

Abstract
Inflammation is a protective response to injury, but it can become chronic, leading to tissue damage and disease. Cigarette smoke causes multiple inflammatory diseases, which account for thousands of deaths and cost billions of dollars annually. Cigarette smoke disrupts the function of immune cells, such as macrophages, by prolonging inflammatory signaling, promoting oxidative stress, and impairing phagocytosis, contributing to increased incidence of infections. Recently, new families of lipid-derived mediators, “specialized proresolving mediators” (SPMs), were identified. SPMs play a critical role in the active resolution of inflammation by counterregulating proinflammatory signaling and promoting resolution pathways. We have identified dysregulated concentrations of lipid mediators in exhaled breath condensate, bronchoalveolar lavage fluid, and serum from patients with chronic obstructive pulmonary disease (COPD). In human alveolar macrophages from COPD and non-COPD patients, D-series resolvins decreased inflammatory cytokines and enhanced phagocytosis. To further investigate the actions of resolvins on human cells, macrophages were differentiated from human blood monocytes and treated with D-series resolvins and then exposed to cigarette smoke extract. Resolvins significantly suppressed macrophage production of proinflammatory cytokines, enzymes, and lipid mediators. Resolvins also increased anti-inflammatory cytokines, promoted an M2 macrophage phenotype, and restored cigarette smoke-induced defects in phagocytosis, highlighting the proresolving functions of these molecules. These actions were receptor-dependent and involved modulation of canonical and noncanonical NF-κB expression, with the first evidence for SPM action on alternative NF-κB signaling. These data show that resolvins act on human macrophages to attenuate cigarette smoke-induced inflammatory effects through proresolving mechanisms and provide new evidence of the therapeutic potential of SPMs.
Keywords: macrophage, resolvin, specialized proresolving mediators, cigarette smoke, chronic obstructive pulmonary disease
cigarette smoke is the leading cause of preventable death, accounting for one in five deaths in the United States and nearly six million deaths annually worldwide, with mortality rates rising (1). Exposure to cigarette smoke causes many diseases, including chronic obstructive pulmonary disease (COPD), comprising chronic bronchitis and emphysema, and an increased susceptibility to bacterial infections, such as those from nontypeable Haemophilus influenzae and Streptococcus pneumoniae (42, 72). Chronic inflammation underlies most cigarette smoke-induced diseases. In particular, chronic activation of macrophages by cigarette smoke promotes tissue destruction and can lead to COPD (60, 72). Macrophages produce cytokines that stimulate excess mucus production and lead to chronic bronchitis. Additionally, macrophages are increased in emphysematous lungs and exhibit increased proteinase activity, reactive oxygen species (ROS) production, and secretion of inflammatory cytokines (60, 72). Despite this chronic inflammatory activation, patients with COPD are also more susceptible to bacterial and viral infections (42, 65) due, at least in part, to an impairment of macrophage phagocytic abilities; these defects in phagocytosis also lead to impaired clearance of apoptotic cells (20, 33, 38, 43, 47). Clearly, the underlying inflammatory mechanisms involved in cigarette smoke exposure and the progression of COPD are complex and inadequately addressed by the current standard treatments, which primarily involve bronchodilators and immunosuppressive steroids.
The resolution of inflammation was thought to be passive. However, it is now known that resolution of inflammation is an active and dynamic process (7). Recent investigations have led to the discovery of specialized proresolving mediators (SPMs). These bioactive lipid mediators, endogenously produced, play a critical role in the active resolution of inflammation by counterregulating proinflammatory actions and promoting resolution pathways and are not immunosuppressive (7, 12, 63). SPMs are formed by enzymatic oxygenation of polyunsaturated fatty acids. They are divided into families, including lipoxins (Lx), resolvins (Rv), protectins, and maresins, on the basis of their metabolic pathway and structures (7). These small molecules are amenable to modification and act via unique receptors, including LxA4 (ALX) receptor and G protein-coupled receptor (GPCR) 32 (GPR32), and new modes of action that give them potential as novel therapeutics (2, 4, 11, 13, 22, 34, 36). Several studies have shown that SPMs are dysregulated in human diseases, and several chronic inflammatory diseases are hypothesized to be a result of failure to resolve. There is a large and important knowledge gap regarding the role of SPMs in COPD and whether SPMs can attenuate the effects of cigarette smoke on human macrophages, as well as the effect of SPMs on human macrophage function in general.
SPMs mediate some of their key actions through modulation of inflammatory signaling pathways, including the mitogen-activated protein kinase (MAPK) and nuclear factor-κ-light-chain enhancer of activated B cells (NF-κB) families (2, 3, 50, 55, 57, 73). NF-κB proteins are involved in a number of cellular responses and are particularly important in promotion and regulation of inflammation (18). Both canonical and noncanonical NF-κB pathways exist, and several members of the alternative NF-κB signaling pathway, specifically RelB, possess anti-inflammatory abilities (5, 18, 68, 70). The actions of SPMs on these signaling pathways in cigarette smoke-exposed cells, and in human cells in general, are of considerable interest, and study of these mechanisms would provide important new insight into the actions of SPMs. In the present study we tested the hypothesis that SPMs attenuate cigarette smoke-induced inflammation via their proresolving and anti-inflammatory actions on human macrophages.
MATERIALS AND METHODS
Materials.
PGE2, PGD2, TxB2, and RvD1 enzyme immunoassay (EIA) kits and all SPMs were purchased from Cayman Chemical (Ann Arbor, MI); antibodies to RelB (catalog no. 4954S), p65 (catalog no. 4764), phosphorylated p65 (catalog no. 3033P), IκBα (catalog no. 4814), p100/p52 (catalog no. 3017), and β-tubulin (catalog no. 2146) from Cell Signaling (Danvers, MA); the ALX/FPR2-specific antagonist Boc-2 from GenScript (Piscataway, NJ); a GPR32-neutralizing antibody (catalog no. GX71225) from GeneTex (Irvine, CA); antibodies to CD11b (catalog no. 560914) and CD14 (catalog no. 555398) and ELISA components for IL-6 (catalog nos. 554543 and 554546) and TNF-α (catalog no. 555212) from BD Biosciences (San Jose, CA); ELISA antibodies for IL-8 (catalog nos. M-801 and M-802-B) from Endogen (Farmingdale, NY); IL-10 ELISA kit (catalog no. 430603) from BioLegend (San Diego, CA); transforming growth factor (TGF)-β ELISA kit (catalog no. DY240) and granulocyte-macrophage colony-stimulating factor (catalog no. 9023305) from R & D Systems (Minneapolis, MN); HO-1 antibody (catalog no. OSA-110) from StressGen (Farmingdale, NY); actin antibody (catalog no. CP-01) from Calbiochem (Darmstadt, Germany); secondary Western blot antibodies (catalog nos. 115-035-146 and 111-035-144) from Jackson Laboratories (Bar Harbor, ME); PBS (catalog no. 14200-075) and RPMI 1640 medium (catalog no. 11875-119) from Gibco (Waltham, MA); and FBS (catalog no. SH30070.03HI) from Hyclone (Pittsburgh, PA).
Assessment of lipid mediator profiles in human samples.
Exhaled breath condensate (EBC), bronchoalveolar lavage fluid (BALF), and human blood and serum were obtained from male and female volunteer donors with informed written consent as approved by the University of Rochester Institutional Review Board and Office for Human Subjects Protection. For EBCs, human volunteer breath was collected in a cold trap for 15 min, separated into 250-μl aliquots, and frozen at −80°C. BALF was obtained from COPD and non-COPD patients undergoing bronchoscopy at the University of Rochester Medical Center. All non-COPD patients are nonsmokers; COPD patients included current and ex-smokers. BALF was strained through sterile gauze to remove mucus and then centrifuged for 5 min at 425 g to pellet cells. Lipid mediator concentrations were determined by mass spectrometry as described by Colas et al. (15) or by EIA according to the manufacturer's protocol [20% cross-reactivity with LxA4, 4.2% cross-reactivity with 17(R)-RvD1, and <1% reactivity for all other SPMs tested].
Human monocyte and macrophage isolation.
Human peripheral blood mononuclear cells were obtained as previously described from male and female healthy, nonsmoker donors (56). Monocytes were purified by incubation with CD14-conjugated DynaBeads according to the manufacturer's protocol (catalog no. 113-67D, Invitrogen, Grand Island, NY). For induction of macrophage differentiation, monocytes were cultured in 1% FBS (to minimize SPM binding by serum components), antimycotic/antimyotic-supplemented RPMI 1640 medium and 10 ng/ml granulocyte-macrophage colony-stimulating factor for 7 days. Blood-derived macrophages were plated in 12-well plates at 1 × 106 cells in serum-free RPMI 1640 medium for treatments unless otherwise described.
BALF was obtained as described above, and the cell pellet was washed with PBS and resuspended in RPMI 1640 medium. Alveolar macrophages were purified by adhesion.
Cigarette smoke extract preparation.
