

Abstract
A method has been developed for one-step ortho-selective ligand-directed H-D exchange, accompanied in some cases by concurrent acid-catalyzed electrophilic deuteration. This method is effective for deuteration of aromatic substrates ranging from ketones to amides and amino acids, including compounds of biological and pharmaceutical interest such as acetaminophen and edaravone. Use of a palladium catalyst featuring an NHC ligand is critical for the observed reactivity. Experimental evidence strongly suggests that palladium facilitates C-H activation of the aromatic substrates, a mechanism seldom observed under strongly acidic conditions. 2015 Elsevier Ltd. All rights reserved.
Keywords: H-D exchange, Palladium, NHC, CF3COOD
Graphical Abstract

Introduction
Development of efficient, practical methods for H-D exchange has accelerated as a result of the rapidly increasing commercial importance of deuterated compounds. Of particular interest are deuterated pharmaceuticals, the commercial value of which is expected to eventually exceed 1 billion USD.1 One deuterated drug, the tetrabenazine derivative SD-809, has shown promising results during phase 3 clinical trials for chorea associated with Huntington’s disease.2 A number of catalytic processes have been investigated to produce these valuable deuterium-labeled compounds, including metal-free conditions as well as both homogeneous and heterogeneous metallic catalysts.3 Of the homogenous transition metal-catalyzed methods for H-D exchange of pharmaceutically relevant compounds, most have focused on the use of rhodium and iridium4 although H-D exchange of other small molecules by homogenous ruthenium,5 palladium5a,6 and platinum5a,6b,7 species has also been reported. These reactions are frequently conducted in deuterated acidic media such as acetic acid-d4 or trifluoroacetic acid-d1 (CF3COOD). However, recent studies have called into question whether certain metal-catalyzed H-D exchange reactions in acidic solvents proceed through C-H activation or electrophilic aromatic substitution mechanisms; in CF3COOD, the latter mechanism has been suggested to predominate.5a,8
Ligand-directed C-H activation by palladium is a versatile method for highly selective functionalization of otherwise unreactive C-H bonds. Numerous challenging transformations have been carried out using this approach, including alkylation, olefination, alkynylation, arylation, and amidation reactions, among others.9 While several different mechanistic pathways are possible depending on the specific reaction, generally the substrate undergoes cyclopalladation with the ligated palladium catalyst to generate the reactive intermediate.9a Weakly-coordinating ligands such as ketones and carboxylic acids have recently emerged as promising directing groups for palladium-catalyzed organic synthesis. Because palladacycles formed using weakly-coordinating directing groups are generally less thermodynamically stable than their tightly-bound counterparts, they are thus more reactive towards functionalization.9c
A few reports of ligand-directed H-D exchange using rhodium10 and ruthenium5b–c catalysts have been published, although most studies employ iridium complexes.11 Recently, a report of ligand-directed palladium-catalyzed ortho-selective H-D exchange of benzoic acids and phenylacetic acids using weak coordination has been published.6a The H-D exchange of benzene and other hydrocarbons in D2O with NHC-amidate palladium catalyst 1 has been previously investigated.6c The use of CF3COOD as a solvent, catalyst, and source of deuterium label for anilines and acetanilides has also been established.12 Herein is reported an H-D exchange methodology that utilizes both of these strategies simultaneously to effect the ortho-selective H-D exchange of aromatic amides, ketones, and amino acids. In several cases, this ligand-directed C-H activation process occurs in conjunction with electrophilic H-D exchange by CF3COOD, allowing for complete H-D exchange of pharmaceutically relevant compounds such as acetaminophen. This distinguished reactivity provides a complimentary strategy for previously inaccessible deuterated substrates.
Results and Discussion
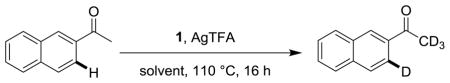
Our previously reported electrophilic H-D exchange method using CF3COOD without any additional metal catalyst was highly effective for aromatic amides and amines.12 However, this method was ineffective for more electron-poor substrates such as acetophenone and its derivatives, and the use of a homogeneous palladium catalyst in conjunction with CF3COOD was considered to improve the extent of deuteration.
