

Abstract
A major goal of cancer research is the identification of tumor-specific vulnerabilities that can be exploited for the development of therapies that are selectively toxic to the tumor. We show here that the transcriptional coactivators peroxisome proliferator-activated receptor gamma coactivator 1β (PGC1β) and estrogen-related receptor α (ERRα) are aberrantly expressed in human colon cell lines and tumors. With kinase suppressor of Ras 1 (KSR1) depletion as a reference standard, we used functional signature ontology (FUSION) analysis to identify the γ1 subunit of AMP-activated protein kinase (AMPK) as an essential contributor to PGC1β expression and colon tumor cell survival. Subsequent analysis revealed that a subunit composition of AMPK (α2β2γ1) is preferred for colorectal cancer cell survival, at least in part, by stabilizing the tumor-specific expression of PGC1β. In contrast, PGC1β and ERRα are not detectable in nontransformed human colon epithelial cells, and depletion of the AMPKγ1 subunit has no effect on their viability. These data indicate that Ras oncogenesis relies on the aberrant activation of a PGC1β-dependent transcriptional pathway via a specific AMPK isoform.
INTRODUCTION
A third of all human cancers, including a substantial percentage of colorectal, lung, and pancreatic cancers, are driven by activating mutations in Ras genes. Activating K-Ras mutations are present in 35 to 40% of colon tumors and are thought to be both drivers of tumorigenesis and determinants of therapeutic regimens (1). Therapeutic disruption of Ras function has been clinically ineffective to date, but investigation of Ras pleiotropy continues to yield a diversity of downstream effectors with obligate roles in the maintenance and adaptation of Ras-driven tumors to changing environments. The Raf–MEK–extracellular signal-regulated kinase (ERK) signaling pathway is essential for the oncogenic properties of mutated K-Ras (2). However, numerous potent and specific MEK inhibitors have been developed yet have failed to demonstrate single-agent efficacy in cancer treatment (3). As a molecular scaffold of the Raf-MEK-ERK kinase cascade (4, 5), kinase suppressor of Ras 1 (KSR1) is necessary and sufficient for RasV12-induced tumorigenesis (4), mouse embryo fibroblast (MEF) transformation (5, 6), and pancreatic cancer growth (7) but dispensable for normal development (4). KSR1 is overexpressed in endometrial carcinoma and is required for both proliferation and anchorage-independent growth of endometrial cancer cells (8). Except for minor defects in hair follicles, KSR1 knockout mice are fertile and develop normally (4).
This observation predicts that small molecules targeting KSR1 and functionally related effectors should preferentially target Ras-driven tumors while leaving normal tissue largely unaffected. More generally, this observation demonstrates that tumor cells, while under selective pressure to adapt to inhospitable environments and proliferate without constraint, will adopt strategies that, while advantageous to that singular purpose, create vulnerabilities that can be exploited by targeted therapies.
We sought to detect and exploit those vulnerabilities in human colon tumor cells using functional signature ontology (FUSION) (9) to identify functional analogs of KSR1. A validated functional analog of KSR1 is required for the survival and tumorigenic properties in Ras-driven cancer cells but is dispensable for survival in normal cells. Applying FUSION analysis to a small interfering RNA (siRNA) screen of genes encoding kinases, phosphatases, and related proteins, a gene expression signature characteristic of KSR1 disruption identified PRKAG1, the gene encoding the γ1 subunit of AMP-activated protein kinase (AMPK), as a component of colon tumor cell survival. Further characterization revealed that a complex of a trimeric AMPK incorporating the α2 and β2 subunits along with the γ1 subunit was critical to human colon tumor cell survival. RNA interference (RNAi)-mediated disruption of these AMPK subunits killed human colon tumor cells without appreciable effect on nontransformed colon epithelial cells. The action of KSR1 and AMPK was linked to the action of transcriptional regulators PGC1β/estrogen-related receptor α (ERRα), whose overexpression is evident in metastatic human colon tumors and whose action is critical in colon tumor cell survival. These results demonstrate the feasibility of using FUSION to identify molecular targets of tumor-specific pathways in K-Ras-driven oncogenic signaling.
MATERIALS AND METHODS
Immunoblotting.
For a complete list of the cell lines, antibodies, and reagents, see the supplemental material. Cells were lysed in cytoplasmic lysis buffer containing 0.5% NP-40, 25 mM HEPES, 5 mM KCl, and 0.5 mM MgCl2, pH 7.4, and a nuclear lysis buffer containing 40 mM Tris-HCl, 150 mM NaCl, 1% Triton X-100, 0.1% SDS, and 0.25% sodium deoxycholate, pH 7.4, with protease and phosphatase inhibitors. Proteins were resolved using SDS-PAGE and transferred to nitrocellulose membranes, blocked in Odyssey blocking buffer (Li-Cor), hybridized with primary and secondary antibodies in Tris-buffered saline (TBS)–0.1% Tween 20, and detected using an Odyssey imaging system (Li-Cor).
Plasmids and shRNA constructs.
A lentiviral p201-green fluorescent protein (GFP) empty construct was a kind gift from Manabu Furukawa. Flag-tagged KSR1 was cloned into this p201 vector, and both the empty vector and Flag-tagged KSR1 were transfected into 293T cells using Lipofectamine 2000 transfection reagent in serum-free medium. Medium was changed after 24 h, and virus was collected at 48 and 72 h posttransfection. Subconfluent HCT116 cells were infected with the virus with 8 μg/ml Polybrene for 48 h. Expression of Flag-tagged KSR1 expression was analyzed by Western blotting. pLKO.1-puro constructs carrying short hairpin RNAs (shRNAs) targeting KSR1 and PGC1β and pGIPZ vectors carrying ERRα shRNA were transfected into HCT116 cells using 10 μl of Lipofectamine 2000 transfection reagent and 4 μg of DNA in serum-free medium. The cells were selected with 10 μg/ml puromycin. Colonies were picked after sterile glass rings were placed around individual colonies. Sequences of shRNAs are provided in Table S1 in the supplemental material. Lentiviral vectors plenti 6 and plenti 6-CA-PGC1β were a kind gift from Donald McDonnell (Duke University). These vectors were transfected into 293T cells using a translentiviral packaging system (Thermo Scientific). Virus was collected and cells were infected as described above. Populations of cells expressing the empty or CA-PGC1β vector were selected using 10 μg/μl puromycin.
siRNA transfection.
All siRNAs and Dharmafect 4 reagent were purchased from Dharmacon (GE-Dharmacon). Individual siRNA duplexes were tested for target knockdown. Validated siRNAs were pooled for further experiments. siRNAs at 50 nM were introduced into HCT116 cells (432,000 cells/35-mm dish) by reverse transfection using Dharmafect 4 transfection reagent in antibiotic-free medium. SW480 cells (600,000 cells/35-mm dish) and SW620 cells (500,000 cells/35-mm dish) were transfected with 100 nM siRNA using 10 μl of RNAiMAX transfection reagent (Invitrogen). Human colonic epithelial cells (HCECs; 250,000 cells/60-mm dish) were transfected with 10 nM siRNA using 4 μl of RNAiMAX. Sequences of siRNAs are provided in Table S2 in the supplemental material.
Gene expression analysis.
Microarray analysis was performed on an Affymetrix chip (HG_U133 Plus 2.0 arrays). Detailed methods are provided in Materials and Methods in the supplemental material.
Quantitative real-time PCR.
