6 | Rolls over, puts both hands together |
| 8 | Socially interactive; babbles; hands show increase in use though fisted, good palmar grasp; tone increased of the left ankle, otherwise normal |
| 10 | Maintains sitting position but tends to lose balance when reaches out with hands; crawls; mild hypertonia & hyperreflexia |
| 11 | Gets into sitting position |
| 12 | Pulls up to standing and cruises along furniture |
| 14 | No pincer grasp; arms and hands flexed; tightened heel cords |
| 17 | Easily distractible; DQ= 77 |
| 22 | 4-word vocabulary; drinks independently from a cup; starts using a spoon. Left hand preference; stands on toes; increased tone right arm |
| 25 | Walks independently |
| 28 | Starts identifying body parts; uses gestures and pointing to communicate; 10–20 poorly intelligible words; clumsy; wide based ataxic gait; spasticity with scissoring; extensor plantar response; DQ=73.6 |
| 31 | Over 20 word vocabulary; decreased stumbling. |
| 36 | Rich single word vocabulary; 2-words utterances; difficulties with concepts of colors, sizes, spatial relationships and the negative; cannot identify animals; improved fine motor control; intermittent worsening ataxia with truncal titubation and intention tremor; drools; head banging behavior. |
| 48 | Puts words in brief phrases; |
| 60 | Short sentences, able to dress and use buttons and zippers; copies a circle; holds to the rail while climbing stairs; less ataxic but developed dystonic and athetoid movements; intermittent urinary retention. Head circumference continued to expand along the 50th percentile. |
| 72 | dysarthria |
| 96 | Difficulties grasping concepts at school; mild head tremor; able to run without difficulties; not able to hop on one foot; difficulties with fast repetitive movements; not able to tie shoelaces; still needs help wiping in the toilet. |
| Age in years | Clinical observations and examination |
|---|---|
| 12 | Cooperative with mild reinforcement but is easily inattentive and distractible; speech with a slurred staccato quality but understandable; rapid alternating hand movements were slow with mild clumsiness when reaching for objects; able to hop on either foot clumsily. Deep tendon reflexes were slightly brisker in the right arm and left leg and plantar response was extensor on the left and equivocal of the right. |
| 22 | No significant change in observations or exam. Cooperative and distractible. Dysarthria, spasticity and ataxia. |
| 25 | No change in observations or exam |
THE G1D SYNDROME
Human GLUT1 deficiency (G1D) due to mutation of the gene SLC2A1 is associated with partial loss-of- function (i.e., GLUT1 activity is never abolished) and results in decreased brain glucose accumulation [6]. G1D manifests as an encephalopathy frequently (but not exclusively) characterized by medication-refractory infantile-onset seizures [14], diminished (n=26 of 86 patients) or normal (n=59 of 86 patients) encephalic mass, intellectual impairment, and complex (i.e., multiform) motor disturbances (spasticity, ataxia, chorea, dystonia and combinations thereof) [2, 3, 20, 21]. This is often accompanied by reduced cerebrospinal fluid (CSF) glucose and lactate concentration. Notably, a series of important neural function defects in G1D are independent of blood-brain barrier GLUT1 dysfunction and can be ameliorated by massively increasing brain tissue glucose concentration (over 10-fold of normal), as we have shown in the G1D mouse [7, 9, 22].
In man, G1D is associated with a reduction in brain glucose transport [23, 24] and, most frequently, drug resistant epilepsy with a ~3 Hz spike-wave EEG pattern suggestive of thalamocortical dysfunction [25] and diminished FDG (18F-deoxyglucose)-PET signal emanating from cerebellum, thalamus and cerebral cortex, highlighting a relatively increased glucose accumulation in the striatum [6, 26], particularly in the caudate nucleus (which exhibits about 117% FDG signal intensity relative to the total intensity arising from the rest of the brain; J.M.P., n=7 patients). In fact, clinically-available FDG-PET can be useful for diagnosis in cases where CSF glucose or genetic testing are close to normal. Movement disorders can be as clinically prominent (Table 1) and may be present in isolation or as part of a broader syndromic combination. G1D does not impact systemic carbohydrate metabolism. Patients develop seizures (often >100/ day, personal observations (n=126 patients) and [25]) in infancy. Epilepsy in G1D patients can be ameliorated by a ketogenic diet [3, 8] or by inducing hyperglycemia [27]. Human cerebral metabolic rate (expressed in mL O2 consumed/100 g tissue/min) also increases after glucose ingestion [16]. However, hyperglycemia is not sustainable nor advisable from a broader medical viewpoint, and the ketogenic diet does not eliminate seizures in many patients nor does it remediate cognitive or other manifestations such as spasticity or ataxia ([8] and J.M.P. and G.M.R., unpublished observations (n=128 patients). No human neuropathological studies have been reported. Brain MRI is often normal, except for prominent Virchow perivascular spaces in the cerebral lobes resembling U-fiber leukoencephalopathy when imaged at 1.5 T magnetic field strength (J.M.P., n=6 patients).
The most commonly observed epileptic syndromes include absence, atypical absence or myoclonic astatic epilepsy, often associated with a spike-wave EEG pattern, ataxia, spasticity, dysarthria and lower limb (predominantly exertion-induced) dystonia. These patients appear normal at birth and some experience a progressively abnormal decrease in the rate of brain growth. Fragmentary seizures typically start at 3 to 6 months of age and coincide with or follow episodic oculomotor crises, first defined here as ‘intermittent involuntary gaze’ (IIG)1. As the infant matures, IIG subsides, but ataxia, dysarthria and spasticity eventually set on and tend to persist indefinitely. Dystonia may become more apparent after the age 3 of years. Movement disorders are often refractory to therapy. Only rarely are seizures controlled by anti-seizure medication alone [G.M.R. and J.M.P., n=128 patients]. In fact, as is also the case with other epilepsies, anti-seizure drugs may exacerbate these manifestations. If left untreated, the severe epilepsy typical of the disorder may eventually follow a relatively benign, age limited course with improvement of seizures following puberty. However, adolescence may herald either the worsening or amelioration of seizures and of obsessive-compulsive traits ([28] and J.M.P. n=28 patients). At the present time, however, the natural history of both the common epileptic form and especially of the movement disorder forms of G1D remain underinvestigated, such that these observations are to be considered preliminary. The effects of life long trajectories are unknown. However, adult manifestations seem to persist indefinitely [28] and there is no reason to expect a diminished lifespan.
A 25-YEAR INVESTIGATION OF THE INDEX PATIENT
We describe the presentation from birth (G.M.R.) and 25 years of continued assessment (G.M.R. and J.M.P.) of an index patient with G1D, who is emblematic of the most common phenotype of the disorder. The early part of this natural history account was first described 25 years ago [2, 3]. This proband was born to non-consanguineous parents of English descent. A detailed family history of 4 generations with 83 identified individuals revealed seizures in three and migraine headaches in five maternal relatives. Pregnancy was associated with excessive nausea. She was born at term with a weight of 3350 grams and head circumference of 35cm. Her Apgar scores were 7 and 9 at 1 and 5 minutes respectively. Her development seemed comparable to her two siblings during the first 2 months of life. From two months onward she had recurring paroxysmal events and presented increasingly with neurodevelopmental impairments. The first witnessed episode occurred at 2-months with sudden onset of pallor, dazed facial expression and unresponsiveness of 5-minutes duration with gradual recovery. The second witnessed episode occurred 2-weeks later and was characterized by roving horizontal eye movements, unresponsiveness, hypotonia and head ‘bobbing’ (which G.M.R. interpreted as possible myoclonus) lasting for 10 minutes and followed by brief postictal phase. Identical episodes were observed every 2-weeks for the following 3-months. By the age of 5-months she was experiencing 8–9 daily events, lasting minutes to hours, of decreased responsiveness associated with a vacant (“sleepy”) facial expression, with her eyes either in a fixed staring ahead position or roving horizontally. During this period four interictal EEGs were normal. Different episodes occurred at 8-months just prior to feeding. She was found unconscious with few respiratory efforts, becoming cyanotic. Gradual but incomplete recovery began after a few minutes though she would neither vocalize nor play for hours. Six hours later she turned again cyanotic, her eyes turned upward to the left and appeared jerking. This was followed by left facial twitching and subsequently by unresponsiveness associated with shallow breathing. Her recovery was gradual, characterized by aimless arm movements and crying. Ninety minutes later she “snapped” back to her normal self.
Sodium valproate (up to 50mg/kg/day) was commenced at 12 months and discontinued two months later for lack of seizure control, nausea and drowsiness. Carbamazepine, which was introduced at 17 months, was also ineffective. Repeated assumed seizures during the second year of life consisted of upward eye deviation and head shaking lasting about 5 seconds each. The frequency of these seizures gradually increased from 2–3 daily at 17-months up to 25 at 22-months and intermittently occurred in clusters. By then she had 10 normal interictal EEGs. However, at 27 months one EEG recording showed brief paroxysmal sharp-slow waves over both frontal head regions clinically associated with upward deviation of the eyes and eyelid blinking.
Investigations from 1989 onwards identified hypoglycorrhachia with mean CSF glucose value of 1.6 mM (range 1.4 to 1.8 mM), mean glucose CSF to blood ratio of 28% (range 20% to 35%) CSF lactate mean value of 1.3 mM (range 0.8 to 1.5 mM) and a range of serum lactate values from 1.0 to 1.4 mM. Causes for secondary hypoglycorrhachia and intermittent hypoglycemia (i.e., primarily common endocrinological disorders) were rigorously explored. Hypoglycorrhachia due to a presumed transport defect was determined in 1989 with a glucagon-loading test where the CSF glucose level remained low at 1.8 and the blood glucose levels increased to 9.4 mM. The clinical diagnosis of G1D was further corroborated while attending the presentation of the other index patient at the Child Neurology Society meeting in 1990 [1]. Glut1 activity was measured in membranes isolated from her erythrocytes using specific D-glucose displaceable 3[H] cytochalasin B binding. The binding to her erythrocytes membrane was 157-pmol/mg protein at a concentration of 1 μM (3-SD below mean for controls= 294, SD= 38). Saturation kinetics showed that the low binding was due to reduced number of binding sites, not lower affinity of binding [3].
