

Abstract
Vascular calcification (VC) is the deposition of calcium/phosphate in the vasculature, which portends a worse clinical outcome and predicts major adverse cardiovascular events. VC is an active process initiated and regulated via a variety of molecular signalling pathways. There are mainly two types of calcifications: the media VC and the intima VC. All major risk factors for cardiovascular disease (CVD) have been linked to the presence/development of VC. Besides the risk factors, a genetic component is also operative to determine arterial calcification. Several events take place before VC is established, including inflammation, trans-differentiation of vascular cells and homing of circulating pro-calcific cells. Diabetes is an important predisposing factor for VC. Compared with non-diabetic subjects, patients with diabetes show increased VC and higher expression of bone-related proteins in the medial layer of the vessels. In this review we will highlight the mechanisms underlying vascular calcification in diabetic patients.
Keywords: Bone, vascular, differentiation, risk factors
Introduction
Vascular calcification (VC) is the deposition of calcium/phosphate crystals in the vascular system. The link between VC and increased mortality is well established: the presence of aortic valve sclerosis, as well VC in other arterial beds, has been associated with increased risk of mortality for cardiovascular disease (CVD) (1). VC portends a worse clinical outcome and predicts major adverse cardiovascular events: the prevalence of calcification increase with age, with evidence of VC present in more than 90% of men and 67% of women over the age of 70 (2). Several conditions such as diabetes mellitus, dyslipidemia and renal diseases are identified as major predisposing factors. However, it is incorrect to consider VC as an equivalent of atherosclerosis. The reason for this is twofold. In ancient cultures, VC has been found in several arterial districts: this not only challenges the fact that that atherosclerosis is a modern disease caused by present day risk factors, but also implies that VC might be associated with other conditions such as certain types of infection (3). An autopsy of an ancient Egyptian teenage male found that he was infected with four parasites (4). Modern day patients with chronic inflammatory diseases experience premature atherosclerosis and VC. These observations suggest that atherosclerosis and, specifically, VC, are the results of interplay between genes and environment.
Types of VC
VC is an active process initiated and regulated by a variety of molecular signaling pathways. There are mainly two types of calcifications: medial VC and the intimal VC (5). Medial VC is characterized by the deposition of hydroxyapatite, mainly localized in the peripheral arteries, deposited along the elastic lamina and extracellular matrix, and is associated with diabetes mellitus and chronic kidney disease (CKD) (6). Recent works suggest a potential role for Receptor activator for nuclear factor κB ligand (RANKL)—Receptor activator for nuclear factor κB (RANK)—osteoprotegerin (OPG) signaling in this type of vascular disease. The clinical outcome of media VC are vascular stiffness, impaired hemodynamic regulation, and increased cardiac post-load. Intimal VC is systemic, mainly determined by the classical CVD risk factors, and it is activated by either oxidative stress or inflammatory pathways (7). In the intima, calcification occurs in two distinct patterns: (I) punctate foci of calcification, which may also undergo osseous metaplasia, including marrow and bone; (II) diffuse calcification, with an undefined biological role of this pattern. The clinical outcomes of intimal VC are arterial stiffness and changes in plaque characteristics, potentially predisposing to vulnerability.
Calcifications can be classified also according to their burden: spotty or granular calcifications (“micro-calcification”), typically <15 nm in diameter, is associated with feature of ruptured plaque while diffuse, homogeneous, or sheet-like calcifications (“macro-calcification”), typically >5 mm, is associated with plaques less prone to rupture (8).
Risk factors for VC
All major risk factors for CVD have been linked to the presence or development of VC (i.e., age, obesity, hypertension, smoking, non-HDL cholesterol, and diabetes). Besides the risk factors, a genetic component is also operative to determine arterial calcification: some rare monogenic disorders have been associated with the pathological development of VC (9). Nevertheless, other investigation showed that up to 40-50% of the variance of aortic and coronary calcium could be also attributed to genetics (10). The 9p21 locus, which has been linked to vascular disease, also associates with calcification (11). A meta-analysis identified 48 single nucleotide polymorphisms at 9p21 near the cyclin genes, cyclin-dependent kinases (CDKN) 2B and CDKN 2AA, that met significance for coronary artery calcium (CAC) score association (12). These genes encode cyclins that may be implicated in cellular senescence and inflammation.
In the presence of dyslipidemia, acetylated LDL increase by 3-fold the osteogenic phenotype of cultured vascular smooth muscle cells (VSMCs) (13). Moreover, it was found that minimally oxidized LDL induced VC and increased alkaline phosphatase (AP), thus promoting osteogenic differentiation of VSMC. Conversely, HDL inhibits osteogenic differentiation pathway in vitro (14). Obesity per se does not seem to be a determinant of VC. However, the epicardial fat seems to be relevant for coronary calcification, which has an intense paracrine activity, probably mediating its propensity to induce calcification (15). It was reported that CAC score correlates with the volume of fat around the heart, and a greater volume of epicardial fat was associated with a higher risk for stenosis on angiography, independent of diabetes status (16). Although epicardial fat is associated with all classical CV risk factors (17), a study has shown that epicardial fat is associated with a 34% increased odds of CAC, independent of visceral fat volume (18). In addition to indicating the presence of calcification, the CAC score per se predicts cardiac events and survival rates when combined with other traditional risk factor scoring systems, as the Framingham risk score (19). For instance, the Diabetes Heart Study, conducted on a population of 1,123 type 2 diabetic patients aged 34-86 years, showed that CAC predicted CVD over 7.4-year follow-up, regardless of the Framingham Risk score (20).
