

Abstract
Inhaled carbon monoxide (CO) gas has therapeutic potential for patients with acute respiratory distress syndrome if a safe, evidence-based dosing strategy and a ventilator-compatible CO delivery system can be developed. In this study, we used a clinically relevant baboon model of Streptococcus pneumoniae pneumonia to 1) test a novel, ventilator-compatible CO delivery system; 2) establish a safe and effective CO dosing regimen; and 3) investigate the local and systemic effects of CO therapy on inflammation and acute lung injury (ALI). Animals were inoculated with S. pneumoniae (108-109 CFU) (n = 14) or saline vehicle (n = 5); in a subset with pneumonia (n = 5), we administered low-dose, inhaled CO gas (100–300 ppm × 60–90 min) at 0, 6, 24, and/or 48 h postinoculation and serially measured blood carboxyhemoglobin (COHb) levels. We found that CO inhalation at 200 ppm for 60 min is well tolerated and achieves a COHb of 6–8% with ambient CO levels ≤ 1 ppm. The COHb level measured at 20 min predicted the 60-min COHb level by the Coburn-Forster-Kane equation with high accuracy. Animals given inhaled CO + antibiotics displayed significantly less ALI at 8 days postinoculation compared with antibiotics alone. Inhaled CO was associated with activation of mitochondrial biogenesis in the lung and with augmentation of renal antioxidative programs. These data support the feasibility of safely delivering inhaled CO gas during mechanical ventilation and provide preliminary evidence that CO may accelerate the resolution of ALI in a clinically relevant nonhuman primate pneumonia model.
Keywords: Streptococcus pneumoniae, pneumonia, carbon monoxide, drug delivery systems, Coburn-Forster-Kane equation
carbon monoxide (co) is now well recognized as a signaling molecule (31) produced by the inducible enzyme heme oxygenase-1 (HO-1) during heme catabolism (60). The HO-1/CO system responds to cellular stress by coupling the cell's anti-inflammatory, antioxidant, and antiapoptotic defenses (1, 7, 22, 51) with the activation of mitochondrial quality control programs such as mitochondrial biogenesis (51). This system can be activated pharmacologically by administering low-dose inhaled CO gas, which is protective against a wide range of pathological conditions, including inflammation (12, 47), oxidative stress (50), ischemia/reperfusion injury (23, 33, 64), sepsis (12, 35, 38, 51), lung inflammation (43), and acute lung injury (ALI) (18, 21, 24, 29, 30, 32, 41, 59, 65). This makes low-dose inhaled CO a potential therapy for critically ill patients, particularly those with sepsis- and pneumonia-induced acute respiratory distress syndrome (ARDS) for which current treatment options are limited. There are practical barriers, however, to implementation of CO therapy in patients, such as the lack of a ventilator-compatible CO delivery system and a safe, evidence-based CO dosing strategy. These require further preclinical work in large animals prior to translation to patients.
We therefore performed a proof-of-concept study using a clinically relevant, established model of Streptococcus pneumoniae pneumonia in baboons (9, 34, 56) to investigate 1) the ability of a novel CO delivery system to safely and predictably deliver inhaled CO to the injured lung during mechanical ventilation; 2) an optimal CO dosing strategy to increase percent carboxyhemoglobin (COHb) above baseline levels but within a safe range with a goal of 6–8% (28, 40, 42, 46, 52, 53, 63); and 3) the effects of inhaled CO on local and systemic cytoprotective programs during experimental pneumonia. Using this baboon model, we have already reported that low-dose inhaled CO reduces proinflammatory urinary cysteinyl leukotrienes (58) and partially restores levels of circulating specialized proresolving lipid mediators (SPMs), which become dysregulated during the pathogenesis of pneumococcal pneumonia (15, 19). We now report that the administration of low-dose inhaled CO to baboons with pneumonia is safely accomplished by use of a novel, ventilator-compatible delivery device and that the rise in COHb can be accurately predicted by using the Coburn-Forster-Kane (CFK) equation to a goal of 6–8%. We also report preliminary evidence of both lung protection and augmentation of renal antioxidant defenses in the CO-treated animals.
MATERIALS AND METHODS
Animal model.
The experimental protocol was submitted to and preapproved by the Duke University Institutional Animal Care and Use Committee. The animals consumed fruit and chow (PMI Nutrition International, St. Louis, MO) ad libitum but fasted overnight prior to the studies.
Adult male colony-bred baboons (Papio cynocephalus) (n = 19), mean ± SD ages 7.4 ± 1.8 yr old and weighing 25.8 ± 2.5 kg, were purchased from Texas Biomedical Research Institute (San Antonio, TX). All animals were housed in the Duke University Vivarium (Durham, NC) and handled in accordance with American Association for Accreditation of Laboratory Animal Care (AAALAC) guidelines. Animals were quarantined for 4 wk and determined by skin testing to be tuberculosis free before use. On day 1, a single animal was deeply sedated with ketamine (20–25 mg/kg; Fort Dodge Animal Health, Fort Dodge, IL) and lorazepam (1–2 mg; Hospira, Lake Forest, IL), intubated, and ventilated with volume-control ventilation (Bear Medical Systems, Riverside, CA) at settings of inspired O2 fraction (FiO2) 0.21, tidal volume 10 ml/kg, positive end-expiratory pressure 2.5–5 cmH2O, and respiratory rate (RR) 12–14 breaths per minute. Blood pressure (BP), heart rate (HR), rectal temperature, pulse oximetry, and telemetry were monitored throughout the experiment. The animal received 1 liter iv 0.9% NaCl to prevent hypovolemia and a warming blanket to prevent hypothermia. By using a fiberoptic bronchoscope (Pentax, Montvale, NJ), a bronchoalveolar lavage (BAL) of the left lower lobe (20 ml 0.9% NaCl) was performed at FiO2 1.0, followed by instillation of 10 ml sterile 0.9% NaCl vehicle (n = 5; “control animals”) or Streptococcus pneumoniae (Serotype 19A-7; American Type Culture Collection, Manassas, VA) (34) at 108-109 CFU (n = 9; “unexposed pneumonia animals”) divided equally between the lingula and left lower lobe (mean ± SD total dose, 1.3 ± 0.7 × 109 CFU; mean ± SD dose per lobe, 6.5 ± 3.4 × 108 CFU). Supplemental oxygen was weaned to room air over 1 h and the animal was recovered, extubated, and placed in isolation. At 24 h postinoculation, the animal was briefly sedated for collection of blood and vital signs. At 48 h postinoculation, the animal was again sedated, intubated, ventilated, and monitored as in day 1. A repeat BAL of the left lower lobe was performed with 20 ml 0.9% NaCl on supplemental oxygen which was quickly weaned to FiO2 0.21. The animal was given 1 g im ceftriaxone (Hospira, Lake Forest, IL) once daily for a total of 3 days starting after the collection of the 48-h samples. The animal was then recovered, extubated, and returned to isolation. At 168 h postinoculation, the animal was euthanized with ketamine (1,000 mg iv) and lorazepam (5 mg iv) followed by saturated KCl for immediate necropsy and tissue harvesting as reported (34).
Clinical definitions.
The diagnosis of pneumonia was established by the presence of all three of the following criteria (34): 1) white blood cell (WBC) >15,000/μl, <4,000/μl, a ≥2-fold change from baseline, or ≥90% neutrophils on differential; 2) isolation of S. pneumoniae from blood or BAL fluid at 48 h; and 3) at least one of the following signs or symptoms at 24 or 48 h: fever >38.2°C, HR >100 beats/min (bpm), RR >25% above baseline, cough, rhinorrhea, decreased oral intake or activity, or infiltrate on CXR. The diagnosis of sepsis was established according to the 2013 Surviving Sepsis Campaign guidelines and defined as the presence of a suspected infection along with tachypnea, tachycardia, fever, and change in WBC count (16).
Experimental groups.
The vital signs, laboratory data, BAL fluid characteristics, and ALI scores in four of the five uninfected, control animals are reported elsewhere (15, 34). The vital signs, laboratory data, BAL fluid characteristics, and ALI scores in eight of the nine unexposed pneumonia animals are reported elsewhere (15, 34). The vital signs, laboratory data, and BAL fluid cell counts from three of the five CO-exposed pneumonia animals are reported elsewhere (15, 58).
Sample collection and laboratory measurements.
Blood samples were collected at 0, 24, 48, and 168 h postinoculation for complete blood counts and arterial blood gas (ABG) measurements, and at 24, 48, and 168 h for cultures as described (34). BAL fluid was collected at 0, 48, and 168 h for total protein, lactate dehydrogenase (LDH) activity, total cell count, and cultures as described (34). HR, temperature, and BP data were collected at 0, 24, 48, and 168 h. RR was recorded prior to sedation at 0, 24, 48, 72, 96, and 168 h.
Delivery of inhaled CO gas.