Ketones have been employed as directing groups for numerous different metal-catalyzed transformations, including ortho-selective H-D exchange with iridium11 and ruthenium.5b To determine the viability of a palladium-catalyzed H-D exchange method using a ketone as a directing group, 2-acetonaphthone was subjected to reaction with CF3COOD in the presence of several different palladium(II) complexes and additives (Table 1).
Table 1.
Effect of palladium salts and AgTFA additive on H-D exchange.a

| ||||
|---|---|---|---|---|
| Entry | Catalyst | Additive | % D (H1) | % D (H2) |
| 1 | none | - | 6 | 2 |
| 2 | PdCl2 | - | 6 | 2 |
| 3 | Pd(MeCN)2Cl2 | - | 6 | 2 |
| 4 | Pd(OAc)2 | - | 7 | 4 |
| 5 | Pd(PPh3)2Cl2 | - | 5 | 1 |
| 6 | Pd(TFA)2 | - | 9 | 9 |
| 7 | 1 | - | 6 | 13 |
| 8 | Pd(PPh3)2Cl2 | AgTFAb | 6 | 1 |
| 9 | none | AgTFA | 6 | 1 |
| 10 | 1 | AgTFA | 11 | 86 |
| 11 | 1 | AgOTf | -c | -c |
H-D exchange of other aromatic positions has been omitted for clarity.
0.04 mmol AgTFA was added.
Significant degradation of the substrate was observed.
In the absence of any palladium catalyst, no significant H-D exchange occurred on the aromatic rings although extensive H-D exchange was observed on the acetyl group (entry 1). Most common palladium salts showed negligible improvement over the control experiment (entries 2 – 5), although Pd(TFA)2 exhibited minor activity (entry 6). While these known catalysts favored regioselective H-D exchange of H1, Pd-NHC catalyst 1 (10 mol%) effected an improved rate of deuteration at H2 preferentially (entry 7). Activation of the palladium salts with AgTFA was then investigated. When activated Pd(PPh3)2Cl2 (generated via addition of two equivalents of AgTFA) and AgTFA alone were used as catalysts, reactivity was unchanged relative to the control experiment (entries 8 – 9).
However, the use of catalyst 1 with AgTFA (10 mol%) resulted in a significant increase in the extent of H-D exchange observed, far superior to any of the other palladium salts tested. Substitution of deuterium was selective, occurring predominantly at one position (H2), ortho to the ketone (entry 10). Adding AgOTf in place of AgTFA resulted in reduced H-D exchange as well as partial degradation of the substrate (entry 11).
In the presence of Pd-NHC catalyst 1, various reaction conditions were then investigated (Table 2). Additional AgTFA resulted in no improvement (entry 1 vs entry 2), and reducing the catalyst loading lowered the extent of H-D exchange somewhat (entry 3). At lower temperatures, very low amounts of deuterium incorporation were observed (entry 4). Increasing or decreasing the volume of CF3COOD gave approximately a 10% reduction in deuterium incorporation (entries 5 – 6). Little or no H-D exchange was observed when deuterated acetic acid or methanol was used in place of CF3COOD as a solvent (entries 7 – 8). Longer reaction times did not result in a proportional increase in deuterium incorporation (entry 9). It was concluded that the original conditions did not require further optimization.
Table 2.
Effect of reaction conditions on H-D exchange.a
| ||||
|---|---|---|---|---|
| Entry | 1 (mol%) | AgTFA (mol%) | solvent | % D |
| 1 | 10 | 10 | CF3COOD | 86 |
| 2 | 10 | 20 | CF3COOD | 82 |
| 3 | 5 | 5 | CF3COOD | 72 |
| 4b | 10 | 10 | CF3COOD | 9 |
| 5c | 10 | 10 | CF3COOD | 77 |
| 6d | 10 | 10 | CF3COOD | 76 |
| 7 | 10 | 10 | DOAc-d3 | 0 |
| 8 | 10 | 10 | MeOD-d3 | 1 |
| 9e | 10 | 10 | CF3COOD | 85 |
H-D exchange of other aromatic positions has been omitted for clarity.