Total RNA was isolated by TRI reagent using an RNeasy kit (catalog number 74106; Qiagen). One microgram of RNA was reverse transcribed into cDNA using iScript Reverse Transcription Supermix (170-8840; Bio-Rad). Reverse transcription-quantitative PCR (RT-qPCR) was performed using 50 ng of cDNA/per reaction mixture using SYBR green reagent (catalog number 172-5265, SsoAdvanced SYBR green Supermix; Bio-Rad) in an Mx3000P qPCR System (Agilent Technologies). The sequences of the primers are given in Table S3 in the supplemental material. Data are shown as the means of three replicates ± 95% confidence intervals.
ISH.
In situ hybridizations (ISH) of PGC1β and ERRα in human colon tumor tissues were performed using a ViewRNA fluorescent RNA in situ hybridization kit (QVT0012; Affymetrics) according to the manufacturer's instructions. Experiments were performed with Institutional Review Board (IRB) approval. Additional details are provided in the supplemental materials.
Anchorage-independent growth in soft agar.
A total of 5 × 103 HCT116 cells/35-mm dish was used to perform anchorage-independent growth assays according to protocols of Kortum and Lewis (5). Colonies greater than 100 μm in diameter were counted, and representative photomicrographs were taken after 14 days of incubation in 37°C and 5% CO2.
Anchorage-independent growth in poly(HEMA)-coated plates.
Ninety-six-well plates were coated in poly-2-hydroxyethyl methacrylate [poly(HEMA)] by evaporating 200 μl of a 10 mg/ml solution in 95% ethanol from each well. Cells were plated in complete medium on poly(HEMA)-coated wells at a concentration of 3.5 × 104 cells/100 μl at 48 h posttransfection. Cell viability was measured by the addition of 100 μl of CellTiter-Glo reagent (Promega) and determining luminescence (POLARstar Optima plate reader).
Cell cycle analysis using propidium iodide staining.
Cells were treated as indicated in the figures and their legends for 72 h. Cells were collected, washed with 1× phosphate-buffered saline (PBS), and then fixed with 70% ice-cold ethanol at −20°C for 1 h to overnight. Cells were pelleted and rehydrated with 1× PBS at 37°C. Cells were again pelleted and mixed with Telford reagent (1% Triton X-100, 33.6 mg/liter EDTA, 26.8 mg/liter RNase A, and 50 mg/ml propidium iodide in 1× PBS) and stored overnight at 4°C before flow cytometry analysis.
Xenograft tumor studies.
A total of 1 × 106 HCT116 cells carrying various shRNA constructs were injected subcutaneously into the left flank of athymic BALB/c nude (nu/nu) female mice. Tumor volume was calculated using the following formula: volume (mm3) = length × (width)2 × 0.5. Survival was defined by the percentage of mice with tumor volumes less than the maximally allowed 500 mm3. All studies were completed following IACUC guidelines and with IACUC approval.
Statistics.
Significance was calculated using analysis of variance (ANOVA) (for the data in Fig. 1E, G, and H; 3A, C, E, and F; 7C and D; and 8B and E) or an unpaired, two-sided t test (for Fig. 3D). Significance in the xenograft studies was calculated using a survival curve comparison based on a log rank (Mantel-Cox) test (n = 10) (for Fig 1I, 8C, and 8F).
FIG 1.

KSR1 regulates anchorage-independent growth and tumor maintenance in colon cancer cell lines. (A) Immunoblot of KSR1 protein expression in multiple colon tumor cell lines compared to expression in HCECs. (B) qPCR analysis of mRNA levels of KSR1 in colon cancer cell lines relative to levels in HCECs normalized relative to glyceraldehyde-3-phosphate dehydrogenase. (C) Validation of individual siRNA duplexes targeting KSR1. (D and E) Immunoblot of KSR1 and analysis of apoptosis following siRNA-mediated depletion of KSR1 in HCECs and HCT116 cells. (F) Immunoblots of HCT116 cells following shRNA-mediated depletion of KSR1. (G) Quantification of anchorage-independent HCT116 cell growth in soft agar following KSR1 RNAi. Representative bright-field pictures are shown (bottom panel). (H) Immunoblots of KSR1 protein expression (top) and quantification of anchorage-independent HCT116 cell growth in soft agar (bottom) following stable knockdown of KSR1 with a rescued phenotype shown after hairpin-resistant mouse Flag-tagged KSR1 was reintroduced. (I) Xenograft tumor growth of HCT116 cells. ***, P < 0.001; ****, P < 0.0001.
FIG 3.

The relative importance of AMPK subunits on colon cancer survival. (A) Viability of HCT116 cells under anchorage-independent conditions was assessed following RNAi of KSR1 or AMPKγ1. (B) Validation of individual siRNA duplexes targeting AMPKγ1. (C) Immunoblotting and apoptosis in HCECs and HCT116 cells following RNAi of AMPKγ1. (D to F) Immunoblotting and apoptosis in HCT116 cells following RNAi of AMPKα1α2 (D), AMPKα1 or AMPKα2 (E), and AMPKβ1, AMPKβ2, or AMPKβ1β2 (F). ****, P < 0.0001. RLU, relative light units.
FIG 7.

In situ hybridization analysis of mRNA expression of PGC1β (A) and ERRα (B) in primary and metastatic human colon cancer in comparison to normal colonic epithelium. Quantification represents the amount of mRNA per nucleus and was performed on 32 normal colon images representing 17 unique patients, 28 primary colon cancer images representing 19 patients, and 37 stage IV liver metastases images representing 15 unique patients for PGC1β (C) and on 16 normal colon images representing 10 unique patients, 16 primary colon images representing 14 unique patients, and 6 stage IV liver metastases images representing 5 unique patients for ERRα (D). Box plots show median, middle two quartiles (box), and minimum and maximum values (tails). **, P < 0.01; ***, P < 0.001. DAPI, 4′,6′-diamidino-2-phenylindole.
FIG 8.

(A and D) Immunoblots of HCT116 cells stably transduced with nontargeting (Cont) shRNA or shRNA targeting PGC1β or ERRα. (B and E) Anchorage-independent growth in HCT116 cells stably transduced with shRNA against PGC1β or ERRα. Representative bright-field pictures are shown (bottom panel). ***, P < 0.001. (C and F) Xenograft tumor growth following implantation of HCT116 cells after shRNA-mediated depletion of PGC1β or ERRα compared to that with a nontargeting shRNA in immunocompromised mice. ****, P < 0.0001.
Microarray data accession number.
Raw CEL and processed RES files were deposited in the NCBI Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/projects/geo/) under accession number GSE65351.
RESULTS
KSR1 regulates anchorage-independent growth and tumor maintenance.
KSR1 regulates the oncogenic potential of activated Ras (5). We analyzed the expression of KSR1 in colon tumor cells and compared its expression to that in human colonic epithelial cells (HCECs). Immunoblotting and qPCR analysis revealed that KSR1 is overexpressed at both the protein (Fig. 1A) and mRNA (Fig. 1B) levels in the colon tumor cell lines tested compared to its expression in HCECs. To assess the feasibility of targeting KSR1 in colorectal cancer therapy, we compared the roles of KSR1 in colorectal cancer cells and HCECs. HCECs were isolated from normal human colon and immortalized with CDK4 and human telomerase reverse transcriptase (hTERT). HCECs maintain wild-type adenomatous polyposis coli (APC) protein, K-RAS, and TP53 and form crypt-like structures in three-dimensional culture. However, HCECs are incapable of anchorage-independent growth or tumor formation in nude mice (10). RNAi of KSR1 did not significantly affect the survival of HCECs but induced significant apoptosis in HCT116 cells (Fig. 1C to E). Further, to determine the role of KSR1 in human colon tumor cell maintenance, we stably depleted KSR1 protein in HCT116 cells with shRNAs that targeted two different sequences of KSR1 or a nontargeting shRNA (Fig. 1F). Cells depleted of KSR1 were unable to form colonies when grown under anchorage-independent conditions in soft agar (Fig. 1G). To determine whether loss of anchorage-independent growth was indeed a specific effect of KSR1 depletion, we reintroduced shRNA-resistant mouse KSR1 (mKSR1) cDNA in HCT116 cell clones depleted of KSR1. Reintroduction of KSR1 rescued the ability of KSR1-depleted clones to grow in soft agar (Fig. 1H). To evaluate the importance of KSR1 for in vivo tumor development, a xenograft study was completed that showed that depletion of KSR1 was sufficient to suppress the growth and increase the latency of tumor formation when HCT116 cells with or without shKSR1 were grafted into immunocompromised mice (Fig. 1I).