A therapeutic ketogenic diet was introduced at 27 months and within 1-week there was a reduction in seizure frequency. She continued to have rare clusters of seizures from 3 - 3.5 years, characterized by pallor or cyanosis, stumbling gait, unresponsiveness, eyes staring upward, limpness and sparse clonic jerks. The last of these seizures happened at age five years. Episodes of hyperactive and aggressive behavior, lethargy, inability to walk and slowed speech ensued approximately 24 hours after violation of the ketogenic diet even in the presence of highly elevated urinary ketones. Furthermore, she continued to have “good days and bad days” of intermittent ataxia without any identifiable triggers. In addition, she experienced severe dehydration during intercurrent infections. From age 4 years onward she displayed identical paroxysmal episodes characterized by autonomic features. She developed dusky coloring and cool skin, decrease in heart rate, gradually slowed down her activities, felt fatigued but retained consciousness and then fell asleep for 15–20 minutes. After sleep she would function at her normal self. These episodes were different than her earlier seizures. A week of video-EEG recording at age 6 years showed no electrographic paroxysmal abnormalities despite a clinical event during sleep where she became pale, felt cool with a regular heart rate of 80/minute, followed by brief eyelid blinking. A cardiac evaluation was also normal. This coincided with the beginning of what would eventually become common and frequent migraines. At 7-years of age, she was easily fatigable and on occasional days she wanted only to sleep. Otherwise she seemed active but affected by adverse effects of the diet such as periodic vomiting, nausea, constipation and abdominal pain. At school she received modified educational programs. At 22 years old, the evolving syndrome had stabilized and the clinical manifestations included spastic ataxia, migraines and brief absence episodes upon nocturnal fasting. Her attitude was cooperative. Major seizures had disappeared. By age 25, poor intellectual, scholastic and training achievement continued to preclude employment. Fasting EEG demonstrated brief generalized 3 Hz spike-wave discharges that occasionally lasted over 3 s. These events coincided with eyelid blinking and absences. Ingestion of 100 g of glucose effectively terminated these discharges in 45 min, resulting in increased attentiveness and comprehension.
The following principles summarize disease pathophysiology to the extent that they inform clinical practice.
BIOCHEMISTRY OF BRAIN METABOLISM
The facilitative transporter GLUT1 is the hexose (predominantly glucose) carrier of blood brain barrier capillaries and astrocytes (Figure 2) [29]. GLUT3 and less abundant homologs allow glucose that has penetrated the brain’s extracellular space flow through neuronal and other membranes (Figure 3)[30]. Facilitative transporters are independent of any co-transport requirements or energy to undergo the conformational changes associated with substrate translocation across the cell membrane. If an unmetabolized glucose molecule were to traverse the brain until it is consumed into pyruvate or stored in glycogen, it could potentially be exposed to a large number of cell membranes arranged in series, which would impart a significant gradient across each permeation step. On the other hand, however, glucose has ample opportunities to be metabolized and resynthesized before being deposited in glycogen-storing cells such as astrocytes [31]. These considerations have rendered any estimation of the metabolic glucose gradient impracticable. Therefore, it is prudent to assert only that GLUT1 deficiency (G1D) may impair both glial and neuronal metabolism, including neurotransmitter metabolism as described below. GLUT1 haploinsufficiency leads to human and murine epilepsy [3, 17, 32], whereas murine GLUT3 haploinsufficiency leads to more modest abnormalities [33]. GLUT1 protein quantification is insufficient to characterize brain metabolism, as G1D leads to regional overexpression of other transporters ([18] and western blot data illustrating 0 to 50% GLUT3 overexpression in mouse G1D cortex, thalamus, striatum or cerebellum; J.M.P., n= 16 mice). After glucose entry in its destination cell, neuronal or glial glycolysis generates most of the brain’s pyruvate, which, together with the oxidation of brain fatty acids and of ketones produced in the liver or in glia [34], leads to the synthesis of acetyl-CoA used by the TCA cycle. A fraction of glucose-derived pyruvate is separately used for brain anaplerosis [35, 36]. Glia can also generate lactate from glucose (Figure 3), which is transferable to neurons via monocarboxylate transporters for pyruvate generation [37]. Most de novo neuronal glutamate synthesis derives from α-ketoglutarate produced in the TCA cycle. Most brain glutamine derives from glutamate via glutamine synthase in glia and can be exchanged during neurotransmission such that neurons release can release and reuptake glutamate, whereas glia can also take it up and convert it into glutamine for potential transfer to neurons in a cycle [38, 39]. A similar cycle can occur in inhibitory synapses [40], where GABA derives from glutamate [41] via an ATP-consuming reaction. Only ~1/5 of the total glucose consumed in neurotransmitter synthesis is related to GABA synthesis [42].
Figure 2.


Expression of phosphorylated GLUT1 in the capillaries of the mouse brain. A: Normal mice. B: G1D mice. G1D is associated with a paucity of immunofluorescence arising from decreased phosphorylated GLUT1 in the context of decreased total GLUT1 protein (not shown). Staining performed with reagents described in [123].
Figure 3.

Schematic of brain glucose flux and metabolism Figure 1. Brain energetic substrate fluxes and glucose transporters. Cells represented include astrocytes (blue), capillaries (red) and a synapse (green). Blood glucose reaches neurons or astrocyes [124], where it is converted to glycogen (not shown), or exported to neurons either intact via GLUT1 (□) and GLUT3 (■), or following conversion into lactate. Ketone bodies readily access the brain through monocarboxylate transporters (not represented). Transporter-independent fluxes (probably of limited magnitude [125, 126]) may also occur.
THE G1D MOUSE
Stable G1D knockdown mouse lines were first generated by antisense transgene incorporation [19], followed by a hemizygous knockout line [17] and a similar model [18]. Many informative studies to date have been obtained in the G1D antisense line [7], which expresses about 50% of total brain GLUT1 brain protein. These mice are spontaneously and visibly epileptic. In contrast, the hemizygous line manifested a modest 34% reduction in total brain GLUT1 protein and seizes only upon fasting [17]. G1D (antisense) mice display frequent spike-wave seizures, ataxia and poor rotarod performance.
The antisense mouse allowed addressing a preliminary question central to G1D: Whether brain glucose influx is significantly reduced in the G1D mouse model. This was confirmed by mouse PET (performed as in [17] with additional normalization to muscle [43]; J.M.P. n=12 mice) and, in greater topographic detail, by emulsion autoradiography via systemic injection of radiolabeled glucose illustrative of deficient cortical and thalamic accumulation as described below. The next question addressed was whether (a) these observations stemmed solely from reduced blood brain barrier (BBB) glucose penetration or (b) BBB-independent astrocyte uptake was impaired. This question could not be addressed a priori because the magnitude of astrocyte glucose transport taking place in normal brain tissue is debated, often in relation to uncertainty about the magnitude of neuronal lactate consumption [44–48]. Moreover, from a therapeutic perspective, disproving (b) would oblige to focus on (a). Consistently with (b), primary G1D astrocyte cultures exhibited diminished glucose uptake (~50% reduction in Vmax; not shown). Scenario (b) was additionally and more pertinently demonstrated in cortical brain slices (which are devoid of BBB), which exhibited profound synaptic dysfunction as described below. This dysfunction depended rapidly and reversibly upon bath glucose or acetate concentration. Of note, acetate is an alternative source of acetyl-coenzyme A.
The third question was that of the potential role of TCA cycle precursor depletion impacting the synapse via reduced neurotransmitter and/or energy generation (a direct consequence of TCA cycle intermediate depletion). To render the latter hypothesis testable, the production of acetyl-CoA, glutamate and glutamine that occurred in the cerebral cortex of the mouse was determined. Glutamate and glutamine are uniquely rich in metabolic information related to the TCA cycle [49–52] and can lend themselves to precise multiplet 13C NMR analysis [53, 54] and to mass spectrometry, accomplished by us for the first time by controlled infusion of 13C-labeled substrates in the awake mouse [54]. Using 13C-glucose, the G1D cortex displayed lower acetyl-CoA abundance and 13C enrichment derived from glucose, which was paralleled by lower 13C-glutamate enrichment. Importantly from a therapeutic point of view, the 13C-glutamate labeling pattern, indicative of TCA cycle integrity, was intact. Therefore, the simplest interpretation is that G1D is associated with both impairment of acetyl-coenzyme A production (due to reduced glucose availability) and reduced glucose-dependent neurotransmitter production, without overall disruption of TCA cycle integrity due to increased consumption of alternative substrates. This militates against energy failure in the G1D brain.
Consistent with a brain acetyl-coenzyme A deficit in G1D, infused odd-carbon fatty acid (13C-heptanoate) replenish G1D acetyl-CoA and TCA-derived glutamine more effectively than in normal mice [15].
Complementary investigations in brain slices reveal additional hyperexcitability mechanisms manifested as spontaneous thalamic electrical oscillations (Figure 4). This is the basis for hypothesizing driven (superimposed onto intrinsic cortical) epileptogenesis in G1D. This observation invokes selective neural network vulnerability, an important theme both in epilepsy and metabolic disorders [55].
Figure 4.
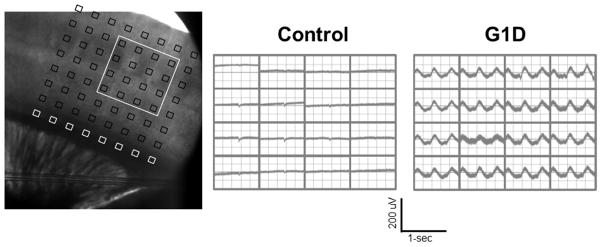
Thalamocortical synchronization in G1D. Left: Thalamocortical slice preparation with 64-channel multi electrode array (MEA) positioned over the sensory barrel cortex. Center: Normal mouse: Field potential recording from MEA array positioned as in the left showing the 16 electrode positions as designated by the white box outlined in left. Right: G1D mouse: note the ~3 Hz oscillation in all the electrodes indicative of thalamocortical hypersynchronization. Slices were bathed in drug-free, 2.5 mM glucose-containing perfusate [9].
The mouse epileptic phenotype is characterized by a spike-wave EEG pattern typical of other models of rodent thalamocortical dysfunction [56]. Several lines of evidence including western blots of GLUT1 and GLUT3 protein, multielectrode array and electrical field potential recordings from thalamocortical slices, and high-resolution emulsion autoradiography using 14C-2-deoxyglucose (2DG) demonstrate thalamic dysfunction in G1D[57]. Autoradiography was performed in young adult (39-day-old, P39) mice by intraperitoneal injection of 2DG in conscious animals: G1D mice exhibit reduced 2DG signal preferentially in the thalamus (53% of normal) and cerebral cortex (full thickness) (61%) relative to other areas such as the cerebellar cortex (70%) [7].
GLUT1 expression (relative to GAPDH, determined as in [23]) in G1D was similarly decreased in thalamus, cortex and whole brain. However, GLUT3/ GAPDH expression was significantly increased in whole brain and cortex but not in thalamus, suggesting that neuronal glucose metabolism may be preferentially impaired in the thalamus (J.M.P. n=28 mice).
MECHANISMS OF G1D ENCEPHALOPATHY
Carbon-deficit
We do not view G1D as an energy failure syndrome, but as a carbon-deficit disorder on the basis of our work cited above and of additional inferences. This clarification is more than terminological: Both of these disease mechanisms can impact synaptic function or the rate of brain growth [9, 22, 24] and, therefore, such (and similar) observations constitute little evidence for or against either mechanism. However, the former mechanism (i.e., a loss of brain energy in G1D) is hardly compatible with the demonstration (in G1D mice) of intact TCA cycle metabolite abundance [7, 15] and flux by NMR, intact brain ATP contents (J.M.P., unpublished), lack of cell death [7, 58], and with the distinct clinical course of G1D [24, 59] relative to all canonical ATP-synthetic (i.e., energy-failure) defects [60, 61]. Carbon-deficit, on the other hand, is supported by depletion of acetyl-CoA [7], abnormal mobilization of fatty acids [7] and kinetically-predictable, rapid overincorporation of carbon-13 in brain glutamine derived from infused heptanoate in the G1D mouse [15]. In summary, if there is no energy failure in G1D and the TCA cycle is preserved, what C7 would stem is the re-routing or overutilization of an array of possible alternative brain metabolic sources normally needed for other aspects of neural function. As proof of principle, we have identified some of these depleted alternative substrate sources [7] (which are replenishable by C7 [62, 63]).