Reaction conducted at 60 °C.
0.5 mL of CF3COOD was added.
2 mL of CF3COOD was added.
Reaction proceeded for 36 h.
The substrate scope of this method was evaluated by testing several different aromatic ketones in the presence of catalyst 1 and AgTFA (Scheme 1). The method was less effective for unsubstituted acetophenone 2 compared to 2-acetonaphthone 6, resulting in 25% total ortho-selective deuterium incorporation. The aliphatic protons adjacent to the carbonyl (acetyl protons) were mostly exchanged with deuterium (>90%); this H-D exchange occurred with all substrates tested independent of palladium catalyst. Similarly, deuterium incorporation in 4′-methoxyacetophenone 3 was marginally improved by the palladium catalyst. Interestingly, while the Pd-NHC catalyst efficiently deuterated highly activated ketone 4 more effectively than CF3COOD, the nitro-substituted ketone 5 was unresponsive to the developed conditions, suggesting a strong dependence on the electronic properties of the aromatic ring. As explained above, 2-acetonaphthone 6 exhibited far superior reactivity and selectivity under the palladium-catalyzed conditions. Other bicyclic ketones such as 7 and 8 were moderately deuterated with a high degree of ortho selectivity. However, when the ketone was distal to the aromatic ring such as in dibenzyl ketone 9, almost no ortho incorporation of deuterium was observed.
Scheme 1.

H-D exchange of aromatic ketones with CF3COOD (condition a) and CF3COOD with catalyst 1 and AgTFA (condition b). The bracketed numbers adjacent to each aromatic position represent percent deuterium incorporation at that position (combined in the case of a symmetrical structure.) Isolated yields are given in parentheses.
Encouraged by these promising results with ketones, we turned out attention to acetanilide derivatives (Scheme 2). Acetanilides have been extensively utilized as directing groups for palladium-catalyzed C-H functionalization reactions and have also been employed to direct H-D exchange reactions with rhodium,10c iridium,11 and ruthenium.5c The palladium-catalyzed ortho-selective H-D exchange of acetanilides was envisaged proceeding in a similar fashion to ketones under identical conditions. Our previous work12 identified acetanilides as substrates well-suited for electrophilic H-D exchange in CF3COOD, so care was taken to identify any improvement in ortho-selective deuterium incorporation by catalyst 1.
Scheme 2.

H-D exchange of aromatic amides and their derivatives with CF3COOD (condition a) and CF3COOD with catalyst 1 and AgTFA (condition b). Results 10a–14a are from our previous work12 using identical conditions and are shown for comparison purposes. No deuterium incorporation was observed at the amide substituents after workup. The bracketed numbers adjacent to each aromatic position represent percent deuterium incorporation at that position (combined in the case of a symmetrical structure.) Isolated yields are given in parentheses. aKeto-enol tautomerism prevents accurate determination of deuterium incorporation at the pyrazolone methylene position.
Electron-rich and electron-neutral acetanilides 10–13 were deuterated efficiently under the described conditions, although electron-poor acetanilide 14 had significantly lower deuterium incorporation. Notably, one-step, nearly complete ring deuteration of pharmaceutically-relevant acetaminophen 13 was achieved, which was a significant improvement over existing literature methods which required multiple inefficient synthetic steps13 or only partially deuterated the aromatic ring.10c,11e,14 As with ketone substrate 9, distal amide substrate 15 was ineffective at promoting H-D exchange. Conversely, pyrazolone-substituted substrate 16, the pharmaceutically-relevant compound edaravone, was selectively ortho-deuterated in good yield in the presence of catalyst. Benzamide 17 was somewhat reactive towards the palladium catalyst but was deuterated much less efficiently than the acetanilides. In summary, the palladium-catalyzed conditions led to a high degree of H-D exchange selectively ortho to the amide group, a significant improvement over the previously developed CF3COOD conditions.
