

Abstract
Although it has been established that effector memory CD4+ T cells play an important role in the protective immunity against chronic infections, little is known about the exact mechanisms responsible for their functioning and maintenance, as well as their effects on innate immune cells. Here we review recent data on the role of IFN-γ priming as a mechanism affecting both innate immune cells and effector memory CD4+ T cells. Suboptimal concentrations of IFN-γ are seemingly crucial for the optimization of innate immune cell functions (including phagocytosis and destruction of reminiscent pathogens), as well as for the survival and functioning of effector memory CD4+ T cells. Thus, IFN-γ priming can thus be considered an important bridge between innate and adaptive immunity.
1. Introduction
The immune system is continually exposed to a great diversity of pathogens. Among them, viruses, bacteria, protozoan parasites, and fungi present unique challenges for the host's immune system. In response to microorganisms, the adaptive immune system develops effector cells and functions capable of counteracting those threats. Among these effector cells, memory CD4+ T (TM) cells are considered a crucial population for the protective immunity against bacterial infections [1], viral infections [2], and protozoan infections such as malaria [3]. CD4+ TM cells participate in the responses against secondary infections by potentiating antipathogen effector mechanisms of innate immunity [4], antibody production, and CD8+ T cell cytotoxicity [2].
In the past decades, however, it has become increasingly clear that the TM population size is not a reliable marker of protective immunity per se. Zinkernagel and Hengartner previously argued that TM cells could not provide protection against fast-dividing pathogens without the maintenance of highly responsive antigen-stimulated lymphocytes [5]. It was suggested that immunity, especially to chronic infection, is the combination of resting memory cells and activated effectors. The description of two distinct TM cell subsets by Sallusto et al. [6] provides an additional basis for this hypothesis. Central memory T (TCM) cells and effector memory T (TEM) cells are classified based on their phenotype and their functional and trafficking characteristics [6, 7]. TCM cells are defined by surface expression of CD62L and CCR7 molecules that allow these cells to circulate between secondary lymphoid tissues, entering the T cell zones [8]. In a T helper 1 (Th1) response, these cells produce IL-2 upon antigen reencounter and, later on, effector cytokines such as IFN-γ. TEM cells, in contrast, have low expression of CD62L and CCR7 and migrate and localize into nonlymphoid, antigen-targeted tissues, where they are capable to quickly produce effector cytokines such as IFN-γ upon antigen reexposure [9, 10].
TEM cells have been considered the predominant population elicited by chronic infections [1, 10]. Therefore, the knowledge about the TEM cell origin, function, and survival is critical for vaccine development. In some infections, TEM cells maintain increased effector function; however, this may require the continued presence of antigen, which can also lead to T cell exhaustion. Alternatively, in the absence of antigen, the TCM population may remain expanded but without prompt functionality [11]. Among the possible mechanisms by which antigen persistence can drive the functioning of TEM cells, the effects of IFN-γ cannot be underestimated. This cytokine, as cited above, is one of the main products secreted by TEM cells in response to secondary antigen encounter [9], and its effects on both TEM cells and the effector branch of the immune system are still to be completely understood. In this review, we describe recent data on the role of IFN-γ on the protective immunity to infectious diseases with a special focus on the importance of the IFN-γ priming.
2. The Concept of IFN Priming and Its Effects on Acute Infectious Diseases
The effects of IFN-γ on the immune system are diverse, and the importance of this cytokine on the functioning of innate immune cells has been previously discussed [19]. Dendritic cells and macrophages are tightly regulated by cytokines to rapidly respond to infections and also to avoid the undesirable effects of excessive activation. Suboptimal concentrations of IFN-γ do not actually activate these cells but make them prepared for a subsequent response to stimuli, which in excess can eventually cause deleterious consequences. This effect is denominated as IFN-γ priming and has been increasingly implicated in the immune response to several infectious diseases such as viral [20, 21], bacterial [15, 22], and parasitical [15] infections. The underlying molecular mechanism for IFN-γ-priming effect involves a complex network of IFN-inducible genes, mostly from the innate immune system [22], whose understanding is still limited [17]. It is presumed that IFN-γ priming induces posttranscriptional and/or epigenetic changes, which are responsible for subsequent Toll-like receptor (TLR) ligand-triggered inflammatory response and classical macrophage activation [20, 21, 23, 24]. Recently, it has been shown that IFN-γ priming downregulates the expression of miR-3473b, a microRNA that suppresses macrophage activation and inflammatory response through directly targeting phosphatase and tensin homolog (PTEN) and promoting IL-10 production [25]. Of note, IL-10 has been shown to prevent the development of immunopathology during acute malaria [26, 27], as well as in Toxoplasma gondii [28] and Trypanosoma cruzi [29] infections. However, IL-10 promotes pathogen survival by downregulating protective immune responses during infections with Mycobacterium tuberculosis [30], Bordetella pertussis [31], and human immunodeficiency virus (HIV) [32]. The dual role of IL-10 is exemplified in Leishmania major infection, where IL-10 from effector Th1 cells is required to control excessive inflammatory response during acute infection [33], but IL-10 from regulatory T cells contributes to parasite persistence by suppressing effector Th1 cells during chronic infection [34, 35].
The IFN-γ priming seems to be particularly involved in several aspects of the immune response to malaria. McCall et al. (2007) showed that Plasmodium falciparum induces enhanced responses to TLR agonists in peripheral blood mononuclear cells [36]. This notion was further corroborated by findings on human subjects and mice, both acutely infected with P. falciparum and Plasmodium chabaudi, respectively [37], which showed an increased innate immune response to unrelated pathogens, in a TLR- and IFN-γ-dependent manner. Besides the effect of IFN-γ priming on TLR signaling, TLR engagement seems to be necessary for the initial IFN-γ production, as described for rodent malaria [38, 39]. Thus, it is likely that TLR signaling mediates initial pathogen recognition, which in turn initiates early IFN-γ production that further boosts the innate response through TLR induction. This mechanism is supported by results with malaria—as previously described [37]—and with several other infections by pathogens such as Listeria monocytogenes [40], L. major [41], Chlamydia pneumonia [42], T. cruzi [43], and Legionella pneumophila [44].