Synaptic dysfunction
The mouse sensory barrel cortex (which is included in the coronal or thalamocortical brain slice) is a pertinent experimental preparation because of high-glucose consumption and robust excitatory and inhibitory currents, ideally suited to directly investigate epileptogenesis by metabolic or pharmacological synaptic manipulation by superfusion [64–66]. At postnatal day 28 (P28), a commonly studied age when electrophysiological slice recordings can be readily performed, GLUT1 expression and cortical glucose metabolism are robust. G1D mice are fully symptomatic (due to ataxia and seizures) at P28, shortly after weaning from maternal (high-fat) milk has been completed [67] and the brain relies primarily on glucose metabolism. Basic, passive neuronal electrical properties are normal in G1D cortex. Generally, in epilepsy, a preserved neuronal membrane potential, such as that found in G1D, leads to consideration of 1) transmitter presynaptic release or 2) postsynaptic action as potential, non-exclusive disease mechanisms assuming that cell structure is preserved (and rapid amelioration by glucose in G1D argues that this is a reasonable assumption). These processes may be disrupted by impaired glucose metabolism. Cortical mouse brain slices maintained in high glucose show a small, significant decrease in mean miniature postsynaptic current (mPSC) amplitude in G1D, indicative of altered spontaneous synaptic transmission onto pyramidal neurons that can be further and rapidly reduced in sustained fashion by decreasing bath glucose closer to physiological concentration [9]. In these preparations, there are no changes in rise or decay current time constants, suggesting either diminished synaptic neurotransmitter release, or impaired postsynaptic receptor action [68]. To address mechanisms, excitatory and inhibitory currents can be further separated pharmacologically. Following block of action potentials to facilitate recordings, excitatory currents (mEPSC) are readily measurable. Under these conditions, mEPSC amplitude is reduced in G1D relative to normal cerebral cortex. This is exacerbated in low glucose without alteration of mEPSC frequency. On the other hand, inhibitory GABAergic current (mIPSC) amplitude is also but more significantly decreased at high and normal glucose concentrations, with an additional decrease in mIPSC frequency, suggesting more pronounced inhibitory transmission impairment. These findings constitute the basis for postulating an excitatory/inhibitory imbalance in G1D cortex [9, 10].
G1D as a modifiable brain functional state
The extent of functional reversibility in G1D is highlighted by patient EEG normalization under hyperglycemia ([26, 27], see also above) and brain-slice patch-clamp in our mouse model. These observations support the postulate that G1D leads to metabolic substrate-dependent synaptic dysfunction. In G1D mice, spontaneous miniature synaptic currents are decreased under low glucose (i.e., inhibitory currents are more reduced than excitatory currents) in the cerebral cortex. These effects, which are pronounced under bath low glucose, are quickly reversed with perfusion chamber solution replacement ([9, 22], J.M.P., n= 25 brain slices). Epileptogenic hyperexcitability also manifests as a similarly reduced evoked (induced) cortical inhibition after stimulation (not shown), and as glucose-responsive thalamocortical hypersynchronization (Figure 4 and[7]). Importantly, ketone bodies also [partly reverse synaptic function abnormalities in G1D (J.M.P., manuscript in preparation). Altogether, these observations are critically important for the design and expectations of any therapy that aims to restore glucose metabolism or synaptic excitability homeostasis in the brain.
Preservation of metabolism in the G1D mouse
Complementing the above results, the capacity for normal brain metabolism is not impaired in G1D as revealed by 13C-NMR and GC-MS studies conducted as in [15, 69, 70]. This is important for any metabolic-substrate related therapy development that relies on the restoration of normal metabolic reactions. We were prompted to conduct these experiments by patient observations [27] illustrating a rapid, favorable EEG response to glucose: Infusion of [U-13C]glucose in G1D and normal mice (n=9 for each group) revealed that: 1) the time course of 13C contents of glutamate and glutamine was equivalent between both animal groups, indicating that glucose oxidation through the TCA cycle is preserved; 2) the concentration of amino acids and TCA cycle intermediates was also no significantly different; 3) the ratio of [1,2-13C]acetyl-CoA:[U-13C]glucose in brain, that is, the fraction of 13C-acetyl-CoA derived from 13C-glucose, was not statistically different either. This is also in accordance with 4) [U-13C]acetoacetate (a ketone body) infusions: the [1,2-13C]acetyl-CoA:[U-13C]acetoacetate ratio in brain was also not significantly different. Therefore, G1D does not irreversibly alter brain metabolism.
Limitations of rodent studies
Because there are important metabolic and ictogenic differences between man and many rodent models, rodent inferences based on the use of ingested C7 in epileptic animals (including ours below) are best understood in context. First, ingested fat metabolism in the mouse is very different from that in man [71], reflecting changes in hepatic mitochondrial metabolism, lipid deposits and their mobilization [72] (for example, mice eat much more frequently than men, and this affects metabolite level stability in blood [73]). These findings derive from diabetes and obesity research. Thus, our mechanistic work on heptanoate effects on G1D mouse brain metabolism [15] was carefully designed to be as relevant as possible to potential therapeutic development while circumventing some of these limitations, because jugular-infused heptanoate [15] initially bypasses the liver. To accomplish this, we devised a method [69] to perform these infusions in the conscious mouse that safeguards brain metabolism integrity (i.e., in a state unsuppressed by anesthesia [74]). Second, in contrast with epilepsy in G1D patients or synaptic dysfunction in the thalamocortical or coronal cortical brain slice, which are largely stereotypic in their own sense, no mouse model of G1D (including our best model [7], used here) has proven a sufficiently controllable epilepsy testbed for preclinical investigation. This applies both to mouse G1D EEG (see data below) and observable absences (which are the predominant seizures human G1D, but cannot be reliably visually rated in mice). These statements are also based on previous work: We and our close collaborators have characterized a variety of G1D mouse lines over the past decade [7, 17, 19], each expressing a different amount of brain GLUT1 protein, including 4 lines studied recently (J.M.P.) for the specific purpose of preclinical investigation. Epilepsy in these mice is either: 1) present only upon fasting [17] or absent in the higher-GLUT1 expressing lines; 2) variable from mouse to mouse and within a mouse for the model used here (~50% residual GLUT1 abundance [7]; EEG data below); or 3) absent for 2 lines exhibiting smaller degrees of GLUT1 protein abundance reduction (unpublished). For all other purposes, our G1D mice [7] represent a faithful disease model because they are fully symptomatic (including epilepsy), display severe ataxia and inability to swim or be rotarod-trained (and are not thus amenable to behavioral testing that requires normal motor performance), all of which resembles common, severe human G1D.
HUMAN G1D OBSERVATIONS
Ictogenesis, spike-waves and absences: epilepsy in G1D
In line with the necessarily reductionist approach outlined above, we have selected EEG parameters as an important outcome for go/no-go decisions. However, there are many non-observable biological aspects in epilepsy and, consequently, the field continues to be driven by the most reliable and accessible indicators, which have been in use for decades (for example, EEG, quality of life and cognitive assessments). Best treatments include those that target causes, as we believe C7 does. But observation need not be synonymous with causation: Absence seizures, such as those noted in G1D, are temporally well-circumscribed impairments of consciousness associated with visually-observable manifestations [75]. During a typical absence, EEG recording reveals repetitive spike-waves [76]. Yet, observable absences, EEG and sustained attention can be differentially impacted by drug treatment [77, 78], supporting the limited-causality clause above. New work expands these considerations: Because of the reliability of fasting spike-waves in G1D, we have conducted simultaneous EEG and fMRI in 6 G1D patients [79] (J.M.P. and cols., manuscript in preparation). Both “activated” and “deactivated” brain regions were identified by BOLD changes. In all subjects, cortical deactivations simultaneous with spike-waves were more prominent than cortical activations. The visual (5 out of 6 patients) and motor areas (6/6) and the cerebellum (5/6) were activated during seizures, while the prefrontal gyrus (6/6), caudate (6/6), the default mode network [80] (5/6), and the spinal cord (4/6) were deactivated. These results suggest that neural-network level phenomena are observable in G1D and constitute a novel paradigm in which interventions may eventually be assessed.
TREATMENTS FOR G1D
Limitations of G1D treatments
The mechanistic understanding and treatment of neurometabolic disorders have suffered from a lack of innovation. Consequently, most G1D research has yielded modest benefits. For example, the finding of apoptosis in cultured G1D mouse embryonic cells [81], led to the search for brain cell death. However, apoptosis seems insignificant in the G1D mouse brain [7, 58] and human MRI scans are generally unremarkable except for mildly diminished myelin abundance [26]. Analogously, the potential role of specific anticonvulsants in G1D was suggested by its spike-wave EEG pattern [25, 82] typical of absence epilepsy [83], but this approach also proved ineffective [22]. Furthermore, regardless of our in vitro cell culture observations [84], most anticonvulsants may paradoxically worsen epilepsy in G1D patients (some, in fact, may inhibit residual glucose transporter function in vitro) and, as a result, are used on a trial and error basis [82, 85–88]. This may propitiate diagnostic biases, since patients whose epilepsy responds well to an antiseizure drug seem less likely to receive metabolic or genetic testing. In contrast, the ketogenic diet, which was first justified for G1D on biochemical grounds [3, 89] and has not been studied in clinical trials in G1D, ameliorates epilepsy in ~2/3 of patients (J.M.P. observations, n=126 patients and [82, 90]). Unfortunately, virtually all other neurological deficits (including cognition and motor performance) persist under the diet in the vast majority of patients, and many others experience reduced tolerability [8]. Thus, at the present time, most therapeutic initiatives for G1D are limited to early diagnosis and initiation of a ketogenic diet or a variant of it [82, 91]. Other interventions such as lipoic acid have not been rigorously evaluated.
Bases for the dietary treatment of G1D: the ketogenic diet
G1D and other metabolic encephalopathies cause seizures that are generally refractory to medication [14, 88]. In these diseases, epilepsy is not unexpected, given the close link between brain glucose influx, the TCA cycle and the metabolism of glutamate and GABA, observed both in vitro and in vivo in man and animals [15, 69, 70, 92, 93]. However, these disorders have remained underexplored, in part owing to their apparent rarity, now contested [20]. They can also elude biochemical principles: For example, excitotoxicity, typical of acute brain glucose deprivation [94], is not observed in G1D [7], while astrocyte-neuron neurotransmitter cycling, postulated to sustain neurotransmission [38], may not occur in all epileptogenic structures [95–97], such that this limits any therapeutic development that may primarily rely on these premises. A ketogenic diet, given with the therapeutic intent of stimulating brain metabolism in G1D, may act through several mechanisms [13, 98], including the production of acetyl coenzyme A (acetyl-CoA) that enters the neural TCA cycle [99, 100], and can abolish seizures in ~2/3 of G1D patients (J.M.P. observations, n= 126 patients and those of two other independent groups [82, 90]). However, ketogenic diets are not universally tolerable [101–105] and do not sufficiently alleviate other incapacitating features of G1D (e.g., intellectual impairment, ataxia, dysarthria [8]), such that only ~5% of young adults with G1D receiving a ketogenic diet are employed or sub-employed (J.M.P., n= 21 patients and [28]).