The H-D exchange of aromatic amino acids using this method was also considered. Various methods for ring deuteration of phenylalanine and tyrosine derivatives, generally employing acid catalysis, have been reported in the literature.7f–g,15 Palladium-catalyzed ortho functionalization of aromatic amino acids such as phenylalanine and tyrosine and their derivatives has been conducted16 but typically uses methyl esters17 or sulfonamides18 to efficiently direct C-H activation. It was found that simple unmodified α-phenylglycine 18 and phenylalanine 19 were deuterated selectively at the ortho positions by the developed catalytic method (Scheme 3). Previously, aromatic deuterium incorporation ortho to the side chain of phenylalanine was achieved by selective protonation of ring-perdeuterated tyrosine followed by reduction to phenylalanine,15c an inefficient and deuterium-uneconomical strategy. However, palladium-catalyzed ortho deuteration was not as effective for tyrosine 20, and electrophilic H-D exchange by CF3COOD was predominant. Varying amounts of deuterium incorporation were observed at the stereocenters, and thus some racemization of the amino acids may have occurred. Interestingly, neither phenylacetic acid 21 nor benzylamine 22 showed selectivity towards H-D exchange by the palladium catalyst, suggesting that efficient ligand direction does not occur in the absence of the amine or carboxylic acid functional groups.
Scheme 3.
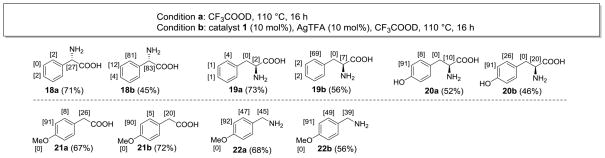
H-D exchange of amino acids and derivatives with CF3COOD (condition a) and CF3COOD with catalyst 1 and AgTFA (condition b). aThe bracketed numbers adjacent to each aromatic position represent percent deuterium incorporation at that position (combined in the case of a symmetrical structure.) Isolated yields are given in parentheses.
Based on the experimental evidence collected, a catalytic cycle was envisioned (Scheme 4) involving coordination of the palladium-NHC complex 1 to the substrate of interest, followed by C-H activation of an accessible aromatic proton. This process may involve formation of an agostic complex with assistance from coordinated trifluoroacetate, as evidenced in several computational studies.19 Subsequent deuterolysis would furnish the deuterated substrate and regenerate the catalyst. This proposed mechanism is conceptually very similar to that suggested by Yu et al. in the palladium-catalyzed H-D exchange of phenylacetic acids and other arenes.6a The active intermediate for such a C-H activation reaction would presumably be a cyclopalladated aryl-Pd(II) complex; however, this proposed intermediate could not be isolated. Acid-catalyzed H-D exchange by CF3COOD, following a simple electrophilic aromatic substitution pathway, would be concomitant with several substrates.
Scheme 4.
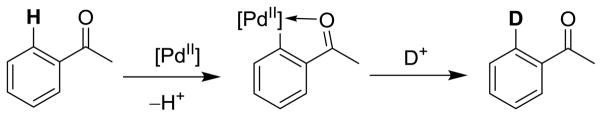
Plausible mechanism of palladium-catalyzed H-D exchange.