In acute infectious diseases, the augmented gene expression of TLR-related molecules induced by IFN-γ likely favors the pathogen recognition by phagocytic cells. Thus, a primed innate immune system can be of utmost importance to prevent or limit aggressive infections, contributing to the host survival. In contrast, a possible deleterious effect of this hypersensitivity can be inferred from the enhanced susceptibility of P. chabaudi-infected mice to LPS treatment [37, 45]. This was in fact demonstrated by the enhanced susceptibility of IFN-γ-primed mice to bacterial sepsis, which showed increased TNF production upon LPS stimulation [46]. Higher sensitivity to secondary infections by bacteria, such as Salmonella, has also been observed in human malaria [47]. Moreover, this hyperactivation of the immune system may contribute for the posterior state of immune paralysis observed in septic patients [48].
CD4+ and CD8+ T cells are also responsive and can be primed by type I IFNs, IFN-α/β, which are produced virtually by any cell type after stimulation [49]. Type I IFNs can be produced in large amounts by myeloid cells upon bacterial infection [50, 51], or by plasmacytoid dendritic cells upon viral stimulation [52, 53]. Production of these cytokines occurs following pathogen recognition by Toll-like receptors (TLRs), RIG-I-like receptors (RLRs), NOD-like receptors (NLRs), and a growing family of intracellular DNA receptors, several of which promote signaling through stimulator of IFN genes (STING) [54, 55]. Similarly to IFN-γ, type I IFNs can drive preferentially the CD4+ T cell differentiation to Th1 phenotype by activating Signal Transducer and Activator of Transcription (STAT) proteins that increase the T cell response to IL-2 [56]. These cytokines also inhibit the Th2 development by epigenetic silencing of GATA3 gene regulatory regions [57]. Interestingly, type I IFN signaling inhibits IL-12 production [58, 59], which contrasts with the type I IFN effects on Th1 cell development. However, type I IFNs themselves also act as signal 3 cytokines during T cell activation—by promoting proliferation, survival, and effector cell differentiation [54]. Different from IFN-γ-induced priming that is always proinflammatory, the effects of type I IFNs on T cell function can be inhibitory in certain cases, especially when cytokine signaling precedes T cell receptor (TCR) engagement on T cells [54]. Optimal cross-priming of CD8+ T cells is seemingly dependent on type I IFN stimulation [60]. Furthermore, type I IFNs activate T cells and sensitize them to Listeria-induced apoptosis [61].
3. IFN-γ Priming in Chronic Infections: Implications for Protective Immunity
IFN-γ is the main cytokine produced by TEM cells committed for the Th1 phenotype (Th1EM cells) upon infection by microorganisms, and it is believed to play a major role in the activation of innate immune response [62]. Thus, it is reasonable to imagine that these TEM cells are a major source of the IFN-γ responsible for priming the innate immune cells during chronic infections, making it an important point of crosstalk between innate and adaptive immunity. Supporting this possibility, we have recently described that the presence of Th1EM cells correlates with the continuous IFN-γ priming of innate immune cells during chronic malaria in mice [14]. This process is crucial for the protective immunity against reinfection with a heterologous strain of the parasite, which is not fully controlled by antibodies generated during primary infection with a different parasite clone. These findings help to explain why the immunity against Plasmodium is rapidly lost when the parasites are eliminated from the hosts, providing a molecular basis from strain-transcending immunity in human malaria [14, 63].
The innate immune effector mechanisms enhanced by IFN-γ priming are diverse, as pointed out by the high number of IFN-inducible genes upregulated in macrophages after in vitro IFN-γ priming [17, 64] or in mouse splenocytes during acute and chronic malaria [14, 37]. The biological significance of this priming is inferred from the genes expressed [14]. For instance, the upregulation of TLR-related and scavenger genes (such as CD36) possibly translates into an enhanced ability of innate immune cells to recognize and phagocytize circulating parasites, leading to an effective control of the disease [12, 45, 65]. It is important to note that an enhanced expression of TLRs facilitates the induction of the phagocytic program in innate immune cells. This was shown in chronic bacterial infections in which TLR3 and TLR9 expression leads to bacterial uptake by macrophages [13]. It is likely that IFN-γ secreted by Th1EM cells also primes the innate immune cells during viral infections in a manner similar to that observed in malaria. A potent Th1EM response is observed during infection with virus such as influenza [2, 66–68]. The IFN-γ priming of innate immune cells may ensure a rapid induction of the inflammatory response, as well as a state of refractoriness against viral proliferation in the surrounding tissues, which are important antiviral effector mechanisms [22, 69].
Besides busting proinflammatory responses, the IFN-γ priming associated with Th1EM cells may trigger feedback inhibitory loops, such as those mediated by IL-10, STAT3, and Suppressor of Cytokine Signaling 1 (SOCS1) [17]. The increased transcription of stat3 gene, another IFN-inducible gene, in mouse splenocytes from chronic malaria indicates a tight control of the innate immune system during continuous IFN-γ priming [14]. The STAT3 is a transcriptional activator of Il10 gene, and a consequent effect of IL-10 production is induction of tolerance mediated by antigen-presenting cells (APCs) [70, 71]. This fine-tuned process seems to be a common feature of TLR-mediated immune responses, since TLR agonists induce a state of late immune tolerance through the inhibition of the corresponding signaling pathways [72, 73]. Of note, mice with chronic malaria display Th1M cells coexpressing IFN-γ and IL-10, which are crucial for both the protective immunity to parasites and the protection against clinical manifestations of the disease [26]. A similar trend appears to happen in human malaria, since tolerance is often observed in patients from holoendemic areas [74]. The IFN-γ priming induced by Th1EM cells, thus, appears to be a fundamental mechanism for an efficient, though tightly regulated, protective immunity against chronic infections.