These data demonstrate that KSR1 is required for human colon tumor cell survival and tumor maintenance but superfluous for nontransformed colonic epithelial cells survival.
FUSION identifies AMPK as a functional analog of KSR1.
Using a gene expression signature that reflected the loss of KSR1, we developed a high-throughput screen using FUSION (9) to identify novel effectors of signaling pathways activated by oncogenic Ras. Gene set enrichment analysis (GSEA) (11) of gene expression profiles from HCT116 cells with or without shRNA-mediated depletion of KSR1 revealed a finite set of pathways regulated by KSR1 (Fig. 2A). From these gene sets, the differential expression of 10 candidate genes was confirmed using multiplexed branched DNA (bDNA) signal amplification (Quantigene, version 2.0; Affymetrix). Six genes (LOXL2, ACSL5, NDRG1, ALDOC, BNIP3L, and BNIP3) demonstrated the most consistent downregulation at 48 and 72 h after siRNA-mediated depletion of KSR1 in HCT116 cells and were selected to represent the loss of the KSR1 phenotype (Fig. 2B). Two invariant housekeeping genes, hypoxanthine phosphoribosyltransferase (HPRT) and peptidylprolyl isomerase B (PPIB), were added to create an eight-probe panel, permitting the normalization of each sample.
FIG 2.

Selection and validation of reporters for FUSION analysis. (A) Hypoxia, glycolysis, and p53 cis binding element gene sets were identified as being downregulated in the HCT116 cell line following stable knockdown of KSR1. (B) Reporters were chosen based on KSR1-dependent changes in expression. (C) Unsupervised hierarchical clustering of reporter gene expression for siRNA gene depletions. The region with a cluster of siRNAs targeting KSR1 is enlarged. (D) Pearson correlation plot of ranked FUSION results with a cutoff P value of 0.01.
To identify functional analogs of KSR1, we chose to interrogate all the known human kinases using an siRNA library (siGenome; Dharmacon/GE Healthcare, Lafayette, CO, US) containing known kinases, phosphatases, and kinase-related genes (“kinome”). We targeted KSR1 or each gene individually (n = 817) using pooled siRNAs and collected gene expression signatures that represent the loss of each gene. Unsupervised hierarchal clustering of the average gene expression signature of the siRNA targeting KRS1 (siKSR1) and each of the 817 kinome siRNAs was performed and demonstrated the tight clustering of positive-control siKSR1-containing wells (Fig. 2C), demonstrating the ability of FUSION to associate independent biological replicate samples based solely on gene expression signatures. The samples were then sorted by their Pearson correlation coefficients, and a 1% P value threshold was used to further identify candidate genes (Fig. 2D).
Anchorage-independent growth on poly(HEMA)-coated tissue culture plates (12, 13) was used to validate hits from FUSION with KSR1-like properties. RNAi of KSR1 significantly decreased the viability of HCT116 cells plated on poly(HEMA)-coated tissue culture plates compared to that of cells transfected with a nontargeting siRNA (Fig. 3A).
5′ AMP-activated protein kinase (AMPK) is a cellular energy sensor that phosphorylates serine/threonine substrates to increase available energy in response to an increased intracellular AMP concentration. KSR1 and KSR2 physically interact with AMPK in nonneoplastic settings to control metabolism (14). AMPK is a trimeric holoenzyme that requires regulatory β and γ subunits for the kinase activity of the α subunit to function efficiently (15, 16). FUSION identified AMPKγ1 as a candidate gene that, similar to KSR1, should be essential for tumor cell survival but is not required for the survival of nontransformed cells. To validate AMPKγ1, HCT116 cells were depleted of AMPKγ1 with a pool of confirmed siRNAs with different sequences than those used in the FUSION analysis (Fig. 3B) and plated in poly(HEMA)-coated wells to assess anchorage-independent growth. RNAi-mediated knockdown of AMPKγ1 significantly decreased viability in a manner comparable to KSR1 depletion (Fig. 3A).
The α2β2γ1 isoform of AMPK promotes colorectal cancer cell survival.
To determine the feasibility of using AMPKγ1 as a selective target in human colorectal cancer cells, AMPKγ1 was knocked down in HCT116 cells and in HCECs (Fig. 3C). RNAi of AMPKγ1 induced significant apoptosis in HCT116 cells but did not affect survival in the HCECs (Fig. 3C). The γ subunit of AMPK stabilizes the catalytic α subunit and is required for effective holoenzyme activity (17). Similar to loss of AMPKγ1, combined RNAi-mediated depletion of AMPKα1 and AMPKα2 isoforms also decreased the viability of the colon tumor cell line HCT116 (Fig. 3D). To further to examine which isoform of the α subunit is responsible for maintaining viability of colorectal cancer cells, we targeted AMPKα1 and AMPKα2 subunits separately by RNAi (Fig. 3E). Depletion of AMPKα2 but not AMPKα1 was sufficient to induce apoptosis (Fig. 3E) in HCT116 cells. These data demonstrate the dependency of colorectal cancer cells on AMPK signaling for their survival. The β subunit of AMPK interacts with both the α and γ subunits and helps in complex formation. To investigate the potential role of AMPKβ subunits in maintaining colon tumor cell viability, we performed RNAi of both AMPKβ1 and -β2 (Fig. 3F). Depletions of AMPKβ2 and AMPKβ1β2 induced similar levels of apoptosis in HCT116 cells, while depletion of AMPKβ1 did not significantly induce apoptosis (Fig. 3F), demonstrating the cancer cells' increased dependency on AMPKβ2 compared to the requirement for AMPKβ1.
PGC1β and ERRα are key downstream effectors of K-Ras, KSR1, and AMPKγ1.
We previously defined the role of PGC1α and ERRα as downstream effectors necessary to support anchorage-independent growth of H-RasV12-transformed mouse embryo fibroblasts (MEFs) (6). PGC1α and PGC1β (PGC1α/β) and ERRα/β/γ isoforms interact in several combinations to form transcriptional coactivator and transcription factor complexes that regulate hundreds of genes and promote a diverse variety of phenotypes (18, 19). Interestingly, we detected PGC1β but no PGC1α in human colon tumor cell lines. To assess the ability of KSR1 and oncogenic K-Ras to modulate PGC1β and ERRα expression in colon cancer cell lines, we inhibited the expression of either protein by RNAi in HCT116 cells. Depletion of K-Ras and KSR1 decreased the expression of both MEK1/2 and ERK1/2 in addition to reducing the expression of both PGC1β and ERRα (Fig. 4A). This observation demonstrates that the effector pathway used by KSR1 and oncogenic K-Ras to control tumorigenesis by regulating PGC1 and ERRα expression in a model system is intact in human cancer cell lines. To evaluate if KSR1 and K-Ras modify the expression of PGC1β and ERRα by modulating MEK and ERK activation, we targeted these downstream effectors individually. siRNA-mediated depletion of ERK1 and ERK2, either alone or in combination (Fig. 4B), or treatment with the MEK inhibitor U0126 or PD0325901 (Fig. 4C) did not decrease the protein expression of PGC1β and ERRα, indicating a MEK/ERK-independent role of KSR1 in the regulation of PGC1β and ERRα.