The contribution of hypertension to VC is still uncertain (21). The renin-angiotensin-aldosterone system is a major pathogenic factor in VSMC apoptosis, growth, and differentiation thus suggesting a possible involvement of the system in arterial calcification (22). Angiotensin-II (Ang-II) can promote VSMC differentiation into osteogenic cells through RANKL activation (23). Calcified arteries showed an up-regulation of angiotensin 1 receptor (AT1) and treatment with an AT1 blocker was able to prevent VC (24). In a rodent model of arterial calcification induced by administration of vitamin D plus oral nicotine, increased calcium content of the arteries was associated with increased levels of Ang-II and aldosterone in the tissue (25). Conversely, treatment with captopril or spironolactone reduced calcium accumulation. Long-standing hypertension can also induce fracturing of elastic fibers (26) that may indirectly predispose to VC.
Inflammation, a hallmark of insulin resistant states, is a potent inducer of VC. Osteogenesis is associated with local inflammation and macrophage infiltration in atherosclerotic plaques of ApoE−/− mice (27). In addition, several cytokines have been shown to induce mineralization of calcifying vascular cells in vitro, mainly through the induction of AP expression (28). These inflammatory processes might be amplified in patients with diabetes, especially in case of concomitant CKD.
Cigarette smoking, a major cause of CVD, is typically associated with VC (29). The effect of smoking can be direct or indirect. Smoking appears to have a multiplicative interaction with the other major risk factors for coronary heart disease. However, cigarette smoke delivers a high level of oxidizing chemicals to smokers, including oxides of nitrogen and many free radicals. Studies indicate that oxidant stress contributes to several potential mechanisms of CVD, including inflammation, endothelial dysfunction, lipid abnormalities such as oxidation of LDL, and platelet activation. All these mechanisms along with alteration in plasma lipids, glucose metabolism, and blood flow can contribute to the ability of smoking to induce VC (30).
Molecular mechanisms leading to VC
Several events take place before VC is established. VSMC transdifferentiate into cells that look like bone-formative cells where smooth muscle-specific genes down-regulate, and genes associated to osteochondrogenesis, such as runt-related transcription factor 2 (Runx2), osterix, osteopontin (OPN), osteocalcin, and AP up-regulate. These cells produce a collagen matrix and form calcium- and phosphorus-rich matrix vesicles (MVs) that are capable of initiating mineralization of the vascular wall. Runx2, a key transcription factor for osteoblast differentiation, is a critical element of this phenotypic change (31,32). Elastin, the most abundant protein in the aortic wall, can be degraded by matrix metalloproteinase (MMP), MMP-2 and MMP-9: the degradation of this protein, increases calcification of VSMC grown in a high-phosphate medium because of its ability to increase the expression of transforming growth factor (TGF)-β, which is involved in osteoblast differentiation (32). An increased calcium-phosphorus product (Ca × P) is important for the mineralization of vascular wall, although the concentration of calcium appears to play a more significant role. Hyperphosphatemia also plays an important role through the activity of type III sodium-dependent phosphate transporters, Pit-1 and Pit-2 (33). The knocking down of Pit-1 in VSMC is associated to significant less sodium-dependent phosphate uptake and calcium deposition. In patients with CKD, parathyroid hormone (PTH) and fibroblast growth factor (FGF)-23 are both implicated in VC (34). In these patients, failure of anti-calcific mechanisms is reported. One of the mechanisms is the matrix Gla protein (MGP), which acts by binding calcium ions and clear excess calcium, as well as binds calcium crystal and inhibits crystal growth (35). MGP expression is down-regulated by vitamin D deficiency (36). γ-Carboxylation converts MGP into its active form, and vitamin K acts as a cofactor for this process. Vitamin K antagonists interfere with the generation of active MGP (37): indeed warfarin use is associated with VC in the hemodialysis population. Fetuin-A is another important inhibitor of calcification: this glycoprotein is present in the circulation, where it binds calcium ions and hydroxyapatite (38). Mice deficient in fetuin-A show increased susceptibility to widespread calcification, and fetuin-A added to bovine VSMC inhibits calcification. OPN inhibits formation of apatite crystals and promotes osteoclast function (39): this phosphoprotein is seen at high levels in calcified arteries, where it counteracts VC (40). OPG, a member of the tumor necrosis factor (TNF)-α receptor superfamily, indirectly inhibits osteoclastogenesis (41). It functions as a soluble “decoy” receptor that binds and inhibits RANKL, which instead promotes calcification (41). Studies in humans have shown an association between OPG levels, presence of CAD and the risk of future CVD events (42).
A progressively increasing role of S100A in calcification is being acknowledged. S100A8 (MRP-8) and S100A9 (MRP-14) are members of the S100 family of calcium-binding proteins and highly expressed in numerous inflammatory conditions (43). Monocytes infiltrating early atherosclerotic lesions, and macrophages within mature plaques of ApoE−/− mice were reported as S100A9+S100A8+ (44). Intracellular S100A8 and S100A9 essentially regulate phagocyte through calcium and mitogen-activated protein kinase transduction pathways affecting the micro-tubular system. S100A8-elicited macrophages exhibit a pro-atherogenic phenotype. Burke et al. found strong expression of S100A12 in human coronary artery VSMC in ruptured plaques associated with sudden cardiac death, with the highest S100A12 expression observed in ruptured plaques of diabetic patients (45). These studies strongly suggest a relationship between the pathological expression of S100A12 in the vasculature and features of plaque instability.