4. IFN-γ Priming Effects on Th1 EM Cells
The population of Th1EM cells declines with time after infection in various experimental models of diseases caused by pathogens, such as Plasmodium [3], Listeria [75], and lymphocytic choriomeningitis virus (LCMV) [76, 77]. This observation suggests that the presence of prosurvival cytokines, such as IL-7 and IL-15, is not sufficient to maintain these cells [1]. On the other hand, large populations of specific Th1EM cells usually persist for long periods of time during phagosomal infections, such as those caused by Salmonella enterica [78], M. tuberculosis [79, 80], and L. major [81]. Likewise, the presence of pathogens ensures the perpetuation of Th1EM cells during polyomavirus infection [82] and malaria [3, 14]. Actually, the decline in the population of Th1EM cells along with chronic malaria is related to the progressive control of residual parasitemia [3]. It has been shown that CD4+ TEM cells have a rapid turnover in both human and mice [18, 83]. Thus, the continuous replenishment of this population may be induced by chronic infection, where pathogen antigens are available together with damage signals from injured tissues. In resume, the molecular signaling that is required for long-term persistence of Th1EM cells seems to be present during active infection and rapidly disappear after its resolution. It is possible that antigen persistence and, consequently, TCR: MHC- (major histocompatibility complex-) peptide complex interactions play a role by itself in the maintenance of CD4+ TM cells, and this has been a subject of interest for malaria [84] as well as for other infections [85, 86]. An interesting study on Salmonella infection in mice showed that peptide: MHC interaction in secondary lymphoid organs harboring bacteria for over 1 year after infection maintained the CD4+ TM cell population stable [78]. IFN-γ priming might further potentiate these interactions—of note, the increased CD4+ T cell proliferation in response to Plasmodium parasites and parasite antigens indicates that IFN-γ priming enhanced antigen presentation during chronic malaria [14]. However, whether this effect was due to higher MHC class II (or costimulatory molecule) expression on APCs was not directly addressed and is still a matter of discussion.
The generation and maintenance of CD4+ and CD8+ TEM cells are facilitated by strong TCR engagement [87, 88], but other signaling pathways may be implicated in these processes. The kinase mammalian target of rapamycin (mTOR) induces in CD8+ T cells a bias toward the glycolytic metabolism and the differentiation to effector functions [89, 90]. The STAT5-mediated IL-2 signaling pathway, a potent inhibitor of the Bcl-6 transcriptional factor and follicular T helper cell differentiation [91], promotes the expression of T-bet transcriptional factor in CD4+ T cells [75]. At this respect, it has been shown that Th1EM cells have sustained expression of T-bet, both in humans [92] and mice [75]. T-bet upregulates IFN-γ production but is also an important target of IFN-γ signaling [93]. Therefore, IFN-γ is believed to induce—in conjunct with IL-12—IFN-γ production by Th1 cells. Thus, it is reasonable to hypothesize that IFN-γ plays a role in the generation and/or maintenance of Th1EM cells during chronic infections.
We have recently addressed the effects of IFN-γ priming on Th1EM cells in mice cured from chronic malaria in which the Th1EM cell response rapidly declines [14]. In these cured mice, administration of suboptimal doses of IFN-γ leads to a shift from TCM cells to TEM cells and restores the proliferative and IFN-γ responses to parasites and TLR agonists. This effect could result from the rescue of cross-reactive TCM cells driven by Plasmodium-unrelated antigens, a phenomenon previously described in human malaria [94]. However, in our study, the shift to TEM cells was specifically observed in previously infected mice, pointing out to a preferential activity of IFN-γ priming on malaria-specific cells [14]. The exact pathways involved in the IFN-γ priming effects on the generation and/or maintenance of Th1EM cells are still not well understood. It is likely that indirect signals derived from IFN-γ-primed innate immune cells play at least a partial role, and this is supported by the observation that TLR signaling is crucial for the maintenance of Th1EM cells [14]. However, a direct effect of IFN-γ priming on Th1EM cells cannot be excluded. Th1EM cells express the IFN receptor (IFNR) on their surface, and IFN-γ signaling helps to maintain the Th1EM phenotype [16]. The direct role of IFN-γ priming on Th1EM cells induced during chronic malaria is currently under investigation by our research group. Preliminary results showed a requirement of IFNR expression on Th1EM cells for their generation and maintenance (Borges da Silva, unpublished data).
Another infectious disease in which IFN-γ priming might be crucial for Th1EM cells is tuberculosis. Evidences from human disease and experimental mouse models show that IFN-γ produced by CD4+ T cells is fundamental for M. tuberculosis control [95]. Importantly, expanded and sustained Th1 responses in the lungs are seemingly crucial for controlling chronic infection [96], making continuous IFN-γ priming possibly beneficial for bacterial clearance. However, in newborns vaccinated with Bacillus Calmette-Guérin (BCG) the IFN-γ production by Th1 cells did not correlate with disease protection [97].
In several infectious diseases, the pathogen persistence maintains short-lived effector T (TEFF) cells alongside TEM cells. This seems to be particularly true for Th1-driving infections. Of note, T-bet+Ly6C+ TEFF cells present during chronic L. major cutaneous infection seem to be crucial for the protective immunity against reinfection and are proposed as major contributors for the state of concomitant immunity observed during chronic infections [98]. It is especially important to consider that the expression of Ly6C in TEFF cells is directly under the control of T-bet, which, as explained above, can be driven by IFN-γ [76, 93]. Thus, it would not be surprising if IFN-γ priming acts also directly on the maintenance of the TEFF cells as well, especially considering the need for infection persistence for their survival [98].
5. Concluding Remarks
The interplay between innate immune cells and Th1EM cells during chronic infections is seemingly complex, involving a crosstalk between innate immune cells and Th1EM cells. In this scenario, IFN-γ seems to play a crucial role as inducers of immune effector mechanisms in both sides, as exemplified in Table 1. Considering our current knowledge, the immune response to chronic infections might be defined as a circuit, where the two arms of the immune system (innate and acquired) constantly communicate with each other in order to achieve a tightly regulated, yet at most cases efficient, control of parasite load (Figure 1). To understand completely this relation, there is still the need to determine what all the “pieces in the puzzle” are, that is, to describe precisely all the aspects of the role of IFN-γ priming communication between TM cells and the innate immune system. In the case of malaria, it will also be crucial to evaluate whether the observations in mouse models also hold true for humans, which are exposed to different degrees of reinfection with heterogeneous parasites. It is likely, though, that the importance of IFN-γ priming in strain-transcending immunity to malaria is a great starting point to explain, among other things, why it is so hard to achieve sterile immunity against Plasmodium; lowering the threshold for the activation of the host immune system could be a promising strategy for the improvement in the protective immunity against this parasite.