To investigate the feasibility, safety, and pharmacokinetics of CO delivery in this model, an additional set of five animals (“CO-exposed pneumonia animals”) underwent a similar experimental pneumonia protocol except that 1) the inoculum for four of the animals was divided equally between one or two lobes bilaterally (mean ± SD total dose, 1.8 ± 1.1 × 109 CFU; mean ± SD dose per lobe, 7.1 ± 5.6 × 108 CFU) to produce a more diffuse lung injury; and 2) the animals were ventilated with a Puritan Bennett 840 ventilator (Mansfield, MA). A novel CO delivery system (12th Man Technologies, Garden Grove, CA) (Fig. 1A) was connected into the breathing circuit (Fig. 1B). This CO delivery system maintains a constant concentration of CO gas in the ventilator circuit by adjusting for changes in minute ventilation and flow (Fig. 1C). A 5,000 ppm CO source gas balanced with air (Airgas, Durham, NC) was injected into the circuit in proportion to the gas flow to provide a constant delivered dose. This was confirmed by downstream measurement of the CO concentration via a calibrated sidestream electrochemical analyzer. Ambient CO levels were monitored continuously with a CO detector (Bacharach Monoxor II, New Kensington, PA).
Fig. 1.
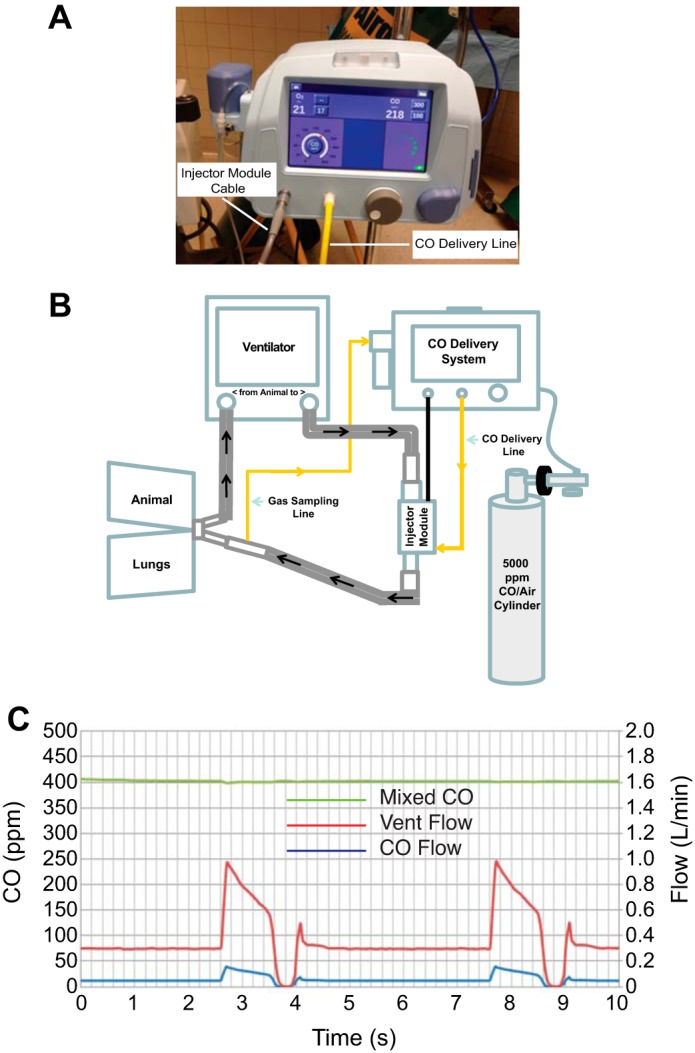
Carbon monoxide (CO) delivery system. A: front view of carbon monoxide delivery system. B: schematic of experimental setup including ventilator, CO delivery system with injector module, and CO gas cylinder. C: volumetric mixing data using the CO delivery system, a mechanical ventilator, and test lung. Dynamic flow-matching graph demonstrates that the instantaneous flow of CO gas (CO flow) matches the patient's inspiratory flow (Vent Flow) to deliver and maintain a precise and constant concentration of CO (Mixed CO) over time, adjusting for the patient's flow rate, respiratory rate, or tidal volume.
Preliminary dose-finding study.
Inhaled CO was administered to these five animals at 0, 6, 24, and/or 48 h postinoculation. In the first two animal experiments, we performed a preliminary dose-finding study to determine the dose of CO needed to achieve our goal COHb after 1 h of inhalation: We administered 100, 200, and/or 300 ppm for 50–70 min at 0, 24, and/or 48 h postinoculation and serially measured COHb. The 200 ppm dose achieved our goal COHb at 1 h and was used for the remaining three animal experiments. The schedule of CO delivery for each animal is shown in Table 1. To characterize CO elimination in this model, animals (n = 4) were given supplemental oxygen (FiO2 1.0) for 60–90 min after CO treatment was completed until COHb levels returned to near baseline levels.
Table 1.
Exact CO dosing schedule for each animal
| Animal Number | Time Point, hours postinoculation | CO Dose, ppm | Duration, min |
|---|---|---|---|
| 28798 | −3 | 100 | 60 |
| −2 | 200 | 30 | |
| 23 | 200 | 60 | |
| 25.5 | 300 | 50 | |
| 29219 | 48 | 200 | 70 |
| 51 | 300 | 40 | |
| 29223 | 49 | 200 | 60 |
| 29697 | 48 | 200 | 90 |
| 29946 | 6 | 200 | 60 |
| 48 | 200 | 60 |
COHb measurement and prediction.
ABG and COHb measurements were drawn before, during, and after CO delivery at 10- to 15-min intervals. In two CO-exposed animals, venous and arterial blood samples were drawn simultaneously to determine agreement between venous and arterial COHb levels. COHb levels were measured in heparinized blood by using the gold standard IL 682 CO-oximeter (Instrumentation Laboratory, Bedford, MA). In two CO-exposed animals, COHb levels were also synchronously measured with an AVOXimeter 4000 CO-oximeter (ITC, Edison, NJ).
We evaluated the accuracy and precision of the CFK equation
(13, 14, 49) to predict the animals' COHb level after 60 min of CO exposure (Table 2). The variables in the CFK equation were calculated as described (49). For all animals, CO production (V̇co) was assumed to be 0.007 ml/min and the affinity constant M was assumed to be 218 (49, 55). To calculate alveolar ventilation (a variable in the CFK equation), a NICO monitor (Philips Respironics, Pittsburgh, PA) was used to measure end-tidal CO2. To estimate DlCO (another variable in the CFK equation), we input into the CFK equation the baseline COHb level (time = 0) and the COHb level measured after a short CO exposure (time = 10 or 20 min) and solved for DlCO by using a computer program generated in MATLAB (MathWorks, Natick, MA). We then input the estimated DlCO and the COHb level measured after 10, 20, 30, 40, or 50 min into the CFK equation and calculated the COHb level predicted after CO exposure for time = 60 min.
Table 2.
Coburn-Forster-Kane equation variables
| Animal no. | Weight, kg | Hb, g/dl | COHb at Baseline, % | V̇e, l/min | Vd/Vt | V̇a, l/min | COHb at 20 min, % | DlCO, ml·min−1·mmHg−1 | Estimated 60-min COHb | Measured 60-min COHb |
|---|---|---|---|---|---|---|---|---|---|---|
| 28798 | 26.1 | 11.6 | 1.3 | 3.7 | 0.29 | 2.6 | 3.3 | 6.2 | 6.8 | 6.3 |
| 29219 | 27.1 | 11.7 | 1.1 | 4.0 | 0.39 | 2.4 | 3.5 | 19.0 | 7.7 | 7.4 |
| 29223 | 24.9 | 11.0 | 1.1 | 4.3 | 0.38 | 2.7 | 3.6 | 8.6 | 7.9 | 7.4 |
| 29697 | 20.7 | 11.1 | 0.8 | 4.3 | 0.31 | 3.0 | 3.4 | 4.9 | 7.9 | 7.8 |
| 29946* | 20.1 | 8.9 | 1.1 | 6.0 | 0.40 | 3.6 | 4.5 | 5.5 | 8.9 | 9.1 |
For all animals: Pb = 760 mmHg; M = 218; V̇CO = 0.007 ml/min; FiO2 = 0.21; CO dose = 200 ppm; treatment duration = 60 min.
FiO2 0.35.
Western analysis.
Western blotting was performed on lung (taken from the area of infection) and kidney tissues taken at necropsy from a subset of control, unexposed pneumonia, and CO-exposed animals (n = 4–5 per group), as described (1). Briefly, tissues were homogenized in RIPA buffer, resolved by SDS-PAGE on gradient or 14% gels (Bio-Rad, Hercules, CA), transferred to polyvinylidene difluoride membranes (Millipore, Billerica, MA), blocked with 4% nonfat dry milk in Tris-buffered saline with Tween 20, and probed overnight at 4°C with polyclonal antibodies against HO-1 (1:1,000; Enzo Life Sciences, Farmingdale, NY), manganese superoxide dismutase (SOD2) (1:5,000; Abcam, Cambridge, MA), mitochondrial transcription factor A (Tfam) (1:1,000; developed in our laboratory), citrate synthase (1:1,000; GeneTex, Irvine, CA), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (1:1,000; GeneTex). After incubation with primary antibody, the membranes were treated with an appropriate horseradish peroxidase-conjugated secondary antibody (1:2,000, Santa Cruz Biotechnology, Dallas, TX) and developed by enhanced chemiluminescence (Western Blotting Luminol Reagent, Santa Cruz Biotechnology). The protein bands were quantified on digitized images in the mid-dynamic range (Image J). Protein loading was confirmed by GAPDH and/or Coomassie blue staining.