To create a microenvironment permissive for calcification, specialized membrane-bound bodies called MVs, serve as nucleation sites for hydroxyapatite. On exposure to high extracellular calcium, or with intracellular calcium release, and when calcification inhibitor levels are low, VSMCs produce mineralization-competent vesicles that contain preformed HA (46). MVs contain apoptotic bodies, which creates a phosphate source by degrading pyrophosphate (47). VSMC as well as leukocyte derived MV may play a role in VC. MV have been identified in both atherosclerotic plaques associated with intimal calcification and in non-atherosclerotic vessels associated with arterial medial calcification (48). Elevation in both calcium and phosphate levels have been shown to induce release of MV from cultured VSMC, whereas elevated calcium levels enhance the mineral formation from these MV (49). MVs, which are found in atherosclerotic plaques and in blood may also contribute to VC: it has been shown that a high number of circulating micro-particles are seen in menopausal women with coronary calcification and the level of micro-particles is directly correlated with the calcium score (50). Table 1 reports an overview of biomarkers and potential modulators of VC, whereas Figure 1 shows the interplay between molecular and cellular mechanisms of VC.
Table 1. Soluble biomarkers and regulators of calcification.
| Biomarker | Mechanism of Action | Effect on VC |
|---|---|---|
| FGF-23 (51) | Requires Klotho as biomarker; promotes phosphate excretion; reduces calcium absorption; decreases PTH release | High levels associate with VC |
| Fetuin-A (52) | Binds calcium phosphate; decreases inflammation | Inhibition of VC |
| MGP (53) | Binds calcium crystals; inhibits crystal growth; inhibits BMP-2 | Inhibition of VC |
| BMP-2 (54) | Regulates bone formation; promotes osteoblast differentiation; decrease miRNA 30b and 30c; promotes mineralization | Promotes VC |
| OPG (55) | Inhibits osteoclast differentiation; interferes with the interaction RANKL/RANk | Inhibits VC |
| OPN (56) | Blocks hydroxyapatite formation; inhibits mineralization of VSMC; pro-inflammatory | Inhibits VC (?) |
| Osteonectin (57) | Binds calcium | Associates with VC |
| OS (58) | Inhibits calcium precipitation; inhibits crystal growth; increased in polarized myeloid procalcific cells | Associates with VC (?) |
| Sclerostin (52) | Inhibits osteoblast-mediated bone formation | Inhibits VC |
?, the mechanism of the biomarker is not established yet. FGF, fibroblast growth factor; PTH, parathyroid hormone; VC, vascular calcification; MGP, matrix Gla protein; BMP-2, bone morphogenetic protein-2; OPG, Osteoprotegerin; RANKL, receptor activator for nuclear factor B ligand; RANK, receptor activator for nuclear factor B; OPN, osteopontin; VSMC, vascular smooth muscle cells; OS, osteocalcin.
Figure 1.
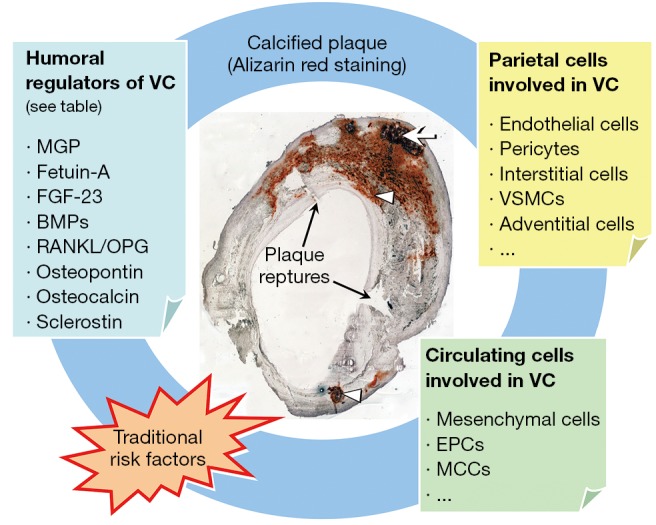
Contributors to plaque calcification. Representative Alizarin red staining of a carotid plaque section is shown with a calcified nodule (arrow) in the medial layer and other calcifications (arrowheads) beneath the endothelial layer; Plaque ruptures are also present where indicated. Multiple mechanisms contribute to plaque calcification and they are interacting mutually and with traditional risk factors. Humoral regulators, parietal as well as circulating cells are depicted. VC, vascular calcification; MGP, matrix Gla protein; FGF, Fibroblast growth factor; BMPs, bone morphogenetic proteins; RANKL, receptor activator for nuclear factor B ligand; OPG, osteoprotegerin; VSMC, vascular smooth muscle cells; EPCs, endothelial progenitor cells; MCCs, myeloid calcifying cells.
Diabetes as a predisposing factor for VC
Diabetes leads to CVD via several mechanisms [reviewed in (59)], and is an important predisposing factor for VC (60). Compared with non-diabetic subjects, patients with diabetes show increased VC and higher expression in the medial layer of the vessels of bone-related proteins, such as OPN, type I collagen, and AP (61). These findings indicate that the activation of an osteogenic program is accelerated within the arterial wall of diabetic patients. Hyperglycemia and insulin resistance are hallmarks of diabetes, and can be actively involved in the pathological processes leading to VC (62). Hyperglycemia influences VC through the production of reactive oxygen species (ROS), major inducers of vascular cell trans-differentiation into osteoblast-like elements (63). Medial calcification occurs preferentially in diabetic patients with neuropathy: this type of calcification is a strong predictor of total CVD mortality, and also a significant predictor of future CHD events, stroke, and lower limb amputation (64). It should also be noted that, in the lower limbs, calcification is typical of intimal plaques leading to femoral artery occlusion (65). High glucose and other potential factors may play an important role by transforming VSMCs into osteoblast-like cells (66). In addition, CKD often complicates long-standing diabetes: this condition is per se associated with accelerated calcification (67). The molecular cues underpinning this calcification pattern remain elusive. Several mechanisms may be involved. One is the formation of advanced glycation end products (AGE). The receptor for these RAGEs colocalizes with inflammatory cells in regions with micro-calcifications (68). In rodents with diet-induced diabetes, RAGE is up-regulated and colocalizes with VSMC undergoing osteochondrogenic differentiation. Diabetes may promote VC by reducing the vitamin K—dependent activation of the inhibitor of calcification, MGP (69). Hyperglycemia, combined with elastin degradation products increases osteogenic markers, such as osteocalcin, and Runx2 in vascular cells (70). In human aortic endothelial cells, high glucose concentration augments expression of BMP-2 and BMP-4, MGP and Noggin, thus suggesting a role also for endothelial cell activation in promoting vascular cell osteogenic activation (71). Diabetic mice and rats showed a dramatic increase in aortic BMP activity, as demonstrated by SMAD1/5/8 phosphorylation. This was associated with increased osteogenesis and calcium accumulation. Such changes were prevented in the Ins2 (Akita/+) mice by breeding them with MGP transgenic mice, which increased aortic BMP inhibition (72). Collectively, these data demonstrate that elevated glucose levels induce the promoters of calcium deposition such as TNFα, IL-6, TGF-β, RANKL, adipokines (leptin), morphogenic proteins (such as BMP-2, Wnt). All these mediators have been shown to induce the osteogenic activity of vascular cell and/or drive the trans-differentiation of VMSCs, and adventitial myofibroblasts into calcifying cells. BMP-2 is known to produce osteogenic differentiation of VSMCs. Endothelial cells produce BMP-2, which is increased by the action of inflammatory mediators; in the presence of an inflammatory milieu endothelial cells release endothelial micro-particles. These micro-particles, which are abundant in BMP-2, are able to stimulate VC through the osteogenic modification of VSMCs (50).