FIG 4.
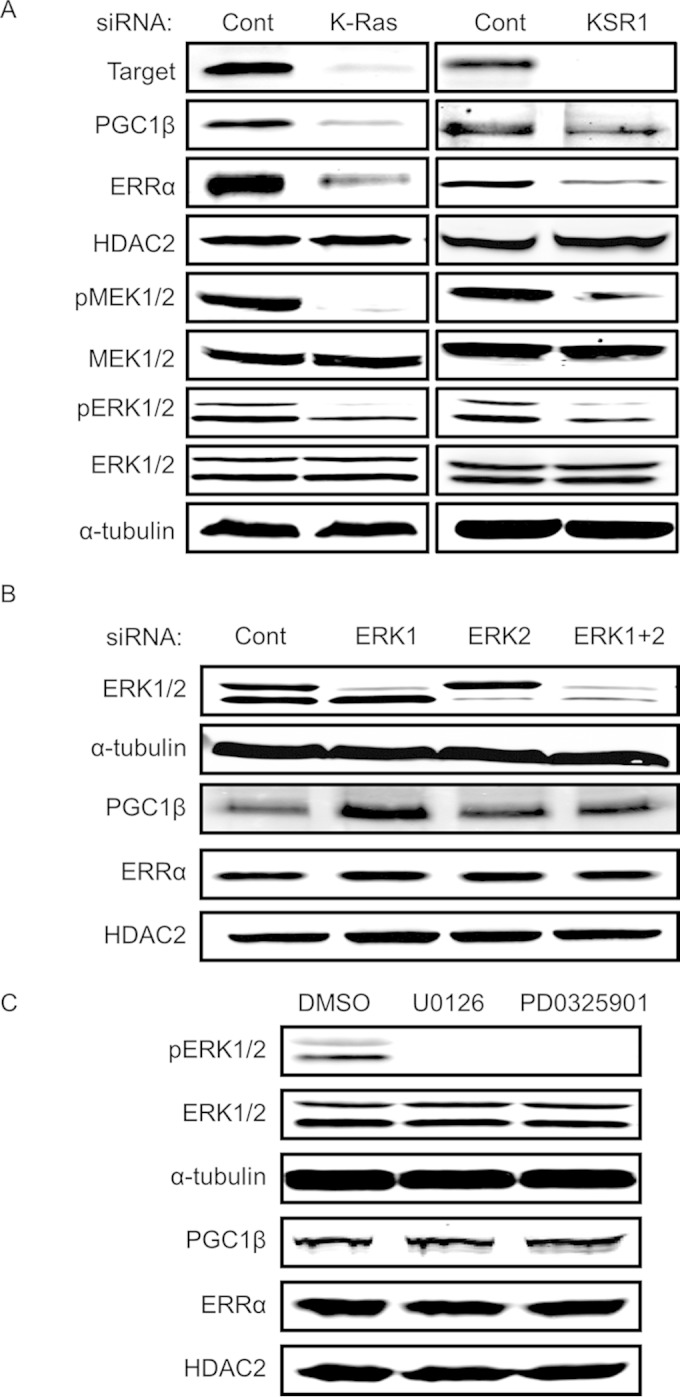
PGC1β and ERRα are key downstream effectors of K-Ras and KSR1 and are regulated through an ERK-independent mechanism. (A) Immunoblots of PGC1β, ERRα, phospho-MEK1/2 (pMEK1/2), total MEK1/2, and phospho (pERK1/2)- and total ERK1/2 in HCT116 cells following RNAi of K-Ras or KSR1. (B and C) ERK1/2, PGC1β, and ERRα immunoblotting following siRNA-mediated depletion of ERK1/2 alone or together (B) or inhibition of MEK activity with 20 μM U0126 or PD0325901 (C) in HCT116 cells.
Similar to depletion of KSR1, RNAi of AMPKγ1 decreased the protein expression of PGC1β and ERRα in HCT116 (Fig. 5A), SW480, and SW620 cells (Fig. 5B). Further depletion of AMPKα2 but not the AMPKα1 subunit decreased PGC1β and ERRα in HCT116 cells (Fig. 5C). However, unlike RNAi of K-Ras or KSR1 (Fig. 4A), RNAi of AMPKγ1 had no effect on MEK/ERK phosphorylation (Fig. 5A), indicating that its effects on PGC1β and ERRα are independent of the MEK/ERK pathway. To assess whether the phosphorylation of AMPK is dependent on K-Ras and KSR1 expression, we individually depleted HCT116 cells of K-Ras, KSR1, or AMPKγ1 by RNAi. As expected, depletion of AMPKγ1 decreased phosphorylation of AMPK at Thr172, likely because the AMPKγ1 subunit is required for AMP binding and activation of the kinase (20). While inhibition of K-Ras did not affect AMPK phosphorylation, inhibition of KSR1 decreased AMPKγ1 levels, which in turn led to a decrease in AMPK activation (Fig. 5D). To assess the levels of AMPK activation in colon tumor cells compared to activation in human colonic epithelial cells, we performed immunoblot analysis for phosphorylation of AMPK at threonine 172 of cytoplasmic lysates prepared from a panel of colon tumor cell lines and compared its level to that observed in HCECs. The level of AMPK phosphorylation at threonine 172 was variable in different cell lines (Fig. 5E).
FIG 5.

AMPK is activated in colon cancer cells and regulates PGC1β and ERRα in an ERK-independent manner. (A) Immunoblots of PGC1β, ERRα, phospho- and total MEK1/2, and phospho- and total ERK1/2 in HCT116 cells following RNAi of AMPKγ1. (B) Immunoblot of PGC1β and ERRα protein expression following siRNA-mediated depletion of AMPKγ1 in SW480 and SW620 cell lines. (C) Immunoblot of PGC1β and ERRα protein expression following siRNA-mediated depletion of AMPKα1 or AMPKα2 in HCT116 cells. (D) Phosphorylation of AMPK at threonine 172 and AMPKα protein expression in HCT116 cells following K-Ras, KSR1, or AMPKγ1 depletion. (E) Phosphorylation of AMPK at threonine 172 in a panel of colon tumor cell lines.
PGC1β and ERRα are overexpressed in colon cancer and are required for colon cancer survival both in vitro and in vivo.
To assess the role of PGC1β and ERRα in colorectal cancer, we determined their expression levels in seven colorectal cancer cell lines and HCECs. Although no PGC1α was detected, all seven colorectal cancer cell lines that were examined showed elevated expression levels of PGC1β and ERRα proteins in contrast to the undetectable levels in HCECs (Fig. 6A). mRNAs encoding PGC1β (Fig. 6B) and ERRα (Fig. 6C) were significantly higher in all colorectal cancer cell lines than in HCECs.
FIG 6.

PGC1β and ERRα are overexpressed in colon cancer. (A) PGC1β and ERRα immunoblots in multiple colon tumor cell lines and HCECs. (B and C) qPCR analysis of the mRNA levels of PGC1β and ERRα in colon cancer cell lines relative to levels in HCECs. K-RasMUT, mutated K-ras.
To determine the extent to which PGC1β and ERRα expression levels are elevated in colorectal cancer, we used in situ hybridization (ViewRNA; Affymetrix) to detect the mRNAs for PGC1β and ERRα in normal colon epithelia, primary colorectal cancer, and liver metastases. In situ hybridization demonstrated that PGC1β and ERRα mRNAs were present at low levels in tissues from normal colonic mucosa, but each transcript was increased in both primary and metastatic colorectal tissue samples (Fig. 7).