Histopathological analyses.
Lung and kidney tissue taken at necropsy was fixed in 10% formalin, embedded in paraffin, cut into 5-μm sections, and mounted on slides. ALI was defined by unilateral or bilateral evidence of histological lung injury (e.g., intra-alveolar WBCs, fibrin, debris, etc., as assessed by standardized scoring), alveolar capillary damage (e.g., elevated BAL fluid protein or lung wet-to-dry weight ratios), inflammation (e.g., elevated BAL fluid cell count), and impaired gas exchange (e.g., hypoxemia) (39). Three randomly selected hematoxylin and eosin-stained slides from the area of lung infection of each animal were examined under light microscopy by a blinded lung pathologist and graded (0–3, absent to severe) for the presence of edema, leukocytes, alveolar filling, fibrin, and necrosis. The mean slide scores for each finding for each animal were summed and reported as the modified ALI score (34). Immunofluorescence was performed on lung slides by probing with primary antibodies against mitochondrial ATP synthase (ATP5A subunit, Abcam) and thyroid transcription factor 1 (TTF-1) (Santa Cruz Biotechnology), and on kidney slides with primary antibodies against cytochrome c (BD Biosciences, San Jose, CA) and SOD2 (Abcam) (all 1:100), followed by the appropriate fluorescent-labeled secondary antibody (1:400) (Thermo Fisher Scientific, Waltham, MA). The nuclei were counterstained with DAPI (Thermo Fisher). The slides were observed under a Nikon Eclipse 50i fluorescence microscope. Wet-to-dry lung weight ratio determinations were made using four randomly selected lung samples from the area of pneumonia.
Statistical analyses.
Grouped data were normally distributed and expressed as means ± SD unless stated otherwise. A subset of the physiological and laboratory data used to define pneumonia in this model is published (15, 34, 58). Statistical analysis was performed with Prism (GraphPad Software, La Jolla, CA). Physiology and laboratory data were analyzed by two-factor ANOVA with Fisher's least significant difference (LSD) post hoc test; COHb measurements were analyzed by Spearman correlations and modeled by Deming Type II linear regression; measured vs. predicted COHb was modeled by linear regression; histological and molecular data were analyzed with one-way ANOVA with Fisher's LSD post hoc test. All P values are two tailed; statistical significance was accepted at P < 0.05 and trends were noted for P values between 0.05 and 0.1 (62).
RESULTS
Development of S. pneumoniae pneumonia, sepsis, and bacteremia.
The vital signs and laboratory data for some of the animals described herein have been reported (see materials and methods) (15, 34, 58). All animals inoculated with S. pneumoniae met previously established criteria for sepsis (16), pneumonia (34), and ALI (39). Baseline temperature, HR, BP, and RR were normal for the species. By 24 to 48 h postinoculation, both unexposed and CO-exposed animals displayed tachypnea, tachycardia, and fever (P < 0.05 compared with uninfected control values) (Fig. 2, A–C). Mean arterial pressure was largely unchanged throughout the experiment (Fig. 2D). Aside from a higher temperature at 48 h in the CO-treated animals, there were no significant differences in vital signs between unexposed animals and CO-exposed animals.
Fig. 2.

Vital signs. A: respiratory rates of both pneumonia groups were significantly elevated above uninfected control animals. B: heart rates of both pneumonia groups were significantly elevated above controls at 24 and 48 h postinoculation. C: temperature of both pneumonia groups was elevated above controls at 24 and 48 h, and the temperature of CO-exposed animals was elevated above control and unexposed pneumonia animals at 48 h. D: mean arterial pressure did not change significantly over the course of the experiment. Gray bars, uninfected, control group mean (n = 5 per time point); white bars, unexposed pneumonia group mean (n = 7–9 per time point); black bars, CO-exposed pneumonia group mean (n = 4–5 per time point); error bars represent standard deviation. *P < 0.05 compared with control, **P < 0.05 compared with control and unexposed pneumonia group. The physiological data for some of these animals have been reported (15, 34, 58).
Laboratory data were similar between unexposed and CO-exposed animals. Both groups of animals displayed variable WBC count responses at 24 and 48 h postinoculation (increased, unchanged, or decreased), but by 168 h WBC counts were near baseline (Fig. 3A). Platelet counts were unremarkable until 168 h when both groups displayed thrombocytosis (P < 0.05 compared with controls) (Fig. 3B). PaO2/PaO2 ratios were normal at baseline but significantly reduced in both unexposed and CO-exposed animals at 48 h (0.52 ± 0.13 and 0.62 ± 0.10, respectively) (P < 0.05 compared with controls) and at 168 h (0.75 ± 0.19 and 0.70 ± 0.15, respectively) (P < 0.05 compared with controls) (Fig. 3C). S. pneumoniae was cultured from BAL fluid at 48 h in the unexposed and CO-exposed animals [median (IQR), CFU/ml: 1.5 (90) × 105 vs. 1.3 (40) × 105, respectively] and from blood at 24 h [median (IQR), CFU/ml: 5 (96) vs. 20 (161), respectively] and 48 h [64 (267) vs. 13 (229), respectively]. Overall, six of nine unexposed animals and all five CO-exposed animals were bacteremic; however, by 168 h, BAL fluid and blood were sterile (Fig. 3, D and E). These values were not significantly different between the two pneumonia groups.
Fig. 3.

Laboratory data. A: white blood cell (WBC) counts of unexposed pneumonia animals were significantly higher than control and CO-exposed animals at 24 h; however, there was significant within-group variability (increased, decreased, or unchanged from baseline) seen in both pneumonia groups at 24–48 h postinoculation. No other significant differences were noted, and by 168 h, the WBC counts of all groups were near baseline. Circles represent individual animals. B: platelet counts (mean ± SD) were significantly elevated in both pneumonia groups relative to controls at 168 h. C: PaO2/PaO2 ratios (mean ± SD) were significantly reduced at 48 and 168 h in both pneumonia groups compared with controls. D and E: the experimental strain of S. pneumoniae was isolated from bronchoalveolar lavage (BAL) fluid (BALF) [median (IQR)] at 48 h (D) and from blood [median (IQR)] at 24 and 48 h (E) in both pneumonia groups but not from controls. Antibiotics were administered to all animals once daily × 3 days starting after collection of the 48 h samples. Gray circles, uninfected, control group, n = 5 per time point; white circles, unexposed pneumonia group, n = 7–9 per time point; black circles, CO-exposed pneumonia group, n = 4–5 per time point. *P < 0.05 compared with control. **P < 0.05 compared with control and CO-exposed pneumonia groups. The laboratory data for some of these animals have been reported (15, 34, 58).
Inhaled CO at 200 ppm for 1 h and COHb% goal.
After 1 h of CO administration at 200 ppm, animals achieved the prespecified goal COHb level of 6–8%. At baseline, arterial COHb levels were 1.1 ± 0.2% and increased linearly to 2.4 ± 0.6, 3.7 ± 0.5, 4.5 ± 0.7, 5.5 ± 0.8, 6.6 ± 0.9, and 7.6 ± 1% at 10, 20, 30, 40, 50, and 60 min of CO administration, respectively (P < 0.0001) (Fig. 4A). One animal was intentionally given a prolonged (90-min) exposure that similarly demonstrated a linear rise in COHb to 7.8, 8.8, 9.7, and 10.3% at 60, 70, 80, and 90 min, respectively (Fig. 4A). Peak COHb levels decreased following administration of FiO2 1.0, returning to near baseline levels after 82 ± 9.5 min (Fig. 4B). Throughout the exposure, ambient CO levels remained ≤1 ppm. The inhaled CO was tolerated well by the animals: there were no significant differences in pre- and post-CO HR, mean arterial pressure, temperature, PaO2, or minute ventilation.
Fig. 4.

Delivery of inhaled CO at 200 ppm for 1 h and carboxyhemoglobin (COHb). A and B: adult male baboons (n = 5) were inoculated with S. pneumoniae (108-109 CFU) and given inhaled CO gas (200 ppm) through the ventilator circuit at 24 or 48 h postinoculation for 60–90 min by use of the CO delivery system. COHb levels were measured in arterial blood every 10–15 min using an IL 682 CO-oximeter. A: arterial COHb levels were 1.08 ± 0.18% at baseline and increased linearly to 7.6 ± 1% after 60 min (P < 0.0001) and 10.3% after 90 min of CO administration. B: peak COHb levels decreased following administration of 1.0 FiO2, returning to near baseline levels after 82 ± 9.5 min.
Venous and arterial COHb correlation.
To determine the correlation between arterial and venous COHb levels, we measured COHb levels from both sites using the IL 682 CO-oximeter and found they were highly correlated (Spearman r = 0.9904; P < 0.0001). Modeling these data with type II linear regression, we found that the regression line was a near-perfect diagonal with a slope of 1.003 [95% CI (0.952–1.05); P < 0.0001] and a y-intercept of −0.064 [95% CI (−0.2941-0.1661)] (Fig. 5A).