In diabetes, pericytes may be particularly prone to osteogenic differentiation, which has been shown to be driven by AGEs (73). Based on their vascular stabilizing activity, it was speculated that the pro-angiogenic and pro-calcific activity of pericytes might be linked (74).
Diabetes and circulating pro-calcific cells
In the recent years, it has been clarified that circulating cells can contribute to the processes that drive ectopic calcification, especially in the vasculature (75,76). It has been reported that about 20% of human peripheral blood CD34+ stem cells express mRNA for OC. Interestingly: the level of such osteoprogenitor in blood appears to be correlated with the extent of the atherosclerotic burden in vivo, in humans. Eghbali-Fatourechi, by using flow cytometry, identified cells positive for osteocalcin and for bone-specific AP in the peripheral blood of adult subjects (77). They also showed that the percentage of OC+ cells correlated with markers of bone formation. In patients undergoing invasive coronary assessment compared with controls, patients with coronary heart disease had significant increase in the percentage of CD34+KDR+ and CD34+CD133+KDR+ endothelial progenitor cells (EPCs) co-staining for OC (78). These findings deserve attention for at least two reasons: first, the levels of osteoprogenitors may determine the extent of atherosclerotic lesions, and, second, important determinants of VC originate from outside the vascular wall. Our group has recently added some important information to the identity and biology of circulating calcifying cells. We identified a population of heterogeneous circulating mononuclear cells expressing osteocalcin and bone AP (OC+BAP+) producing high amounts of spotty areas of calcification, which do not include bone or cartilage (79). These OC+BAP+ cells, called myeloid calcifying cells (MCCs), are distinct from hematopoietic stem cells, but the expression of CD45, CD14, and CD68 suggest that they derived from monocyte/macrophage lineage. The myeloid origin was confirmed using lineage tracing of the BCR-ABL transcript in naïve chronic myeloid leukemia patients. We also found that these cells, called MCCs, were significantly increased in the bloodstream of patients with either CVD or diabetes. In diabetic versus non-diabetic patients, MCCs were higher in the presence and in the absence of CVD. In parallel, it seems that EPCs cultured from diabetic patients with CAD occasionally formed structures highly suggestive of calcified nodules, and the expression of osteogenic markers by EPCs from control subjects was significantly increased in response to the toll-like receptor agonist LPS (80). In parallel to this finding, it should be noted that Yao et al. reported that, in models of diabetes and in response to hyperglycemia, the endothelium undergoes loss of MGP expression, dedifferentiation to an oligopotent mesenchymal-like state, invasion of the subintimal space and induction of calcification (81). Further to this, Albiero et al. tested whether MCCs truly promote atherosclerotic calcification in vivo (82). They show that the murine spleen contains OC+BAP+ cells with a phenotype similar to human MCCs, a high expression of adhesion molecules and CD11b, and capacity to calcify in vitro and in vivo. Finally, Menegazzo et al. reported that MCCs have anti-angiogenic activity in vitro and in vivo, by virtue of their intense paracrine secretion (83). The integration of calcification with angiostasis might be seen in the framework of chronic inflammation as an extreme attempt to confine a chronic inflammatory stimulus, which, in the vascular wall, can be represented by cholesterol crystals (84).
Glycemic control and VC
In this setting, the aim of a treatment is either to avoid or to delay VC, especially in diabetic patients, who are much more prone than non-diabetic ones to suffer from an exaggerated calcification burden. It is clinically important to determine whether the correction of HbA1c is associated with a reduction of calcification. Anand and colleagues have reported that, in a multivariate model, baseline CAC, and an HbA1c >7%, are strong predictors of CAC progression in type 2 diabetic patients (85). Very recently Carson and colleagues have observed that during the 5-year follow-up period, higher HbA1c was associated with incident CAC [risk ratio (RR) =1.45; 95% CI, 1.02-2.06], any CAC progression (RR =1.51; 95% CI, 1.16-1.96), and advanced CAC progression (RR =2.42; 95% CI, 1.47-3.99) after adjustment for sociodemographic factors (86). Jorgensen and colleagues have found that the odd ratio for hard plaques versus no plaques was 5.8 in the highest HbA1c group (>6.4%) compared with subjects in the lowest group (<5.0%) after adjustment for several possible confounders (87). Dayan and colleagues have found that there is a significant relationship between CAC score, albuminuria and inflammation in patients with type 2 diabetes (88). Flammer and colleagues have shown that patients with elevated HbA1c compared with those with normal HbA1c had a significantly higher percentage of circulating OC+ mononuclear cells, higher numbers of OC+ EPCs, thus suggesting an association between the osteogenic drift of EPCs and an HbA1c in the pre-diabetic range (89). As described before, Fadini and colleagues have also shown that in diabetic versus non-diabetic patients, MCCs were higher in the presence and in the absence of CVD, and that an HbA1c decreased by 1.15% was associated with a significant decrease of circulating MCC levels (79). Higher HbA1c level was found to have a modest and independent association with subclinical coronary atherosclerosis, even in metabolically healthy individuals (90). These reports suggest that not only overly but also slightly elevated glucose levels are associated with increased VC. However, to what extent pursuing glycemic goals will reduce the VC burden and its effect on VC outcomes remains to be elucidated.
