

Abstract
The enzyme catalase converts solar radiation into reactive oxidant species (ROS). In this study, we report that several bacterial catalases (hydroperoxidases, HP), including Escherichia coli HP-I and HP-II also generate reactive oxidants in response to ultraviolet B light (UVB). HP-I and HP-II are identical except for the presence of NADPH. We found that only one of the catalases, HPI, produces oxidants in response to UVB light, indicating a potential role for the nucleotide in ROS production. This prompts us to speculate that NADPH may act as a cofactor regulating ROS generation by mammalian catalases. Structural analysis of the NADPH domains of several mammalian catalases revealed that the nucleotide is bound in a constrained conformation and that UVB irradiation induces NADPH oxidation and positional changes. Biochemical and kinetic analysis indicate that ROS formation by the enzyme is enhanced by oxidation of the cofactor. Conformational changes following absorption of UVB light by catalase NADPH have the potential to facilitate ROS production by the enzyme.
Keywords: catalase, UVB, ROS, NADPH, light
Reactive oxygen species (ROS) are ubiquitous in mammalian and nonmammalian species, generated as by-products of aerobic metabolism and through enzymatic activity1–4 Recent studies indicate that the stable reactive oxygen moiety hydrogen peroxide (H2O2) can act as a second messenger in signal transduction pathways regulating an array of physiological processes including wound healing, immune cell activation, inflammation, cell proliferation and apoptosis.5–9 In unicellular organisms, H2O2 principally acts to stimulate the production of antioxidants as well as ROS-removing and repairing enzymes, whereas the principle physiological role of H2O2 in animals is activation of signaling pathways.10–13 Meanwhile, the production and accumulation of excessive hydrogen peroxide and its decomposition products, including peroxynitrite and hydroxyl radicals, is harmful for most cellular components.10 Therefore, rapid and efficient removal of ROS is of essential importance for all aerobically living cells.11
Tissue peroxide levels are largely regulated by the activity of hydroperoxidases, a group of enzymes comprised of both catalases and peroxidases. These oxidoreductases are capable of the heterolytic cleavage of the peroxidic bond. In this reaction, hydrogen peroxide most commonly is the substrate, however, in some circumstances, small organic peroxides or electron rich azo-molecules can also undergo the redox reactions catalyzed by these enzymes.1,12–14 Catalases generate molecular oxygen as a byproduct of the oxidation of hydrogen peroxide in a process referred to as disproportionation. The activity of the enzyme is dependent on the structural conformation of three essential domains, the heme moiety at the active site, a reduced NADPH bound in the NADPH-binding domain and a complex secondary structure formed by the threading and intertwining of long peptide loops during tetramerization.1,13,15 The activity of the enzyme is dependent on the structural conformation of three essential domains, the heme moiety at the active site, a reduced NADPH bound in the NADPH-binding domain and a complex secondary structure formed by the threading and intertwining of long peptide loops during tetramerization.1,13,15 Active mammalian catalase is a homotetramer and individual protein monomers have no peroxide degrading activity. Mammalian catalase is known for its facile ability to convert hydrogen peroxide into water and oxygen (catalatic activity), and its activity oxidizing low molecular weight alcohols in the presence of low concentrations of hydrogen peroxide (peroxidatic activity). The enzyme is one of the best characterized best characterized of all of the antioxidant enzymes and belongs to a family of Fe-protoporphyrin IX containing proteins that include a variety of cytochromes, globins, and peroxidases.1,14,16 The conversion of hydrogen peroxide to water and oxygen by catalase is a well understood two-step process whereby catalase heme Fe+3 reduces one molecule of hydrogen peroxide to water, generating a covalent Fe+4O oxyferryl species and a porphyrin cation radical. This reaction intermediate, referred to as compound I, then oxidizes a second hydrogen peroxide molecule forming molecular oxygen and another molecule of water16–18 The peroxidatic activity of catalase results from the ability of compound I to oxidize alcohols to aldehydes and water when levels of hydrogen peroxide sufficient for completing the catalytic cycle are unavailable.19–21 Each catalase monomer contains a single heme subunit; the tetrameric holoenzyme also binds NADPH. In enzymatically active mammalian catalase, only two of the four identical NADPH binding sites are occupied by the nucleotide (1/monamer). However, because hydrogen peroxide provides both oxidative and reductive potential during catalysis, the precise role of this cofactor in enzyme activity remains open to speculation.