Fig. 5.

Correlation between venous and arterial COHb. Animals were given inhaled CO at 200 ppm through the ventilator for 60 min by using the CO delivery system. A and B: in select experiments (n = 2), arterial and venous blood samples were drawn simultaneously for comparison of venous and arterial COHb levels, and comparison of the IL 682 and AVOXimeter 4000 (Avox) CO-oximeters. A: there was excellent correlation between arterial and venous COHb levels obtained by using the IL 682 CO-oximeter (Spearman r, 0.9904; P < 0.0001). Deming Type II linear regression modeling demonstrated a near-perfect diagonal regression line (slope 1.003, y-intercept −0.064; P < 0.0001). B: measurements of COHb levels using the point-of-care AVOXimeter 4000 CO-oximeter correlated poorly with those obtained using the gold standard IL 682 CO-oximeter, especially at lower COHb. The AVOXimeter 4000 CO-oximeter did not perform as accurately when compared with the gold standard IL 682 CO-oximeter.
To determine the accuracy and precision of the point-of-care AVOXimeter 4000 CO-oximeter relative to the gold-standard IL 682 CO-oximeter, we measured COHb levels using both devices on synchronously drawn arterial blood. Compared with the IL 682 CO-oximeter, the AVOXimeter 4000 CO-oximeter was substantially less sensitive since COHb was not detected until levels had reached 4–5%. Furthermore, the AVOXimeter consistently reported COHb levels that were four percentage points lower than the gold standard (Fig. 5B).
CFK equation predictions of COHb%.
The CFK equation has been shown to accurately predict the rise in COHb following CO exposure in humans with normal lung function (13, 14, 49). To determine whether the CFK equation can predict the COHb level after 60 min of CO inhalation in an injured lung model, we first input the 10-min measured COHb level and found a good correlation between the measured COHb levels and the COHb levels predicted by the CFK equation (Spearman r = 0.9038; P < 0.0001) (Fig. 6A). However, we found a better correlation between the measured and predicted COHb levels by using the 20-min measured COHb level (Spearman r = 0.9828; P < 0.0001) (Fig. 6B). Modeling the 20-min data with linear regression, we found that the regression line was a near-perfect diagonal with a slope of 0.9373 [95% CI (0.8885–0.9860)], a y-intercept of 0.7291 [95% CI (0.3976–1.061)], and goodness-of-fit R2 = 0.9864 (P < 0.0001). Furthermore, the 20-min measured COHb was highly accurate in predicting the 60-min COHb with a difference between predicted and measured COHb of 0.24 ± 0.33% [95% CI (−0.17–0.66)] (Fig. 6C). Taken together, by inputting the 20-min measured COHb level into the CFK equation, we can predict with high accuracy the COHb level after 60 min of CO exposure in baboons with ALI.
Fig. 6.

Coburn-Forster-Kane (CFK) equation and COHb prediction at 60 min. Animals (n = 5) were given inhaled CO at 200 ppm for 60–90 min and arterial COHb levels were measured every 10–15 min with an IL 682 CO-oximeter. The CFK equation was used to predict COHb levels by using the measured COHb level at 10 (A) and 20 (B) min. A: correlation between measured COHb and predicted COHb levels by using the 10-min COHb level and CFK equation (Spearman r, 0.9038, P < 0.0001; goodness-of-fit R2, 0.735, P < 0.0001). B: correlation between measured COHb and predicted COHb levels by use of the 20-min COHb level and CFK equation (Spearman r, 0.9828, P < 0.0001; goodness-of-fit R2, 0.9864, P < 0.0001). C: accuracy of the 10, 20, 30, 40, and 50-min measured COHb levels to predict the 60-min COHb by use of the CFK equation. The 20-min COHb was highly accurate in predicting the 60-min COHb, with a difference between predicted and actual COHb of 0.24 ± 0.33% [95% CI (−0.17–0.66)].
Inhaled CO and ALI resolution.
To investigate the effects of inhaled CO on pneumonia-induced ALI, we first measured BAL fluid LDH, total protein, and total cell counts at 0, 48, and 168 h. The BAL fluid characteristics for some of these animals are published (15, 34). In contrast to the control group, which remained stable throughout, the BAL fluid LDH, total protein, and total cell counts in both pneumonia groups peaked at 48 h, but there was no significant difference between unexposed pneumonia and CO-exposed pneumonia animals (Fig. 7). We then scored hematoxylin and eosin-stained lung sections in a blinded fashion for the presence of edema, leukocytes, alveolar filling, fibrin, and necrosis (Fig. 8A). Some of the lung injury scores for the uninfected, procedural control animals and the unexposed pneumonia animals are published (34) and redisplayed in Fig. 8, B and C. Lung inflammation, when present, was generally diffuse for both unexposed and CO-exposed animals. In CO-exposed animals, however, we found significantly lower lung injury scores compared with unexposed animals (1.9 ± 2.7 vs. 4.5 ± 2.2, respectively; P < 0.05) (Fig. 8B). The edema scores in particular were significantly lower in CO-exposed animals than in unexposed animals (0 ± 0 vs. 0.6 ± 0.7, respectively; P < 0.05) (Fig. 8C). Moreover, lung wet-to-dry weight ratios in CO-exposed animals were similar to control but significantly lower than in unexposed animals (4.6 ± 0.5 vs. 5.3 ± 0.4; P < 0.05) (Fig. 8D). Together these data indicate that low-dose inhaled CO is associated with improved resolution of pneumonia-induced ALI, and in particular the reduction of extravascular lung water.
Fig. 7.

Bronchoalveolar fluid studies. Lactate dehydrogenase (LDH) (A), total protein (B), and cell counts (C) in uninfected, control animals (gray circles; n = 3–5 for cell counts and n = 5 for LDH and total protein), unexposed pneumonia animals (white circles; n = 7–9) and CO-exposed pneumonia animals (black circles; n = 4–5) at 0, 48, and 168 h postinoculation. BAL fluid indexes peaked at 48 h in both pneumonia groups and were at or near baseline by 168 h. There were no significant differences between the two pneumonia groups. Bars represent means. *P < 0.05 compared with control values; #P < 0.1 compared with control values. The BAL fluid characteristics for some of these animals are reported (15, 34).
Fig. 8.

Inhaled CO and resolution of pneumonia-induced acute lung injury (ALI). A: representative histopathology at 168 h from a control animal, an unexposed pneumonia animal, and a CO-exposed pneumonia animal. Control photomicrograph demonstrates empty alveoli with delicate septa. Pneumonia photomicrograph demonstrates severe intra-alveolar filling due to edema, leukocytes, erythrocytes, and other debris. Pneumonia + CO photomicrograph demonstrates significantly less alveolar filling with leukocytes and fibrin. Original magnification is ×400. B: ALI scores (reflecting leukocytes, edema, alveolar filling, fibrin, and necrosis) were significantly lower in the CO-exposed pneumonia (PNA + CO) animals (1.9 ± 2.7) than in the unexposed pneumonia (PNA) animals (4.5 ± 2.5) (P < 0.05). C: edema scores were significantly lower in the CO-exposed pneumonia animals (0 ± 0) than in the unexposed animals (0.6 ± 0.7) (P < 0.05). D: wet-to-dry weight ratios of lung tissue taken at 168 h were significantly lower in the CO-exposed pneumonia animals (4.6 ± 0.5) than in the unexposed pneumonia animals (5.3 ± 0.4) (P < 0.05). *P < 0.05; bars represent means. Some of the ALI scores for the uninfected control animals and unexposed pneumonia animals are published (34) and redisplayed in B and C. ALI scores for images shown in A are 0, 6, and 1.3, respectively.
Inhaled CO and mitochondrial biogenesis in the lungs post S. pneumoniae pneumonia.
In murine pneumonia, resolution is related to the activation of mitochondrial biogenesis in the lung, of which inhaled CO is a potent stimulus (1); hence we investigated the effects of inhaled CO on mitochondrial biogenesis in baboon pneumonia. Total lung protein obtained at 168 h was resolved by SDS-PAGE and Western blots were performed for SOD2, HO-1, mitochondrial transcription factor A (Tfam), and the mitochondrial matrix protein citrate synthase (Fig. 9A). In pneumonia animals without CO, there was a trend for increased SOD2 and HO-1 relative to controls (P < 0.1). In CO-exposed animals there was a trend for increased Tfam expression relative to controls (P < 0.1). The CO-exposed animals did, however, display significantly higher citrate synthase expression relative to the unexposed animals, indicative of increased mitochondrial mass.
Fig. 9.