Conclusions
The evidence herein summarized clearly indicates that diabetes and its associated complications represent a preferential milieu for the development of VC. It is well recognized that diabetes induces VC and knowledge of the mechanisms and consequences of this phenomenon is expanding. The clinical significance of excess VC in terms of cardiovascular risk is still a matter of debate. While medical calcification increases arterial stiffness, blood pressure and cardiac post-load and impairs vascular reactivity/adaptivity, the role of intimal atherosclerotic calcification is less clear. Extensive calcification is supposed to stabilize atherosclerotic plaques, while spotty micro-calcification, especially in the shoulder or subendothelial space is expected to increase plaque instability and vulnerability (91,92). Future studies should unveil the relationships between aging pathways and VC, to identify potential common targets of prevention or intervention.
Acknowledgements
None.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Pradelli D, Faden G, Mureddu G, et al. Impact of aortic or mitral valve sclerosis and calcification on cardiovascular events and mortality: a meta-analysis. Int J Cardiol 2013;170:e51-5. [DOI] [PubMed] [Google Scholar]
- 2.Bild DE, Detrano R, Peterson D, et al. Ethnic differences in coronary calcification: the Multi-Ethnic Study of Atherosclerosis (MESA). Circulation 2005;111:1313-20. [DOI] [PubMed] [Google Scholar]
- 3.Finch CE. Atherosclerosis is an old disease: Summary of the Ruffer Centenary Symposium, The Paleocardiology of Ancient Egypt, a meeting report of the Horus Study team. Exp Gerontol 2011;46:843-6. [DOI] [PubMed] [Google Scholar]
- 4.Clarke EM, Thompson RC, Allam AH, et al. Is atherosclerosis fundamental to human aging? Lessons from ancient mummies. J Cardiol 2014;63:329-34. [DOI] [PubMed] [Google Scholar]
- 5.Abedin M, Tintut Y, Demer LL. Vascular calcification: mechanisms and clinical ramifications. Arterioscler Thromb Vasc Biol 2004;24:1161-70. [DOI] [PubMed] [Google Scholar]
- 6.Lanzer P, Boehm M, Sorribas V, et al. Medial vascular calcification revisited: review and perspectives. Eur Heart J 2014;35:1515-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Virmani R, Joner M, Sakakura K. Recent highlights of ATVB: calcification. Arterioscler Thromb Vasc Biol 2014;34:1329-32. [DOI] [PubMed] [Google Scholar]
- 8.Ehara S, Kobayashi Y, Yoshiyama M, et al. Spotty calcification typifies the culprit plaque in patients with acute myocardial infarction: an intravascular ultrasound study. Circulation 2004;110:3424-9. [DOI] [PubMed] [Google Scholar]
- 9.Rutsch F, Nitschke Y, Terkeltaub R. Genetics in arterial calcification: pieces of a puzzle and cogs in a wheel. Circ Res 2011;109:578-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hofmann Bowman MA, McNally EM. Genetic pathways of vascular calcification. Trends Cardiovasc Med 2012;22:93-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Assimes TL, Knowles JW, Basu A, et al. Susceptibility locus for clinical and subclinical coronary artery disease at chromosome 9p21 in the multi-ethnic ADVANCE study. Hum Mol Genet 2008;17:2320-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.O'Donnell CJ, Kavousi M, Smith AV, et al. Genome-wide association study for coronary artery calcification with follow-up in myocardial infarction. Circulation 2011;124:2855-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Proudfoot D, Davies JD, Skepper JN, et al. Acetylated low-density lipoprotein stimulates human vascular smooth muscle cell calcification by promoting osteoblastic differentiation and inhibiting phagocytosis. Circulation 2002;106:3044-50. [DOI] [PubMed] [Google Scholar]
- 14.Parhami F, Basseri B, Hwang J, et al. High-density lipoprotein regulates calcification of vascular cells. Circ Res 2002;91:570-6. [DOI] [PubMed] [Google Scholar]
- 15.Nakanishi R, Rajani R, Cheng VY, et al. Increase in epicardial fat volume is associated with greater coronary artery calcification progression in subjects at intermediate risk by coronary calcium score: a serial study using non-contrast cardiac CT. Atherosclerosis 2011;218:363-8. [DOI] [PubMed] [Google Scholar]
- 16.Gorter PM, de Vos AM, van der Graaf Y, et al. Relation of epicardial and pericoronary fat to coronary atherosclerosis and coronary artery calcium in patients undergoing coronary angiography. Am J Cardiol 2008;102:380-5. [DOI] [PubMed] [Google Scholar]
- 17.Katsiki N, Mikhailidis DP, Wierzbicki AS. Epicardial fat and vascular risk: a narrative review. Curr Opin Cardiol 2013;28:458-63. [DOI] [PubMed] [Google Scholar]
- 18.Yerramasu A, Dey D, Venuraju S, et al. Increased volume of epicardial fat is an independent risk factor for accelerated progression of sub-clinical coronary atherosclerosis. Atherosclerosis 2012;220:223-30. [DOI] [PubMed] [Google Scholar]