While evidence suggests metal-catalyzed H-D exchange reactions of arenes in strong acids such as CF3COOD, especially with additives such as AgTFA, are in many cases predominantly electrophilic,5a,8 a Lewis-acid type H-D exchange mechanism under the described conditions is unlikely for several reasons. First, Lewis-acid catalyzed H-D exchange is typically restricted to nonpolar arenes3a without functional groups that the Lewis acid could coordinate to preferentially. Moreover, simple palladium salts were not nearly as effective catalytically as the NHC-palladium complex 1, which exhibits increased thermal stability and electron density on palladium due to strong σ donation from the NHC ligand. This complex has previously been utilized for H-D exchange reactions under neutral conditions.6c In this case, the role of the AgTFA is thus to abstract chloride from palladium to generate an open coordination site, not to increase the Lewis acidity of the metal center. Additionally, the high degree of ortho selectivity observed for H-D exchange of ketones, without comparable meta or para reactivity, further suggests a C-H activation mechanism. Finally, when AlCl3 was utilized in place of palladium with 2-acetonapthone 6, no improvement over simple acid-catalyzed conditions were seen, both with and without stoichiometric AgTFA additive.
Conclusion
Using a palladium-NHC catalyst in deuterated trifluoroacetic acid, one-step ligand-directed ortho-selective H-D exchange of aromatic ketones, amides, and amino acids has been conducted. The Pd-NHC complex tested showed distinguished reactivity compared to other palladium salts, which were ineffective deuteration catalysts. In general, acetanilides were more reactive than ketones, and a strong correlation of reactivity with the electronic properties of the aromatic ring was observed. Direct ortho deuteration of amino acids was possible without prior functionalization at nitrogen or oxygen. Experimental data supports the intermediacy of an aryl-palladium species generated by C-H activation of the coordinated substrate rather than Lewis acid-type catalysis. With highly activated substrates, substantial concurrent acid-catalyzed electrophilic H-D exchange also occurred, resulting in nearly complete ring deuteration of several acetanilides, including pharmaceutically-relevant acetaminophen. Ortho-selective aromatic functionalization with other electrophiles besides deuterium cation may be possible using a variant of this method. Further work will explore this possibility and investigate synthetic modification of the catalyst scaffold and development of additional NHC ligands to improve catalytic activity and selectivity.
Supplementary Material
Figure 1.
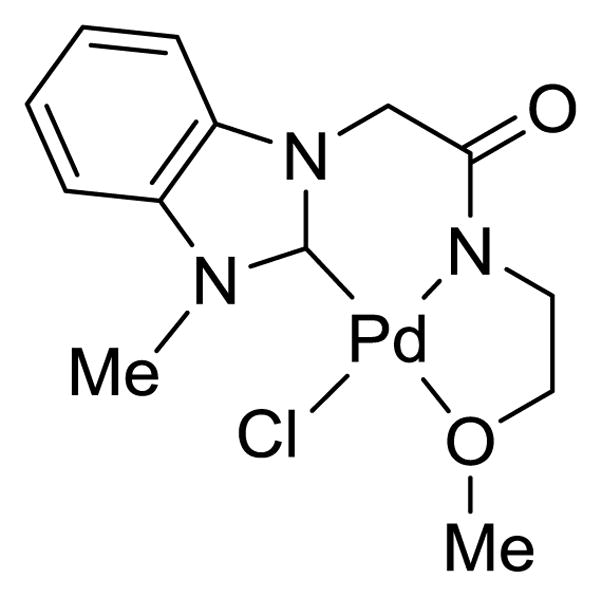
NHC-amidate palladium catalyst 1.
Acknowledgments
The authors acknowledge Joo Ho Lee and Nima Zargari for their assistance with collection of FTIR data. We also acknowledge generous financial support from the Hydrocarbon Research Foundation and the National Institute of Health (S10 RR025432).