Inhaled CO and mitochondrial biogenesis induction in the lung. A: lung tissue was taken from control (gray circles; n = 5), unexposed pneumonia (white circles; n = 5), and CO-exposed pneumonia (black circles; n = 4) animals at necropsy (168 h) and homogenized, and total protein was probed by Western blot for SOD2, HO-1, Tfam, and citrate synthase. Protein expression is relative to Coomassie blue staining. Bars represent means. #P < 0.1 relative to control, *P < 0.05. B: immunofluorescence of nuclear TTF-1 (alveolar type 2 cell marker; green) and ATP synthase (mitochondrial marker; red) in inflation-fixed lung sections from control, unexposed pneumonia, and CO-exposed pneumonia animals. Nuclei are counterstained with DAPI. The distribution of ATP synthase staining is greater in alveolar type 2 cells (arrowheads) and alveolar macrophages (asterisks) in the CO-exposed animals, indicative of increased mitochondrial mass in these cell types. Some of the macrophages in the unexposed pneumonia group appear to stain positively for cytoplasmic TTF-1 (green), which may reflect phagocytosis of alveolar type 2 cell debris. Bars are 100 μM.
To localize mitochondrial mass in the lung, we performed immunofluorescence microscopy for ATP synthase (red), a mitochondrial enzyme, and for TTF-1 (green), a marker for alveolar type 2 cells (Fig. 9B). Compared with control lung tissue, lung tissue from unexposed pneumonia animals demonstrated alveolar type 2 cell hyperplasia; however, the staining for ATP synthase in these cells was less, suggesting mitochondrial loss. In contrast, lung tissue from CO-exposed pneumonia animals displayed abundant alveolar type 2 cells with significantly more ATP synthase staining. ATP synthase staining was also more evident in the alveolar macrophages. Taken together, CO exposure during pneumonia is associated with increased mitochondrial mass localized to the alveolar type 2 cells and to alveolar macrophages.
Inhaled CO and sepsis-induced antioxidant defenses in the kidney.
To investigate the systemic effects of inhaled CO during S. pneumoniae pneumonia, we performed Western analysis on kidney sections from control animals, unexposed animals with pneumonia, and CO-exposed animals with pneumonia (Fig. 10A). In contrast to HO-1 expression in the lung, renal HO-1 expression was decreased in all but one of the unexposed pneumonia animals and was preserved in many of the CO-exposed animals; however, this was not statistically significant because of one outlier in the unexposed pneumonia group. And while there was no discernable pattern in Tfam, there was a trend for increased citrate synthase expression in both unexposed and CO-exposed pneumonia animals relative to controls (P < 0.1), suggesting that S. pneumoniae sepsis stimulates renal mitochondrial biogenesis. S. pneumoniae sepsis also induced mitochondrial SOD2 expression which was induced even further by CO exposure (P < 0.05). In total, inhaled CO leads to augmentation of the renal antioxidant defenses during S. pneumoniae sepsis.
Fig. 10.

Inhaled CO and sepsis-induced renal antioxidant defenses. A: kidney tissue was taken from control (gray circles; n = 4), unexposed pneumonia (white circles; n = 5), and CO-exposed pneumonia (black circles; n = 5) animals at necropsy (168 h), homogenized, and total protein was probed by Western blot for Tfam, HO-1, citrate synthase, and SOD2. No discernable pattern was appreciated for Tfam expression. Mean HO-1 expression is similar in control and CO-exposed animals and lower in unexposed pneumonia animals. Both unexposed and CO-exposed pneumonia animals displayed a trend for higher citrate synthase relative to controls, suggestive of sepsis-mediated activation of mitochondrial biogenesis. Sepsis also increased SOD2 expression relative to controls, which was further increased by CO exposure. Protein expression is relative to GAPDH. Bars represent means. #P < 0.1 relative to control, **P < 0.05 relative to control and unexposed pneumonia groups. B: immunofluorescence microscopy of SOD2 (green) and cytochrome c (red) in formalin-fixed kidney sections from control, pneumonia, and CO-exposed pneumonia animals. Compared with controls, SOD2/cytochrome c colocalization (yellow) is decreased in pneumonia animals, indicative of mitochondrial injury and release of cytochrome c into the cytoplasm, whereas colocalization is maintained with inhaled CO, consistent with a mitochondrial protective effect. Original magnification is ×600 for control and PNA and ×400 for PNA + CO.
To further explore the effects of S. pneumoniae sepsis ± CO exposure on renal mitochondrial quality control, we costained kidney sections from each group for SOD2 (green) and cytochrome c (red) and performed immunofluorescence microscopy (Fig. 10B). The colocalization of cytochrome c and SOD2 (illustrated by yellow) in the cortical proximal tubules was significantly less in animals with pneumonia compared with control animals, implying cytochrome c release into the cytoplasm as a consequence of mitochondrial injury. Furthermore, colocalization of cytochrome c and SOD2 was significantly increased in the CO-exposed animals, consistent with attenuated mitochondrial injury and cytochrome c retention in intact mitochondria. In summary, S. pneumoniae sepsis causes in situ renal mitochondrial injury, which is attenuated with low-dose, inhaled CO and linked to the CO-mediated augmentation of renal antioxidant defenses.
DISCUSSION
We have developed and tested 1) a novel, ventilator-compatible CO delivery system that safely administers inhaled CO gas; and 2) a CO dosing strategy utilizing the CFK equation to achieve a COHb level of 6–8% in an established baboon model of S. pneumoniae pneumonia (15, 34, 58). We also found that animals receiving inhaled CO displayed significantly less pneumonia-induced ALI and renal tubular cell mitochondrial injury, which was associated with the activation of lung mitochondrial biogenesis and augmentation of renal antioxidant defenses, respectively. These findings provide proof of principle for testing inhaled CO gas as a therapy in critically ill patients with ARDS.
Translation of inhaled CO therapy into the intensive care unit has been limited in part by the lack of a safe, ventilator-compatible CO delivery system. Preliminary work on CO delivery during mechanical ventilation comprised serial, manual injections of gas into the ventilator circuit, which produced inconsistent CO dosing and, in one animal, an overdose (17). Our device uses automated CO injections and real-time monitoring to deliver a constant concentration of CO, adjusting for fluctuations in flow and minute ventilation. It is connected in series with the ventilator circuit and results in no detectable rise in ambient CO (≤1 ppm). These ambient values are far below the OSHA-permissible exposure limit of 50 ppm as an 8-h time-weighted average (44) and indicate that this device can be safely used in a clinical setting without risk to healthcare providers.
COHb is a readily measurable biomarker for CO uptake that has been consistently utilized in human CO therapy studies (4, 40, 54) including a recently completed clinical trial in idiopathic pulmonary fibrosis (NCT01214187). Although direct exposure of lung epithelial cells to CO gas may evoke lung cytoprotective effects, it seems unlikely that systemic effects (e.g., renal protection) would be achieved without some elevation of COHb and tissue CO levels. We targeted a COHb level of 6–8%, a range that has been previously shown to be safe in humans (28, 40, 42, 46, 52, 53, 63) and consistently achieved this range by administering CO at 200 ppm for 60 min. Whether this regimen will be sufficient to reach the target COHb in patients with ARDS, who may have lower diffusion capacities than the animals in this study, is unknown and will require further clinical study. It is also unknown whether a target COHb of 6–8% will be sufficient to treat ARDS in patients, or whether inhaled CO would be efficacious in the presence of full intensive care therapies such as antibiotics, nutrition, and active fluid and pressor management. Such issues will require further work before proper CO dosing can be established in critically ill patients. We did, however, find that the CFK equation can predict the 60-min COHb level with high accuracy, even in the setting of an injured lung, based on the COHb measured after 20 min of CO exposure. This represents a novel application of the CFK equation, which had previously been used only for physiological or toxicological monitoring (13), and suggests that the dose of CO might be tailored individually to patients with pneumonia- or sepsis-induced ARDS to reach a target COHb level.
Others have found close agreement between COHb levels measured in peripheral venous, central venous, mixed venous, and arterial blood (27, 37, 61). In our study, COHb levels measured from peripheral venous blood were as accurate and reliable as arterial measurements. Overall, these measurements support the use of either venous or arterial blood to monitor COHb levels during inhaled CO treatment. We also demonstrated that COHb measurements made by the AVOXimeter 4000 CO-oximeter were not as accurate as those of a gold standard optical device, the IL 682 CO-oximeter. The reason for this discrepancy is not known, but it was clearly most evident at lower COHb levels, suggesting a low-sensitivity detection method. These findings underscore the need for rigorous quality control standards for devices that measure COHb and have implications for future clinical studies and for patient care.
Prior murine and porcine experiments have often found reduced ALI with inhaled CO (18, 21, 23, 24, 29, 30, 32, 33, 41, 59, 64, 65) and preliminary nonhuman primate (NHP) work in macaques by Mitchell et al. (43) found that peak lung inflammation (e.g., BAL fluid total cell count) following LPS challenge was modestly reduced with inhaled CO. Although our study found no discernable reduction in lung inflammation with inhaled CO, we administered doses lower than Mitchell et al., who similarly found no effect on inflammation at CO doses less than 500 ppm (43). However, we did find that animals given low-dose CO displayed lung protection. Specifically, we found significantly lower histological ALI scores and lung wet-to-dry weight ratios at 168 h, indicative of improved ALI resolution. Although the BAL fluid total protein and LDH activity were not reduced in the CO-exposed animals, this may reflect their inherent variability and dependence on lung sampling and BAL fluid return or could reflect a CO-mediated reduction in lung water, thereby raising alveolar protein and LDH concentrations (6).