Table 1.
Effects of IFN-γ priming on innate immune cells and TEM cells. The table summarizes the effects of IFN-γ priming on innate immune cells and on TEM cells. The references relative to each function induced by priming are in parenthesis.
Figure 1.
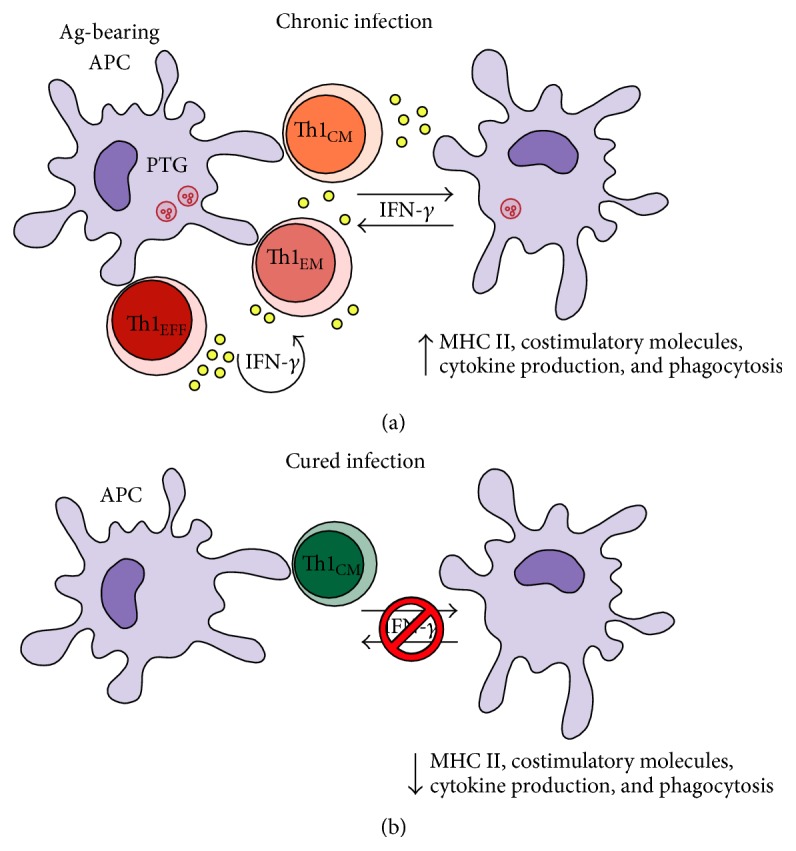
Schematic illustration to explain the IFN-γ priming effects on APCs and Th1EFF/Th1EM cells during chronic infections. This figure explains how IFN-γ produced by Th1EM cells act on APCs (usually DCs) and directly on CD4+ T cells during chronic infections. (a) When the pathogen is still present, antigen- (Ag-) bearing APCs activate CD4+ T cells that produce small amounts of IFN-γ. These small amounts of IFN-γ are enough to maintain APCs poised for function, for example, phagocytosis, cytokine production, and antigen presentation. At the same time, IFN-γ acts directly on CD4+ T cells and maintains the pool of Th1EFF/Th1EM cells. Both effects culminate in enhanced immune system activation, cytokine production, and pathogen clearance. (b) After complete pathogen elimination, the IFN-γ priming on APCs and Th1EFF/Th1EM cells ceases and, in consequence, these effector populations rapidly decline. The remaining Th1CM cells are important to control a secondary infection. However, in some infectious diseases such as malaria, continuous IFN-γ priming, and persistence of Th1EFF/Th1EM cells seem to be required to protect against reinfection.
Acknowledgments
This study was supported by Grants from the São Paulo Research Foundation (FAPESP) 2013/07140-2 (Maria Regina D'Império Lima) and 2014/00810-5 (Henrique Borges da Silva) and from National Council for Scientific and Technological Development (CNPq) 303676/2014-0 (Maria Regina D'Império Lima) and 448765/2014-4 (Maria Regina D'Império Lima).
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper. The funders had no role in the preparation of the paper.
References
- 1.Tubo N. J., Jenkins M. K. CD4+ T Cells: guardians of the phagosome. Clinical Microbiology Reviews. 2014;27(2):200–213. doi: 10.1128/cmr.00097-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Strutt T. M., Mckinstry K. K., Marshall N. B., Vong A. M., Dutton R. W., Swain S. L. Multipronged CD4+ T-cell effector and memory responses cooperate to provide potent immunity against respiratory virus. Immunological Reviews. 2013;255(1):149–164. doi: 10.1111/imr.12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Freitas do Rosário A. P., Muxel S. M., Rodríguez-Málaga S. M., et al. Gradual decline in malaria-specific memory T cell responses leads to failure to maintain long-term protective immunity to Plasmodium chabaudi AS despite persistence of B cell memory and circulating antibody. Journal of Immunology. 2008;181(12):8344–8355. doi: 10.4049/jimmunol.181.12.8344. [DOI] [PubMed] [Google Scholar]
- 4.Zens K. D., Farber D. L. Influenza Pathogenesis and Control—Volume II. Vol. 386. Berlin, Germany: Springer; 2015. Memory CD4 T cells in influenza; pp. 399–421. (Current Topics in Microbiology and Immunology). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zinkernagel R. M., Hengartner H. Protective ‘immunity’ by pre-existent neutralizing antibody titers and preactivated T cells but not by so-called 'immunological memory'. Immunological Reviews. 2006;211:310–319. doi: 10.1111/j.0105-2896.2006.00402.x. [DOI] [PubMed] [Google Scholar]
- 6.Sallusto F., Lenig D., Förster R., Lipp M., Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401(6754):708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 7.Jameson S. C., Masopust D. Diversity in T cell memory: an embarrassment of riches. Immunity. 2009;31(6):859–871. doi: 10.1016/j.immuni.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mora J. R., von Andrian U. H. T-cell homing specificity and plasticity: new concepts and future challenges. Trends in Immunology. 2006;27(5):235–243. doi: 10.1016/j.it.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 9.Reinhardt R. L., Khoruts A., Merica R., Zell T., Jenkins M. K. Visualizing the generation of memory CD4 T cells in the whole body. Nature. 2001;410(6824):101–105. doi: 10.1038/35065111. [DOI] [PubMed] [Google Scholar]
- 10.Pepper M., Jenkins M. K. Origins of CD4+ effector and central memory T cells. Nature Immunology. 2011;12(6):467–471. doi: 10.1038/ni.2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Opata M. M., Stephens R. Early decision: effector and effector memory T cell differentiation in chronic infection. Current Immunology Reviews. 2013;9(3):190–206. doi: 10.2174/1573395509666131126231209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Serghides L., Smith T. G., Patel S. N., Kain K. C. CD36 and malaria: friends or foes? Trends in Parasitology. 2003;19(10):461–469. doi: 10.1016/j.pt.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 13.Doyle S. E., O'Connell R. M., Miranda G. A., et al. Toll-like receptors induce a phagocytic gene program through p38. Journal of Experimental Medicine. 2004;199(1):81–90. doi: 10.1084/jem.20031237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.da Silva H. B., de Salles É. M., Panatieri R. H., et al. IFN-γ-induced priming maintains long-term strain-transcending immunity against blood-stage Plasmodium chabaudi malaria. Journal of Immunology. 2013;191(10):5160–5169. doi: 10.4049/jimmunol.1300462. [DOI] [PubMed] [Google Scholar]
- 15.Kalis C., Gumenscheimer M., Freudenberg N., et al. Requirement for TLR9 in the immunomodulatory activity of Propionibacterium acnes . Journal of Immunology. 2005;174(7):4295–4300. doi: 10.4049/jimmunol.174.7.4295. [DOI] [PubMed] [Google Scholar]