Cigarette smoke extract (CSE) was prepared in RPMI 1640 medium as previously described by our lab (30). CSE was diluted to 30 U (U/ml) unless otherwise described. Serum-free RPMI 1640 medium was used as a vehicle control.
ELISA and EIA.
Macrophages were incubated with SPMs at 1–100 nM or vehicle (0.1% ethanol in PBS) for 24 h and then exposed to CSE. Supernatants were removed 24 h after CSE exposure, and cytokines and lipid concentrations were determined by ELISA (IL-6, IL-8, IL-10, TNF-α, and TGF-β) or EIA (PGE2, PGD2, and TxB2) according to the manufacturer's protocols. Viability of the cells was determined by Trypan blue exclusion. For alveolar macrophages, cells were incubated with SPMs for 24 h; then supernatants were removed and assayed for inflammatory mediators by ELISA.
Western blot and OxyBlot analysis.
Macrophages were treated as described above. Cells were washed twice with 1× PBS and lysed with CW buffer (50 mM Tris·HCl and 2% SDS). Total protein was quantified by bicinchoninic acid assay (Thermo Scientific, Waltham, MA). Western blot analysis was performed as described previously (40). For OxyBlot analysis, samples were derivatized with a 1× 2,4-dinitrophenylhydrazine solution; then a neutralization buffer was added, and carbonylated proteins were detected according to the manufacturer's protocol (Millipore, Darmstadt, Germany).
Quantification of ROS.
Macrophages were incubated with 1–100 nM SPM or vehicle (0.01% ethanol in PBS) for 1 h and then with 1 μM 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA; Invitrogen, Waltham, MA) for 30 min. The cells were washed and exposed to CSE. ROS were quantified by flow cytometry.
Determination of macrophage phagocytosis.
Macrophages were treated with SPMs or vehicle (0.01% ethanol in PBS) for 1 h and then exposed to CSE for 2 h. Cells were washed and incubated with FITC-labeled Escherichia coli (Molecular Probes, Waltham, MA) for 1 h at a 1:5 cell-to-E. coli ratio. Cells were detached from plates using a release buffer (PBS with 0.5 mM EDTA) and resuspended in phosphate-azide-bovine serum albumin buffer with Trypan blue to quench extracellular fluorescence. Macrophages were washed with phosphate-azide-bovine serum albumin buffer, fixed with 4% paraformaldehyde, and stained with anti-CD14 and anti-CD11b antibodies. The percentage of E. coli-positive macrophages and E. coli mean fluorescence intensity were assessed by flow cytometry.
Inhibition of SPM receptors.
Macrophages were treated with Boc-2 (1 μM) or anti-GPR32 (10 μg/ml) for 30 min prior to SPM treatment and CSE exposure as described above. Specificity for Boc-2 and anti-GPR32 has been established previously (31, 45, 67). Supernatants were removed, and lysates were collected. ELISA and EIA were performed as described above.
Statistical analysis.
Values are means ± SE. Statistical analyses on normally distributed data were performed using a t-test or one- or two-way ANOVA with Bonferroni's posttest correction for multiple comparisons using GraphPad Prism software (San Diego, CA).
RESULTS
COPD patients have dysregulated lipid mediator levels.
The chronic inflammatory causes of COPD can be viewed as a failure to resolve inflammation. To explore this idea, we first investigated the levels of proresolving lipid mediators in COPD and non-COPD human EBC and BALF. Using lipid-mediator metabolipidomics (15), we identified, by matching criteria and MS/MS spectra, bioactive lipid mediator, RvD1, and pathway markers 17-hydroxydocosahexaenoic acid (17-HDHA) and 14-HDHA in healthy and COPD EBCs (Fig. 1A, Table 1). Increases in HDHAs (14-HDHA and 17-HDHA) and hydroxyeicosatetraenoic acids (12-HETE and 15-HETE) from arachidonic acid were also identified in BALF of COPD patients, with hydroxyeicosapentaenoic acids (5-HEPE and 12-HEPE) trending toward an increase that was not statistically significant (Fig. 1, B–D). Additionally, RvD1 concentration was decreased in BALF and serum of COPD patients (Fig. 1, E and F). LxA4, a potential confounder due to cross-reactivity in the ELISA, was not detected in our BALF and has been found at extremely low levels in human serum, making the likelihood of cross-reactivity very low (52, 53). This is the first evidence of dysregulated levels of the ω-3-derived SPMs in COPD patients.
Fig. 1.

Lipid mediators are dysregulated in patients with chronic obstructive pulmonary disease (COPD). Lipid mediators were identified in exhaled breath condensates (EBCs), as shown with representative multiple-reaction monitoring, along with MS-MS spectra used for identification of D-series resolvin (RvD1, A), and in bronchoalveolar lavage fluid (BALF, B–D) of COPD and non-COPD or healthy human subjects by lipid-mediator metabolipidomics. COPD patients had increased concentrations of the pathway markers 17-hydroxydocosahexaenoic acid (17-HDHA) and 14-HDHA and 12-hydroxyeicosatetraenoic acid (12-HETE) and 15-HETE in BALF, as well as other dysregulated lipid signaling. E and F: enzyme immunoassay of RvD1 concentration in BALF and serum. Values in B–D are means ± SE (n = 3–6). Each symbol in E and F represents an individual donor. Dashed arrows in A represent retention time of compounds below the limit of detection, which is ∼0.1 pg. m/z, Mass-to-charge ratio. *P < 0.05 vs. non-COPD (by t-test).
Table 1.
Lipid mediators and pathway markers levels in human exhaled breath condensate from healthy volunteers and COPD patients
| Lipid Mediator Levels, pg/ml |
||||
|---|---|---|---|---|
| Q1 | Q3 | Healthy volunteers (n = 7) | COPD patients (n = 10) | |
| DHA bioactive metabolome and pathway markers | ||||
| RvD1 | 375 | 141 | 14.3 ± 9.2 | 11.8 ± 4.6 |
| RvD2 | 375 | 141 | * | * |
| RvD3 | 375 | 147 | * | * |
| RvD5 | 359 | 199 | * | * |
| RvD6 | 359 | 159 | * | * |
| PD1 | 359 | 153 | * | * |
| MaR1 | 359 | 250 | * | * |
| 17-HDHA | 343 | 245 | 2.8 ± 1.1 | 4.8 ± 2.6 |
| 14-HDHA | 343 | 205 | 45.0 ± 17.8 | 39.7 ± 8.5 |
| 7-HDHA | 343 | 141 | 7.5 ± 0.9 | 12.5 ± 3.6 |
| 4-HDHA | 343 | 101 | 29.5 ± 4.6 | 35.9 ± 17.1 |
| DHA | 327 | 283 | 307.0 ± 29.3 | 1,271.2 ± 525.0† |
| EPA bioactive metabolome and pathway markers | ||||
| RvE1 | 349 | 195 | * | * |
| RvE2 | 333 | 199 | * | * |
| RvE3 | 333 | 201 | * | * |
| 18-HEPE | 317 | 259 | * | * |
| 15-HEPE | 317 | 219 | * | * |
| 12-HEPE | 317 | 179 | 11.3 ± 1.4 | 4.4 ± 1.6‡ |
| 5-HEPE | 317 | 115 | 6.3 ± 2.9 | 2.4 ± 1.1 |
| EPA | 301 | 257 | 142.0 ± 18.5 | 205.0 ± 60.4 |
| AA bioactive metabolome and pathway markers | ||||
| LXA4 | 235 | * | * | |
| LXB4 | 351 | 221 | 53.2 ± 15.4 | 18.8 ± 7.7† |
| 5(S),15(S)-diHETE | 335 | 235 | * | * |
| LTB4 | 335 | 195 | * | 0.8 ± 0.8 |
| 20-OH-LTB4 | 351 | 195 | * | * |
| 20-COOH-LTB4 | 351 | 195 | * | * |
| PGD2 | 351 | 233 | * | * |
| PGE2 | 351 | 189 | 10.3 ± 2.0 | 9.4 ± 2.9 |
| PGF2α | 353 | 193 | * | * |
| TxB2 | 369 | 169 | 17.4 ± 10.6 | 128.0 ± 62.7† |
| 15-HETE | 319 | 219 | 17.4 ± 4.8 | 12.1 ± 2.4 |
| 12-HETE | 319 | 179 | 131.4 ± 31.7 | 60.6 ± 20.4† |
| 5-HETE | 319 | 115 | 32.9 ± 15.4 | 20.3 ± 6.5 |
| AA | 303 | 257 | 173.4 ± 25.1 | 372.8 ± 174.4 |
Values are means ± SE. Human exhaled breath condensate was collected and immediately frozen. Lipid mediators (LM) were assessed using LM-metabolipidomics. Q1, M-H (parent ion); Q3, diagnostic ion in the MS-MS (daughter ion); COPD, chronic obstructive pulmonary disease; DHA, docosahexaenoic acid; RvD, D-series resolvin; PD, protectin; MaR, maresin; EPA, eicosapentaenoic acid; lipoxin; HDHA, hydroxyl-DHA; HETE, hydroxyeicosatetraenoic acid; HEPE, hydroxyeicosapentaenoic acid.; PG, prostaglandin; LT, leukotriene; AA, arachidonic acid.