Footnotes
Supplementary material, including 1H NMR spectral data, is available online in PDF format.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Timmins GS. Expert Opin Ther Patents. 2014;24:1067–1075. doi: 10.1517/13543776.2014.943184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bates GP, Dorsey R, Gusella JF, Hayden MR, Kay C, Leavitt BR, Nance M, Ross CA, Scahill RI, Wetzel R, Wild EJ, Tabrizi SJ. Nat Rev Dis Primers. 2015:15005. doi: 10.1038/nrdp.2015.5. [DOI] [PubMed] [Google Scholar]
- 3.(a) Di Giuseppe A, Castarlenas R, Oro LA. Comptes Rendus Chimie. 2015;18:713–741. [Google Scholar]; (b) Lockley WJS, Heys JR. J Labelled Comp Rad. 2010;53:635–644. [Google Scholar]; (c) Atzrodt J, Derdau V, Fey T, Zimmermann J. Angew Chem Int Ed. 2007;46:7744–7765. doi: 10.1002/anie.200700039. [DOI] [PubMed] [Google Scholar]; (d) Junk T, Catallo WJ. Chem Soc Rev. 1997;26:401–406. [Google Scholar]
- 4.Allen PH, Hickey MJ, Kingston LP, Wilkinson DJ. J Labelled Comp Rad. 2010;53:731–738. [Google Scholar]
- 5.(a) Munz D, Webster-Gardiner M, Fu R, Strassner T, Goddard WA, III, Gunnoe TB. ACS Catal. 2015;5:769–775. [Google Scholar]; (b) Piola L, Fernández-Salas JA, Manzinia S, Nolan SP. Org Biomol Chem. 2014;12:8683–8688. doi: 10.1039/c4ob01798f. [DOI] [PubMed] [Google Scholar]; (c) Ackermann L, Wang L, Wolfram R, Lygin AV. Org Lett. 2012;14:728–731. doi: 10.1021/ol203251s. [DOI] [PubMed] [Google Scholar]; (d) Lockley WJS, Hesk D. J Labelled Comp Rad. 2010;53:704–715. (and references therein) [Google Scholar]
- 6.(a) Ma S, Villa G, Thuy-Boun PS, Homs A, Yu JQ. Angew Chem Int Ed. 2014;126:753–756. doi: 10.1002/anie.201305388. [DOI] [PubMed] [Google Scholar]; (b) Emmert MH, Gary JB, Villalobos JM, Sanford MS. Angew Chem Int Ed. 2010;49:5884–5886. doi: 10.1002/anie.201002351. [DOI] [PubMed] [Google Scholar]; (c) Lee JH, Yoo KS, Park CP, Olsen JM, Sakaguchi S, Prakash GKS, Mathew T, Jung KW. Adv Synth Catal. 2009;351:563–568. doi: 10.1002/adsc.200800698. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Anderson GK, Phipps AK. J Organomet Chem. 1984;266:C15–C18. [Google Scholar]; (e) Anderson GK, Saum SE, Cross RJ, Morris SA. Organometallics. 1983;2:780–782. [Google Scholar]
- 7.(a) Hickman AJ, Cismesia MA, Sanford MS. Organometallics. 2012;31:1761–1766. [Google Scholar]; (b) Villalobos JM, Hickman AJ, Sanford MS. Organometallics. 2010;29:257–262. [Google Scholar]; (c) Hickman AJ, Villalobos JM, Sanford MS. Organometallics. 2009;28:5316–5322. [Google Scholar]; (d) Ilyas T, Davies O, McNeill A, Smith DI. J Label Compd Radiopharm. 2007;50:477–479. [Google Scholar]; (e) Kański R, Kańska M. J Radioanal Nucl Chem Lett. 2003;257:385–390. and references cited therein. [Google Scholar]; (f) Kanska M. J Radioanal Nucl Chem Lett. 1988;125:183–188. [Google Scholar]; (g) Kanska M. J Radioanal Nucl Chem Lett. 1984;87:95–100. [Google Scholar]; (h) Shilov AE, Shteinman AA. Coord Chem Rev. 1977;24:97–143. [Google Scholar]; (i) Garnett JL, Hodges RJ. J Am Chem Soc. 1967;89:4546–4547. [Google Scholar]
- 8.Lehman MC, Gary JB, Boyle PD, Sanford MS, Ison EA. ACS Catal. 2013;3:2304–2310. [Google Scholar]
- 9.For recent reviews of this subject area, see: Shi G, Zhang Y. Adv Synth Catal. 2014;356:1419–1442. doi: 10.1002/adsc.201301033.Neufeldt SR, Sanford MS. Acc Chem Res. 2012;45:936–946. doi: 10.1021/ar300014f.Engle KM, Mei TS, Wasa M, Yu JQ. Acc Chem Res. 2012;45:788–802. doi: 10.1021/ar200185g.Lyons TW, Sanford MS. Chem Rev. 2010;110:1147–1169. doi: 10.1021/cr900184e.