- 16.Hu X., Ivashkiv L. B. Cross-regulation of signaling pathways by IFN-γ: implications for immune responses and autoimmune diseases. Immunity. 2009;31(4):539–550. doi: 10.1016/j.immuni.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu X., Chakravarty S. D., Ivashkiv L. B. Regulation of interferon and Toll-like receptor signaling during macrophage activation by opposing feedforward and feedback inhibition mechanisms. Immunological Reviews. 2008;226(1):41–56. doi: 10.1111/j.1600-065x.2008.00707.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Macallan D. C., Wallace D., Zhang Y., et al. Rapid turnover of effector-memory CD4+ T cells in healthy humans. Journal of Experimental Medicine. 2004;200(2):255–260. doi: 10.1084/jem.20040341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boehm U., Klamp T., Groot M., Howard J. C. Cellular responses to interferon-γ . Annual Review of Immunology. 1997;15:749–795. doi: 10.1146/annurev.immunol.15.1.749. [DOI] [PubMed] [Google Scholar]
- 20.Nansen A., Christensen J. P., Marker O., Thomsen A. R. Sensitization to lipopolysaccharide in mice with asymptomatic viral infection: role of T cell-dependent production of interferon-γ . Journal of Infectious Diseases. 1997;176(1):151–157. doi: 10.1086/514017. [DOI] [PubMed] [Google Scholar]
- 21.Fejér G., Szalay K., Gyory I., et al. Adenovirus infection dramatically augments lipopolysaccharide-induced TNF production and sensitizes to lethal shock. Journal of Immunology. 2005;175(3):1498–1506. doi: 10.4049/jimmunol.175.3.1498. [DOI] [PubMed] [Google Scholar]
- 22.Schroder K., Hertzog P. J., Ravasi T., Hume D. A. Interferon-γ: an overview of signals, mechanisms and functions. Journal of Leukocyte Biology. 2004;75(2):163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- 23.Chen J., Ivashkiv L. B. IFN-γ abrogates endotoxin tolerance by facilitating Toll-like receptor-induced chromatin remodeling. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(45):19438–19443. doi: 10.1073/pnas.1007816107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bowdridge S., Gause W. C. Regulation of alternative macrophage activation by chromatin remodeling. Nature Immunology. 2010;11(10):879–881. doi: 10.1038/ni1010-879. [DOI] [PubMed] [Google Scholar]
- 25.Wu C., Xue Y., Wang P., et al. IFN-γ primes macrophage activation by increasing phosphatase and tensin homolog via downregulation of miR-3473b. Journal of Immunology. 2014;193(6):3036–3044. doi: 10.4049/jimmunol.1302379. [DOI] [PubMed] [Google Scholar]
- 26.do Rosário A. P. F., Lamb T., Spence P., et al. IL-27 promotes IL-10 production by effector Th1 CD4+ T cells: a critical mechanism for protection from severe immunopathology during malaria infection. Journal of Immunology. 2012;188(3):1178–1190. doi: 10.4049/jimmunol.1102755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kurtzhals J. A. L., Adabayeri V., Goka B. Q., et al. Low plasma concentrations of interleukin 10 in severe malarial anaemia compared with cerebral and uncomplicated malaria. The Lancet. 1998;351(9118):1768–1772. doi: 10.1016/s0140-6736(97)09439-7. [DOI] [PubMed] [Google Scholar]
- 28.Gazzinelli R. T., Wysocka M., Hieny S., et al. In the absence of endogenous IL-10, mice acutely infected with Toxoplasma gondii succumb to a lethal immune response dependent on CD4+ T cells and accompanied by overproduction of IL-12, IFN-γ, and TNF-α . Journal of Immunology. 1996;157(2):798–805. [PubMed] [Google Scholar]
- 29.Hunter C. A., Ellis-Neyes L. A., Slifer T., et al. IL-10 is required to prevent immune hyperactivity during infection with Trypanosoma cruzi . The Journal of Immunology. 1997;158(7):3311–3316. [PubMed] [Google Scholar]
- 30.Gong J.-H., Zhang M., Modlin R. L., et al. Interleukin-10 downregulates Mycobacterium tuberculosis-induced Th1 responses and CTLA-4 expression. Infection and Immunity. 1996;64(3):913–918. doi: 10.1128/iai.64.3.913-918.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nagamatsu K., Kuwae A., Konaka T., et al. Bordetella evades the host immune system by inducing IL-10 through a type III effector, BopN. The Journal of Experimental Medicine. 2009;206(13):3073–3088. doi: 10.1084/jem.20090494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alter G., Kavanagh D., Rihn S., et al. IL-10 induces aberrant deletion of dendritic cells by natural killer cells in the context of HIV infection. Journal of Clinical Investigation. 2010;120(6):1905–1913. doi: 10.1172/jci40913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anderson C. F., Oukka M., Kuchroo V. J., Sacks D. CD4+CD25−Foxp3− Th1 cells are the source of IL-10-mediated immune suppression in chronic cutaneous leishmaniasis. The Journal of Experimental Medicine. 2007;204(2):285–297. doi: 10.1084/jem.20061886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Belkaid Y., Piccirillo C. A., Mendez S., Shevach E. M., Sacks D. L. CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature. 2002;420(6915):502–507. doi: 10.1038/nature01152. [DOI] [PubMed] [Google Scholar]
- 35.Suffia I. J., Reckling S. K., Piccirillo C. A., Goldszmid R. S., Belkaid Y. Infected site-restricted Foxp3+ natural regulatory T cells are specific for microbial antigens. Journal of Experimental Medicine. 2006;203(3):777–788. doi: 10.1084/jem.20052056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McCall M. B. B., Netea M. G., Hermsen C. C., et al. Plasmodium falciparum infection causes proinflammatory priming of human TLR responses. Journal of Immunology. 2007;179(1):162–171. doi: 10.4049/jimmunol.179.1.162. [DOI] [PubMed] [Google Scholar]