- 19.Youssef G, Budoff MJ. Coronary artery calcium scoring, what is answered and what questions remain. Cardiovasc Diagn Ther 2012;2:94-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Agarwal S, Cox AJ, Herrington DM, et al. Coronary calcium score predicts cardiovascular mortality in diabetes: diabetes heart study. Diabetes Care 2013;36:972-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rattazzi M, Bertacco E, Puato M, et al. Hypertension and vascular calcification: a vicious cycle? J Hypertens 2012;30:1885-93. [DOI] [PubMed] [Google Scholar]
- 22.Savoia C, Burger D, Nishigaki N, et al. Angiotensin II and the vascular phenotype in hypertension. Expert Rev Mol Med 2011;13:e11. [DOI] [PubMed] [Google Scholar]
- 23.Osako MK, Nakagami H, Shimamura M, et al. Cross-talk of receptor activator of nuclear factor-kappaB ligand signaling with renin-angiotensin system in vascular calcification. Arterioscler Thromb Vasc Biol 2013;33:1287-96. [DOI] [PubMed] [Google Scholar]
- 24.Armstrong ZB, Boughner DR, Drangova M, et al. Angiotensin II type 1 receptor blocker inhibits arterial calcification in a pre-clinical model. Cardiovasc Res 2011;90:165-70. [DOI] [PubMed] [Google Scholar]
- 25.Wu SY, Yu YR, Cai Y, et al. Endogenous aldosterone is involved in vascular calcification in rat. Exp Biol Med (Maywood) 2012;237:31-7. [DOI] [PubMed] [Google Scholar]
- 26.Arribas SM, Hinek A, Gonzalez MC. Elastic fibres and vascular structure in hypertension. Pharmacol Ther 2006;111:771-91. [DOI] [PubMed] [Google Scholar]
- 27.Aikawa E, Nahrendorf M, Sosnovik D, et al. Multimodality molecular imaging identifies proteolytic and osteogenic activities in early aortic valve disease. Circulation 2007;115:377-86. [DOI] [PubMed] [Google Scholar]
- 28.Deuell KA, Callegari A, Giachelli CM, et al. RANKL enhances macrophage paracrine pro-calcific activity in high phosphate-treated smooth muscle cells: dependence on IL-6 and TNF-alpha. J Vasc Res 2012;49:510-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Iribarren C, Sidney S, Sternfeld B, et al. Calcification of the aortic arch: risk factors and association with coronary heart disease, stroke, and peripheral vascular disease. JAMA 2000;283:2810-5. [DOI] [PubMed] [Google Scholar]
- 30.Rasmussen T, Frestad D, Kober L, et al. Development and progression of coronary artery calcification in long-term smokers: adverse effects of continued smoking. J Am Coll Cardiol 2013;62:255-7. [DOI] [PubMed] [Google Scholar]
- 31.Speer MY, Li X, Hiremath PG, et al. Runx2/Cbfa1, but not loss of myocardin, is required for smooth muscle cell lineage reprogramming toward osteochondrogenesis. J Cell Biochem 2010;110:935-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun Y, Byon CH, Yuan K, et al. Smooth muscle cell-specific runx2 deficiency inhibits vascular calcification. Circ Res 2012;111:543-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shanahan CM, Crouthamel MH, Kapustin A, et al. Arterial calcification in chronic kidney disease: key roles for calcium and phosphate. Circ Res 2011;109:697-711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shroff R, Long DA, Shanahan C. Mechanistic insights into vascular calcification in CKD. J Am Soc Nephrol 2013;24:179-89. [DOI] [PubMed] [Google Scholar]
- 35.Price PA, Urist MR, Otawara Y. Matrix Gla protein, a new gamma-carboxyglutamic acid-containing protein which is associated with the organic matrix of bone. Biochem Biophys Res Commun 1983;117:765-71. [DOI] [PubMed] [Google Scholar]
- 36.Chatrou ML, Winckers K, Hackeng TM, et al. Vascular calcification: the price to pay for anticoagulation therapy with vitamin K-antagonists. Blood Rev 2012;26:155-66. [DOI] [PubMed] [Google Scholar]
- 37.Proudfoot D, Shanahan CM. Molecular mechanisms mediating vascular calcification: role of matrix Gla protein. Nephrology (Carlton) 2006;11:455-61. [DOI] [PubMed] [Google Scholar]
- 38.Brylka L, Jahnen-Dechent W. The role of fetuin-A in physiological and pathological mineralization. Calcif Tissue Int 2013;93:355-64. [DOI] [PubMed] [Google Scholar]
- 39.Scatena M, Liaw L, Giachelli CM. Osteopontin: a multifunctional molecule regulating chronic inflammation and vascular disease. Arterioscler Thromb Vasc Biol 2007;27:2302-9. [DOI] [PubMed] [Google Scholar]
- 40.Kaartinen MT, Murshed M, Karsenty G, et al. Osteopontin upregulation and polymerization by transglutaminase 2 in calcified arteries of Matrix Gla protein-deficient mice. J Histochem Cytochem 2007;55:375-86. [DOI] [PubMed] [Google Scholar]
- 41.Hofbauer LC, Schoppet M. Clinical implications of the osteoprotegerin/RANKL/RANK system for bone and vascular diseases. JAMA 2004;292:490-5. [DOI] [PubMed] [Google Scholar]
- 42.Lomashvili KA, Narisawa S, Millan JL, et al. Vascular calcification is dependent on plasma levels of pyrophosphate. Kidney Int 2014;85:1351-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kraus C, Rohde D, Weidenhammer C, et al. S100A1 in cardiovascular health and disease: closing the gap between basic science and clinical therapy. J Mol Cell Cardiol 2009;47:445-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McCormick MM, Rahimi F, Bobryshev YV, et al. S100A8 and S100A9 in human arterial wall. Implications for atherogenesis. J Biol Chem 2005;280:41521-9. [DOI] [PubMed] [Google Scholar]