Below limit of detection, which was ∼0.1 pg.
P < 0.05 and ††P < 0.01 vs. healthy volunteers.
RvDs act on human alveolar macrophages to decrease inflammatory mediators and enhance phagocytosis.
Macrophages are involved in the initiation, propagation, and resolution phases of inflammation and are major producers of inflammatory cytokines and lipid mediators. Smokers have increased susceptibility to bacterial infections, in part because of impaired macrophage phagocytic abilities (20, 38, 43, 47). On the basis of this key role of macrophages in mediating COPD pathology, we investigated whether SPMs could have a therapeutic impact on human alveolar macrophages. In initial studies we evaluated 1–100 nM RvD1 and RvD2 in several donors. RvD1 and RvD2 dampened spontaneous production of IL-6 and TNF-α, with the most consistent effect at 100 nM (Fig. 2, A and B). We next investigated the efficacy of 100 nM RvD1 and RvD2 in COPD and non-COPD patients. RvD1 and RvD2 dampened spontaneous production of TNF-α, but not IL-8; RvD2 additionally dampened production of IL-6 and enhanced phagocytic uptake of E. coli by unstimulated human alveolar macrophages (Fig. 2, C–F). We also examined COPD and non-COPD populations separately (Table 2). RvD1 and RvD2 were effective in COPD and non-COPD alveolar macrophages, with stronger potency in non-COPD patients, demonstrating further their potential efficacy in humans as potential therapeutics (Table 2).
Fig. 2.

Specialized proresolving mediators (SPMs) dampen cytokine release and enhance phagocytosis in unstimulated alveolar macrophages from COPD and non-COPD patients. A and B: alveolar macrophages from human subjects were incubated with 1–100 nM RvD1 or RvD2 for 24 h, and supernatants were assessed for IL-6 and TNF-α by ELISA. C–E: alveolar macrophages from COPD (closed symbols) and non-COPD (open symbols) human subjects were incubated with 100 nM RvD1 or RvD2 for 24 h, and supernatants were assessed for concentrations of IL-6, IL-8, and TNF-α by ELISA. F: cells were incubated with fluorescently labeled E. coli, and phagocytosis was determined by flow cytometry. Each symbol represents an individual donor (n = 4–8). *P < 0.05, **P < 0.01 vs. vehicle alone [by 1-way ANOVA (A and B) or paired t-test (C–F)].
Table 2.
Comparison of alveolar macrophages from COPD and non-COPD patients
| COPD |
Non-COPD |
|||||
|---|---|---|---|---|---|---|
| Vehicle | RvD1 | RvD2 | Vehicle | RvD1 | RvD2 | |
| IL-6, pg/ml | 487 ± 279 | 442 ± 289 | 392 ± 228 | 543 ± 137 | 428 ± 137 | 212 ± 62* |
| IL-8, pg/ml | 2,792 ± 745 | 3,017 ± 690 | 2,712 ± 1,007 | 1,761 ± 307 | 1,766 ± 289 | 1,528 ± 234 |
| TNF-α, pg/ml | 966 ± 414 | 764 ± 46† | 592 ± 38† | 961 ± 291 | 385 ± 60* | 202 ± 87.6* |
| %Phagocytosis | 12.40 | 15.40† | 16.66† | 4.20 | 8.87† | 10.40† |
Values are means ± SE. Specialized proresolving mediators dampen cytokine release and enhance phagocytosis in unstimulated alveolar macrophages from COPD and non-COPD patients. Alveolar macrophages from COPD and non-COPD human subjects were incubated with 100 nM RvD1 or RvD2 for 24 h. Supernatants were assessed for concentrations of IL-6, IL-8, and TNF-α by ELISA, and phagocytosis was determined by flow cytometry in cells incubated with fluorescently labeled E. coli. Data are the same as those shown in Fig. 2, C–F, but effects of RvDs were analyzed separately for COPD and non-COPD populations.
P < 0.05 vs. vehicle in the same disease group (by paired t-test).
Sample size insufficient for statistical analysis (n = 2–5).
RvDs dampen cigarette smoke-induced production of proinflammatory cytokines and promote anti-inflammatory cytokines and the M2 macrophage phenotype.
To characterize the effects of SPMs on human macrophages, we conducted further experiments using macrophages derived from peripheral blood monocytes. Blood-derived monocytes from multiple healthy human donors were isolated to >95% purity (Fig. 3A), differentiated to macrophages, and incubated with 100 nM SPMs for 24 h. On the basis of our human lipid profiles, effect on alveolar macrophages, and screening of multiple SPMs and 17-HDHA (Fig. 3, B and C), RvD1 and RvD2 were chosen for these studies; on the basis of preliminary dose-curve studies in blood-derived macrophages (data not shown) and our results with macrophage bronchoalveolar lavage, we used SPMs at 1 and 100 nM (Fig. 2, A and B). After SPM treatment, cells were exposed to CSE for 24 h. Neither SPMs nor CSE at the indicated doses affected cell viability (Fig. 3D). CSE increased production of the proinflammatory mediators IL-6, IL-8, and TNF-α, cytokines important in propagating an immune response, attracting neutrophils, enhancing phagocytosis, and promoting cell death. RvDs completely attenuated increases in IL-6 (Fig. 4A) and dose-dependently dampened IL-8. RvD1 at 1 nM and RvD2 at 1 and 100 nM additionally dampened TNF-α expression (Fig. 4, B and C). In addition to preventing proinflammatory cytokine release, resolvins promote a resolution phenotype in macrophages and the production of anti-inflammatory cytokines. CSE decreased production of the anti-inflammatory cytokine IL-10 (Fig. 4D), but this effect was not attenuated by SPMs. Conversely, RvD1 increased concentrations of total TGF-β, and RvD2 increased concentrations of both active and total TGF-β and increased the ratio of active to total TGF-β, an anti-inflammatory cytokine important in tissue repair (Fig. 4, E–G). We hypothesized that the reduction in cytokines and the increase in the ratio of active to total TGF-β might indicate a skewing of the macrophage phenotype from a classically activated, proinflammatory “M1” phenotype to an alternatively activated, anti-inflammatory and proresolving “M2” phenotype (39). To assess this idea, we analyzed surface expression of CD80 (an M1 marker) and CD206 (an M2 marker) by flow cytometry. CSE alone drove alveolar macrophages toward a proinflammatory M1 macrophage profile, indicated by a decreased CD206-to-CD80 ratio (Fig. 4H). RvD1 at 100 nM prevented this M1 skewing, while RvD2 dose-dependently prevented M1 skewing and increased the alternative M2 phenotype compared with the vehicle group (Fig. 4H).
Fig. 3.

RvDs are effective in reducing cigarette smoke extract (CSE)-induced inflammatory mediator production. A: human monocyte purity was confirmed by staining with anti-CD14 and anti-CD62 antibodies and assessed by flow cytometry. B and C: monocytes were differentiated to macrophages and treated with SPMs at 100 nM for 24 h, cells were exposed to CSE for 24 h, and supernatant levels of the proinflammatory cytokines IL-6 and IL-8 were evaluated by ELISA. D: viability was determined by Trypan blue exclusion. ##P < 0.01, ###P < 0.001 vs. vehicle (veh)/veh; *P < 0.05, **P < 0.01, ***P < 0.001 vs. CSE alone (by 2-way ANOVA with Bonferroni's posttest). Each symbol represents an individual donor (n = 3–4).
Fig. 4.

Resolvins dampen CSE-induced increases in inflammatory cytokines and promote anti-inflammatory cytokine production. A–G: blood-derived macrophages were incubated with RvD1 or RvD2 for 24 h and then exposed to CSE for 24 h. RvDs reduced concentrations of proinflammatory (IL-6, IL-8, and TNF-α) and increased concentrations of anti-inflammatory (IL-10 and active and total TGF-β) cytokines and chemokines, as determined by ELISA. H: ratio of CD206+ to CD80+ macrophages. Values are means ± SE (n = 6 individual donors). #P < 0.05, ##P < 0.01, ###P < 0.001 vs. veh/veh; *P < 0.05, **P < 0.01, ***P < 0.001 vs. CSE alone (by 2-way ANOVA with Bonferroni's posttest); ns, not significant.
The generation of certain pro- and anti-inflammatory cytokines and bioactive lipid mediators is regulated by the cyclooxygenase-2 (Cox-2) enzyme (46). Cigarette smoke can increase expression of Cox-2, and elevated Cox-2 expression is associated with several smoking-related diseases, including COPD and cancer (5, 37, 48, 51). CSE increased expression of Cox-2 in human macrophages, and treatment with RvDs significantly attenuated Cox-2 expression (Fig. 5, A and B). Macrophages produce lipid mediators, including Cox-2-regulated prostaglandins, such as PGE2 and PGD2 (29). PGD2 and PGE2 are important prostaglandins with both pro- and anti-inflammatory roles (25, 27, 29, 32). At 24 h, CSE significantly increased PGE2 production, and this increase was dose-dependently diminished by RvDs (Fig. 5C). CSE similarly increased the concentration of PGD2, but, in contrast to PGE2, RvD2 further potentiated this increase (Fig. 5D). We also evaluated the concentration of TxB2, the inactive metabolite of TxA2, an important prothrombotic signaling molecule involved in tissue injury and inflammation (44). TxB2 was unaffected by CSE or RvD exposure (Fig. 5E), indicating that RvDs may regulate specific lipid mediator pathways.