- 37.Franklin B. S., Parroche P., Ataíde M. A., et al. Malaria primes the innate immune response due to interferon-γ induced enhancement of toll-like receptor expression and function. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(14):5789–5794. doi: 10.1073/pnas.0809742106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Franklin B. S., Rodrigues S. O., Antonelli L. R., et al. MyD88-dependent activation of dendritic cells and CD4+ T lymphocytes mediates symptoms, but is not required for the immunological control of parasites during rodent malaria. Microbes and Infection. 2007;9(7):881–890. doi: 10.1016/j.micinf.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 39.Coban C., Ishii K. J., Uematsu S., et al. Pathological role of Toll-like receptor signaling in cerebral malaria. International Immunology. 2007;19(1):67–79. doi: 10.1093/intimm/dxl123. [DOI] [PubMed] [Google Scholar]
- 40.Edelson B. T., Unanue E. R. MyD88-dependent but Toll-like receptor 2-independent innate immunity to Listeria: no role for either in macrophage listericidal activity. The Journal of Immunology. 2002;169(7):3869–3875. doi: 10.4049/jimmunol.169.7.3869. [DOI] [PubMed] [Google Scholar]
- 41.Muraille E., De Trez C., Brait M., De Baetselier P., Leo O., Carlier Y. Genetically resistant mice lacking MyD88-adapter protein display a high susceptibility to Leishmania major infection associated with a polarized Th2 response. Journal of Immunology. 2003;170(8):4237–4241. doi: 10.4049/jimmunol.170.8.4237. [DOI] [PubMed] [Google Scholar]
- 42.Netea M. G., Kullberg B. J., Jacobs L. E. H., et al. Chlamydia pneumoniae stimulates IFN-γ synthesis through MyD88-dependent, TLR2- and TLR4-independent induction, of IL-18 release. Journal of Immunology. 2004;173(2):1477–1482. doi: 10.4049/jimmunol.173.2.1477. [DOI] [PubMed] [Google Scholar]
- 43.Bafica A., Santiago H. C., Goldszmid R., Ropert C., Gazzinelli R. T., Sher A. Cutting edge: TLR9 and TLR2 signaling together account for MyD88-dependent control of parasitemia in Trypanosoma cruzi infection. Journal of Immunology. 2006;177(6):3515–3519. doi: 10.4049/jimmunol.177.6.3515. [DOI] [PubMed] [Google Scholar]
- 44.Spörri R., Joller N., Albers U., Hilbi H., Oxenius A. MyD88-dependent IFN-γ production by NK cells is key for control of Legionella pneumophila infection. Journal of Immunology. 2006;176(10):6162–6171. doi: 10.4049/jimmunol.176.10.6162. [DOI] [PubMed] [Google Scholar]
- 45.Parroche P., Lauw F. N., Goutagny N., et al. Malaria hemozoin is immunologically inert but radically enhances innate responses by presenting malaria DNA to Toll-like receptor 9. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(6):1919–1924. doi: 10.1073/pnas.0608745104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heremans H., Van Damme J., Dillen C., Dijkmans R., Billiau A. IFN-γ, a mediator of lethal lipopolysaccharide-induced shwartzman-like shock reactions in mice. Journal of Experimental Medicine. 1990;171(6):1853–1869. doi: 10.1084/jem.171.6.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cunnington A. J., de Souza J. B., Walther M., Riley E. M. Malaria impairs resistance to Salmonella through heme- and heme oxygenase-dependent dysfunctional granulocyte mobilization. Nature Medicine. 2012;18(1):120–127. doi: 10.1038/nm.2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gilroy D. W., Yona S. HIF1α allows monocytes to take a breather during sepsis. Immunity. 2015;42(3):397–399. doi: 10.1016/j.immuni.2015.02.016. [DOI] [PubMed] [Google Scholar]
- 49.Le Bon A., Tough D. F. Type I interferon as a stimulus for cross-priming. Cytokine and Growth Factor Reviews. 2008;19(1):33–40. doi: 10.1016/j.cytogfr.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 50.Stockinger S., Kastner R., Kernbauer E., et al. Characterization of the interferon-producing cell in mice infected with Listeria monocytogenes . PLoS Pathogens. 2009;5(3) doi: 10.1371/journal.ppat.1000355.e1000355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Solodova E., Jablonska J., Weiss S., Lienenklaus S. Production of IFN-β during listeria monocytogenes infection is restricted to monocyte/macrophage lineage. PLoS ONE. 2011;6(4) doi: 10.1371/journal.pone.0018543.e18543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nakano H., Yanagita M., Gunn M. D. CD11c+B220+Gr-1+ cells in mouse lymph nodes and spleen display characteristics of plasmacytoid dendritic cells. The Journal of Experimental Medicine. 2001;194(8):1171–1178. doi: 10.1084/jem.194.8.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Asselin-Paturel C., Boonstra A., Dalod M., et al. Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nature Immunology. 2001;2(12):1144–1150. doi: 10.1038/ni736. [DOI] [PubMed] [Google Scholar]
- 54.Crouse J., Kalinke U., Oxenius A. Regulation of antiviral T cell responses by type I interferons. Nature Reviews Immunology. 2015;15(4):231–242. doi: 10.1038/nri3806. [DOI] [PubMed] [Google Scholar]
- 55.Cavlar T., Ablasser A., Hornung V. Induction of type I IFNs by intracellular DNA-sensing pathways. Immunology and Cell Biology. 2012;90(5):474–482. doi: 10.1038/icb.2012.11. [DOI] [PubMed] [Google Scholar]
- 56.Matikainen S., Sareneva T., Ronni T., Lehtonen A., Koskinen P. J., Julkunen I. Interferon-α activates multiple STAT proteins and upregulates proliferation-associated IL-2Rα, c-myc, and pim-1 genes in human T cells. Blood. 1999;93(6):1980–1991. [PubMed] [Google Scholar]