Overall, our findings are limited by the lack of CO-only controls, but it is unlikely that administering CO alone to control, uninfected animals would impact our conclusions, as such low-dose CO exposures do not affect markers of lung injury in lower species (18, 30, 32). Another limitation is the variable CO dosing and delivery schedule, which was designed to study CO pharmacokinetics rather than treatment effects. Despite these limitations, our findings do suggest a reduction in the severity of pneumonia-induced ALI, and especially in extravascular lung water, in animals given inhaled CO.
One explanation for this apparent lung protection is the CO-mediated activation of the mitochondrial quality control program. This cytoprotective program includes mitochondrial biogenesis, which is critical for postinfection lung and other organ recovery (1, 3, 8, 10, 26, 38, 57) and is activated by inhaled CO (1, 38, 54). In this study we found evidence supporting the activation of mitochondrial biogenesis in the lungs of CO-exposed animals, including increased expression of the mitochondrial matrix protein citrate synthase. On the basis of the immunofluorescence staining of ATP synthase, another mitochondrial matrix protein, this was localized mainly to alveolar macrophages and alveolar type 2 cells. Alveolar type 2 cells are essential for alveolar epithelial barrier reconstitution after ALI (1, 2), and their activation by CO may explain the reduced edema we found in the CO-exposed animals. Although these animals did not display a statistically significant increase in lung Tfam protein, a mitochondrial DNA-bound transcription factor necessary for mitochondrial biogenesis, the trend for higher Tfam expression in this group relative to controls is suggestive of benefit and may only reflect that the study was underpowered to detect significant differences in these genetically unrelated NHPs. Additionally, we did not find relative HO-1 overexpression in the lung after CO exposure, and, in fact, there was a trend for higher HO-1 expression in the unexposed pneumonia animals. This may reflect ongoing oxidative stress and continued activation of the HO-1 system in the unexposed pneumonia animals, which displayed a different stage of lung injury resolution than did the animals that received CO. Alternatively, the lack of HO-1 increase could indicate that its induction by inhaled CO is short lived. Furthermore, lung HO-1 expression in both pneumonia groups differed markedly from renal HO-1 expression, indicating the organ-specific activation of the HO-1/CO system in response to infection.
Another mechanism for lung protection is CO-mediated augmentation of SPM levels. We have reported that SPMs become acutely dysregulated during pneumonia but are partially restored by administering inhaled CO (15). SPMs actively accelerate the resolution of inflammation by reducing further leukocyte influx, promoting macrophage efferocytosis, restoring epithelial barrier integrity, and enhancing microbial containment (11, 12, 19, 36), and SPM induction provides another plausible mechanism for this apparent lung protection. Taken together, these findings suggest that low-dose CO safely activates cytoprotective programs such as mitochondrial biogenesis and SPM production that can accelerate ALI resolution.
Acute kidney injury is a common complication of pneumonia and sepsis and likely involves mitochondrial injury in the tubular epithelial cells (48). In our study, the renal proximal tubule cells of animals with S. pneumoniae pneumonia and sepsis displayed more cytoplasmic cytochrome c staining than did control animals, suggestive of sepsis-induced mitochondrial injury. However, this injury was significantly attenuated in animals given inhaled CO. Low-dose inhaled CO has known beneficial effects in the kidney. In rodent and porcine studies, CO attenuated acute kidney injury and was linked to activation of antiapoptotic and antioxidant programs (5, 20, 25, 45). One such antioxidant protein is mitochondrial SOD2, which is critical for organ protection during sepsis (10). We found that renal SOD2 expression was stimulated by S. pneumoniae sepsis but was significantly augmented in animals given low-dose inhaled CO. These findings advance prior mouse work to NHPs and suggest that low-dose inhaled CO may be a beneficial therapy for sepsis-induced acute kidney injury.
In summary, we report a novel, ventilator-compatible CO delivery system that safely delivers inhaled CO to ventilated baboons with S. pneumoniae pneumonia and sepsis. We achieved our goal COHb of 6–8% by administering 200 ppm for 60 min and predicted the 60-min COHb level with high accuracy using the CFK equation. Furthermore, we present preliminary evidence of both local and systemic protective effects of inhaled CO, including improved resolution of ALI that was associated with activation of lung mitochondrial biogenesis and a reduction in renal mitochondrial injury that was associated with augmented antioxidant defenses. These results strongly support further testing of the CO delivery system and the CFK equation in a Phase I clinical trial that is currently underway (NCT02425579) and provide proof of principle for testing of inhaled CO as a therapy for critically ill patients.
GRANTS
This work was supported by the Gates Foundation Global Health Institute and the National Institutes of Health grant P01 HL108801.
DISCLOSURES
Dean Hess (Philips Respironics, Bayer, McGraw Hill, Jones and Bartlett, and UpToDate); Alex Stenzler (12th Man Technologies, Proterris); Augustine Choi (Proterris).
AUTHOR CONTRIBUTIONS
L.E.F., B.D.K., D.R.H., K.E.W.-W., and C.A.P. conception and design of research; L.E.F., B.D.K., D.R.H., M.A.W., J.D.D., K.E.W.-W., and C.A.P. performed experiments; L.E.F., B.D.K., D.R.H., R.S.H., M.A.W., H.B.S., V.L.R., T.W., A.S., K.E.W.-W., and C.A.P. analyzed data; L.E.F., B.D.K., D.R.H., R.S.H., H.B.S., V.L.R., T.W., R.M.B., B.T.T., A.M.C., K.E.W.-W., and C.A.P. interpreted results of experiments; L.E.F. and B.D.K. prepared figures; L.E.F. and B.D.K. drafted manuscript; L.E.F., B.D.K., D.R.H., R.S.H., A.S., R.M.B., B.T.T., K.E.W.-W., and C.A.P. edited and revised manuscript; L.E.F., B.D.K., D.R.H., R.S.H., M.A.W., H.B.S., V.L.R., J.D.D., T.W., A.S., R.M.B., B.T.T., A.M.C., K.E.W.-W., and C.A.P. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Paul Nuccio, Aaron Walker, Sonny Boso, Craig Marshall, Emeka Ifedigbo, and Olga Better for technical assistance.
Glossary
- A
Pco2/M[HbO2]
- B
1/DlCO + Pl/V̇a
- DlCO
Diffusivity of the lung for CO, ml·min−1·mmHg−1
- exp(x)
Exponential function ex, where e is the base of the natural logarithm
- FiO2
Inspired O2 fraction
- [HbO2]
O2 volume per milliliter of blood, ml
- [HbCO]t
CO volume per milliliter of blood at time t, ml
- [HbCO]0
CO volume per milliliter of blood at the beginning of the exposure interval, ml
- M
Ratio of the affinity of blood for CO to that for O2
- Pb
Barometric pressure
- Pco2
Average partial pressure of oxygen in lung capillaries, mmHg
- PiCO
Partial pressure of CO in the inhaled air, mmHg
- Pl
Barometric pressure minus the vapor pressure of water at body temperature, mmHg
- t
Exposure duration, min
- V̇a
Alveolar ventilation, l/m
- Vb
Blood volume, ml
- V̇co
Rate of endogenous CO production, ml/min
- Vd/Vt
Ratio of dead space to tidal volume
- V̇e
Minute ventilation, l/min
REFERENCES
- 1.Athale J, Ulrich A, Chou Macgarvey N, Bartz RR, Welty-Wolf KE, Suliman HB, Piantadosi CA. Nrf2 promotes alveolar mitochondrial biogenesis and resolution of lung injury in Staphylococcus aureus pneumonia in mice. Free Radic Biol Med 53: 1584–1594, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barkauskas CE, Cronce MJ, Rackley CR, Bowie EJ, Keene DR, Stripp BR, Randell SH, Noble PW, Hogan BL. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest 123: 3025–3036, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bartz RR, Fu P, Suliman HB, Crowley SD, MacGarvey NC, Welty-Wolf K, Piantadosi CA. Staphylococcus aureus sepsis induces early renal mitochondrial DNA repair and mitochondrial biogenesis in mice. PloS One 9: e100912, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bathoorn E, Slebos DJ, Postma DS, Koeter GH, van Oosterhout AJ, van der Toorn M, Boezen HM, Kerstjens HA. Anti-inflammatory effects of inhaled carbon monoxide in patients with COPD: a pilot study. Eur Respir J 30: 1131–1137, 2007. [DOI] [PubMed] [Google Scholar]