Fig. 5.

RvDs prevent CSE-induced increases in cyclooxygenase-2 (Cox-2) and proinflammatory lipid mediators. Macrophages were incubated with RvD1 or RvD2 for 24 h and then exposed to CSE for 24 h. A and B: cells were lysed, and Cox-2 protein expression levels were assessed by Western blotting. Blots are representative of results from 3 donors. C–E: supernatants from treated macrophages were also assessed for PGE2, PGD2, and TxB2 by enzyme immunoassay. Values are means ± SE (n = 5–6 individual donors). #P < 0.05 vs. veh/veh; *P < 0.05 vs. CSE alone (by 2-way ANOVA with Bonferroni's posttest).
RvDs dampen cigarette smoke-induced oxidative stress.
In addition to production of inflammatory mediators, macrophages are important players in the regulation of oxidative stress. Cigarette smoke induces the generation of ROS, and chronic smoke exposure leads to oxidative stress and tissue damage (19, 54). Little is known about the effect of SPMs on oxidative stress, particularly in human cells. To evaluate the ability of resolvins to attenuate oxidative stress, we investigated several markers of oxidative damage. CSE increased ROS, as indicated by levels of H2DCFDA in exposed macrophages (Fig. 6, A and B). RvD treatment, however, did not dampen this initial oxidative burst 20 min (data not shown) or 1 h (Fig. 6, A and B) after CSE exposure. We additionally investigated RvD1 and RvD2 actions on carbonylated proteins; ROS can induce the modification of native amino acids to carbonyl derivatives. RvDs did not significantly affect levels of carbonyl groups at 1 h after smoke exposure (Fig. 6, C and D). We next evaluated longer-duration CSE exposure to determine if RvDs were acting to shorten resolution time or reduce propagation of oxidative stress, rather than to prevent initiation. RvDs did not affect ROS at 24 h (data not shown), but 1 nM RvD1 and 100 nM RvD2 did act to decrease levels of carbonylated proteins (Fig. 6, E and F), the first evidence for RvDs acting on protein carbonylation.
Fig. 6.

Resolvins prevent propagation of oxidative stress, but not induction. Macrophages were incubated with RvD1 or RvD2 for 24 h, incubated with 2′,7′-dichlorodihydrofluorescein diacetate, and exposed to CSE. A and B: ROS levels were determined by flow cytometry 1 h after CSE exposure. MFI, mean fluorescence intensity. C–F: presence of carbonylated groups in RvD- and CSE-exposed macrophages assessed by OxyBlot at 1 h (C and D) and 24 h (E and F). Blots are representative of results from 3 donors. Values are means ± SE (n = 3–4 individual donors). #P < 0.05, ##P < 0.01 vs. veh/veh; *P < 0.05, **P < 0.01 vs. CSE alone.
RvDs rescue cigarette smoke-induced defects in phagocytosis.
On the basis of our data showing that SPMs enhanced phagocytosis in alveolar macrophages, we wanted to evaluate if these molecules could rescue or enhance cigarette smoke-induced phagocytic defects in blood-derived macrophages. Macrophages were incubated with RvD1 and RvD2 and exposed to CSE. CSE impaired uptake of fluorescently labeled E. coli (Fig. 7, A and B). Incubation with RvDs prevented these decreases and restored macrophage phagocytic abilities (Fig. 7, A and B). In addition, CSE decreased the number of macrophages that phagocytized bacteria, and this decrease was rescued by RvDs (Fig. 7C).
Fig. 7.
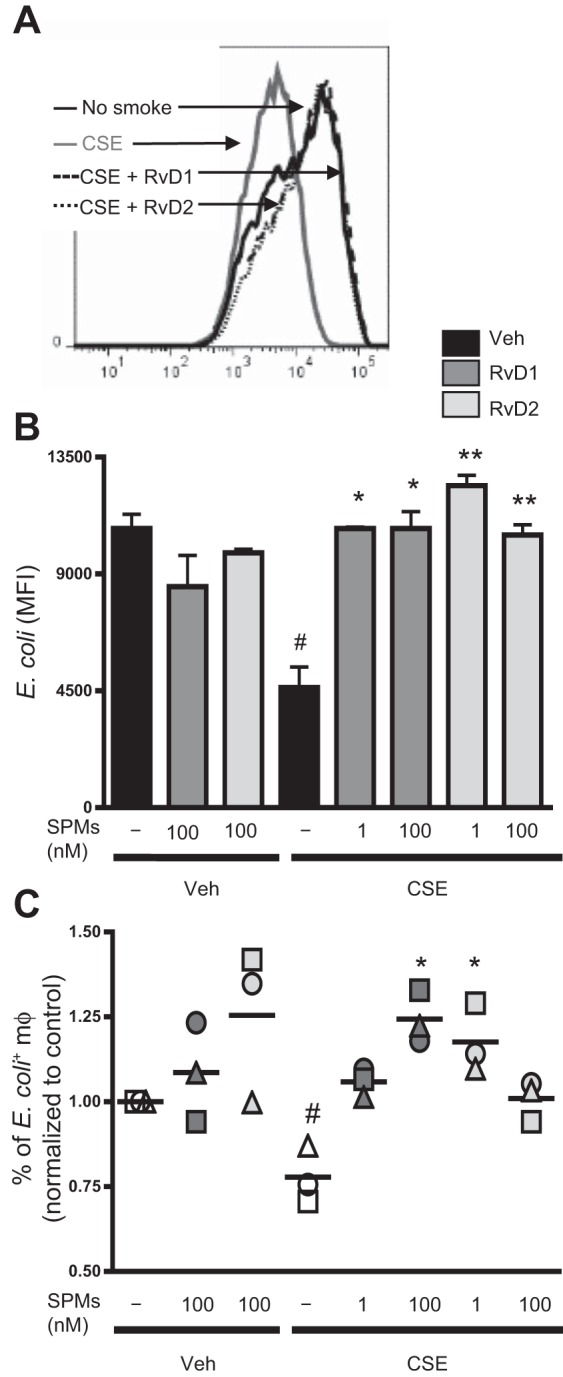
Resolvins attenuate CSE-induced decreases in phagocytosis. Macrophages were incubated with RvD1 or RvD2 for 24 h and then exposed to CSE for 24 h. Cells were incubated with fluorescently labeled E. coli, and phagocytosis was determined by flow cytometry. A and B: mean fluorescence intensity as a measure of E. coli uptake. Results from 1 representative donor are shown in A. Values are means ± SE (n = 4). C: percentage of macrophages that phagocytized E. coli. Each symbol represents an individual human donor. #P < 0.05 vs. veh/veh; *P < 0.05, **P < 0.01 vs. CSE alone (by 2-way ANOVA with Bonferroni's posttest).
RvD1 signals through GPCRs to mediate proresolving effects.
RvD1 is known to signal through two receptors, ALX and GPR32; the receptors for RvD2 are unknown (4, 34, 45). We used the ALX inhibitor Boc-2 and an anti-GPR32 neutralizing antibody to investigate the role of these receptors. Boc-2 partially prevented the effects of RvD1 on IL-6 and completely blocked dampening of TNF-α. Complementarily, anti-GPR32 blocked the effects of RvD1 on IL-6 and partially prevented dampening of TNF-α (Fig. 8).
Fig. 8.
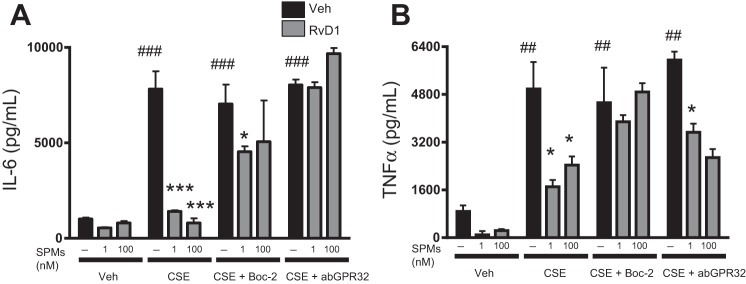
RvD1 signals through G protein-coupled receptors (GPCRs) to mediate proresolving effects. Macrophages were treated with Boc-2 or neutralizing anti-GPR32 antibody for 30 min, incubated with RvD1 and RvD2, and exposed to CSE. Effects of ALX (A) and GPR32 (B) inhibition were assessed by evaluating concentrations of IL-6 and TNF-α by ELISA. Values are means ± SE (n = 3 individual donors). ##P < 0.01, ###P < 0.001 vs. veh/veh; *P < 0.05, ***P < 0.001 vs. CSE or CSE with inhibitor for each respective group (by 2-way ANOVA with Bonferroni's posttest).