To determine the importance of PGC1β and ERRα to the tumorigenic properties of human colon tumor cells, we depleted PGC1β and ERRα using RNAi in the human colon tumor cell line HCT116. Three shRNAs targeting different regions of the PGC1β transcript and two shRNAs targeting different regions of the ERRα transcript were used to stably deplete PGC1β and ERRα protein expression in HCT116 cells. shRNA targeting PGC1β (Fig. 8A) resulted in a reduction in PGC1β protein expression, a concomitant reduction in ERRα protein expression, and suppressed anchorage-independent growth in soft agar (Fig. 8B). shRNA-mediated depletion of ERRα (Fig. 8D) also suppressed anchorage-independent growth (Fig. 8E), suggesting that maintenance of ERRα expression by PGC1β is contributing to the tumorigenic properties of colon cancer cells. RNAi-mediated depletion of PGC1β or ERRα also suppressed tumor formation of HCT116 cells grafted into immunodeficient mice (Fig. 8C and F). However, a constitutively active form of PGC1β (CA-PGC1β) expressed from an exogenous promoter robustly increased ERRα expression (Fig. 9A) but was unable to rescue the effects of K-Ras, KSR1, and AMPKγ1 depletion in an anchorage-independent cell viability assay (Fig. 9B and C). These results reveal that although PGC1β and ERRα play a central role in colon tumor cell growth and tumor maintenance, transient overexpression of CA-PGC1β is insufficient to rescue the loss of cell viability observed following K-Ras, KSR1, or AMPKγ1 depletion.
FIG 9.
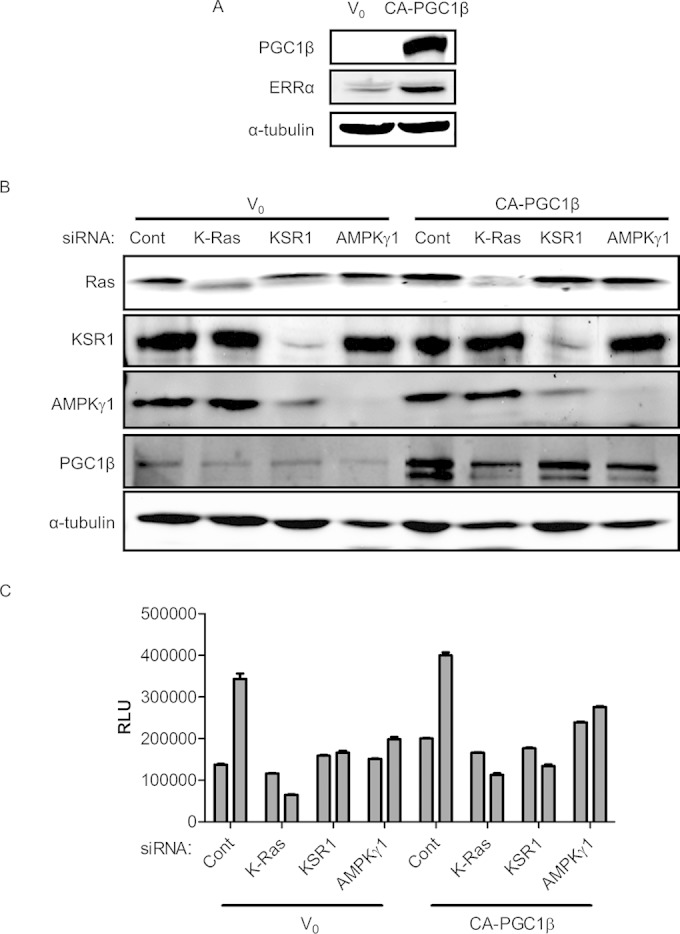
Constitutively active PGC1β (CA-PGC1β) does not rescue cell viability of HCT116 cells following depletion of K-Ras, KSR1, or AMPKγ1. (A) Immunoblot showing ERRα protein levels with either an empty vector control (V0) or CA-PGC1β. (B and C) Immunoblot showing K-Ras, KSR1, AMPKγ1, and PGC1β levels (B) corresponding to the viability data in control (V0) and CA-PGC1β-overexpressing cells (C). RLU, relative light units.
PGC1β and ERRα are key downstream effectors of AMPKγ1.
To investigate the potential mechanism of regulation of PGC1β by AMPK, we quantified the mRNA levels of PGC1β and ERRα after RNAi of AMPKγ1. qPCR analysis suggests that depletion of AMPK does not affect the mRNA levels of PGC1β and ERRα, indicating that the regulation is posttranscriptional (Fig. 10A and B). To investigate this mechanism, HCT116 cells were treated with the translational inhibitor cycloheximide (CHX) in the presence or in the absence of the proteosomal inhibitor MG132. Treatment with CHX decreased the protein level of PGC1β, which could be rescued with MG132 (Fig. 10C). These data suggest that PGC1β is modified at the posttranscriptional level and is degraded through the proteosome. To investigate the possible role of AMPK in this process, RNAi of AMPKγ1 was performed for 72 h, after which cells were treated for 6 h with either a dimethyl sulfoxide (DMSO) control or MG132. Depletion of AMPKγ1 decreased the protein levels of PGC1β, which was rescued by the addition of the proteosomal inhibitor MG132 (Fig. 10D). Similar to AMPKγ1 depletion, RNAi of K-Ras also decreased the PGC1β protein level, which could be rescued upon MG132 treatment (Fig. 10E). The effect of KSR1 depletion on PGC1β protein levels is consistent with K-Ras and AMPKγ1 depletion; however, the restoration of PGC1β protein levels with MG132 treatment in KSR1-depleted cells is modest (Fig. 10F). These data suggest that AMPK, K-Ras, and, to a lesser extent, KSR1 regulate the stability of PGC1β, revealing a distinct Ras-driven and KSR1-dependent signaling pathway in human cancer cells that is also modulated by AMPK.
FIG 10.

PGC1β and ERRα are key downstream effectors of AMPKγ1. (A and B) qPCR analysis of PGC1β and ERRα mRNA levels after RNAi of AMPKγ1. (C) Immunoblot of PGC1β protein expression in cells treated with 100 μM cycloheximide for 0 to 120 min with and without 10 μM MG132 proteasome inhibitor treatment for 6 h. (D to F) Immunoblots of PGC1β protein expression in AMPKγ1-, K-Ras-, or KSR1-depleted cells with and without 10 μM MG132 proteasome inhibitor treatment for 5 h.
DISCUSSION
In human colon tumor cells dependent upon oncogenic Ras, KSR1 is required for anchorage-independent proliferation, survival, and in vivo growth. In nontransformed human colon epithelial cells (HCECs), KSR1 is dispensable for survival, consistent with the observation that KSR1 is not required for normal development (4). Transient depletion of KSR1 results in about 30% cell death in Ras-driven cancer cells. Additionally, cells that survive KSR1 knockdown lose their ability to form anchorage-independent colonies in soft agar. These in vivo and in vitro analyses demonstrate that tumor-specific pathways are activated by oncogenic Ras in a KSR1-dependent manner.
With KSR1 as a reference standard, we used FUSION (9) to identify AMPKγ1 as a gene that is required for human colon tumor cell survival. Subsequent analyses showed that the α2 isoform of the AMPK kinase subunit and the β2 isoform of the AMPK regulatory subunit are also essential components. This observation suggests that a specific composition of the trimeric enzyme regulates effector pathways critical to tumor cell survival.