- 45.Burke AP, Kolodgie FD, Zieske A, et al. Morphologic findings of coronary atherosclerotic plaques in diabetics: a postmortem study. Arterioscler Thromb Vasc Biol 2004;24:1266-71. [DOI] [PubMed] [Google Scholar]
- 46.Reynolds JL, Joannides AJ, Skepper JN, et al. Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: a potential mechanism for accelerated vascular calcification in ESRD. J Am Soc Nephrol 2004;15:2857-67. [DOI] [PubMed] [Google Scholar]
- 47.Johnson RC, Leopold JA, Loscalzo J. Vascular calcification: pathobiological mechanisms and clinical implications. Circ Res 2006;99:1044-59. [DOI] [PubMed] [Google Scholar]
- 48.Tanimura A, McGregor DH, Anderson HC. Matrix vesicles in atherosclerotic calcification. Proc Soc Exp Biol Med 1983;172:173-7. [DOI] [PubMed] [Google Scholar]
- 49.Kapustin AN, Shanahan CM. Calcium regulation of vascular smooth muscle cell-derived matrix vesicles. Trends Cardiovasc Med 2012;22:133-7. [DOI] [PubMed] [Google Scholar]
- 50.Buendía P, Montes de Oca A, Madueno JA, et al. Endothelial microparticles mediate inflammation-induced vascular calcification. FASEB J 2015;29:173-81. [DOI] [PubMed] [Google Scholar]
- 51.Prié D, Beck L, Urena P, et al. Recent findings in phosphate homeostasis. Curr Opin Nephrol Hypertens 2005;14:318-24. [DOI] [PubMed] [Google Scholar]
- 52.Evrard S, Delanaye P, Kamel S, et al. Vascular calcification: from pathophysiology to biomarkers. Clin Chim Acta 2015;438:401-14. [DOI] [PubMed] [Google Scholar]
- 53.Schurgers LJ, Uitto J, Reutelingsperger CP. Vitamin K-dependent carboxylation of matrix Gla-protein: a crucial switch to control ectopic mineralization. Trends Mol Med 2013;19:217-26. [DOI] [PubMed] [Google Scholar]
- 54.Balderman JA, Lee HY, Mahoney CE, et al. Bone morphogenetic protein-2 decreases microRNA-30b and microRNA-30c to promote vascular smooth muscle cell calcification. J Am Heart Assoc 2012;1:e003905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu M, Rementer C, Giachelli CM. Vascular calcification: an update on mechanisms and challenges in treatment. Calcif Tissue Int 2013;93:365-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hunter GK. Role of osteopontin in modulation of hydroxyapatite formation. Calcif Tissue Int 2013;93:348-54. [DOI] [PubMed] [Google Scholar]
- 57.Moe SM, Chen NX. Pathophysiology of vascular calcification in chronic kidney disease. Circ Res 2004;95:560-7. [DOI] [PubMed] [Google Scholar]
- 58.Vasuri F, Fittipaldi S, Pasquinelli G. Arterial calcification: Finger-pointing at resident and circulating stem cells. World J Stem Cells 2014;6:540-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Paneni F, Costantino S, Cosentino F. Molecular mechanisms of vascular dysfunction and cardiovascular biomarkers in type 2 diabetes. Cardiovasc Diagn Ther 2014;4:324-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Avogaro A, Rattazzi M, Fadini GP. Ectopic calcification in diabetic vascular disease. Expert Opin Ther Targets 2014;18:595-609. [DOI] [PubMed] [Google Scholar]
- 61.Chen NX, Moe SM. Arterial calcification in diabetes. Curr Diab Rep 2003;3:28-32. [DOI] [PubMed] [Google Scholar]
- 62.Fadini GP, Pauletto P, Avogaro A, et al. The good and the bad in the link between insulin resistance and vascular calcification. Atherosclerosis 2007;193:241-4. [DOI] [PubMed] [Google Scholar]
- 63.Li H, Jiang LS, Dai LY. High glucose potentiates collagen synthesis and bone morphogenetic protein-2-induced early osteoblast gene expression in rat spinal ligament cells. Endocrinology 2010;151:63-74. [DOI] [PubMed] [Google Scholar]
- 64.Ostrom MP, Gopal A, Ahmadi N, et al. Mortality incidence and the severity of coronary atherosclerosis assessed by computed tomography angiography. J Am Coll Cardiol 2008;52:1335-43. [DOI] [PubMed] [Google Scholar]
- 65.Ohana M, El Ghannudi S, Girsowicz E, et al. Detailed cross-sectional study of 60 superficial femoral artery occlusions: morphological quantitative analysis can lead to a new classification. Cardiovasc Diagn Ther 2014;4:71-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tavridou A, Petridis I, Vasileiadis M, et al. Association of VKORC1 -1639 G>A polymorphism with carotid intima-media thickness in type 2 diabetes mellitus. Diabetes Res Clin Pract 2011;94:236-41. [DOI] [PubMed] [Google Scholar]
- 67.Schiffrin EL, Lipman ML, Mann JF. Chronic kidney disease: effects on the cardiovascular system. Circulation 2007;116:85-97. [DOI] [PubMed] [Google Scholar]
- 68.Kim HS, Chung W, Kim AJ, et al. Circulating levels of soluble receptor for advanced glycation end product (sRAGE) are inversely associated with vascular calcification in patients on hemodialysis independent of S100A12 (EN-RAGE) levels. Nephrology (Carlton) 2013;18:777-82. [DOI] [PubMed] [Google Scholar]