In this regard, the ketogenic diet, which is used for the treatment of G1D solely on the basis of biochemical assumptions and empirical observations and has not been the subject of a controlled clinical trial in G1D [24], ameliorates some movement disorders and epilepsy in 2/3 of patients (JMP, unpublished, and [82, 90]). Unfortunately, neurological deficits, particularly those centered on other aspects of movement coordination such as ataxia and dysarthria, and cognition tend to persist under the diet, with a significant fraction of patients experiencing recurrent or incompletely treated abnormalities [8], whereas the remaining 1/3 of incompletely-treated or diet-unresponsive patients face an even more problematic clinical course. The diet, given with the therapeutic intent of stimulating brain metabolism in G1D, may act as an anti-seizure treatment through several potential mechanisms [13, 98], including the production of acetyl-coenzyme A that can stimulate the neural TCA cycle [99, 100], but its mode of action has not been investigated in G1D. Importantly, in addition to insufficiently alleviating all incapacitating features of G1D [8], ketogenic diets are not universally tolerable [101–105], and few adults receive them, whereas other interventions, such as the modified Atkins diet, have not been subject to rigorous investigation in G1D. Thus, most therapeutic efforts have been limited to early diagnosis and initiation of a ketogenic diet during childhood [82, 91].
There is some redundancy between fat metabolism and carbohydrate metabolism in many human tissues, including the brain [106]: All food-derived fatty acids and their metabolic products (the ketone bodies beta-hydroxybutyrate and acetoacetate) contain an even number of carbons and are sources of acetyl-coA, which is fully consumed in the brain TCA cycle and, in doing so, they match an important physiological role of glucose. We exploited, for therapeutic development purposes, the fundamental biochemical question of whether that degree of redundancy is synonymous with equivalence, as generally assumed, or whether interchangeability is, in fact, not possible. Furthermore, treatment development stemming from this question has been hindered because cell culture systems do not recapitulate brain metabolism, few useful animal models of these disorders are available, and brain metabolism remains underinvestigated in the conscious state (i.e., unsuppressed by anesthesia [69, 74]). The answer resides, to a great extent, in flux measurements in brain tissue: We established a systematic program of metabolic flux analysis using 13C-labeled metabolic substrates in the conscious state of animal models harboring central neurometabolic disorders (G1D, pyruvate dehydrogenase deficiency and pyruvate carboxylase deficiency) and glioblastoma (work in progress and [7, 15, 69, 70, 93, 107–109]). We have also elucidated the relevance of many of these metabolic measurements to neural excitability with EEG, brain-slice patch-clamp and field potential electrophysiological recordings [7, 9, 22]. This has led us to postulate that a carbon deficit exists in G1D, which is not amenable to restoration by natural fat metabolism (i.e., including the ketone bodies beta-hydroxybutyrate and acetoacetate) in lieu of proper brain glucose influx. In contrast, odd-carbon ketones (beta-ketopentanoate and beta-hydroxypentanoate) generated by hepatic metabolism of artificial odd-carbon fatty acids such as heptanoate are anaplerotic: a fraction is also fully consumed in the TCA cycle, but another replenishes TCA cycle precursors effectively, substituting for glucose in this critically important anabolic process. Thus, they may support neural function more effectively than food-derived natural ketones when glucose availability is decreased. Importantly, 1) no glucose metabolism disease is known or expected to impair ketone body utilization [35, 110], and 2) TCA cycle flux augmentation by a broad variety of exogenous carbon sources (glucose, palmitate, lactate and octanoate) is not linked to disruption or excess of mitochondrial respiration [111].
ANAPLEROTIC THERAPY OF G1D
Glucose supports brain metabolism and neurometabolic diseases are often associated with intractable seizures [14]. Additional molecules, such as ketogenic-diet fatty acids and their derivative ketone bodies can partially substitute for glucose, except that: 1) euglycemia or postprandrial increases in glycemia can interfere with ketosis, such that canonical ketogenic diets are –paradoxically for brain states associated with decreased glucose flux [112]- carbohydrate-restricted; 2) mitochondrial fatty acid oxidation can compete with glucose metabolism [113]; and 3) all food-derived fats and ketones contain an even number of carbons and are thus fully consumed in the tricarboxylic acid (TCA) cycle and excreted as water and CO2, yielding no net carbon to compensate for the natural loss of metabolites [35, 110, 114]. This compensation is normally provided by anaplerosis, which, in brain, stems principally from glucose via carbon-donor reactions [115]. Thus, the widespread notion of ketone bodies as alternative brain fuels is incomplete and has, in our view, delayed therapeutic development. In contrast with ketogenic diets, the synthetic 7-carbon fatty acid heptanoate (a constituent of the edible triglyceride oil triheptanoin (C7)) generates both even-carbon and 5-carbon ketones (beta-hydroxypentanoate, beta-ketopentanoate) in the liver, and the latter are anaplerotic [35, 62, 63], potentially affording superior benefit.
What is triheptanoin?
Triheptanoin (C7) is a naturally-occurring fat [116] that is readily synthesized from castor bean oil for use in the human food industry as an additive to dairy products or as an emollient in cosmetics [117]. In the U.S., food-grade triheptanoin (used as a medical food) first received orphan designation for the treatment of fatty-acid oxidation disorders after work performed under IND 59303 and, more recently (2012), it achieved an equivalent legal status in the European Union for the treatment of very-long-chain 3-hydroxyacyl-CoA-dehydrogenase (VLCAD) deficiency (designation EU/3/12/1081).
In our recent study, G1D participants received open-label, adjunctive dietary supplementation with food-grade triheptanoin, a medium chain triglyceride of heptanoic acid that is colorless and odorless [118]. In agreement with the requirements of a medical food, triheptanoin was consumed enterally under the supervision of a physician and was intended for the specific dietary management of a disease or condition for which distinctive nutritional requirements, based on recognized scientific principles, were established by medical evaluation. Thus, from the very inception of the concept of anaplerotic therapy and the derived first human studies, C7 was intended for investigation and use as a medical food.
Clinical outcomes
As indicated by blood general analytical values (blood glucose, beta-hydroxybutyrare, lactate, cholesterol (HDL, LDL and total), triglycerides, electrolytes, blood urea nitrogen, creatinine, CK, transaminases (AST, ALT, GGT) and blood cell count), there was no change in glucose levels or in lipid levels. This indicates that, despite the addition of triheptanoin to the diet, the food supplement is safe and metabolically neutral as assessed by these analyses.
Because we identified a prohibitively small detection rate of absences by mere observation, EEG recordings served as the most appropriate quantifiable outcome. EEG spike-wave seizure frequency and duration and normalized scored neuropsychological results of receptive and expressive vocabulary were favorably impacted by triheptanoin [118]. Other non-clinical data and non-systematic evidence support the clinical improvement noticed by many patients as well. The voluntary continuation rate (11 out of 11 at 3 months, 10 out of 11 at 6 months) was indicative of ongoing benefit. Several parents spontaneously reported that other caregivers (school teachers, speech and physical therapists) who were unaware of study participation noted marked improvement, both motor and cognitive. This is consistent with improvement on the vocabulary tests, which measure not only language ability, but also general cognitive function, as patients are required to maintain attention and concentration for the duration of the task. Perhaps most striking, the very young participants (age 2 and 4 years), who were significantly delayed in all aspects of intellectual and motor function, made rapid improvements in developmental milestones, meeting them at age-appropriate intervals.
Cerebral metabolic rate CMRO2
The CMRO2 can be measured accurately and non-invasively and is diminished in G1D [119]. In contrast with the cerebral metabolic rate characteristic of normal subjects of the same age than those studied by us [120], G1D patients demonstrate decreased but uniquely different CMRO2 values. This phenomenon may reflect the broad phenotypic diversity of our case series, of which the CMRO2 is but one aspect. Because G1D is a life-long genetic disorder that impacts the brain in early infancy, it is also unknown what the maximum achievable CMRO2 level is in G1D should patients experience a full restoration of glucose influx after the disorder has proven symptomatic for an extended developmental period of their lives, or even if the age-normal CMRO2 can be exceeded as the consequence of a favorable therapeutic effect. Of note, it is not plausible – nor was it attempted - to safely advance general correlations between CMRO2 and spike-wave frequency or neuropsychological indices, as this outside the scope and inferential power of our study. Therefore, only individual observations deserve remarks: Of 5 subjects who received CMRO2 determination, three demonstrated rapid (i.e., consistent with the cerebral metabolism of triheptanoin metabolites), significant increases. The CMRO2 of two subjects did not increase after acute triheptanoin ingestion. One of them exhibited spike-wave seizures that were completely abolished by triheptanoin and the other did not exhibit spike-wave seizures neither before nor after triheptanoin. The robustness of the CMRO2 measurements prompts an explanatory framework for these single-subject observations: The simplest interpretation of these results, should they prove a general feature of G1D, is that triheptanoin metabolism may lead to increased oxygen consumption only while the brain undergoes a reduction of ictogenesis. In parallel to this contention, we hypothesize that, when ictogenesis is abolished by triheptanoin or absent at baseline, triheptanoin does exert little or no effect on CMRO2. Because the relationship between ictogenesis and whole-brain oxygen consumption is unknown (as determined by the global encephalic CMRO2 reported here), and because G1D spike-wave seizures may be caused by aberrant thalamocortical synchronization rather than global or multifocal epileptogenesis [7], further work currently in progress will aim to elucidate CMRO2 changes in regional, ictogenic G1D tissue. Also notable is the significant increase in CMRO2 for one subject after 6 months of treatment. Despite the absence of epilepsy as detected by observation and by EEG, this finding is compatible with triheptanoin –induced long-term changes in the non-epileptic G1D brain.
CONCLUSIONS
Table 4 reflects some longstanding assumptions that are often stated and are now called into question. Each assumption carries practical diagnostic or therapeutic implications and is linked to particular pre-conceived notions that are not experimentally supported or which are directly contradicted by the evidence. Ignoring these statements primarily weakens patient treatment opportunities and therapeutic development, such that treatments based on these uncorrected principles would have led to modest results (at best) or, simply, to confusion, as is sometimes the case for neurological therapeutic development. Thus, it seems clear that vigorous scientific scrutiny is a necessary prerequisite before embarking in any acceleration of therapies subordinate to deficient biological assumptions.
TABLE 4.