AMPK has been shown to both support and inhibit the transformed phenotype (21–23), and one of its activating kinases, LKB1, is a well-known tumor suppressor (24, 25). However, LKB1 has multiple targets (26) that may contribute to tumorigenesis. Indirect evidence supporting a tumor-suppressive role for AMPK comes from the inverse correlation of cancer rates with the use of the antidiabetic drug metformin, an electron transport complex I inhibitor that indirectly activates AMPK via suppression of electron transport-driven ATP synthesis. Metformin was associated with a 30% reduction in solid cancer incidence compared to results with other diabetic treatments (27). However, metformin may inhibit cancer susceptibility via its general effect on mitochondrial function (28) or through other mechanisms of action distinct from its effect on AMPK (29). On the other hand, disruption of the AMPKα1 subunit inhibits aerobic glycolysis and suppresses lymphomagenesis (23). Further, AMPK has additional effects that potently impair cell growth and proliferation. AMPK has been reported to phosphorylate B-Raf, impairing its interaction with KSR1 and the subsequent activation of MEK and ERK (30). AMPK promotes cell cycle arrest through phosphorylation of p53, and inhibition of the mTORC1 complex through phosphorylation of the TSC2 complex and Raptor (31–33). AMPK activation inhibits fatty acid and cholesterol synthesis, which is essential to meet the increased demand in tumor cells for lipid membranes (34, 35).
Recent evidence, however, has pointed to the tumor-promoting role of AMPK under certain circumstances. RasV12-activated AMPKα1/α2 null MEFs are resistant to tumor formation in vivo, with increased susceptibility to anoikis (36, 37), and KSR2-dependent MEF transformation requires AMPK activation (38). AMPK activity is increased in early stages of gliomas (39). AMPK sustains lung cancer cell viability under conditions of cellular and metabolic stress by maintenance of NADPH homeostasis (40). In these lung carcinoma cells, AMPK stimulates fatty acid oxidation by phosphorylating and inhibiting acetyl coenzyme A (acetyl-CoA) carboxylase to maintain NADPH homeostasis, which is required for anchorage-independent survival of the tumor cells (40). This observation mirrors our observation that the depletion of a specific AMPK subunit composition impairs anchorage-independent survival of HCT116 colon cancer cells. This observation is also consistent with the suggestion that the contribution of AMPK to tumor maintenance may be a function of tumor stage (41). Additionally, recent evidence has demonstrated the dichotomy in the apparent role of the α1 and α2 AMPK subunits. AMPKα1−/− mice have a normal metabolic phenotype, while AMPKα2−/− mice demonstrate a phenotype similar to that of type II diabetes (42). These apparent differences suggest that the potential for AMPK to act as a tumor suppressor or oncogene may be, at a minimum, a function of the α catalytic subunit.
While FUSION did not identify AMPKα2, FUSION is limited by the efficacy of the siRNAs present in the library and was designed to limit false positives preferentially over false negatives. It is likely other KSR1-like genes were not identified. The selective dependence of HCT116 cell survival on the AMPKα2 catalytic subunit suggests that pathways promoting tumor cell survival prefer trimeric AMPK enzymes containing the α2 subunit. The expression and activity of specific AMPK subunits in cell proliferation and cancer have been recognized recently. An RNAi screen demonstrated that in prostate cancer the AMPKβ1 subunit was required for tumor maintenance and found that it was elevated in metastatic tissues compared to levels in the primary tumor (43). A chemical genetic screen detected 28 novel substrates for AMPKα2, several of which were essential for mitotic progression (44). A careful examination of AMPK substrates may yield insight into AMPK-dependent pathways that are selectively required for Ras-driven tumor cell survival.
Our data reveal a previously unreported function of AMPK to support the expression of PGC1β and ERRα and confer a survival advantage to colon tumor cell lines. We previously identified PGC1α and ERRα as genes upregulated by KSR1 in an H-RasV12-dependent manner (6). Here, we show K-Ras-mediated and KSR1-dependent upregulation of PGC1β and ERRα in colon tumor cells and tissues. Our data indicate a previously unappreciated role of KSR1 and oncogenic K-Ras in promoting the expression of PGC1β and ERRα in human colorectal cancer. These data also indicate that AMPK confers a growth advantage to colon tumor cell lines by signaling through metabolic regulators PGC1β and ERRα. In situ hybridization shows that PGC1β and ERRα are expressed at very low levels in primary human colon epithelial cells but markedly elevated in colorectal cancer metastases. Furthermore, suppressing PGC1β and ERRα expression decreased colon tumor cell viability and anchorage-independent growth and delayed tumor formation in nude mice, making them potential targets in cancer therapy. PGC1β and ERRα have been studied extensively as metabolic regulators that promote tumorigenesis in breast cancer (45–49). Breast cancer has a low incidence of activating Ras mutations but are often driven by the overexpression of ERBB2, which signals through Ras (50). Thus, pathways regulating the expression of PGC1 isoforms and ERRα are of increasing relevance to the etiology of cancer in general.
Very little is known about the posttranslational modifications of PGC1β. PGC1 proteins undergo extensive posttranslational modification, which has been well documented for PGC1α. PGC1α has been shown to be phosphorylated by AMPK at T177 and S538, which increases the activity of PGC1α at its own promoter, inducing its transcription (51). Recent evidence indicates that the E3 ligase synoviolin ubiquitinates and targets PGC1β for degradation (52). We demonstrate that not only AMPK but also K-Ras and, to a lesser extent, KSR1, stabilize PGC1β protein, which, based on its rescued expression in the presence of a proteosomal inhibitor (Fig. 10), appears to be degraded by the proteosome. Further studies will define the mechanisms that lead to AMPK-dependent effects on PGC1β stability. Further, it will be interesting to see whether the stability of PGC1β as conferred by K-Ras and KSR1 is dependent or independent of AMPK. The modest rescue of PGC1β in the presence of the proteasomal inhibitor in KSR1-depleted cells may point toward a broader role played by KSR1 in the regulation of PGC1β, potentially at the level of PGC1β posttranscriptional modification or translation.
The degree to which the aberrant expression of PGC1β and ERRα persists in additional tumors bearing oncogenic Ras alleles will reveal the importance of these transcriptional regulators in the creation of tumor cells and the promotion of their survival. Elucidation of the detailed mechanisms that control their expression and their action may expose novel vulnerabilities in Ras-driven tumors, creating new avenues for therapy in a broad spectrum of cancers.
Supplementary Material
ACKNOWLEDGMENTS
We thank Amy Wells from the University of Nebraska Medical Center (UNMC) Eppley Institute high-throughput screening facility for her assistance in performing the genome-scale screen, the UNMC Cell Analysis Facility for performing the flow cytometry analysis, and the UNMC Tissue Sciences Facility for providing us with normal colon and colon tumor tissue microarrays. We acknowledge Premila Leiphrakpam for her support throughout this project. We thank Rob Livergood for his contribution in the quantification of the in situ hybridization.
This work was supported by grants from NCI (CA157774 to R.E.L.), NIH (CA071443 to M.A.W and CA1499833 to J.M.), and the Nebraska Department of Health and Human Services (LB506 to R.E.L.) and by an Eppley Institute Cancer Biology training grant (NCI T32CA009476) and Fred and Pamela Buffett Cancer Center support grant (P30CA036727).
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication. We declare that we have no conflicts of interests.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00528-15.