- 57.Huber J. P., Horn S. R. G.-V., Roybal K. T., Gill M. A., Farrar J. D. IFN-α suppresses GATA3 transcription from a distal exon and promotes H3K27 trimethylation of the CNS-1 enhancer in human Th2 cells. The Journal of Immunology. 2014;192(12):5687–5694. doi: 10.4049/jimmunol.1301908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cousens L. P., Orange J. S., Su H. C., Biron C. A. Interferon-α/β inhibition of interleukin 12 and interferon-γ production in vitro and endogenously during viral infection. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(2):634–639. doi: 10.1073/pnas.94.2.634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Byrnes A. A., Ma X., Cuomo P., et al. Type I interferons and IL-12: convergence and cross-regulation among mediators of cellular immunity. European Journal of Immunology. 2001;31(7):2026–2034. doi: 10.1002/1521-4141(200107)31:7lt;2026::aid-immu202662;3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 60.Le Bon A., Durand V., Kamphuis E., et al. Direct stimulation of T cells by type I IFN enhances the CD8+ T cell response during cross-priming. Journal of Immunology. 2006;176(8):4682–4689. doi: 10.4049/jimmunol.176.8.4682. [DOI] [PubMed] [Google Scholar]
- 61.Carrero J. A., Calderon B., Unanue E. R. Type I interferon sensitizes lymphocytes to apoptosis and reduces resistance to Listeria infection. Journal of Experimental Medicine. 2004;200(4):535–540. doi: 10.1084/jem.20040769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Soudja S. M., Chandrabos C., Yakob E., Veenstra M., Palliser D., Lauvau G. Memory-T-cell-derived interferon-γ instructs potent innate cell activation for protective immunity. Immunity. 2014;40(6):974–988. doi: 10.1016/j.immuni.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Doolan D. L., Dobaño C., Baird J. K. Acquired immunity to Malaria. Clinical Microbiology Reviews. 2009;22(1):13–36. doi: 10.1128/cmr.00025-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schroder K., Sweet M. J., Hume D. A. Signal integration between IFNγ and TLR signalling pathways in macrophages. Immunobiology. 2006;211(6–8):511–524. doi: 10.1016/j.imbio.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 65.Naik R. S., Branch O. H., Woods A. S., et al. Glycosylphosphatidylinositol anchors of Plasmodium falciparum: molecular characterization and naturally elicited antibody response that may provide immunity to malaria pathogenesis. Journal of Experimental Medicine. 2000;192(11):1563–1576. doi: 10.1084/jem.192.11.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Graham M. B., Braciale V. L., Braciale T. J. Influenza virus-specific CD4+ T helper type 2 T lymphocytes do not promote recovery from experimental virus infection. The Journal of Experimental Medicine. 1994;180(4):1273–1282. doi: 10.1084/jem.180.4.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brown D. M., Dilzer A. M., Meents D. L., Swain S. L. CD4+T cell-mediated protection from lethal influenza: perform and antibody-mediated mechanisms give a one-two punch. Journal of Immunology. 2006;177(5):2888–2898. doi: 10.4049/jimmunol.177.5.2888. [DOI] [PubMed] [Google Scholar]
- 68.Brown D. M., Lee S., Garcia-Hernandez M. D. L. L., Swain S. L. Multifunctional CD4 cells expressing gamma interferon and perforin mediate protection against lethal influenza virus infection. Journal of Virology. 2012;86(12):6792–6803. doi: 10.1128/jvi.07172-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Swain S. L., McKinstry K. K., Strutt T. M. Expanding roles for CD4+ T cells in immunity to viruses. Nature Reviews Immunology. 2012;12(2):136–148. doi: 10.1038/nri3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cheng F., Wang H.-W., Cuenca A., et al. A critical role for Stat3 signaling in immune tolerance. Immunity. 2003;19(3):425–436. doi: 10.1016/s1074-7613(03)00232-2. [DOI] [PubMed] [Google Scholar]
- 71.Saraiva M., O'Garra A. The regulation of IL-10 production by immune cells. Nature Reviews Immunology. 2010;10(3):170–181. doi: 10.1038/nri2711. [DOI] [PubMed] [Google Scholar]
- 72.Favorite G. O., Morgan H. R. Effects produced by the intravenous injection in man of a toxic antigenic material derived from Eberthella typhosa: clinical, hematological, chemical and serological studies. Journal of Clinical Investigation. 1942;21(5):589–599. doi: 10.1172/jci101337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Miller S. I., Ernst R. K., Bader M. W. LPS, TLR4 and infectious disease diversity. Nature Reviews Microbiology. 2005;3(1):36–46. doi: 10.1038/nrmicro1068. [DOI] [PubMed] [Google Scholar]
- 74.Boutlis C. S., Yeo T. W., Anstey N. M. Malaria tolerance—for whom the cell tolls? Trends in Parasitology. 2006;22(8):371–377. doi: 10.1016/j.pt.2006.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pepper M., Pagán A. J., Igyártó B. Z., Taylor J. J., Jenkins M. K. Opposing signals from the Bcl6 transcription factor and the interleukin-2 receptor generate T helper 1 central and effector memory cells. Immunity. 2011;35(4):583–595. doi: 10.1016/j.immuni.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Marshall H. D., Chandele A., Jung Y. W., et al. Differential expression of Ly6C and T-bet distinguish effector and memory Th1 CD4+ cell properties during viral infection. Immunity. 2011;35(4):633–646. doi: 10.1016/j.immuni.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Homann D., Teyton L., Oldstone M. B. A. Differential regulation of antiviral T-cell immunity results in stable CD8+ but declining CD4+ T-cell memory. Nature Medicine. 2001;7(8):913–919. doi: 10.1038/90950. [DOI] [PubMed] [Google Scholar]