Commonly held assumptions and facts in G1D
| Assumption | Fact | References |
|---|---|---|
| The blood-brain barrier is defective | Synaptic abnormalities are independent of the blood-brain barrier | [9, 22] |
| G1D is an energy failure syndrome | The tricarboxylic acid cycle is preserved in the mouse G1D brain | [7] |
| CSF/blood glucose ratio is diagnostic | The denominator is excessively dependent upon non-disease related factors | |
| CSF glucose derives from brain tissue | CSF glucose is primarily excreted by the choroid plexus | [47] |
| Genotype – phenotype correlations indicate a continuum of a single syndrome | Phenotypes diverge widely and constitute in effect separable syndromes | |
| Seizures are manifest | EEG can reveal a large number of nonapparent seizures | [118] |
| Ketone bodies (betahydroxybutyric and acetoacetic acids) are a complete alternative energetic source | These ketone bodies supply energy but do not refill tricarboxylic acid precursors lost in the course of normal metabolism | [15] |
| FDG-PET reflects glucose uptake | FDG-PET represents radioactive atom accumulation (influx minus effux) |
As a result of the evidence presented in this article, several postulates are in order. The purpose of these considerations is to catalyze debate and, more importantly, experimental dismissal or confirmation. Thus, we expect that the following statements will be followed by vigorous discussion and meaningful research. This is the framework for future investigation that we propose:
One gene, multiple phenotypes, multiple disease mechanisms?
The variety of phenotypes in G1D is often interpreted as manifestations along a continuum of disease severity. However, several principles argue against the legitimacy of conflating disparate symptoms: First, G1D can be associated with isolated symptoms in the absence of other disease characteristics. For example, it is possible to identify pure absence epilepsy or hemolysis without the rest of syndromic manifestations; second, G1D manifestations involve disparate brain functional systems. For example, dystonia, ataxia, epilepsy reflect the differential implication of separate, functionally semi-autonomous brain properties; Third, the response to available treatments (i.e., a ketogenic diet) depends on the symptom being considered, as the diet significantly ameliorates certain manifestations but not others. In contrast with this notion, it is possible that GLUT1 acts as a regulator on several independent brain functions that are intrinsically differentially vulnerable, exerting, when mutated, a permissive effect on other dormant functional abnormalities that become de-repressed in G1D.
Cerebellar ictogenesis?
G1D is associated with a prominent cerebellar syndrome in many patients. IIG, among other G1D manifestations, is suggestive of cerebellar dysfunction. Ample precedent for ictal cerebellar discharges exists for genetic absence thalamocortical epilepsy [121, 122]. Consistent with these observations is our fMRI evidence of robust cerebellar activation detected only during G1D seizures. In this context, IIG may represent cerebellar seizures during early infancy. The role of the cerebellum in G1D thus remains an important target for mechanistic investigation and potential therapeutic development.
Incomplete metabolic alternatives or irreversible developmental effects?
A premise of metabolic therapies is the potential reversibility of functional deficits as illustrated by glucose responsiveness of EEG and electrophysiological brain slice studies. However, many neurological deficits persist under a ketogenic or a triheptanoin diet in G1D. It is unknown whether glucose plays a critical role in functions separate from – simply - brain fueling. For example, the magnitude of flux via the pentose phosphate and other important pathways during development and their full function is unknown. This uncertainty is anecdotally illustrated by a case of congenital hypoglycemia that resembled G1D in adolescence despite the resolution of the former in childhood [26]. This case raises the possibility that G1D or subtle intermittent hypoglycemia lead to permanent circuit or system abnormalities when they are untreated during critical developmental stages.
The value of diagnosis and treatment
Despite this state of affairs, an incomplete treatment is superior to no treatment or deleterious treatment. Figure 5 illustrates the impact of diagnosis of G1D on medical intervention obtained from the voluntary, patient-driven G1D patient registry. The data illustrate that, despite the inadequacies of current treatments, diagnosing G1D has strong repercussions on patients, families and society. It is incumbent upon all – and urgently imperative – to lend an opportunity to the many patients who remain almost undoubtedly mis- or undiagnosed.
Figure 5.
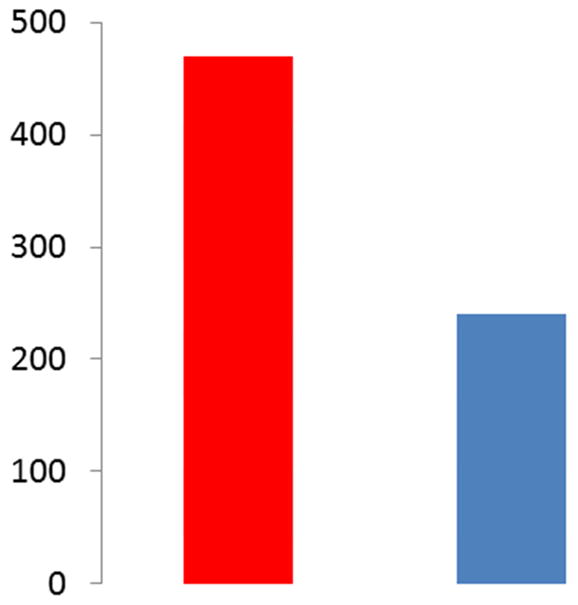
Impact of G1D diagnosis. Left: number of emergency visits before the diagnosis of G1D. Right: Emergency department visits following the diagnosis and treatment of G1D. Data from the G1D patient registry (G1DRegistry.org, n= 82 patients).
Acknowledgments
J.M.P. and G.M.R. had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. J.M.P. would like to acknowledge the generous support of the Glut1 Deficiency Foundation, the Dallas Women’s Foundation (Billingsley Fund). He also thanks Drs. Levi Good, Qian Ma and Richard Wang for generating some of the figures. He is also supported by NIH grants NS077015, NS067015, NS078059, MH084021, RR002584, RR024982 and by the NIH Office of Rare Diseases Research Glucose transporter type I deficiency syndrome (G1D) collaboration, education, and test translation (CETT) program for rare genetic diseases. The antibody used in Figure 2 was kindly provided by Dr. Richard Wang. None of the acknowledged persons or institutions participated in the design and conduct of the study; collection, management, analysis, and interpretation of the data; or preparation, review, or approval of the manuscript; or decision to submit the manuscript for publication.
Footnotes
Intermittent involuntary gaze (IIG), which is to be distinguished from opsoclonus, are episodes of isolated, impersistent conjugate (and less frequently dysconjugate) alternating pendular oculomotor deviation (usually horizontal) lasting less than 1 second per swing, in association with neck deviation that may follow the direction of gaze. Vestibulo ocular maneuvres such as head repositioning have little effect on IIG, which can last several minutes. The infant appears aware of and engaged in - though surprised by - these episodes. In contrast, opsoclonus exhibits more rapid eye deviations with impaired conjugation.
Author contributions:
JMP: Design, data collection and interpretation, manuscript draft
GMR: Design, data collection and interpretation, manuscript draft
Competing interests: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.De Vivo D, Trifiletti R, Jacobson R, Harik S. Glucose transporter deficiency causing persistent Hypoglycorrhachia: a unique cause of infantile seizures and acquired microcephaly. Ann Neurol. 1990;29:414–5. [Google Scholar]
- 2.Ronen GM, De Vivo DC, Harik SI. A second case of defective glucose transport at the blood-brain barrier. Emergence of a novel clinical syndrome. Ann Neurol. 1991;30:452. [Google Scholar]
- 3.De Vivo DC, Trifiletti RR, Jacobson RI, Ronen GM, Behmand RA, Harik SI. Defective glucose transport across the blood-brain barrier as a cause of persistent hypoglycorrhachia, seizures, and developmental delay. N Engl J Med. 1991;325:703–9. doi: 10.1056/NEJM199109053251006. [DOI] [PubMed] [Google Scholar]
- 4.Seidner G, Alvarez MG, Yeh JI, O’Driscoll KR, Klepper J, Stump TS, et al. GLUT-1 deficiency syndrome caused by haploinsufficiency of the blood-brain barrier hexose carrier. Nat Genet. 1998;18:188–91. doi: 10.1038/ng0298-188. [DOI] [PubMed] [Google Scholar]
- 5.Wang D, Pascual JM, De Vivo D. Glucose Transporter Type 1 Deficiency Syndrome. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews® [Internet] Seattle (WA): University of Washington, Seattle; 2002. Jul 30, [Updated 2015 Jan 22] 1993–2015. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1430/ [PubMed] [Google Scholar]
- 6.Pascual JM, Van Heertum RL, Wang D, Engelstad K, De Vivo DC. Imaging the metabolic footprint of Glut1 deficiency on the brain. Ann Neurol. 2002;52:458–64. doi: 10.1002/ana.10311. [DOI] [PubMed] [Google Scholar]
- 7.Marin-Valencia I, Good LB, Ma Q, Duarte J, Bottiglieri T, Sinton CM, et al. Glut1 deficiency (G1D): epilepsy and metabolic dysfunction in a mouse model of the most common human phenotype. Neurobiol Dis. 2012;48:92–101. doi: 10.1016/j.nbd.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klepper J. Glucose transporter deficiency syndrome (GLUT1DS) and the ketogenic diet. Epilepsia. 2008;49(Suppl 8):46–9. doi: 10.1111/j.1528-1167.2008.01833.x. [DOI] [PubMed] [Google Scholar]
- 9.Good LB, Espinosa F, Ma Q, Heilig CW, Kavalali ET, Pascual JM. Epilepsia; A neuronal excitability defect in a prototypic energy metabolism disorder American Epilepsy Society annual meeting; Seattle, WA, USA. 2008. p. 364. [Google Scholar]
- 10.Good LB, Ma Q, Kavalali ET, Heilig CW, Pascual JM. Diminished synaptic quantal amplitudes in a brain energy metabolic disorder. Society for Neuroscience Annual Meeting; Chicago: Society for Neuroscience; 2009. p. 330.8/H27. [Google Scholar]
- 11.Marin-Valencia I, Good LB, Ma Q, Arning E, Bottiglieri T, Jeffrey FM, et al. Glycolysis, citric acid cycle flux and neurotransmitter synthesis in mouse brain by 13C NMR spectroscopy. Society for Neuroscience Annual Meeting; Chicago. 2009. p. 406.1. [Google Scholar]