REFERENCES
- 1.Stephen Andrew G, Esposito D, Bagni Rachel K, McCormick F. 2014. Dragging Ras back in the ring. Cancer Cell 25:272–281. doi: 10.1016/j.ccr.2014.02.017. [DOI] [PubMed] [Google Scholar]
- 2.Dhillon AS, Hagan S, Rath O, Kolch W. 2007. MAP kinase signalling pathways in cancer. Oncogene 26:3279–3290. doi: 10.1038/sj.onc.1210421. [DOI] [PubMed] [Google Scholar]
- 3.Roberts PJ, Der CJ. 2007. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 26:3291–3310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- 4.Nguyen A, Burack WR, Stock JL, Kortum R, Chaika OV, Afkarian M, Muller WJ, Murphy KM, Morrison DK, Lewis RE, McNeish J, Shaw AS. 2002. Kinase suppressor of Ras (KSR) is a scaffold which facilitates mitogen-activated protein kinase activation in vivo. Mol Cell Biol 22:3035–3045. doi: 10.1128/MCB.22.9.3035-3045.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kortum RL, Lewis RE. 2004. The molecular scaffold KSR1 regulates the proliferative and oncogenic potential of cells. Mol Cell Biol 24:4407–4416. doi: 10.1128/MCB.24.10.4407-4416.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fisher KW, Das B, Kortum RL, Chaika OV, Lewis RE. 2011. Kinase suppressor of ras 1 (KSR1) regulates PGC1α and estrogen-related receptor alpha to promote oncogenic Ras-dependent anchorage-independent growth. Mol Cell Biol 31:2453–2461. doi: 10.1128/MCB.05255-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xing HR, Cordon-Cardo C, Deng X, Tong W, Campodonico L, Fuks Z, Kolesnick R. 2003. Pharmacologic inactivation of kinase suppressor of ras-1 abrogates Ras-mediated pancreatic cancer. Nat Med 9:1266–1268. [DOI] [PubMed] [Google Scholar]
- 8.Llobet D, Eritja N, Domingo M, Bergada L, Mirantes C, Santacana M, Pallares J, Macia A, Yeramian A, Encinas M, Moreno-Bueno G, Palacios J, Lewis RE, Matias-Guiu X, Dolcet X. 2011. KSR1 is overexpressed in endometrial carcinoma and regulates proliferation and TRAIL-induced apoptosis by modulating FLIP levels. Am J Pathol 178:1529–1543. doi: 10.1016/j.ajpath.2010.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Potts MB, Kim HS, Fisher KW, Hu Y, Carrasco YP, Bulut GB, Ou YH, Herrera-Herrera ML, Cubillos F, Mendiratta S, Xiao G, Hofree M, Ideker T, Xie Y, Huang LJ, Lewis RE, MacMillan JB, White MA. 2013. Using functional signature ontology (FUSION) to identify mechanisms of action for natural products. Sci Signal 6:ra90. doi: 10.1126/scisignal.2004657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roig AI, Eskiocak U, Hight SK, Kim SB, Delgado O, Souza RF, Spechler SJ, Wright WE, Shay JW. 2010. Immortalized epithelial cells derived from human colon biopsies express stem cell markers and differentiate in vitro. Gastroenterology 138:1012–1021.e1–5. doi: 10.1053/j.gastro.2009.11.052. [DOI] [PubMed] [Google Scholar]
- 11.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawada M, Fukazawa H, Mizuno S, Uehara Y. 1997. Inhibition of anchorage-independent growth of ras-transformed cells on polyHEMA surface by antisense oligodeoxynucleotides directed against K-ras. Biochem Biophys Res Commun 231:735–737. doi: 10.1006/bbrc.1997.6179. [DOI] [PubMed] [Google Scholar]
- 13.Xu L-H, Yang X, Bradham CA, Brenner DA, Baldwin AS, Craven RJ, Cance WG. 2000. The focal adhesion kinase suppresses transformation-associated, anchorage-independent apoptosis in human breast cancer cells. J Biol Chem 275:30597–30604. doi: 10.1074/jbc.M910027199. [DOI] [PubMed] [Google Scholar]
- 14.Costanzo-Garvey DL, Pfluger PT, Dougherty MK, Stock JL, Boehm M, Chaika O, Fernandez MR, Fisher K, Kortum RL, Hong EG, Jun JY, Ko HJ, Schreiner A, Volle DJ, Treece T, Swift AL, Winer M, Chen D, Wu M, Leon LR, Shaw AS, McNeish J, Kim JK, Morrison DK, Tschop MH, Lewis RE. 2009. KSR2 is an essential regulator of AMP kinase, energy expenditure, and insulin sensitivity. Cell Metab 10:366–378. doi: 10.1016/j.cmet.2009.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao G, Fernandez CS, Stapleton D, Auster AS, Widmer J, Dyck JRB, Kemp BE, Witters LA. 1996. Non-catalytic beta- and gamma-subunit isoforms of the 5′-AMP-activated protein kinase. J Biol Chem 271:8675–8681. doi: 10.1074/jbc.271.15.8675. [DOI] [PubMed] [Google Scholar]
- 16.Woods A, Cheung PCF, Smith FC, Davison MD, Scott J, Beri RK, Carling D. 1996. Characterization of AMP-activated protein kinase and subunits. J Biol Chem 271:10282–10290. doi: 10.1074/jbc.271.17.10282. [DOI] [PubMed] [Google Scholar]
- 17.Adams J, Chen ZP, Van Denderen BJ, Morton CJ, Parker MW, Witters LA, Stapleton D, Kemp BE. 2004. Intrasteric control of AMPK via the γ1 subunit AMP allosteric regulatory site. Protein Sci 13:155–165. doi: 10.1110/ps.03340004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Villena JA, Kralli A. 2008. ERRα: a metabolic function for the oldest orphan. Trends Endocrinol Metab 19:269–276. doi: 10.1016/j.tem.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spiegelman BM. 2007. Transcriptional control of mitochondrial energy metabolism through the PGC1 coactivators. Novartis Found Symp 287:60–63. doi: 10.1002/9780470725207.ch5. [DOI] [PubMed] [Google Scholar]
- 20.Cheung PC, Salt IP, Davies SP, Hardie DG, Carling D. 2000. Characterization of AMP-activated protein kinase gamma-subunit isoforms and their role in AMP binding. Biochem J 346:659–669. doi: 10.1042/bj3460659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jeon SM, Hay N. 2012. The dark face of AMPK as an essential tumor promoter. Cell Logist 2:197–202. doi: 10.4161/cl.22651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liang J, Mills GB. 2013. AMPK: a contextual oncogene or tumor suppressor? Cancer Res 73:2929–2935. doi: 10.1158/0008-5472.CAN-12-3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Faubert B, Boily G, Izreig S, Griss T, Samborska B, Dong Z, Dupuy F, Chambers C, Fuerth BJ, Viollet B, Mamer OA, Avizonis D, DeBerardinis RJ, Siegel PM, Jones RG. 2013. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab 17:113–124. doi: 10.1016/j.cmet.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hemminki A, Markie D, Tomlinson I, Avizienyte E, Roth S, Loukola A, Bignell G, Warren W, Aminoff M, Hoglund P, Jarvinen H, Kristo P, Pelin K, Ridanpaa M, Salovaara R, Toro T, Bodmer W, Olschwang S, Olsen AS, Stratton MR, de la Chapelle A, Aaltonen LA. 1998. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 391:184–187. doi: 10.1038/34432. [DOI] [PubMed] [Google Scholar]
- 25.Jenne DE, Reimann H, Nezu J, Friedel W, Loff S, Jeschke R, Muller O, Back W, Zimmer M. 1998. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet 18:38–43. doi: 10.1038/ng0198-38. [DOI] [PubMed] [Google Scholar]