- 5.Bernhardt WM, Campean V, Kany S, Jurgensen JS, Weidemann A, Warnecke C, Arend M, Klaus S, Gunzler V, Amann K, Willam C, Wiesener MS, Eckardt KU. Preconditional activation of hypoxia-inducible factors ameliorates ischemic acute renal failure. J Am Soc Nephrol 17: 1970–1978, 2006. [DOI] [PubMed] [Google Scholar]
- 6.Berthiaume Y, Broaddus VC, Gropper MA, Tanita T, Matthay MA. Alveolar liquid and protein clearance from normal dog lungs. J Appl Physiol 65: 585–593, 1988. [DOI] [PubMed] [Google Scholar]
- 7.Brouard S, Otterbein LE, Anrather J, Tobiasch E, Bach FH, Choi AM, Soares MP. Carbon monoxide generated by heme oxygenase 1 suppresses endothelial cell apoptosis. J Exp Med 192: 1015–1026, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carraway MS, Suliman HB, Kliment C, Welty-Wolf KE, Oury TD, Piantadosi CA. Mitochondrial biogenesis in the pulmonary vasculature during inhalational lung injury and fibrosis. Antioxid Redox Signal 10: 269–275, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cevey-Macherel M, Galetto-Lacour A, Gervaix A, Siegrist CA, Bille J, Bescher-Ninet B, Kaiser L, Krahenbuhl JD, Gehri M. Etiology of community-acquired pneumonia in hospitalized children based on WHO clinical guidelines. Eur J Pediatr 168: 1429–1436, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cherry AD, Suliman HB, Bartz RR, Piantadosi CA. Peroxisome proliferator-activated receptor gamma co-activator 1-alpha as a critical co-activator of the murine hepatic oxidative stress response and mitochondrial biogenesis in Staphylococcus aureus sepsis. J Biol Chem 289: 41–52, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chiang N, Fredman G, Backhed F, Oh SF, Vickery T, Schmidt BA, Serhan CN. Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature 484: 524–528, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chiang N, Shinohara M, Dalli J, Mirakaj V, Kibi M, Choi AM, Serhan CN. Inhaled carbon monoxide accelerates resolution of inflammation via unique proresolving mediator-heme oxygenase-1 circuits. J Immunol 190: 6378–6388, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coburn RF. Carbon monoxide uptake and excretion: testing assumptions made in deriving the Coburn-Forster-Kane equation. Respir Physiol Neurobiol 187: 224–233, 2013. [DOI] [PubMed] [Google Scholar]
- 14.Coburn RF, Forster RE, Kane PB. Considerations of the physiological variables that determine the blood carboxyhemoglobin concentration in man. J Clin Invest 44: 1899–1910, 1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dalli J, Kraft BD, Colas RA, Shinohara M, Fredenburgh LE, Hess DR, Chiang N, Welty-Wolf KE, Choi AM, Piantadosi CA, Serhan CN. Proresolving lipid mediator profiles in baboon pneumonia are regulated by inhaled carbon monoxide. Am J Respir Cell Mol Biol. 2015 Jan 8. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dellinger RP, Levy MM, Rhodes A, Annane D, Gerlach H, Opal SM, Sevransky JE, Sprung CL, Douglas IS, Jaeschke R, Osborn TM, Nunnally ME, Townsend SR, Reinhart K, Kleinpell RM, Angus DC, Deutschman CS, Machado FR, Rubenfeld GD, Webb SA, Beale RJ, Vincent JL, Moreno R; Surviving Sepsis Campaign Guidelines Committee including the Pediatric Subgroup. Surviving sepsis campaign: international guidelines for management of severe sepsis and septic shock: 2012. Crit Care Med 41: 580–637, 2013. [DOI] [PubMed] [Google Scholar]
- 17.Dingley J, Foex BA, Swart M, Findlay G, DeSouza PR, Wardrop C, Willis N, Smithies M, Little RA. Blood volume determination by the carbon monoxide method using a new delivery system: accuracy in critically ill humans and precision in an animal model. Crit Care Med 27: 2435–2441, 1999. [DOI] [PubMed] [Google Scholar]
- 18.Dolinay T, Szilasi M, Liu M, Choi AM. Inhaled carbon monoxide confers antiinflammatory effects against ventilator-induced lung injury. Am J Respir Crit Care Med 170: 613–620, 2004. [DOI] [PubMed] [Google Scholar]
- 19.Eickmeier O, Seki H, Haworth O, Hilberath JN, Gao F, Uddin M, Croze RH, Carlo T, Pfeffer MA, Levy BD. Aspirin-triggered resolvin D1 reduces mucosal inflammation and promotes resolution in a murine model of acute lung injury. Mucosal Immunol 6: 256–266, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Faleo G, Neto JS, Kohmoto J, Tomiyama K, Shimizu H, Takahashi T, Wang Y, Sugimoto R, Choi AM, Stolz DB, Carrieri G, McCurry KR, Murase N, Nakao A. Carbon monoxide ameliorates renal cold ischemia-reperfusion injury with an upregulation of vascular endothelial growth factor by activation of hypoxia-inducible factor. Transplantation 85: 1833–1840, 2008. [DOI] [PubMed] [Google Scholar]
- 21.Faller S, Foeckler M, Strosing KM, Spassov S, Ryter SW, Buerkle H, Loop T, Schmidt R, Hoetzel A. Kinetic effects of carbon monoxide inhalation on tissue protection in ventilator-induced lung injury. Lab Invest 92: 999–1012, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fredenburgh LE, Perrella MA, Mitsialis SA. The role of heme oxygenase-1 in pulmonary disease. Am J Respir Cell Mol Biol 36: 158–165, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fujita T, Toda K, Karimova A, Yan SF, Naka Y, Yet SF, Pinsky DJ. Paradoxical rescue from ischemic lung injury by inhaled carbon monoxide driven by derepression of fibrinolysis. Nat Med 7: 598–604, 2001. [DOI] [PubMed] [Google Scholar]
- 24.Goebel U, Siepe M, Mecklenburg A, Stein P, Roesslein M, Schwer CI, Schmidt R, Doenst T, Geiger KK, Pahl HL, Schlensak C, Loop T. Carbon monoxide inhalation reduces pulmonary inflammatory response during cardiopulmonary bypass in pigs. Anesthesiology 108: 1025–1036, 2008. [DOI] [PubMed] [Google Scholar]
- 25.Goebel U, Siepe M, Schwer CI, Schibilsky D, Foerster K, Neumann J, Wiech T, Priebe HJ, Schlensak C, Loop T. Inhaled carbon monoxide prevents acute kidney injury in pigs after cardiopulmonary bypass by inducing a heat shock response. Anesth Analg 111: 29–37, 2010. [DOI] [PubMed] [Google Scholar]
- 26.Haden DW, Suliman HB, Carraway MS, Welty-Wolf KE, Ali AS, Shitara H, Yonekawa H, Piantadosi CA. Mitochondrial biogenesis restores oxidative metabolism during Staphylococcus aureus sepsis. Am J Respir Crit Care Med 176: 768–777, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hampson NB, Piantadosi CA, Thom SR, Weaver LK. Practice recommendations in the diagnosis, management, and prevention of carbon monoxide poisoning. Am J Respir Crit Care Med 186: 1095–1101, 2012. [DOI] [PubMed] [Google Scholar]
- 28.Hausberg M, Somers VK. Neural circulatory responses to carbon monoxide in healthy humans. Hypertension 29: 1114–1118, 1997. [DOI] [PubMed] [Google Scholar]
- 29.Hoetzel A, Dolinay T, Vallbracht S, Zhang Y, Kim HP, Ifedigbo E, Alber S, Kaynar AM, Schmidt R, Ryter SW, Choi AM. Carbon monoxide protects against ventilator-induced lung injury via PPAR-gamma and inhibition of Egr-1. Am J Respir Crit Care Med 177: 1223–1232, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kanagawa F, Takahashi T, Inoue K, Shimizu H, Omori E, Morimatsu H, Maeda S, Katayama H, Nakao A, Morita K. Protective effect of carbon monoxide inhalation on lung injury after hemorrhagic shock/resuscitation in rats. J Trauma 69: 185–194, 2010. [DOI] [PubMed] [Google Scholar]
- 31.Kim HP, Ryter SW, Choi AM. CO as a cellular signaling molecule. Annu Rev Pharmacol Toxicol 46: 411–449, 2006. [DOI] [PubMed] [Google Scholar]
- 32.Kim HP, Wang X, Zhang J, Suh GY, Benjamin IJ, Ryter SW, Choi AM. Heat shock protein-70 mediates the cytoprotective effect of carbon monoxide: involvement of p38 beta MAPK and heat shock factor-1. J Immunol 175: 2622–2629, 2005. [DOI] [PubMed] [Google Scholar]
- 33.Kohmoto J, Nakao A, Kaizu T, Tsung A, Ikeda A, Tomiyama K, Billiar TR, Choi AM, Murase N, McCurry KR. Low-dose carbon monoxide inhalation prevents ischemia/reperfusion injury of transplanted rat lung grafts. Surgery 140: 179–185, 2006. [DOI] [PubMed] [Google Scholar]