- 10.(a) Rhinehart JL, Manbeck KA, Buzak SK, Lippa GM, Brennessel WW, Goldberg KI, Jones WD. Organometallics. 2012;31:1943–1952. [Google Scholar]; (b) Gary JB, Carter TJ, Sanford MS. Top Catal. 2012;55:565–570. [Google Scholar]; (c) Lockley WJS. J Labelled Comp Rad. 1985;22:623–630. [Google Scholar]
- 11.(a) Cochrane AR, Idziak C, Kerr WJ, Mondal B, Paterson LC, Tuttle T, Andersson S, Nilsson GN. Org Biomol Chem. 2014;12:3598–3603. doi: 10.1039/c4ob00465e. [DOI] [PubMed] [Google Scholar]; (b) Brown JA, Cochrane AR, Irvine S, Kerr WJ, Mondal B, Parkinson JA, Paterson LC, Reid M, Tuttle T, Andersson S, Nilsson GN. Adv Synth Catal. 2014;356:3551–3562. [Google Scholar]; (c) Parmentier M, Hartung T, Pfaltz A, Muri D. Chem Eur J. 2014;20:11496–11504. doi: 10.1002/chem.201402078. [DOI] [PubMed] [Google Scholar]; (d) Cochrane AR, Irvine S, Kerr WJ, Reid M, Andersson S, Nilsson GN. J Labelled Comp Rad. 2013;56:451–454. doi: 10.1002/jlcr.3084. [DOI] [PubMed] [Google Scholar]; (e) Hesk D, Das PR, Evans B. J Labelled Comp Rad. 1995;36:497–502. [Google Scholar]
- 12.Giles R, Lee A, Jung E, Kang A, Jung KW. Tetrahedron Lett. 2015;56:747–749. doi: 10.1016/j.tetlet.2014.12.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Johnston D, Elder D. J Labelled Comp Rad. 1988;25:1315–1318. [Google Scholar]; (b) Forte AJ, Wilson JM, Slattery JT, Nelson SD. Drug Metab Dispos. 1984;12:484–491. [PubMed] [Google Scholar]; (c) Freed CR, Murphy RC. J Labelled Comp Rad. 1978;15:637–643. [Google Scholar]
- 14.Tuck KL, Tan H-W, Hayball PJ. J Labelled Comp Rad. 2000;43:817–823. [Google Scholar]
- 15.(a) Wang L, Murai Y, Yoshida T, Okamoto M, Masuda K, Sakihama Y, Hashidoko Y, Hatanaka Y, Hashimoto M. Biosci Biotech Bioch. 2014;78:1129–1134. doi: 10.1080/09168451.2014.917267. [DOI] [PubMed] [Google Scholar]; (b) Berthomieu C, Boussac A. Biospectroscopy. 1995;1:187–206. [Google Scholar]; (c) Nishiyama K, Oba M, Ueno R, Morita A, Nakamura Y, Kainosho M. J Labelled Comp Rad. 1994;34:831–837. [Google Scholar]; (d) Wishart DS, Sykes BD, Richards FM. Biochim Biophys Acta. 1993;1164:36–46. doi: 10.1016/0167-4838(93)90109-5. [DOI] [PubMed] [Google Scholar]; (e) Feeney J, Birdsall B, Akiboye J, Tendler SJB, Jiménez Barbero J, Ostler G, Arnold JRP, Roberts GCK, Kühn A, Roth K. FEBS Lett. 1989:57–61. [Google Scholar]; (f) Dollinger G, Eisenstein L, Lin S-L, Nakanishi K, Termini J. Biochemistry. 