RvD2 acts via modulation of NF-κB signaling pathways.
Resolvins can mediate their effects through canonical inflammatory pathways, including NF-κB signaling (9, 35, 55). To investigate SPM actions in human macrophages, we focused on the mechanistic actions of RvD2. Total expression of p65 was unaffected by CSE or RvD2. However, expression of phosphorylated p65 was increased 30 min after CSE exposure (Fig. 9, A and B). This increase in phosphorylated p65 was dampened with RvD2. Phosphorylated IκBα expression was also increased with CSE and attenuated by RvD2 (Fig. 9, C and D).
Fig. 9.

RvD2 dampens classical and promotes alternative activation of the NF-κB pathway. Macrophages were incubated with RvD2 and then exposed to CSE. A–H: protein expression levels of total p65 and phosphorylated p65 (p-p65), total IκBα and phosphorylated IκBα (p-IκBα), p100/p52, and RelB were determined by Western blotting and quantified by densitometry. Blots are representative of results from 4 individual donors. Values are means ± SE (n = 4 individual donors). #P < 0.05 vs. veh/veh; *P < 0.05, **P < 0.01 vs. CSE alone (by 2-way ANOVA with Bonferroni's posttest).
Along with the canonical NF-κB signaling pathway, there is an alternative NF-κB signaling pathway that has several anti-inflammatory effects (68). Our lab has shown the importance of RelB in mediating anti-inflammatory responses to cigarette smoke (5, 40, 70). Thus we decided to investigate several members of the alternative NF-κB signaling pathway. CSE had no effect on p100 expression but increased expression of the cleaved p52 product (Fig. 9, E and F). This increase in p52 was reduced by RvD2, although p100 expression remained unchanged. Interestingly, CSE caused a dramatic loss of RelB expression, which was significantly prevented by RvD2 (Fig. 9, G and H). These studies support changes in NF-κB expression as a possible mechanism for resolvins and, for the first time, include evidence supporting involvement of the alternative NF-κB pathway.
DISCUSSION
SPMs represent a novel class of lipid mediators with high clinical potential for treatment of inflammatory diseases. We have shown that RvDs dampen inflammatory effects induced by either cigarette smoke or chronic disease states in human blood-derived and human alveolar macrophages. RvDs dampened production of key proinflammatory cytokines, enzymes, and lipid mediators. RvDs also acted through antioxidant mechanisms to dampen the effects of cigarette smoke. In addition to these anti-inflammatory and proresolving actions, RvD1 and RvD2 promoted the production of anti-inflammatory cytokines and enhanced proresolving macrophage phagocytosis in both macrophage types. We have presented evidence that these actions are mediated through changes in NF-κB expression. Taken together, these data present new evidence for the role of SPMs as novel therapeutics in attenuating human cigarette smoke-induced inflammation.
Previous studies have used targeted lipidomic analysis to evaluate the presence of a few particular lipids in COPD patients and have shown dysregulated levels of PGE2, leukotriene B4, and several isoprostanes (17, 21, 24, 69). Derivatives of ω-3 and ω-6 were also detected in the urine of smoker and nonsmoker volunteers, with smokers having decreased concentrations of SPMs (61). The lipid-mediator metabolipidomics methodology used here can unambiguously identify >50 pro- and anti-inflammatory bioactive lipid mediators and their pathway markers. Our analysis further demonstrates dysregulated levels of HDHA and HETE intermediates, as well as dampened concentrations of RvD1, indicating that COPD may certainly be seen as a “failure-to-resolve” inflammation and that bioactive lipid mediators play a key role in this process. We previously showed increased expression of a key SPM receptor in COPD lungs, which correlates with the increases in HDHA and HETE shown here; these increases are likely a compensatory mechanism as cells attempt to respond to a chronic inflammatory stimulus (31). Alternatively, cigarette smoke may be dampening enzymatic activity that promotes SPM formation (resulting in decreased RvD1) while promoting autoxidation that contributes to increased HDHA and HETEs (41, 64, 77). Multiple other SPMs, including other resolvins, such as RvD3 and RvD5, were not tested here and may play important roles in cigarette smoke-associated diseases and in future investigations. Together, these data provide novel insight into endogenous inflammatory and resolving signals in the lung, as well as in the context of cigarette smoke exposure.
SPMs have both anti-inflammatory and proresolving effects. They have been shown not only to reduce the production of proinflammatory proteins, but also to induce a phenotypic shift in cells to promote anti-inflammatory effects and alternative M2 macrophage activation (7, 16, 49, 57, 71). Particularly in the context of an inflammatory microenvironment, the collective changes in these pro- and anti-inflammatory cytokines may have a combined potent effect and act in concert to promote resolution. These experiments are critical, because they reinforce the idea that resolvins are not only anti-inflammatory, but also proresolving, and are not fully immunosuppressive, a crucial characteristic for their advancement as therapeutics. In addition to changes in protein production, SPMs have been shown to increase uptake of apoptotic neutrophils by human cells and to enhance macrophage phagocytic abilities in a mouse model of acute lung exposure (23, 30, 62, 66). Our studies here demonstrate, for the first time, that RvD1 and RvD2 prevent cigarette smoke-induced loss of human macrophage phagocytic abilities and promote increased uptake of bacteria. Additionally, we showed that RvDs enhance phagocytosis in human alveolar macrophages from patients with chronic pulmonary diseases. In both cases, actions of RvDs on macrophages were noted in cells from many different human donors. By using multiple donors, we were able to account for interhuman variations and to gauge the efficacy of these molecules across a broader human population with multiple different underlying inflammatory conditions than would be seen in a cell line. This further strengthens the role of SPMs as potential therapeutics in human smoking-related diseases.
Alternatively, activated macrophages that promote resolution and phagocytosis, rather than proinflammatory signaling, have recently become a key area of interest and investigation (8). Here we show that macrophages treated with RvDs not only dampen inflammatory cytokine production, but also produce anti-inflammatory cytokines, such as TGF-β and IL-10, and increase the ratio M2 to M1 macrophages. In addition, RvD treatment alone increased expression of Cox-2. While this enzyme is commonly categorized as proinflammatory (37, 51), it can also produce precursors of SPMs (14, 27, 59) and can be temporally regulated by SPMs (76). Furthermore, several prostaglandins can have pro- or anti-inflammatory actions, depending on concentration and cell type. PGD2, for example, signals through two different GPCRs in macrophages with differential effects, which may account for increases in expression with both smoke and RvDs (25, 28). PGD2 also counterregulates expression of PGE2 and is converted to 15-deoxyPGJ2, which has proresolution properties that may result from inhibition of NF-κB (26, 32, 58, 59). Although PGE2 is often considered a proinflammatory mediator, it also has anti-inflammatory properties, including the ability to reduce IL-6 levels and suppress allergic airway inflammation (6, 32, 37, 79). PGE2 also upregulates IL-10 (32). It is interesting in this context that RvD1 and RvD2 inhibit IL-6 production but do not upregulate IL-10; we cannot rule out the possibility that the absence of IL-10 is due to reduced PGE2 production. The mechanisms by which SPMs regulate other lipids appear to be complicated and merit in-depth investigation. Overall, Cox-2 is capable of producing a milieu of lipid mediators with counteracting and concentration-dependent actions, and investigation of the role of Cox-2 in resolution is an intriguing and ongoing area of research.
In addition to production of pro- and anti-inflammatory proteins, macrophages have key roles in oxidative stress and antioxidant activities. Oxidative stress is a hallmark of COPD and most other smoking-associated diseases and, when left unchecked, can cause excessive tissue damage. In our study, RvDs did not alter CSE-induced production of ROS. Other SPM precursors, such as docosahexaenoic acid, have been shown to reduce ROS induction in THP-1 cells, a monocytic cell line (14). This discrepancy may be due to a number of differences in methodology, as well as inherent differences between primary human cells and immortalized cell lines. We did observe changes in carbonylated proteins at 24 h, indicating that some time is required for both carbonylated proteins to appear and for the RvDs to have an effect. Since RvDs have not been shown to be direct antioxidants, they may be acting to increase antioxidative proteins or enzymes such as superoxide dismutase or glutathione, rather than to prevent ROS generation or enhance clearance and degradation of oxidatively damaged proteins. More comprehensive studies are needed to fully elucidate the effects of RvDs as antioxidant molecules and the mechanisms through which they act.
There are several mechanistic pathways through which SPMs promote the resolution of inflammation. Human macrophages have been shown to have multiple SPM receptors, including ALX and GPR32 (34). We showed that Boc-2 and anti-GPR32 antibodies can attenuate the effects of RvD1, with the inhibitors/antibodies affecting release of different cytokines. This implies complementary roles for the different RvD1 receptors. The receptor for RvD2 is unknown, and further investigations are needed to determine its binding partners.