- 12.Klepper J. GLUT1 deficiency syndrome in clinical practice. Epilepsy Res. 2012;100:272–7. doi: 10.1016/j.eplepsyres.2011.02.007. [DOI] [PubMed] [Google Scholar]
- 13.Kim do Y, Rho JM. The ketogenic diet and epilepsy. Curr Opin Clin Nutr Metab Care. 2008;11:113–20. doi: 10.1097/MCO.0b013e3282f44c06. [DOI] [PubMed] [Google Scholar]
- 14.Pascual JM, Campistol J, Gil-Nagel A. Epilepsy in inherited metabolic disorders. Neurologist. 2008;14:S2–S14. doi: 10.1097/01.nrl.0000340787.30542.41. [DOI] [PubMed] [Google Scholar]
- 15.Marin-Valencia I, Good LB, Ma Q, Malloy CR, Pascual JM. Heptanoate as a neural fuel: energetic and neurotransmitter precursors in normal and glucose transporter I-deficient (G1D) brain. J Cereb Blood Flow Metab. 2013;33:175–82. doi: 10.1038/jcbfm.2012.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pascual JM, Good LB, Liu P, Marin-Valencia I, Ma Q, Tondo M, et al. Curing the Epilepsies 2013: Pathways Forward. Bethesda, MD, USA: National Institute of Neurological Disorders and Stroke, Bethesda, MD; 2013. Synaptic excitation-inhibition imbalance in glucose transporter I deficiency (G1D) and first treatment of its associated human epilepsy with triheptanoin. [Google Scholar]
- 17.Wang D, Pascual JM, Yang H, Engelstad K, Mao X, Cheng J, et al. A mouse model for Glut-1 haploinsufficiency. Hum Mol Genet. 2006;15:1169–79. doi: 10.1093/hmg/ddl032. [DOI] [PubMed] [Google Scholar]
- 18.Ohtsuki S, Kikkawa T, Hori S, Terasaki T. Modulation and compensation of the mRNA expression of energy related transporters in the brain of glucose transporter 1-deficient mice. Biol Pharm Bull. 2006;29:1587–91. doi: 10.1248/bpb.29.1587. [DOI] [PubMed] [Google Scholar]
- 19.Heilig CW, Saunders T, Brosius FC, 3rd, Moley K, Heilig K, Baggs R, et al. Glucose transporter-1-deficient mice exhibit impaired development and deformities that are similar to diabetic embryopathy. Proc Natl Acad Sci U S A. 2003;100:15613–8. doi: 10.1073/pnas.2536196100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arsov T, Mullen SA, Rogers S, Phillips AM, Lawrence KM, Damiano JA, et al. Glucose transporter 1 deficiency in the idiopathic generalized epilepsies. Ann Neurol. 2012;72:807–15. doi: 10.1002/ana.23702. [DOI] [PubMed] [Google Scholar]
- 21.Mullen SA, Suls A, De Jonghe P, Berkovic SF, Scheffer IE. Absence epilepsies with widely variable onset are a key feature of familial GLUT1 deficiency. Neurology. 2010;75:432–40. doi: 10.1212/WNL.0b013e3181eb58b4. [DOI] [PubMed] [Google Scholar]
- 22.Good LB, Ma Q, Kavalali ET, Heilig CW, Pascual JM. Diminished synaptic quantal amplitudes in a brain energy metabolic disorder. Society for Neuroscience Annual Meeting; Chicago: Society for Neuroscience; 2009. p. 330.8/H27. [Google Scholar]
- 23.Pascual JM, Wang D, Yang R, Shi L, Yang H, De Vivo DC. Structural signatures and membrane helix 4 in GLUT1: inferences from human blood-brain glucose transport mutants. J Biol Chem. 2008;283:16732–42. doi: 10.1074/jbc.M801403200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang D, Pascual JM, Yang H, Engelstad K, Jhung S, Sun RP, et al. Glut-1 deficiency syndrome: clinical, genetic, and therapeutic aspects. Ann Neurol. 2005;57:111–8. doi: 10.1002/ana.20331. [DOI] [PubMed] [Google Scholar]
- 25.Leary LD, Wang D, Nordli DR, Jr, Engelstad K, De Vivo DC. Seizure characterization and electroencephalographic features in Glut-1 deficiency syndrome. Epilepsia. 2003;44:701–7. doi: 10.1046/j.1528-1157.2003.05302.x. [DOI] [PubMed] [Google Scholar]
- 26.Pascual JM, Wang D, Hinton V, Engelstad K, Saxena CM, Van Heertum RL, et al. Brain glucose supply and the syndrome of infantile neuroglycopenia. Arch Neurol. 2007;64:507–13. doi: 10.1001/archneur.64.4.noc60165. [DOI] [PubMed] [Google Scholar]
- 27.Akman CI, Engelstad K, Hinton VJ, Ullner P, Koenigsberger D, Leary L, et al. Acute hyperglycemia produces transient improvement in glucose transporter type 1 deficiency. Ann Neurol. 2010;67:31–40. doi: 10.1002/ana.21797. [DOI] [PubMed] [Google Scholar]
- 28.Leen WG, Taher M, Verbeek MM, Kamsteeg EJ, van de Warrenburg BP, Willemsen MA. GLUT1 deficiency syndrome into adulthood: a follow-up study. J Neurol. 2014 doi: 10.1007/s00415-014-7240-z. [DOI] [PubMed] [Google Scholar]
- 29.Vannucci SJ, Clark RR, Koehler-Stec E, Li K, Smith CB, Davies P, et al. Glucose transporter expression in brain: relationship to cerebral glucose utilization. Dev Neurosci. 1998;20:369–79. doi: 10.1159/000017333. [DOI] [PubMed] [Google Scholar]
- 30.Simpson IA, Dwyer D, Malide D, Moley KH, Travis A, Vannucci SJ. The facilitative glucose transporter GLUT3: 20 years of distinction. Am J Physiol Endocrinol Metab. 2008;295:E242–53. doi: 10.1152/ajpendo.90388.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Heeswijk RB, Morgenthaler FD, Xin L, Gruetter R. Quantification of brain glycogen concentration and turnover through localized 13C NMR of both the C1 and C6 resonances. NMR Biomed. 2010;23:270–6. doi: 10.1002/nbm.1460. [DOI] [PubMed] [Google Scholar]
- 32.Pascual JM, Wang D, Lecumberri B, Yang H, Mao X, Yang R, et al. GLUT1 deficiency and other glucose transporter diseases. Eur J Endocrinol. 2004;150:627–33. doi: 10.1530/eje.0.1500627. [DOI] [PubMed] [Google Scholar]
- 33.Zhao Y, Fung C, Shin D, Shin BC, Thamotharan S, Sankar R, et al. Neuronal glucose transporter isoform 3 deficient mice demonstrate features of autism spectrum disorders. Mol Psychiatry. 15:286–99. doi: 10.1038/mp.2009.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guzman M, Blazquez C. Ketone body synthesis in the brain: possible neuroprotective effects. Prostaglandins, leukotrienes, and essential fatty acids. 2004;70:287–92. doi: 10.1016/j.plefa.2003.05.001. [DOI] [PubMed] [Google Scholar]
- 35.Brunengraber H, Roe CR. Anaplerotic molecules: current and future. J Inherit Metab Dis. 2006;29:327–31. doi: 10.1007/s10545-006-0320-1. [DOI] [PubMed] [Google Scholar]
- 36.Mason GF, Petersen KF, de Graaf RA, Shulman GI, Rothman DL. Measurements of the anaplerotic rate in the human cerebral cortex using 13C magnetic resonance spectroscopy and [1-13C] and [2-13C] glucose. J Neurochem. 2007;100:73–86. doi: 10.1111/j.1471-4159.2006.04200.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Magistretti PJ, Pellerin L. Cellular mechanisms of brain energy metabolism and their relevance to functional brain imaging. Philos Trans R Soc Lond B Biol Sci. 1999;354:1155–63. doi: 10.1098/rstb.1999.0471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci U S A. 1994;91:10625–9. doi: 10.1073/pnas.91.22.10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mason GF, Gruetter R, Rothman DL, Behar KL, Shulman RG, Novotny EJ. Simultaneous determination of the rates of the TCA cycle, glucose utilization, alpha-ketoglutarate/glutamate exchange, and glutamine synthesis in human brain by NMR. J Cereb Blood Flow Metab. 1995;15:12–25. doi: 10.1038/jcbfm.1995.2. [DOI] [PubMed] [Google Scholar]
- 40.Chowdhury GM, Patel AB, Mason GF, Rothman DL, Behar KL. Glutamatergic and GABAergic neurotransmitter cycling and energy metabolism in rat cerebral cortex during postnatal development. J Cereb Blood Flow Metab. 2007;27:1895–907. doi: 10.1038/sj.jcbfm.9600490. [DOI] [PubMed] [Google Scholar]
- 41.Roberts E, Frankel S. gamma-Aminobutyric acid in brain: its formation from glutamic acid. J Biol Chem. 1950;187:55–63. [PubMed] [Google Scholar]
- 42.Patel AB, de Graaf RA, Mason GF, Rothman DL, Shulman RG, Behar KL. The contribution of GABA to glutamate/glutamine cycling and energy metabolism in the rat cortex in vivo. Proc Natl Acad Sci U S A. 2005;102:5588–93. doi: 10.1073/pnas.0501703102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Logan J. Graphical analysis of PET data applied to reversible and irreversible tracers. Nucl Med Biol. 2000;27:661–70. doi: 10.1016/s0969-8051(00)00137-2. [DOI] [PubMed] [Google Scholar]
- 44.Barros LF, Bittner CX, Loaiza A, Porras OH. A quantitative overview of glucose dynamics in the gliovascular unit. Glia. 2007;55:1222–37. doi: 10.1002/glia.20375. [DOI] [PubMed] [Google Scholar]
- 45.Simpson IA, Carruthers A, Vannucci SJ. Supply and demand in cerebral energy metabolism: the role of nutrient transporters. J Cereb Blood Flow Metab. 2007;27:1766–91. doi: 10.1038/sj.jcbfm.9600521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jolivet R, Allaman I, Pellerin L, Magistretti PJ, Weber B. Comment on recent modeling studies of astrocyte-neuron metabolic interactions. J Cereb Blood Flow Metab. 30:1982–6. doi: 10.1038/jcbfm.2010.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mangia S, Simpson IA, Vannucci SJ, Carruthers A. The in vivo neuron-to-astrocyte lactate shuttle in human brain: evidence from modeling of measured lactate levels during visual stimulation. J Neurochem. 2009;109(Suppl 1):55–62. doi: 10.1111/j.1471-4159.2009.06003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lund-Andersen H. Transport of glucose from blood to brain. Physiol Rev. 1979;59:305–52. doi: 10.1152/physrev.1979.59.2.305. [DOI] [PubMed] [Google Scholar]
- 49.Hassel B, Sonnewald U, Fonnum F. Glial-neuronal interactions as studied by cerebral metabolism of [2-13C]acetate and [1-13C]glucose: an ex vivo 13C NMR spectroscopic study. J Neurochem. 1995;64:2773–82. doi: 10.1046/j.1471-4159.1995.64062773.x. [DOI] [PubMed] [Google Scholar]
- 50.Brenner E, Sonnewald U, Schweitzer A, Andrieux A, Nehlig A. Hypoglutamatergic activity in the STOP knockout mouse: a potential model for chronic untreated schizophrenia. J Neurosci Res. 2007;85:3487–93. doi: 10.1002/jnr.21200. [DOI] [PubMed] [Google Scholar]