- 69.Liabeuf S, Bourron O, Vemeer C, et al. Vascular calcification in patients with type 2 diabetes: the involvement of matrix Gla protein. Cardiovasc Diabetol 2014;13:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sinha A, Vyavahare NR. High-glucose levels and elastin degradation products accelerate osteogenesis in vascular smooth muscle cells. Diab Vasc Dis Res 2013;10:410-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Boström K, Tsao D, Shen S, et al. Matrix GLA protein modulates differentiation induced by bone morphogenetic protein-2 in C3H10T1/2 cells. J Biol Chem 2001;276:14044-52. [DOI] [PubMed] [Google Scholar]
- 72.Boström KI, Jumabay M, Matveyenko A, et al. Activation of vascular bone morphogenetic protein signaling in diabetes mellitus. Circ Res 2011;108:446-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yamagishi S, Fujimori H, Yonekura H, et al. Advanced glycation endproducts accelerate calcification in microvascular pericytes. Biochem Biophys Res Commun 1999;258:353-7. [DOI] [PubMed] [Google Scholar]
- 74.Collett G, Wood A, Alexander MY, et al. Receptor tyrosine kinase Axl modulates the osteogenic differentiation of pericytes. Circ Res 2003;92:1123-9. [DOI] [PubMed] [Google Scholar]
- 75.Fadini GP, Rattazzi M, Matsumoto T, et al. Emerging role of circulating calcifying cells in the bone-vascular axis. Circulation 2012;125:2772-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Albiero M, Avogaro A, Fadini GP. Circulating cellular players in vascular calcification. Curr Pharm Des 2014;20:5889-96. [DOI] [PubMed] [Google Scholar]
- 77.Eghbali-Fatourechi GZ, Lamsam J, Fraser D, et al. Circulating osteoblast-lineage cells in humans. N Engl J Med 2005;352:1959-66. [DOI] [PubMed] [Google Scholar]
- 78.Gössl M, Modder UI, Atkinson EJ, et al. Osteocalcin expression by circulating endothelial progenitor cells in patients with coronary atherosclerosis. J Am Coll Cardiol 2008;52:1314-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fadini GP, Albiero M, Menegazzo L, et al. Widespread increase in myeloid calcifying cells contributes to ectopic vascular calcification in type 2 diabetes. Circ Res 2011;108:1112-21. [DOI] [PubMed] [Google Scholar]
- 80.Fadini GP, Albiero M, Menegazzo L, et al. Procalcific phenotypic drift of circulating progenitor cells in type 2 diabetes with coronary artery disease. Exp Diabetes Res 2012;2012:921685. [DOI] [PMC free article] [PubMed]
- 81.Yao Y, Jumabay M, Ly A, et al. A role for the endothelium in vascular calcification. Circ Res 2013;113:495-504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Albiero M, Rattazzi M, Menegazzo L, et al. Myeloid calcifying cells promote atherosclerotic calcification via paracrine activity and allograft inflammatory factor-1 overexpression. Basic Res Cardiol 2013;108:368. [DOI] [PubMed] [Google Scholar]
- 83.Menegazzo L, Albiero M, Millioni R, et al. Circulating myeloid calcifying cells have antiangiogenic activity via thrombospondin-1 overexpression. FASEB J 2013;27:4355-65. [DOI] [PubMed] [Google Scholar]
- 84.Freigang S, Ampenberger F, Weiss A, et al. Fatty acid-induced mitochondrial uncoupling elicits inflammasome-independent IL-1alpha and sterile vascular inflammation in atherosclerosis. Nat Immunol 2013;14:1045-53. [DOI] [PubMed] [Google Scholar]
- 85.Anand DV, Lim E, Darko D, et al. Determinants of progression of coronary artery calcification in type 2 diabetes role of glycemic control and inflammatory/vascular calcification markers. J Am Coll Cardiol 2007;50:2218-25. [DOI] [PubMed] [Google Scholar]
- 86.Carson AP, Steffes MW, Carr JJ, et al. Hemoglobin a1c and the progression of coronary artery calcification among adults without diabetes. Diabetes Care 2015;38:66-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jørgensen L, Jenssen T, Joakimsen O, et al. Glycated hemoglobin level is strongly related to the prevalence of carotid artery plaques with high echogenicity in nondiabetic individuals: the Tromso study. Circulation 2004;110:466-70. [DOI] [PubMed] [Google Scholar]
- 88.Dayan A, Narin B, Biteker M, et al. Coronary calcium score, albuminuria and inflammatory markers in type 2 diabetic patients: associations and prognostic implications. Diabetes Res Clin Pract 2012;98:98-103. [DOI] [PubMed] [Google Scholar]
- 89.Flammer AJ, Gossl M, Li J, et al. Patients with an HbA1c in the prediabetic and diabetic range have higher numbers of circulating cells with osteogenic and endothelial progenitor cell markers. J Clin Endocrinol Metab 2012;97:4761-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chang Y, Yun KE, Jung HS, et al. A1C and coronary artery calcification in nondiabetic men and women. Arterioscler Thromb Vasc Biol 2013;33:2026-31. [DOI] [PubMed] [Google Scholar]
- 91.Kataoka Y, Puri R, Hammadah M, et al. Spotty calcification and plaque vulnerability in vivo: frequency-domain optical coherence tomography analysis. Cardiovasc Diagn Ther 2014;4:460-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Murray SW, Patel B, Stables RH, et al. Site-specific intravascular ultrasound analysis of remodelling index and calcified necrosis patterns reveals novel blueprints for coronary plaque instability. Cardiovasc Diagn Ther 2014;4:287-98. [DOI] [PMC free article] [PubMed] [Google Scholar]