Additionally, multiple inflammatory stimuli, including cigarette smoke, can induce activation of the NF-κB pathway; SPMs have additionally been shown to act by affecting expression of this pathway (9, 35, 55). We have shown here that, in human macrophages, RvD2 acts, in part, by preventing the phosphorylation and, thereby, degradation of IκBα and activation of p65. In addition, RvD2 acts through the noncanonical NF-κB pathway to mediate its effects. Alternative NF-κB signaling and, in particular, expression of RelB have recently been shown by our lab to be anti-inflammatory (5, 40, 70, 78). Expression of RelB is decreased in BALF of cigarette smoke-exposed mice, and overexpression of RelB can attenuate the effects of cigarette smoke exposure in human fibroblasts in vitro (40). Our new data here show that cigarette smoke exposure causes a loss of RelB expression in human macrophages and that this loss can be partially prevented by RvD2. This is the first evidence that SPMs alter alternative NF-κB signaling and present a novel pathway through which these bioactive lipid mediators may act. Other signaling pathways, including STAT3, cAMP response element-binding protein, and MAPK signaling pathways, may also play important roles in mediating SPM effects. SPMs, including RvD1 and LxA4, have been shown to decrease cytokines by decreasing STAT3 signaling (10, 74, 75). Multiple SPMs have also been shown to decrease members of MAPK pathways (31, 50, 73). Clearly, the signaling mechanisms of resolution are multifaceted and merit extensive investigation.
In conclusion, we have shown that RvDs effectively dampen and attenuate the effects of cigarette smoke exposure on human macrophages. Our data showing the proresolving effects of RvDs on human alveolar macrophages particularly highlight the potential of these SPMs in dampening inflammation associated with chronic diseases, including COPD, as well as the novel identification of the action of SPMs on alternative NF-κB signaling. Because of their dual anti-inflammatory and proresolving actions and their nonimmunosuppressive nature, these bioactive lipid mediators are important candidates for treatment of inflammatory diseases, including those induced by cigarette smoke exposure. Future investigation of the mechanisms of action and specific targets of these novel endogenous mediators will allow for translation into a clinical setting and development of SPMs as therapeutics.
GRANTS
This work was supported by National Institutes of Health Grants T32 ES-007026, P30 ES-01247, RO1 HL-120908, T32 HL-066988, and P01 GM-095467 (to C. N. Serhan) and Clinical and Translational Science Institute Incubator Grants UL1 RR-024160 and 8UL1 TR-000042 and by the PhRMA Foundation.
DISCLOSURES
P. J. Sime received personal fees from Boehringer Ingelheim outside the submitted work during the time frame of the conducted research. C. N. Serhan is an inventor on patents [on resolvins and their analogs] assigned to Brigham and Women's Hospital and licensed to Resolvyx Pharmaceuticals for clinical development. He was the scientific founder of Resolvyx Pharmaceuticals and owns equity (stock currently of unknown value) in the company. His interests were reviewed and are managed by the Brigham and Women's Hospital and Partners HealthCare in accordance with their conflict of interest policies.
AUTHOR CONTRIBUTIONS
A.C., R.M.K., R.A.C., J.D., C.N.S., P.J.S., and R.P.P. developed the concept and designed the research; A.C. and R.A.C. performed the experiments; A.C., T.H.T., R.M.K., R.A.C., and J.D. analyzed the data; A.C., T.H.T., R.A.C., J.D., C.N.S., P.J.S., and R.P.P. interpreted the results of the experiments; A.C., T.H.T., and R.A.C. prepared the figures; A.C. drafted the manuscript; A.C., T.H.T., R.A.C., J.D., C.N.S., P.J.S., and R.P.P. edited and revised the manuscript; A.C., T.H.T., R.M.K., R.A.C., J.D., C.N.S., P.J.S., and R.P.P. approved the final version of the manuscript.
ACKNOWLEDGMENTS
We thank the University of Rochester Medical Center Blood Bank and Ann Casey for assistance with collection of blood donations, Lindsay Zehr and Elizabeth Lyda for assistance with collection of serum, EBC, and broncholavage from human patients, and the University of Rochester Medical Center Flow Cytometry Core.
REFERENCES
- 1.Anonymous. Surgeon General's Report. How Tobacco Smoke Causes Disease: The Biology and Behavioral Basis for Smoking-Attributable Disease. Atlanta, GA: Centers for Disease Control, 2010. [PubMed] [Google Scholar]
- 2.Arita M, Bianchini F, Aliberti J, Sher A, Chiang N, Hong S, Yang R, Petasis NA, Serhan CN. Stereochemical assignment, antiinflammatory properties, and receptor for the ω-3 lipid mediator resolvin E1. J Exp Med 201: 713–722, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arita M, Ohira T, Sun YP, Elangovan S, Chiang N, Serhan CN. Resolvin E1 selectively interacts with leukotriene B4 receptor BLT1 and ChemR23 to regulate inflammation. J Immunol 178: 3912–3917, 2007. [DOI] [PubMed] [Google Scholar]
- 4.Back M, Powell WS, Dahlen SE, Drazen JM, Evans JF, Serhan CN, Shimizu T, Yokomizo T, Rovati GE. Update on leukotriene, lipoxin and oxoeicosanoid receptors: IUPHAR Review 7. Br J Pharmacol 171: 3551–3574, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baglole CJ, Maggirwar SB, Gasiewicz TA, Thatcher TH, Phipps RP, Sime PJ. The aryl hydrocarbon receptor attenuates tobacco smoke-induced cyclooxygenase-2 and prostaglandin production in lung fibroblasts through regulation of the NF-κB family member RelB. J Biol Chem 283: 28944–28957, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bagole CJ, Bushinsky SM, Garcia TM, Kode A, Rahman I, Sime PJ, Phipps RP. Differential induction of apoptosis by cigarette smoke extract in primary human lung fibroblast strains: implications for emphysema. Am J Physiol Lung Cell Mol Physiol 291: L19–L29, 2006. [DOI] [PubMed] [Google Scholar]
- 7.Buckley CD, Gilroy DW, Serhan CN. Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity 40: 315–327, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cassetta L, Cassol E, Poli G. Macrophage polarization in health and disease. Sci World J 11: 2391–2402, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen F, Fan XH, Wu YP, Zhu JL, Wang F, Bo LL, Li JB, Bao R, Deng XM. Resolvin D1 improves survival in experimental sepsis through reducing bacterial load and preventing excessive activation of inflammatory response. Eur J Clin Microbiol Infect Dis 33: 457–464, 2014. [DOI] [PubMed] [Google Scholar]
- 10.Chen J, Shetty S, Zhang P, Gao R, Hu Y, Wang S, Li Z, Fu J. Aspirin-triggered resolvin D1 down-regulates inflammatory responses and protects against endotoxin-induced acute kidney injury. Toxicol Appl Pharmacol 277: 118–123, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chiang N, Arita M, Serhan CN. Anti-inflammatory circuitry: lipoxin, aspirin-triggered lipoxins and their receptor ALX. Prostaglandins Leukot Essent Fatty Acids 73: 163–177, 2005. [DOI] [PubMed] [Google Scholar]
- 12.Chiang N, Fredman G, Backhed F, Oh SF, Vickery T, Schmidt BA, Serhan CN. Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature 484: 524–528, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chiang N, Serhan CN, Dahlen SE, Drazen JM, Hay DW, Rovati GE, Shimizu T, Yokomizo T, Brink C. The lipoxin receptor ALX: potent ligand-specific and stereoselective actions in vivo. Pharmacol Rev 58: 463–487, 2006. [DOI] [PubMed] [Google Scholar]
- 14.Cipollina C, Di Vincenzo S, Gerbino S, Siena L, Gjomarkaj M, Pace E. Dual anti-oxidant and anti-inflammatory actions of the electrophilic cyclooxygenase-2-derived 17-oxo-DHA in lipopolysaccharide- and cigarette smoke-induced inflammation. Biochim Biophys Acta 1840: 2299–2309, 2014. [DOI] [PubMed] [Google Scholar]