- 78.Nelson R. W., McLachlan J. B., Kurtz J. R., Jenkins M. K. CD4+ T cell persistence and function after infection are maintained by low-level peptide:MHC class II presentation. The Journal of Immunology. 2013;190(6):2828–2834. doi: 10.4049/jimmunol.1202183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Reiley W. W., Shafiani S., Wittmer S. T., et al. Distinct functions of antigen-specific CD4+T cells during murine Mycobacterium tuberculosis infection. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(45):19408–19413. doi: 10.1073/pnas.1006298107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kaveh D. A., Carmen Garcia-Pelayo M., Hogarth P. J. Persistent BCG bacilli perpetuate CD4 T effector memory and optimal protection against tuberculosis. Vaccine. 2014;32(51):6911–6918. doi: 10.1016/j.vaccine.2014.10.041. [DOI] [PubMed] [Google Scholar]
- 81.Pagán A. J., Peters N. C., Debrabant A., et al. Tracking antigen-specific CD4+ T cells throughout the course of chronic Leishmania major infection in resistant mice. European Journal of Immunology. 2013;43(2):427–438. doi: 10.1002/eji.201242715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lin E., Kemball C. C., Hadley A., et al. Heterogeneity among viral antigen-specific CD4+ T cells and their de novo recruitment during persistent polyomavirus infection. Journal of Immunology. 2010;185(3):1692–1700. doi: 10.4049/jimmunol.0904210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Surh C. D., Sprent J. Homeostasis of naive and memory T cells. Immunity. 2008;29(6):848–862. doi: 10.1016/j.immuni.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 84.Langhorne J., Ndungu F. M., Sponaas A.-M., Marsh K. Immunity to malaria: more questions than answers. Nature Immunology. 2008;9(7):725–732. doi: 10.1038/ni.f.205. [DOI] [PubMed] [Google Scholar]
- 85.McKinstry K. K., Strutt T. M., Swain S. L. The potential of CD4 T-cell memory. Immunology. 2010;130(1):1–9. doi: 10.1111/j.1365-2567.2010.03259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Taylor J. J., Jenkins M. K. CD4+ memory T cell survival. Current Opinion in Immunology. 2011;23(3):319–323. doi: 10.1016/j.coi.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 87.Catron D. M., Rusch L. K., Hataye J., Itano A. A., Jenkins M. K. CD4+ T cells that enter the draining lymph nodes after antigen injection participate in the primary response and become central-memory cells. Journal of Experimental Medicine. 2006;203(4):1045–1054. doi: 10.1084/jem.20051954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sarkar S., Teichgräber V., Kalia V., et al. Strength of stimulus and clonal competition impact the rate of memory CD8+ T cell differentiation. Journal of Immunology. 2007;179(10):6704–6714. doi: 10.4049/jimmunol.179.10.6704. [DOI] [PubMed] [Google Scholar]
- 89.Araki K., Turner A. P., Shaffer V. O., et al. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460(7251):108–112. doi: 10.1038/nature08155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pearce E. L., Walsh M. C., Cejas P. J., et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. 2009;460(7251):103–107. doi: 10.1038/nature08097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Johnston R. J., Choi Y. S., Diamond J. A., Yang J. A., Crotty S. STAT5 is a potent negative regulator of TFH cell differentiation. The Journal of Experimental Medicine. 2012;209(2):243–250. doi: 10.1084/jem.20111174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yu S.-F., Zhang Y.-N., Yang B.-Y., Wu C.-Y. Human memory, but not naive, CD4+ T cells expressing transcription factor T-bet might drive rapid cytokine. Journal of Biological Chemistry. 2014;289(51):35561–35569. doi: 10.1074/jbc.m114.608745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Schulz E. G., Mariani L., Radbruch A., Höfer T. Sequential polarization and imprinting of type 1 T helper lymphocytes by interferon-gamma and interleukin-12. Immunity. 2009;30(5):673–683. doi: 10.1016/j.immuni.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 94.Artavanis-Tsakonas K., Tongren J. E., Riley E. M. The war between the malaria parasite and the immune system: immunity, immunoregulation and immunopathology. Clinical and Experimental Immunology. 2003;133(2):145–152. doi: 10.1046/j.1365-2249.2003.02174.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.O'Garra A., Redford P. S., McNab F. W., Bloom C. I., Wilkinson R. J., Berry M. P. R. The immune response in tuberculosis. Annual Review of Immunology. 2013;31:475–527. doi: 10.1146/annurev-immunol-032712-095939. [DOI] [PubMed] [Google Scholar]
- 96.Feng C. G., Jankovic D., Kullberg M., et al. Maintenance of pulmonary Th1 effector function in chronic tuberculosis requires persistent IL-12 production. Journal of Immunology. 2005;174(7):4185–4192. doi: 10.4049/jimmunol.174.7.4185. [DOI] [PubMed] [Google Scholar]
- 97.Kagina B. M. N., Abel B., Scriba T. J., et al. Specific T cell frequency and cytokine expression profile do not correlate with protection against tuberculosis after bacillus Calmette-Guérin vaccination of newborns. American Journal of Respiratory and Critical Care Medicine. 2010;182(8):1073–1079. doi: 10.1164/rccm.201003-0334OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Peters N. C., Pagán A. J., Lawyer P. G., et al. Chronic parasitic infection maintains high frequencies of short-lived Ly6C+CD4+ effector T cells that are required for protection against re-infection. PLoS Pathogens. 2014;10(12) doi: 10.1371/journal.ppat.1004538.e1004538 [DOI] [PMC free article] [PubMed] [Google Scholar]