1986;25:6524–6533. doi: 10.1021/bi00369a028. [DOI] [PubMed] [Google Scholar]; (g) Kinsey RA, Kintanar A, Oldfield E. J Biol Chem. 1981;256:9028–9036. [PubMed] [Google Scholar]; (h) Matthews HR, Matthews KS, Opella SJ. Biochim Biophys Acta. 1977;497:1–13. doi: 10.1016/0304-4165(77)90134-9. [DOI] [PubMed] [Google Scholar]; (i) Griffiths DB, Feeney J, Roberts GCK, Burgen ASV. Biochim Biophys Acta. 1976;446:479–485. doi: 10.1016/0005-2795(76)90014-3. [DOI] [PubMed] [Google Scholar]
- 16.Noisier AFM, Brimble MA. Chem Rev. 2014;114:8775–8806. doi: 10.1021/cr500200x. [DOI] [PubMed] [Google Scholar]
- 17.(a) Albert J, Ariza X, Calvet T, Font-Bardia M, Garcia J, Granell J, Lamela A, López B, Martinez M, Ortega L, Rodriguez A, Santos D. Organometallics. 2013;32:649–659. [Google Scholar]; (b) López B, Rodriguez A, Santos D, Albert J, Ariza X, Garcia J, Granell J. Chem Commun. 2011;47:1054–1056. doi: 10.1039/c0cc03478a. [DOI] [PubMed] [Google Scholar]; (c) Nieto S, Arnau P, Serrano E, Navarro R, Soler T, Cativiela C, Urriolabeitia EP. Inorg Chem. 2009;48:11963–11975. doi: 10.1021/ic901941s. [DOI] [PubMed] [Google Scholar]; (d) Vicente J, Saura-Llamas I, García-López JA, Calmuschi-Cula B, Bautista D. Organometallics. 2007;26:2768–2776. [Google Scholar]
- 18.(a) García-Rubia A, Laga E, Cativiela C, Urriolabeitia EP, Gómez-Arrayás R, Carretero JC. J Org Chem. 2015;80:3321–3331. doi: 10.1021/jo502912m. [DOI] [PubMed] [Google Scholar]; (b) He G, Zhao Y, Zhang S, Lu C, Chen G. J Am Chem Soc. 2012;134:3–6. doi: 10.1021/ja210660g. [DOI] [PubMed] [Google Scholar]; (c) Vickers CJ, Mei TS, Yu JQ. Org Lett. 2010;12:2511–2513. doi: 10.1021/ol1007108. [DOI] [PubMed] [Google Scholar]; (d) Mei TS, Wang X, Yu JQ. J Am Chem Soc. 2009;131:10806–10807. doi: 10.1021/ja904709b. [DOI] [PubMed] [Google Scholar]; (e) Li JJ, Mei TS, Yu JQ. Angew Chem Int Ed. 2008;47:6452–6455. doi: 10.1002/anie.200802187. [DOI] [PubMed] [Google Scholar]
- 19.(a) Jiang J, Yu JQ, Morokuma K. ACS Catal. 2015;5:3648–3661. [Google Scholar]; (b) Munz D, Strassner T. Chem Eur J. 2014;20:14872–14879. doi: 10.1002/chem.201403910. [DOI] [PubMed] [Google Scholar]; (c) Aullón G, Chat R, Favier I, Font-Bardia M, Gómez M, Granell J, Martínez M, Solans X. Dalton Trans. 2009:8292–8300. doi: 10.1039/b905134a. [DOI] [PubMed] [Google Scholar]; (d) Davies DL, Donald SMA, Macgregor SA. J Am Chem Soc. 2005;127:13754–13755. doi: 10.1021/ja052047w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.