- 15.Colas RA, Shinohara M, Dalli J, Chiang N, Serhan CN. Identification and signature profiles for pro-resolving and inflammatory lipid mediators in human tissue. Am J Physiol Cell Physiol 307: C39–C54, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dalli J, Zhu M, Vlasenko NA, Deng B, Haeggstrom JZ, Petasis NA, Serhan CN. The novel 13S,14S-epoxy-maresin is converted by human macrophages to maresin1 (MaR1), inhibits leukotriene A4 hydrolase (LTA4H), and shifts macrophage phenotype. FASEB J 27: 2573–2583, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Batlle J, Sauleda J, Balcells E, Gomez FP, Mendez M, Rodriguez E, Barreiro E, Ferrer JJ, Romieu I, Gea J, Anto J, Garcia-Aymerich J. Association between ω-3 and ω-6 fatty acid intakes and serum inflammatory markers in COPD. J Nutr Biochem 23: 817–821, 2012. [DOI] [PubMed] [Google Scholar]
- 18.DiDonato JA, Mercurio F, Karin M. NF-κB and the link between inflammation and cancer. Immunol Rev 246: 379–400, 2012. [DOI] [PubMed] [Google Scholar]
- 19.Domej W, Oettl K, Renner W. Oxidative stress and free radicals in COPD—implications and relevance for treatment. Int J Chron Obstruct Pulm Dis 9: 1207–1224, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Drannik AG, Pouladi MA, Robbins CS, Goncharova SI, Kianpour S, Stampfli MR. Impact of cigarette smoke on clearance and inflammation after Pseudomonas aeruginosa infection. Am J Respir Crit Care Med 170: 1164–1171, 2004. [DOI] [PubMed] [Google Scholar]
- 21.Drozdovszky O, Barta I, Antus B. Sputum eicosanoid profiling in exacerbations of chronic obstructive pulmonary disease. Respir Int Rev Thorac Dis 87: 408–415, 2014. [DOI] [PubMed] [Google Scholar]
- 22.Fiore S, Ryeom SW, Weller PF, Serhan CN. Lipoxin recognition sites. Specific binding of labeled lipoxin A4 with human neutrophils. J Biol Chem 267: 16168–16176, 1992. [PubMed] [Google Scholar]
- 23.Fredman G, Oh SF, Ayilavarapu S, Hasturk H, Serhan CN, Van Dyke TE. Impaired phagocytosis in localized aggressive periodontitis: rescue by resolvin E1. PLos One 6: 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fritscher LG, Post M, Rodrigues MT, Silverman F, Balter M, Chapman KR, Zamel N. Profile of eicosanoids in breath condensate in asthma and COPD. J Breath Res 6: 026001, 2012. [DOI] [PubMed] [Google Scholar]
- 25.Giles H, Leff P. The biology and pharmacology of PGD2. Prostaglandins 35: 277–300, 1988. [DOI] [PubMed] [Google Scholar]
- 26.Gilroy DW, Colville-Nash PR, McMaster S, Sawatzky DA, Willoughby DA, Lawrence T. Inducible cyclooxygenase-derived 15-deoxy-Δ12,14-PGJ2 brings about acute inflammatory resolution in rat pleurisy by inducing neutrophil and macrophage apoptosis. FASEB J 17: 2269–2271, 2003. [DOI] [PubMed] [Google Scholar]
- 27.Gilroy DW, Colville-Nash PR, Willis D, Chivers J, Paul-Clark MJ, Willoughby DA. Inducible cyclooxygenase may have anti-inflammatory properties. Nat Med 5: 698–701, 1999. [DOI] [PubMed] [Google Scholar]
- 28.Gosset P, Bureau F, Angeli V, Pichavant M, Faveeuw C, Tonnel AB, Trottein F. Prostaglandin D2 affects the maturation of human monocyte-derived dendritic cells: consequence on the polarization of naive Th cells. J Immunol 170: 4943–4952, 2003. [DOI] [PubMed] [Google Scholar]
- 29.Harizi H, Corcuff JB, Gualde N. Arachidonic-acid-derived eicosanoids: roles in biology and immunopathology. Trends Mol Med 14: 461–469, 2008. [DOI] [PubMed] [Google Scholar]
- 30.Hsiao HM, Sapinoro RE, Thatcher TH, Croasdell A, Levy EP, Fulton RA, Olsen KC, Pollock SJ, Serhan CN, Phipps RP, Sime PJ. A novel anti-inflammatory and pro-resolving role for resolvin D1 in acute cigarette smoke-induced lung inflammation. PLos One 8: e58258, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hsiao HM, Thatcher TH, Levy EP, Fulton RA, Owens KM, Phipps RP, Sime PJ. Resolvin D1 attenuates polyinosinic-polycytidylic acid-induced inflammatory signaling in human airway epithelial cells via TAK1. J Immunol 193: 4980–4987, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kalinski P. Regulation of immune responses by prostaglandin E2. J Immunol 188: 21–28, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kirkham PA, Spooner G, Rahman I, Rossi AG. Macrophage phagocytosis of apoptotic neutrophils is compromised by matrix proteins modified by cigarette smoke and lipid peroxidation products. Biochem Biophys Res Commun 318: 32–37, 2004. [DOI] [PubMed] [Google Scholar]
- 34.Krishnamoorthy S, Recchiuti A, Chiang N, Yacoubian S, Lee CH, Yang R, Petasis NA, Serhan CN. Resolvin D1 binds human phagocytes with evidence for proresolving receptors. Proc Natl Acad Sci USA 107: 1660–1665, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kure I, Nishiumi S, Nishitani Y, Tanoue T, Ishida T, Mizuno M, Fujita T, Kutsumi H, Arita M, Azuma T, Yoshida M. Lipoxin A4 reduces lipopolysaccharide-induced inflammation in macrophages and intestinal epithelial cells through inhibition of nuclear factor-κB activation. J Pharmacol Exp Ther 332: 541–548, 2010. [DOI] [PubMed] [Google Scholar]
- 36.Maddox JF, Hachicha M, Takano T, Petasis NA, Fokin VV, Serhan CN. Lipoxin A4 stable analogs are potent mimetics that stimulate human monocytes and THP-1 cells via a G-protein-linked lipoxin A4 receptor. J Biol Chem 272: 6972–6978, 1997. [DOI] [PubMed] [Google Scholar]
- 37.Martey CA, Pollock SJ, Turner CK, O'Reilly KM, Baglole CJ, Phipps RP, Sime PJ. Cigarette smoke induces cyclooxygenase-2 and microsomal prostaglandin E2 synthase in human lung fibroblasts: implications for lung inflammation and cancer. Am J Physiol Lung Cell Mol Physiol 287: L981–L991, 2004. [DOI] [PubMed] [Google Scholar]
- 38.Marti-Lliteras P, Regueiro V, Morey P, Hood DW, Saus C, Sauleda J, Agusti AG, Bengoechea JA, Garmendia J. Nontypeable Haemophilus influenzae clearance by alveolar macrophages is impaired by exposure to cigarette smoke. Infect Immun 77: 4232–4242, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol 27: 451–483, 2009. [DOI] [PubMed] [Google Scholar]
- 40.McMillan DH, Baglole CJ, Thatcher TH, Maggirwar S, Sime PJ, Phipps RP. Lung-targeted overexpression of the NF-κB member RelB inhibits cigarette smoke-induced inflammation. Am J Pathol 179: 125–133, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mobley A, Tanizawa H, Iwanaga T, Tai CL, Tai HH. Selective inhibition of 5-lipoxygenase pathway in rat pulmonary alveolar macrophages by cigarette smoking. Biochim Biophys Acta 918: 115–119, 1987. [DOI] [PubMed] [Google Scholar]
- 42.Moghaddam SJ, Ochoa CE, Sethi S, Dickey BF. Nontypeable Haemophilus influenzae in chronic obstructive pulmonary disease and lung cancer. Int J Chron Obstruct Pulm Dis 6: 113–123, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Monick MM, Powers LS, Walters K, Lovan N, Zhang M, Gerke A, Hansdottir S, Hunninghake GW. Identification of an autophagy defect in smokers' alveolar macrophages. J Immunol 185: 5425–5435, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nakahata N. Thromboxane A2: physiology/pathophysiology, cellular signal transduction and pharmacology. Pharmacol Ther 118: 18–35, 2008. [DOI] [PubMed] [Google Scholar]
- 45.Norling LV, Dalli J, Flower RJ, Serhan CN, Perretti M. Resolvin D1 limits polymorphonuclear leukocyte recruitment to inflammatory loci: receptor-dependent actions. Arterioscler Thromb Vasc Biol 32: 1970–1978, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O'Banion MK, Winn VD, Young DA. cDNA cloning and functional activity of a glucocorticoid-regulated inflammatory cyclooxygenase. Proc Natl Acad Sci USA 89: 4888–4892, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Phipps JC, Aronoff DM, Curtis JL, Goel RD, O'Brien E, Mancuso P. Cigarette smoke exposure impairs pulmonary bacterial clearance and alveolar macrophage complement-mediated phagocytosis of Streptococcus pneumoniae. Infect Immun 78: 1214–1220, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pietras T, Szemraj J, Panek M, Witusik A, Banasiak M, Antczak A, Gorski P. Functional polymorphism of cyclooxygenase-2 gene (G-765C) in chronic obstructive pulmonary disease patients. Mol Biol Rep 39: 2163–2167, 2012. [DOI] [PubMed] [Google Scholar]
- 49.Prescott D, McKay DM. Aspirin-triggered lipoxin enhances macrophage phagocytosis of bacteria while inhibiting inflammatory cytokine production. Am J Physiol Gastrointest Liver Physiol 301: G487–G497, 2011. [DOI] [PubMed] [Google Scholar]