- 51.Yudkoff M, Daikhin Y, Nissim I, Horyn O, Lazarow A, Luhovyy B, et al. Response of brain amino acid metabolism to ketosis. Neurochem Int. 2005;47:119–28. doi: 10.1016/j.neuint.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 52.Bogen IL, Risa O, Haug KH, Sonnewald U, Fonnum F, Walaas SI. Distinct changes in neuronal and astrocytic amino acid neurotransmitter metabolism in mice with reduced numbers of synaptic vesicles. J Neurochem. 2008;105:2524–34. doi: 10.1111/j.1471-4159.2008.05344.x. [DOI] [PubMed] [Google Scholar]
- 53.Malloy CR, Sherry AD, Jeffrey FM. Carbon flux through citric acid cycle pathways in perfused heart by 13C NMR spectroscopy. FEBS Lett. 1987;212:58–62. doi: 10.1016/0014-5793(87)81556-9. [DOI] [PubMed] [Google Scholar]
- 54.Marin-Valencia I, Good LB, Ma Q, Jeffrey FM, Malloy CR, Pascual JM. High-resolution detection of (13)C multiplets from the conscious mouse brain by ex vivo NMR spectroscopy. J Neurosci Methods. 2011 doi: 10.1016/j.jneumeth.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Holopainen IE. Seizures in the developing brain: cellular and molecular mechanisms of neuronal damage, neurogenesis and cellular reorganization. Neurochem Int. 2008;52:935–47. doi: 10.1016/j.neuint.2007.10.021. [DOI] [PubMed] [Google Scholar]
- 56.Huguenard JR, Prince DA. Intrathalamic rhythmicity studied in vitro: nominal T-current modulation causes robust antioscillatory effects. J Neurosci. 1994;14:5485–502. doi: 10.1523/JNEUROSCI.14-09-05485.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sokoloff L. The deoxyglucose method for the measurement of local glucose utilization and the mapping of local functional activity in the central nervous system. Int Rev Neurobiol. 1981;22:287–333. doi: 10.1016/s0074-7742(08)60296-2. [DOI] [PubMed] [Google Scholar]
- 58.Ullner PM, Di Nardo A, Goldman JE, Schobel S, Yang H, Engelstad K, et al. Murine Glut-1 transporter haploinsufficiency: postnatal deceleration of brain weight and reactive astrocytosis. Neurobiol Dis. 2009;36:60–9. doi: 10.1016/j.nbd.2009.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang D, Pascual JM, De Vivo D. Glucose Transporter Type 1 Deficiency Syndrome. 2012. [PubMed] [Google Scholar]
- 60.Smeitink J, van den Heuvel L, DiMauro S. The genetics and pathology of oxidative phosphorylation. Nat Rev Genet. 2001;2:342–52. doi: 10.1038/35072063. [DOI] [PubMed] [Google Scholar]
- 61.Dimauro S. A history of mitochondrial diseases. J Inherit Metab Dis. 2011;34:261–76. doi: 10.1007/s10545-010-9082-x. [DOI] [PubMed] [Google Scholar]
- 62.Kinman RP, Kasumov T, Jobbins KA, Thomas KR, Adams JE, Brunengraber LN, et al. Parenteral and enteral metabolism of anaplerotic triheptanoin in normal rats. Am J Physiol Endocrinol Metab. 2006;291:E860–6. doi: 10.1152/ajpendo.00366.2005. [DOI] [PubMed] [Google Scholar]
- 63.Gu L, Zhang GF, Kombu RS, Allen F, Kutz G, Brewer WU, et al. Parenteral and enteral metabolism of anaplerotic triheptanoin in normal rats. II. Effects on lipolysis, glucose production, and liver acyl-CoA profile. Am J Physiol Endocrinol Metab. 2010;298:E362–71. doi: 10.1152/ajpendo.00384.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cholet N, Pellerin L, Welker E, Lacombe P, Seylaz J, Magistretti P, et al. Local injection of antisense oligonucleotides targeted to the glial glutamate transporter GLAST decreases the metabolic response to somatosensory activation. J Cereb Blood Flow Metab. 2001;21:404–12. doi: 10.1097/00004647-200104000-00009. [DOI] [PubMed] [Google Scholar]
- 65.Giaume C, Maravall M, Welker E, Bonvento G. The barrel cortex as a model to study dynamic neuroglial interaction. Neuroscientist. 2009;15:351–66. doi: 10.1177/1073858409336092. [DOI] [PubMed] [Google Scholar]
- 66.Gibson JR, Bartley AF, Hays SA, Huber KM. Imbalance of neocortical excitation and inhibition and altered UP states reflect network hyperexcitability in the mouse model of fragile X syndrome. J Neurophysiol. 2008;100:2615–26. doi: 10.1152/jn.90752.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nehlig A. Brain uptake and metabolism of ketone bodies in animal models. Prostaglandins Leukot Essent Fatty Acids. 2004;70:265–75. doi: 10.1016/j.plefa.2003.07.006. [DOI] [PubMed] [Google Scholar]
- 68.Hestrin S, Sah P, Nicoll RA. Mechanisms generating the time course of dual component excitatory synaptic currents recorded in hippocampal slices. Neuron. 1990;5:247–53. doi: 10.1016/0896-6273(90)90162-9. [DOI] [PubMed] [Google Scholar]
- 69.Marin-Valencia I, Good LB, Ma Q, Jeffrey FM, Malloy CR, Pascual JM. High-resolution detection of (1)(3)C multiplets from the conscious mouse brain by ex vivo NMR spectroscopy. J Neurosci Methods. 2012;203:50–5. doi: 10.1016/j.jneumeth.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Marin-Valencia I, Good LB, Ma Q, Malloy CR, Patel MS, Pascual JM. Cortical metabolism in pyruvate dehydrogenase deficiency revealed by ex vivo multiplet (13)C NMR of the adult mouse brain. Neurochem Int. 2012;61:1036–43. doi: 10.1016/j.neuint.2012.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chegary M, Brinke H, Ruiter JP, Wijburg FA, Stoll MS, Minkler PE, et al. Mitochondrial long chain fatty acid beta-oxidation in man and mouse. Biochim Biophys Acta. 2009;1791:806–15. doi: 10.1016/j.bbalip.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yin W, Carballo-Jane E, McLaren DG, Mendoza VH, Gagen K, Geoghagen NS, et al. Plasma lipid profiling across species for the identification of optimal animal models of human dyslipidemia. J Lipid Res. 2012;53:51–65. doi: 10.1194/jlr.M019927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bergen WG, Mersmann HJ. Comparative aspects of lipid metabolism: impact on contemporary research and use of animal models. J Nutr. 2005;135:2499–502. doi: 10.1093/jn/135.11.2499. [DOI] [PubMed] [Google Scholar]
- 74.Brunner EA, Cheng SC, Berman ML. Effects of anesthesia on intermediary metabolism. Annu Rev Med. 1975;26:391–401. doi: 10.1146/annurev.me.26.020175.002135. [DOI] [PubMed] [Google Scholar]
- 75.Penry JK, Dreifuss FE. Automatisms associated with the absence of petit mal epilepsy. Arch Neurol. 1969;21:142–9. doi: 10.1001/archneur.1969.00480140042004. [DOI] [PubMed] [Google Scholar]
- 76.Tenney JR, Glauser TA. The current state of absence epilepsy: can we have your attention? Epilepsy Curr. 2013;13:135–40. doi: 10.5698/1535-7511-13.3.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Glauser TA, Cnaan A, Shinnar S, Hirtz DG, Dlugos D, Masur D, et al. Ethosuximide, valproic acid, and lamotrigine in childhood absence epilepsy. N Engl J Med. 2010;362:790–9. doi: 10.1056/NEJMoa0902014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Masur D, Shinnar S, Cnaan A, Shinnar RC, Clark P, Wang J, et al. Pretreatment cognitive deficits and treatment effects on attention in childhood absence epilepsy. Neurology. 2013;81:1572–80. doi: 10.1212/WNL.0b013e3182a9f3ca. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pascual JM, Good LB, Liu P, Marin-Valencia I, Ma Q, Kelly D, et al. Stroke NIoNDa, editor. Curing the Epilepsies 2013: Pathways Forward. Bethesda, MD: 2013. Synaptic excitation-inhibition imbalance in glucose transporter I deficiency (G1D) and first treatment of its associated human epilepsy with triheptanoin. [Google Scholar]
- 80.Gusnard DA, Akbudak E, Shulman GL, Raichle ME. Medial prefrontal cortex and self-referential mental activity: relation to a default mode of brain function. Proc Natl Acad Sci U S A. 2001;98:4259–64. doi: 10.1073/pnas.071043098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Heilig C, Brosius F, Siu B, Concepcion L, Mortensen R, Heilig K, et al. Implications of glucose transporter protein type 1 (GLUT1)-haplodeficiency in embryonic stem cells for their survival in response to hypoxic stress. Am J Pathol. 2003;163:1873–85. doi: 10.1016/S0002-9440(10)63546-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pong AW, Geary BR, Engelstad KM, Natarajan A, Yang H, De Vivo DC. Glucose transporter type I deficiency syndrome: epilepsy phenotypes and outcomes. Epilepsia. 2012;53:1503–10. doi: 10.1111/j.1528-1167.2012.03592.x. [DOI] [PubMed] [Google Scholar]
- 83.Posner E. Pharmacological treatment of childhood absence epilepsy. Expert Rev Neurother. 2006;6:855–62. doi: 10.1586/14737175.6.6.855. [DOI] [PubMed] [Google Scholar]
- 84.Kim SK, Yang H, Pascual JM, De Vivo DC. Valproic acid enhances glucose transport in the cultured brain astrocytes of glucose transporter 1 heterozygous mice. J Child Neurol. 2013;28:70–6. doi: 10.1177/0883073812440044. [DOI] [PubMed] [Google Scholar]
- 85.Klepper J, Florcken A, Fischbarg J, Voit T. Effects of anticonvulsants on GLUT1-mediated glucose transport in GLUT1 deficiency syndrome in vitro. Eur J Pediatr. 2003;162:84–9. doi: 10.1007/s00431-002-1112-8. [DOI] [PubMed] [Google Scholar]
- 86.Klepper J, Fischbarg J, Vera JC, Wang D, De Vivo DC. GLUT1-deficiency: barbiturates potentiate haploinsufficiency in vitro. Pediatr Res. 1999;46:677–83. doi: 10.1203/00006450-199912000-00006. [DOI] [PubMed] [Google Scholar]
- 87.Ho YY, Yang H, Klepper J, Fischbarg J, Wang D, De Vivo DC. Glucose transporter type 1 deficiency syndrome (Glut1DS): methylxanthines potentiate GLUT1 haploinsufficiency in vitro. Pediatr Res. 2001;50:254–60. doi: 10.1203/00006450-200108000-00015. [DOI] [PubMed] [Google Scholar]
- 88.Wang D, Pascual JM, De Vivo D. Glucose Transporter Type 1 Deficiency Syndrome. GeneReviews [Internet] Resource. 2012;1993 available on line at http://wwwncbinlmnihgov/pubmed/20301603. [Google Scholar]
- 89.DeVivo DC, Leckie MP, Ferrendelli JS, McDougal DB., Jr Chronic ketosis and cerebral metabolism. Ann Neurol. 1978;3:331–37. doi: 10.1002/ana.410030410. [DOI] [PubMed] [Google Scholar]
- 90.Klepper J, Scheffer H, Leiendecker B, Gertsen E, Binder S, Leferink M, et al. Seizure control and acceptance of the ketogenic diet in GLUT1 deficiency syndrome: a 2- to 5-year follow-up of 15 children enrolled prospectively. Neuropediatrics. 2005;36:302–8. doi: 10.1055/s-2005-872843. [DOI] [PubMed] [Google Scholar]