- 26.Lizcano JM, Goransson O, Toth R, Deak M, Morrice NA, Boudeau J, Hawley SA, Udd L, Makela TP, Hardie DG, Alessi DR. 2004. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J 23:833–843. doi: 10.1038/sj.emboj.7600110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DeCensi A, Puntoni M, Goodwin P, Cazzaniga M, Gennari A, Bonanni B, Gandini S. 2010. Metformin and cancer risk in diabetic patients: a systematic review and meta-analysis. Cancer Prev Res 3:1451–1461. doi: 10.1158/1940-6207.CAPR-10-0157. [DOI] [PubMed] [Google Scholar]
- 28.Andrzejewski S, Gravel S-P, Pollak M, St-Pierre J. 2014. Metformin directly acts on mitochondria to alter cellular bioenergetics. Cancer Metab 2:12. doi: 10.1186/2049-3002-2-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miller RA, Chu Q, Xie J, Foretz M, Viollet B, Birnbaum MJ. 2013. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature 494:256–260. doi: 10.1038/nature11808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shen CH, Yuan P, Perez-Lorenzo R, Zhang Y, Lee SX, Ou Y, Asara JM, Cantley LC, Zheng B. 2013. Phosphorylation of BRAF by AMPK impairs BRAF-KSR1 association and cell proliferation. Mol Cell 52:161–172. doi: 10.1016/j.molcel.2013.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB. 2005. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell 18:283–293. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 32.Inoki K, Zhu T, Guan KL. 2003. TSC2 mediates cellular energy response to control cell growth and survival. Cell 115:577–590. doi: 10.1016/S0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 33.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. 2008. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang C, Rajput S, Watabe K, Liao DF, Cao D. 2010. Acetyl-CoA carboxylase-a as a novel target for cancer therapy. Front Biosci (Schol Ed) 2:515–526. [DOI] [PubMed] [Google Scholar]
- 35.Hardie DG. 2007. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol 8:774–785. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- 36.Laderoute KR, Amin K, Calaoagan JM, Knapp M, Le T, Orduna J, Foretz M, Viollet B. 2006. 5′-AMP-activated protein kinase (AMPK) is induced by low-oxygen and glucose deprivation conditions found in solid-tumor microenvironments. Mol Cell Biol 26:5336–5347. doi: 10.1128/MCB.00166-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ng TL, Leprivier G, Robertson MD, Chow C, Martin MJ, Laderoute KR, Davicioni E, Triche TJ, Sorensen PH. 2012. The AMPK stress response pathway mediates anoikis resistance through inhibition of mTOR and suppression of protein synthesis. Cell Death Differ 19:501–510. doi: 10.1038/cdd.2011.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fernandez MR, Henry MD, Lewis RE. 2012. Kinase suppressor of Ras 2 (KSR2) regulates tumor cell transformation via AMPK. Mol Cell Biol 32:3718–3731. doi: 10.1128/MCB.06754-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu X, Chhipa RR, Pooya S, Wortman M, Yachyshin S, Chow LM, Kumar A, Zhou X, Sun Y, Quinn B, McPherson C, Warnick RE, Kendler A, Giri S, Poels J, Norga K, Viollet B, Grabowski GA, Dasgupta B. 2014. Discrete mechanisms of mTOR and cell cycle regulation by AMPK agonists independent of AMPK. Proc Natl Acad Sci U S A 111:E435–E444. doi: 10.1073/pnas.1311121111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jeon SM, Chandel NS, Hay N. 2012. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 485:661–665. doi: 10.1038/nature11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Svensson RU, Shaw RJ. 2012. Cancer metabolism: Tumour friend or foe. Nature 485:590–591. doi: 10.1038/485590a. [DOI] [PubMed] [Google Scholar]
- 42.Viollet B, Athea Y, Mounier R, Guigas B, Zarrinpashneh E, Horman S, Lantier L, Hebrard S, Devin-Leclerc J, Beauloye C, Foretz M, Andreelli F, Ventura-Clapier R, Bertrand L. 2009. AMPK: lessons from transgenic and knockout animals. Front Biosci 14:19–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ros S, Santos CR, Moco S, Baenke F, Kelly G, Howell M, Zamboni N, Schulze A. 2012. Functional metabolic screen identifies 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 4 as an important regulator of prostate cancer cell survival. Cancer Discov 2:328–343. doi: 10.1158/2159-8290.CD-11-0234. [DOI] [PubMed] [Google Scholar]
- 44.Banko MR, Allen JJ, Schaffer BE, Wilker EW, Tsou P, White JL, Villén J, Wang B, Kim SR, Sakamoto K, Gygi SP, Cantley LC, Yaffe MB, Shokat KM, Brunet A. 2011. Chemical genetic screen for AMPKα2 substrates uncovers a network of proteins involved in mitosis. Mol Cell 44:878–892. doi: 10.1016/j.molcel.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chang CY, Kazmin D, Jasper JS, Kunder R, Zuercher WJ, McDonnell DP. 2011. The metabolic regulator ERRα, a downstream target of HER2/IGF-1R, as a therapeutic target in breast cancer. Cancer Cell 20:500–510. doi: 10.1016/j.ccr.2011.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ariazi EA, Clark GM, Mertz JE. 2002. Estrogen-related receptor alpha and estrogen-related receptor gamma associate with unfavorable and favorable biomarkers, respectively, in human breast cancer. Cancer Res 62:6510–6518. [PubMed] [Google Scholar]
- 47.Wang L, Liu Q, Li F, Qiu J, Fan H, Ma H, Zhu Y, Wu L, Han X, Yang Z, Jiang H, Wei J, Xia H. 2013. Apoptosis induced by PGC-1β in breast cancer cells is mediated by the mTOR pathway. Oncol Rep 30:1631–1638. doi: 10.3892/or.2013.2628. [DOI] [PubMed] [Google Scholar]
- 48.Li Y, Li Y, Wedren S, Li G, Charn TH, Desai KV, Bonnard C, Czene K, Humphreys K, Darabi H, Einarsdottir K, Heikkinen T, Aittomaki K, Blomqvist C, Chia KS, Nevanlinna H, Hall P, Liu ET, Liu J. 2011. Genetic variation of ESR1 and its co-activator PPARGC1B is synergistic in augmenting the risk of estrogen receptor-positive breast cancer. Breast Cancer Res 13:R10. doi: 10.1186/bcr2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eichner LJ, Perry MC, Dufour CR, Bertos N, Park M, St-Pierre J, Giguere V. 2010. miR-378(*) mediates metabolic shift in breast cancer cells via the PGC-1β/ERRγ transcriptional pathway. Cell Metab 12:352–361. doi: 10.1016/j.cmet.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 50.Deblois G, Chahrour G, Perry MC, Sylvain-Drolet G, Muller WJ, Giguere V. 2010. Transcriptional control of the ERBB2 amplicon by ERRα and PGC-1β promotes mammary gland tumorigenesis. Cancer Res 70:10277–10287. doi: 10.1158/0008-5472.CAN-10-2840. [DOI] [PubMed] [Google Scholar]
- 51.Jager S, Handschin C, St-Pierre J, Spiegelman BM. 2007. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. Proc Natl Acad Sci U S A 104:12017–12022. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fujita H, Yagishita N, Aratani S, Saito-Fujita T, Morota S, Yamano Y, Hansson MJ, Inazu M, Kokuba H, Sudo K, Sato E, Kawahara KI, Nakajima F, Hasegawa D, Higuchi I, Sato T, Araya N, Usui C, Nishioka K, Nakatani Y, Maruyama I, Usui M, Hara N, Uchino H, Elmer E, Nishioka K, Nakajima T. 2015. The E3 ligase synoviolin controls body weight and mitochondrial biogenesis through negative regulation of PGC-1β. EMBO J 34:1042–1055. doi: 10.15252/embj.201489897. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.