- 50.Prieto P, Cuenca J, Traves PG, Fernandez-Velasco M, Martin-Sanz P, Bosca L. Lipoxin A4 impairment of apoptotic signaling in macrophages: implication of the PI3K/Akt and the ERK/Nrf-2 defense pathways. Cell Death Differ 17: 1179–1188, 2010. [DOI] [PubMed] [Google Scholar]
- 51.Profita M, Sala A, Bonanno A, Riccobono L, Ferraro M, La Grutta S, Albano GD, Montalbano AM, Gjomarkaj M. Chronic obstructive pulmonary disease and neutrophil infiltration: role of cigarette smoke and cyclooxygenase products. Am J Physiol Lung Cell Mol Physiol 298: L261–L269, 2010. [DOI] [PubMed] [Google Scholar]
- 52.Psychogios N, Hau DD, Peng J, Guo AC, Mandal R, Bouatra S, Sinelnikov I, Krishnamurthy R, Eisner R, Gautam B, Young N, Xia J, Knox C, Dong E, Huang P, Hollander Z, Pedersen TL, Smith SR, Bamforth F, Greiner R, McManus B, Newman JW, Goodfriend T, Wishart DS. The human serum metabolome. PLos One 6: e16957, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Quehenberger O, Armando AM, Brown AH, Milne SB, Myers DS, Merrill AH, Bandyopadhyay S, Jones KN, Kelly S, Shaner RL, Sullards CM, Wang E, Murphy RC, Barkley RM, Leiker TJ, Raetz CR, Guan Z, Laird GM, Six DA, Russell DW, McDonald JG, Subramaniam S, Fahy E, Dennis EA. Lipidomics reveals a remarkable diversity of lipids in human plasma. J Lipid Res 51: 3299–3305, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rahman I, Morrison D, Donaldson K, MacNee W. Systemic oxidative stress in asthma, COPD, and smokers. Am J Respir Crit Care Med 154: 1055–1060, 1996. [DOI] [PubMed] [Google Scholar]
- 55.Ramon S, Bancos S, Serhan CN, Phipps RP. Lipoxin A modulates adaptive immunity by decreasing memory B-cell responses via an ALX/FPR2-dependent mechanism. Eur J Immunol 44: 357–369, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ramon S, Gao F, Serhan CN, Phipps RP. Specialized proresolving mediators enhance human B cell differentiation to antibody-secreting cells. J Immunol 189: 1036–1042, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rogerio AP, Haworth O, Croze R, Oh SF, Uddin M, Carlo T, Pfeffer MA, Priluck R, Serhan CN, Levy BD. Resolvin D1 and aspirin-triggered resolvin D1 promote resolution of allergic airways responses. J Immunol 189: 1983–1991, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rossi A, Kapahi P, Natoli G, Takahashi T, Chen Y, Karin M, Santoro MG. Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IκB kinase. Nature 403: 103–108, 2000. [DOI] [PubMed] [Google Scholar]
- 59.Sandig H, Pease JE, Sabroe I. Contrary prostaglandins: the opposing roles of PGD2 and its metabolites in leukocyte function. J Leukoc Biol 81: 372–382, 2007. [DOI] [PubMed] [Google Scholar]
- 60.Sarir H, Henricks PA, van Houwelingen AH, Nijkamp FP, Folkerts G. Cells, mediators and Toll-like receptors in COPD. Eur J Pharmacol 585: 346–353, 2008. [DOI] [PubMed] [Google Scholar]
- 61.Sasaki A, Fukuda H, Shiida N, Tanaka N, Furugen A, Ogura J, Shuto S, Mano N, Yamaguchi H. Determination of ω-6 and ω-3 PUFA metabolites in human urine samples using UPLC/MS/MS. Anal Bioanal Chem 407: 1625–1639, 2015. [DOI] [PubMed] [Google Scholar]
- 62.Scannell M, Maderna P. Lipoxins and annexin-1: resolution of inflammation and regulation of phagocytosis of apoptotic cells. Sci World J 6: 1555–1573, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol 8: 349–361, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sethi S. Inhibition of leukocyte-endothelial interactions by oxidized ω-3 fatty acids: a novel mechanism for the anti-inflammatory effects of ω-3 fatty acids in fish oil. Redox Rep Commun Free Radic Res 7: 369–378, 2002. [DOI] [PubMed] [Google Scholar]
- 65.Shang S, Ordway D, Henao-Tamayo M, Bai X, Oberley-Deegan R, Shanley C, Orme IM, Case S, Minor M, Ackart D, Hascall-Dove L, Ovrutsky AR, Kandasamy P, Voelker DR, Lambert C, Freed BM, Iseman MD, Basaraba RJ, Chan ED. Cigarette smoke increases susceptibility to tuberculosis—evidence from in vivo and in vitro models. J Infect Dis 203: 1240–1248, 2011. [DOI] [PubMed] [Google Scholar]
- 66.Spite M, Norling LV, Summers L, Yang R, Cooper D, Petasis NA, Flower RJ, Perretti M, Serhan CN. Resolvin D2 is a potent regulator of leukocytes and controls microbial sepsis. Nature 461: 1287–1291, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stenfeldt AL, Karlsson J, Wenneras C, Bylund J, Fu H, Dahlgren C. Cyclosporin H, Boc-MLF and Boc-FLFLF are antagonists that preferentially inhibit activity triggered through the formyl peptide receptor. Inflammation 30: 224–229, 2007. [DOI] [PubMed] [Google Scholar]
- 68.Sun SC. The noncanonical NF-κB pathway. Immunol Rev 246: 125–140, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Telenga ED, Hoffmann RF, Ruben TK, Hoonhorst SJ, Willemse BW, van Oosterhout AJ, Heijink IH, van den Berge M, Jorge L, Sandra P, Postma DS, Sandra K, ten Hacken NH. Untargeted lipidomic analysis in chronic obstructive pulmonary disease Uncovering sphingolipids. Am J Respir Crit Care Med 190: 155–164, 2014. [DOI] [PubMed] [Google Scholar]
- 70.Thatcher TH, Maggirwar SB, Baglole CJ, Lakatos HF, Gasiewicz TA, Phipps RP, Sime PJ. Aryl hydrocarbon receptor-deficient mice develop heightened inflammatory responses to cigarette smoke and endotoxin associated with rapid loss of the nuclear factor-κB component RelB. Am J Pathol 170: 855–864, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Titos E, Rius B, Gonzalez-Periz A, Lopez-Vicario C, Moran-Salvador E, Martinez-Clemente M, Arroyo V, Claria J. Resolvin D1 and its precursor docosahexaenoic acid promote resolution of adipose tissue inflammation by eliciting macrophage polarization toward an M2-like phenotype. J Immunol 187: 5408–5418, 2011. [DOI] [PubMed] [Google Scholar]
- 72.Tuder RM, Petrache I. Pathogenesis of chronic obstructive pulmonary disease. J Clin Invest 122: 2749–2755, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang YP, Wu Y, Li LY, Zheng J, Liu RG, Zhou JP, Yuan SY, Shang Y, Shang-Long Y. Aspirin-triggered lipoxin A4 attenuates LPS-induced pro-inflammatory responses by inhibiting activation of NF-κB and MAPKs in BV-2 microglial cells. J Neuroinflamm 8: 95, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang Z, Cheng Q, Tang K, Sun Y, Zhang K, Zhang Y, Luo S, Zhang H, Ye D, Huang B. Lipid mediator lipoxin A4 inhibits tumor growth by targeting IL-10-producing regulatory B (Breg) cells. Cancer Lett 364: 118–124, 2015. [DOI] [PubMed] [Google Scholar]
- 75.Wang ZF, Li Q, Liu SB, Mi WL, Hu S, Zhao J, Tian Y, Mao-Ying QL, Jiang JW, Ma HJ, Wang YQ, Wu GC. Aspirin-triggered lipoxin A4 attenuates mechanical allodynia in association with inhibiting spinal JAK2/STAT3 signaling in neuropathic pain in rats. Neuroscience 273: 65–78, 2014. [DOI] [PubMed] [Google Scholar]
- 76.Wu D, Zheng S, Li W, Yang L, Liu Y, Zheng X, Yang Y, Yang L, Wang Q, Smith FG, Jin S. Novel biphasic role of resolvin D1 on expression of cyclooxygenase-2 in lipopolysaccharide-stimulated lung fibroblasts is partly through PI3K/AKT and ERK2 pathways. Mediators Inflamm 2013: 964012, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yin H, Brooks JD, Gao L, Porter NA, Morrow JD. Identification of novel autoxidation products of the ω-3 fatty acid eicosapentaenoic acid in vitro and in vivo. J Biol Chem 282: 29890–29901, 2007. [DOI] [PubMed] [Google Scholar]
- 78.Zago M, Rico de Souza A, Hecht E, Rousseau S, Hamid Q, Eidelman DH, Baglole CJ. The NF-κB family member RelB regulates microRNA miR-146a to suppress cigarette smoke-induced COX-2 protein expression in lung fibroblasts. Toxicol Lett 226: 107–116, 2014. [DOI] [PubMed] [Google Scholar]
- 79.Zaslona Z, Okunishi K, Bourdonnay E, Domingo-Gonzalez R, Moore BB, Lukacs NW, Aronoff DM, Peters-Golden M. Prostaglandin E2 suppresses allergic sensitization and lung inflammation by targeting the E prostanoid 2 receptor on T cells. J Allergy Clin Immunol 133: 379–387, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]