- 34.Kraft BD, Piantadosi CA, Benjamin AM, Lucas JE, Zaas AK, Betancourt-Quiroz M, Woods CW, Chang AL, Roggli VL, Marshall CD, Ginsburg GS, Welty-Wolf K. Development of a novel preclinical model of pneumococcal pneumonia in nonhuman primates. Am J Respir Cell Mol Biol 50: 995–1004, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee S, Lee SJ, Coronata AA, Fredenburgh LE, Chung SW, Perrella MA, Nakahira K, Ryter SW, Choi AM. Carbon monoxide confers protection in sepsis by enhancing beclin 1-dependent autophagy and phagocytosis. Antioxid Redox Signal 20: 432–442, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Levy BD, Serhan CN. Resolution of acute inflammation in the lung. Annu Rev Physiol 76: 467–492, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lopez DM, Weingarten-Arams JS, Singer LP, Conway EE Jr. Relationship between arterial, mixed venous, and internal jugular carboxyhemoglobin concentrations at low, medium, and high concentrations in a piglet model of carbon monoxide toxicity. Crit Care Med 28: 1998–2001, 2000. [DOI] [PubMed] [Google Scholar]
- 38.MacGarvey NC, Suliman HB, Bartz RR, Fu P, Withers CM, Welty-Wolf KE, Piantadosi CA. Activation of mitochondrial biogenesis by heme oxygenase-1-mediated NF-E2-related factor-2 induction rescues mice from lethal Staphylococcus aureus sepsis. Am J Respir Crit Care Med 185: 851–861, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matute-Bello G, Downey G, Moore BB, Groshong SD, Matthay MA, Slutsky AS, Kuebler WM; Acute Lung Injury in Animals Study Group. An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol 44: 725–738, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mayr FB, Spiel A, Leitner J, Marsik C, Germann P, Ullrich R, Wagner O, Jilma B. Effects of carbon monoxide inhalation during experimental endotoxemia in humans. Am J Respir Crit Care Med 171: 354–360, 2005. [DOI] [PubMed] [Google Scholar]
- 41.Mazzola S, Forni M, Albertini M, Bacci ML, Zannoni A, Gentilini F, Lavitrano M, Bach FH, Otterbein LE, Clement MG. Carbon monoxide pretreatment prevents respiratory derangement and ameliorates hyperacute endotoxic shock in pigs. FASEB J 19: 2045–2047, 2005. [DOI] [PubMed] [Google Scholar]
- 42.McFarland RA. Low level exposure to carbon monoxide and driving performance. Arch Environ Health 27: 355–359, 1973. [DOI] [PubMed] [Google Scholar]
- 43.Mitchell LA, Channell MM, Royer CM, Ryter SW, Choi AM, McDonald JD. Evaluation of inhaled carbon monoxide as an anti-inflammatory therapy in a nonhuman primate model of lung inflammation. Am J Physiol Lung Cell Mol Physiol 299: L891–L897, 2010. [DOI] [PubMed] [Google Scholar]
- 44.National Institute for Occupational Safety and Health Administration. Carbon Monoxide (by COHb) (Online). https://www.osha.gov/dts/chemicalsampling/data/CH_225610.html [Updated September 6, 2012. Accessed September 7, 2015]. [Google Scholar]
- 45.Neto JS, Nakao A, Kimizuka K, Romanosky AJ, Stolz DB, Uchiyama T, Nalesnik MA, Otterbein LE, Murase N. Protection of transplant-induced renal ischemia-reperfusion injury with carbon monoxide. Am J Physiol Renal Physiol 287: F979–F989, 2004. [DOI] [PubMed] [Google Scholar]
- 46.O'Donnell RD, Mikulka P, Heinig P, Theodore J. Low level carbon monoxide exposure and human psychomotor performance. Toxicol Appl Pharmacol 18: 593–602, 1971. [DOI] [PubMed] [Google Scholar]
- 47.Otterbein LE, Bach FH, Alam J, Soares M, Tao Lu H, Wysk M, Davis RJ, Flavell RA, Choi AM. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med 6: 422–428, 2000. [DOI] [PubMed] [Google Scholar]
- 48.Parikh SM. Therapeutic targeting of the mitochondrial dysfunction in septic acute kidney injury. Curr Opin Crit Care 19: 554–559, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Peterson JE, Stewart RD. Predicting the carboxyhemoglobin levels resulting from carbon monoxide exposures. J Appl Physiol 39: 633–638, 1975. [DOI] [PubMed] [Google Scholar]
- 50.Piantadosi CA, Carraway MS, Babiker A, Suliman HB. Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circ Res 103: 1232–1240, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Piantadosi CA, Withers CM, Bartz RR, MacGarvey NC, Fu P, Sweeney TE, Welty-Wolf KE, Suliman HB. Heme oxygenase-1 couples activation of mitochondrial biogenesis to anti-inflammatory cytokine expression. J Biol Chem 286: 16374–16385, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Raub JA, Benignus VA. Carbon monoxide and the nervous system. Neurosci Biobehav Rev 26: 925–940, 2002. [DOI] [PubMed] [Google Scholar]
- 53.Ren X, Dorrington KL, Robbins PA. Respiratory control in humans after 8 h of lowered arterial Po2, hemodilution, or carboxyhemoglobinemia. J Appl Physiol 90: 1189–1195, 2001. [DOI] [PubMed] [Google Scholar]
- 54.Rhodes MA, Carraway MS, Piantadosi CA, Reynolds CM, Cherry AD, Wester TE, Natoli MJ, Massey EW, Moon RE, Suliman HB. Carbon monoxide, skeletal muscle oxidative stress, and mitochondrial biogenesis in humans. Am J Physiol Heart Circ Physiol 297: H392–H399, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rodkey FL, O'Neal JD, Collison HA. Oxygen and carbon monoxide equilibria of human adult hemoglobin at atmospheric and elevated pressure. Blood 33: 57–65, 1969. [PubMed] [Google Scholar]
- 56.Said MA, Johnson HL, Nonyane BAS, Deloria-Knoll M, O′Brien KL; AGEDD Adult Pneumococcal Burden Study Team, Andreo F, Beovic B, Blanco S, Boersma WG, Boulware DR, Butler JC, Carratalà J, Chang FY, Charles PG, Diaz AA, Domínguez J, Ehara N, Endeman H, Falcó V, Falguera M, Fukushima K, Garcia-Vidal C, Genne D, Guchev IA, Gutierrez F, Hernes SS, Hoepelman AI, Hohenthal U, Johansson N, Kolek V, Kozlov RS, Lauderdale TL, Mareković I, Masiá M, Matta MA, Miró Ò, Murdoch DR, Nuermberger E, Paolini R, Perelló R, Snijders D, Pleèko V, Sordé R, Strålin K, van der Eerden MM, Vila-Corcoles A, Watt JP. Estimating the burden of pneumococcal pneumonia among adults: a systematic review and meta-analysis of diagnostic techniques. PloS One 8: e60273, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schumacker PT, Gillespie MN, Nakahira K, Choi AM, Crouser ED, Piantadosi CA, Bhattacharya J. Mitochondria in lung biology and pathology: more than just a powerhouse. Am J Physiol Lung Cell Mol Physiol 306: L962–L974, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shinohara M, Kibi M, Riley IR, Chiang N, Dalli J, Kraft BD, Piantadosi CA, Choi AM, Serhan CN. Cell-cell interactions and bronchoconstrictor eicosanoid reduction with inhaled carbon monoxide and resolvin D1. Am J Physiol Lung Cell Mol Physiol 307: L746–L757, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Song R, Kubo M, Morse D, Zhou Z, Zhang X, Dauber JH, Fabisiak J, Alber SM, Watkins SC, Zuckerbraun BS, Otterbein LE, Ning W, Oury TD, Lee PJ, McCurry KR, Choi AM. Carbon monoxide induces cytoprotection in rat orthotopic lung transplantation via anti-inflammatory and anti-apoptotic effects. Am J Pathol 163: 231–242, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tenhunen R, Marver HS, Schmid R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc Natl Acad Sci USA 61: 748–755, 1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Touger M, Gallagher EJ, Tyrell J. Relationship between venous and arterial carboxyhemoglobin levels in patients with suspected carbon monoxide poisoning. Ann Emerg Med 25: 481–483, 1995. [DOI] [PubMed] [Google Scholar]
- 62.Welty-Wolf KE, Carraway MS, Ortel TL, Ghio AJ, Idell S, Egan J, Zhu X, Jiao JA, Wong HC, Piantadosi CA. Blockade of tissue factor-factor X binding attenuates sepsis-induced respiratory and renal failure. Am J Physiol Lung Cell Mol Physiol 290: L21–L31, 2006. [DOI] [PubMed] [Google Scholar]
- 63.Zevin S, Saunders S, Gourlay SG, Jacob P, Benowitz NL. Cardiovascular effects of carbon monoxide and cigarette smoking. J Am Coll Cardiol 38: 1633–1638, 2001. [DOI] [PubMed] [Google Scholar]
- 64.Zhang X, Shan P, Alam J, Davis RJ, Flavell RA, Lee PJ. Carbon monoxide modulates Fas/Fas ligand, caspases, and Bcl-2 family proteins via the p38alpha mitogen-activated protein kinase pathway during ischemia-reperfusion lung injury. J Biol Chem 278: 22061–22070, 2003. [DOI] [PubMed] [Google Scholar]
- 65.Zhou H, Liu J, Pan P, Jin D, Ding W, Li W. Carbon monoxide inhalation decreased lung injury via anti-inflammatory and anti-apoptotic effects in brain death rats. Exp Biol Med (Maywood) 235: 1236–1243, 2010. [DOI] [PubMed] [Google Scholar]