- 91.Klepper J, Leiendecker B, Bredahl R, Athanassopoulos S, Heinen F, Gertsen E, et al. Introduction of a ketogenic diet in young infants. J Inherit Metab Dis. 2002;25:449–60. doi: 10.1023/a:1021238900470. [DOI] [PubMed] [Google Scholar]
- 92.Sibson NR, Shen J, Mason GF, Rothman DL, Behar KL, Shulman RG. Functional energy metabolism: in vivo 13C-NMR spectroscopy evidence for coupling of cerebral glucose consumption and glutamatergic neuronalactivity. Dev Neurosci. 1998;20:321–30. doi: 10.1159/000017327. [DOI] [PubMed] [Google Scholar]
- 93.Jeffrey FM, Marin-Valencia I, Good LB, Shestov AA, Henry PG, Pascual JM, et al. Modeling of brain metabolism and pyruvate compartmentation using C NMR in vivo: caution required. J Cereb Blood Flow Metab. 2013 doi: 10.1038/jcbfm.2013.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Auer RN, Wieloch T, Olsson Y, Siesjo BK. The distribution of hypoglycemic brain damage. Acta Neuropathol. 1984;64:177–91. doi: 10.1007/BF00688108. [DOI] [PubMed] [Google Scholar]
- 95.Kam K, Nicoll R. Excitatory synaptic transmission persists independently of the glutamate-glutamine cycle. J Neurosci. 2007;27:9192–200. doi: 10.1523/JNEUROSCI.1198-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bryant AS, Li B, Beenhakker MP, Huguenard JR. Maintenance of thalamic epileptiform activity depends on the astrocytic glutamate-glutamine cycle. J Neurophysiol. 2009;102:2880–8. doi: 10.1152/jn.00476.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Liang SL, Carlson GC, Coulter DA. Dynamic regulation of synaptic GABA release by the glutamate-glutamine cycle in hippocampal area CA1. J Neurosci. 2006;26:8537–48. doi: 10.1523/JNEUROSCI.0329-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lutas A, Yellen G. The ketogenic diet: metabolic influences on brain excitability and epilepsy. Trends Neurosci. 2013;36:32–40. doi: 10.1016/j.tins.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yudkoff M, Daikhin Y, Melo TM, Nissim I, Sonnewald U, Nissim I. The ketogenic diet and brain metabolism of amino acids: relationship to the anticonvulsant effect. Annu Rev Nutr. 2007;27:415–30. doi: 10.1146/annurev.nutr.27.061406.093722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bough K. Energy metabolism as part of the anticonvulsant mechanism of the ketogenic diet. Epilepsia. 2008;49(Suppl 8):91–3. doi: 10.1111/j.1528-1167.2008.01846.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kielb S, Koo HP, Bloom DA, Faerber GJ. Nephrolithiasis associated with the ketogenic diet. J Urol. 2000;164:464–6. [PubMed] [Google Scholar]
- 102.Stewart WA, Gordon K, Camfield P. Acute pancreatitis causing death in a child on the ketogenic diet. J Child Neurol. 2001;16:682. doi: 10.1177/088307380101600910. [DOI] [PubMed] [Google Scholar]
- 103.Best TH, Franz DN, Gilbert DL, Nelson DP, Epstein MR. Cardiac complications in pediatric patients on the ketogenic diet. Neurology. 2000;54:2328–30. doi: 10.1212/wnl.54.12.2328. [DOI] [PubMed] [Google Scholar]
- 104.Berry-Kravis E, Booth G, Taylor A, Valentino LA. Bruising and the ketogenic diet: evidence for diet-induced changes in platelet function. Ann Neurol. 2001;49:98–103. doi: 10.1002/1531-8249(200101)49:1<98::aid-ana13>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 105.Hoyt CS, Billson FA. Optic neuropathy in ketogenic diet. Br J Ophthalmol. 1979;63:191–4. doi: 10.1136/bjo.63.3.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Owen OE, Morgan AP, Kemp HG, Sullivan JM, Herrera MG, Cahill GF., Jr Brain metabolism during fasting. J Clin Invest. 1967;46:1589–95. doi: 10.1172/JCI105650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Marin-Valencia I, Cho SK, Rakheja D, Hatanpaa KJ, Kapur P, Mashimo T, et al. Glucose metabolism via the pentose phosphate pathway, glycolysis and Krebs cycle in an orthotopic mouse model of human brain tumors. NMR Biomed. 2012;25:1177–86. doi: 10.1002/nbm.2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Maher EA, Marin-Valencia I, Bachoo RM, Mashimo T, Raisanen J, Hatanpaa KJ, et al. Metabolism of [U-13 C]glucose in human brain tumors in vivo. NMR Biomed. 2012;25:1234–44. doi: 10.1002/nbm.2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Marin-Valencia I, Yang C, Mashimo T, Cho S, Baek H, Yang XL, et al. Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo. Cell Metab. 2012;15:827–37. doi: 10.1016/j.cmet.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Marin-Valencia I, Roe CR, Pascual JM. Pyruvate carboxylase deficiency: mechanisms, mimics and anaplerosis. Mol Genet Metab. 2010;101:9–17. doi: 10.1016/j.ymgme.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 111.From AH, Zimmer SD, Michurski SP, Mohanakrishnan P, Ulstad VK, Thoma WJ, et al. Regulation of the oxidative phosphorylation rate in the intact cell. Biochemistry. 1990;29:3731–43. doi: 10.1021/bi00467a020. [DOI] [PubMed] [Google Scholar]
- 112.Klepper J, Leiendecker B. Glut1 deficiency syndrome and novel ketogenic diets. J Child Neurol. 2013;28:1045–8. doi: 10.1177/0883073813487600. [DOI] [PubMed] [Google Scholar]
- 113.Houten SM, Wanders RJ. A general introduction to the biochemistry of mitochondrial fatty acid beta-oxidation. J Inherit Metab Dis. 2010;33:469–77. doi: 10.1007/s10545-010-9061-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kovac S, Abramov AY, Walker MC. Energy depletion in seizures: anaplerosis as a strategy for future therapies. Neuropharmacology. 2013;69:96–104. doi: 10.1016/j.neuropharm.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 115.Gruetter R. In vivo 13C NMR studies of compartmentalized cerebral carbohydrate metabolism. Neurochem Int. 2002;41:143–54. doi: 10.1016/s0197-0186(02)00034-7. [DOI] [PubMed] [Google Scholar]
- 116.Addison RF, Ackman RG. Exceptional occurrence of odd-chain fatty acids in smelt (Osmerus mordax) from Jeddore Harbour, Nova Scotia. Lipids. 1970;5:554–7. doi: 10.1007/BF02532744. [DOI] [PubMed] [Google Scholar]
- 117.Johnson W., Jr Final report on the safety assessment of trilaurin, triarachidin, tribehenin, tricaprin, tricaprylin, trierucin, triheptanoin, triheptylundecanoin, triisononanoin, triisopalmitin, triisostearin, trilinolein, trimyristin, trioctanoin, triolein, tripalmitin, tripalmitolein, triricinolein, tristearin, triundecanoin, glyceryl triacetyl hydroxystearate, glyceryl triacetyl ricinoleate, and glyceryl stearate diacetate. Int J Toxicol. 2001;20(Suppl 4):61–94. [PubMed] [Google Scholar]
- 118.Pascual JM, Liu P, Mao D, Kelly DI, Hernandez A, Sheng M, et al. Triheptanoin for glucose transporter type I deficiency (G1D): modulation of human ictogenesis, cerebral metabolic rate, and cognitive indices by a food supplement. JAMA Neurol. 2014;71:1255–65. doi: 10.1001/jamaneurol.2014.1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Xu F, Liu P, Pascual JM, Xiao G, Huang H, Lu H. Acute effect of glucose on cerebral blood flow, blood oxygenation, and oxidative metabolism. Human brain mapping. 2015;36:707–16. doi: 10.1002/hbm.22658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chugani HT, Phelps ME, Mazziotta JC. Positron emission tomography study of human brain functional development. Ann Neurol. 1987;22:487–97. doi: 10.1002/ana.410220408. [DOI] [PubMed] [Google Scholar]
- 121.Maejima T, Wollenweber P, Teusner LU, Noebels JL, Herlitze S, Mark MD. Postnatal loss of P/Q-type channels confined to rhombic-lip-derived neurons alters synaptic transmission at the parallel fiber to purkinje cell synapse and replicates genomic Cacna1a mutation phenotype of ataxia and seizures in mice. J Neurosci. 2013;33:5162–74. doi: 10.1523/JNEUROSCI.5442-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Matsushita K, Wakamori M, Rhyu IJ, Arii T, Oda S, Mori Y, et al. Bidirectional alterations in cerebellar synaptic transmission of tottering and rolling Ca2+ channel mutant mice. J Neurosci. 2002;22:4388–98. doi: 10.1523/JNEUROSCI.22-11-04388.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lee EE, Ma J, Sacharidou A, Mi W, Salato VK, Nguyen N, et al. A Protein Kinase C Phosphorylation Motif in GLUT1 Affects Glucose Transport and is Mutated in GLUT1 Deficiency Syndrome. Mol Cell. 2015;58:845–53. doi: 10.1016/j.molcel.2015.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Nehlig A, Wittendorp-Rechenmann E, Lam CD. Selective uptake of [14C]2-deoxyglucose by neurons and astrocytes: high-resolution microautoradiographic imaging by cellular 14C-trajectography combined with immunohistochemistry. J Cereb Blood Flow Metab. 2004;24:1004–14. doi: 10.1097/01.WCB.0000128533.84196.D8. [DOI] [PubMed] [Google Scholar]
- 125.Giaume C, Tabernero A, Medina JM. Metabolic trafficking through astrocytic gap junctions. Glia. 1997;21:114–23. [PubMed] [Google Scholar]
- 126.Forsyth RJ. Astrocytes and the delivery of glucose from plasma to neurons. Neurochem Int. 1996;28:231–41. doi: 10.1016/0197-0186(95)00094-1. [DOI] [PubMed] [Google Scholar]
- 127.Friedman JR, Thiele EA, Wang D, Levine KB, Cloherty EK, Pfeifer HH, De Vivo DC, Carruthers A, Natowicz MR. Atypical GLUT1 deficiency with prominent movement disorder responsive to ketogenic diet. Mov Disord. 2006;21:241–5. doi: 10.1002/mds.20660. [DOI] [PubMed] [Google Scholar]
- 128.Perez-Duenas B, Prior C, Ma Q, Fernandez-Alvarez E, Setoain X, Artuch R, Pascual JM. Childhood chorea with cerebral hypotrophy: a treatable GLUT1 energy failure syndrome. Arch Neurol. 2009;66:1410–4. doi: 10.1001/archneurol.2009.236. [DOI] [PubMed] [Google Scholar]
- 129.Roubergue A, Apartis E, Mesnage V, Doummar D, Trocello JM, Roze E, Taieb G, De Villemeur TB, Vuillaumier-Barrot S, Vidailhet M, Levy R. Dystonic tremor caused by mutation of the glucose transporter gene GLUT1. J Inherit Metab Dis. 2011;34:483–8. doi: 10.1007/s10545-010-9264-6. [DOI] [PubMed] [Google Scholar]