Catalase was one of the first proteins to be crystallized in studies conducted by Sumner and Dounce in 1937. Since then, 14 3D structures of typical heme catalases in native and oxidized form have been solved.22–25 These crystal structures have been integral to revealing the mechanism of hydrogen peroxide degradation. In all catalytically active enzymes, the architecture of the substrate channels leading to the deeply buried active center has been shown to play a crucial role in catalysis. A main, highly restrictive access channel, located perpendicular to the plain of the heme, has been identified as the preferred route for H2O2 access to the active site in mammalian catalase structures.22,26 Interestingly, the rate of dismutation of hydrogen peroxide by catalase is extremely high, with the substrate turnover number exceeding that of any other enzymatic reaction. The maximal turnover number for hydrogen peroxide is estimated to be 16,000,000–44,000,000 s-1 per folded tetra-heme molecule.1,26,27 However, the affinity of catalase for hydrogen peroxide is surprisingly low. Due to the extreme rate of substrate turnover, and deleterious oxidation of the enzyme in the presence of high concentrations of hydrogen peroxide, the exact rate is undetermined; pseudokinetics studies indicate that the km is approximately 10–30 mM.1,28–30 In contrast, other peroxide degrading enzymes, including glutathione peroxidase, exhibit binding affinities for peroxide in the low micromolar range.
The role of NADPH in catalase activity has long been debated. Whereas full enzymatic activity of the holoenzyme requires the presence of two reduced molecules of the nucleotide, the cofactor is bound in a peculiar constrained conformation near the protein surface, approximately 19Å from the active site. Oxidation of NADPH is clearly associated with reduced substrate turnover, but the mechanism underlying this effect has not been defined. It has been speculated that during peroxide degradation, catalase is subject to establishing an Fe(IV)-oxo (ferryl) species with saturated ð-orbitals, a compound II intermediate. A transient tyrosyl radical, first observed in bovine liver catalase (BLC) and generated by the spontaneous decay of compound I, is suspected to be involved in this process. NADPH is believed to deter compound II formation by donating a reducing equivalent via an electron tunneling or charge relay network.11,15,18 Clearly, the severe structural limitations that modulate access to the active site and the low affinity of the enzyme for hydrogen peroxide are inconsistent with the high rate of enzymatic activity. These, in conjunction with the unresolved determination of the role of enzyme NADPH, present perplexing inconsistencies. An additional concern arises when considering that when exposed to the low peroxide concentrations often found in many mammalian cell types, the degradation of hydrogen peroxide is hindered. As there is no strong evidence of high catalase peroxidase activity in most mammalian cells, the physiological role of the enzyme is less clear.
To begin to address the problem of determining the physiological functions of catalase, we examined the responses of catalase in epidermal cells exposed to ultraviolet light. It is well recognized that keratinocytes, skin stromal cells, generate high levels of ROS when irradiated with UV light, and we reasoned that catalase may play a role in maintaining oxidant levels in these cells. We determined that in response to ultraviolet B light (UVB), catalase itself generates reactive oxygen intermediates.5 This finding was highly divergent from the well recognized antioxidant functions of catalase and indicated that, depending on the intracellular oxidant status, UVB light-induced catalase activity can be either protective, degrading peroxide, or toxic, by generating ROS. As strongly reflected in the scientific literature, the net response generated by catalase activity in response to ultraviolet light is profoundly protective. The absorption of high-energy, DNA-damaging short wave UVB light by the enzyme protects nucleic material from light-mediated disruption. Potentially, catalase acts to protect DNA by converting damaging UV radiation into ROS species that can be further metabolized and detoxified by cellular antioxidant enzymes. Disruption of this system, resulting in the excessive or inappropriate accumulation of ROS, may result in oxidative stress, damage to critical cellular molecules, and contribute to the development of pathophysio-logical conditions including skin cancer.5
A question arises as to the mechanisms by which catalase converts UVB light energy into ROS. To address this problem, we investigated the effects of UVB light on ROS generation by bacterial hydroperoxidases. In these studies, Escherichia coli HP-I and HP-II, bacterial enzymes that are highly homologous with mammalian catalase but divergent in NADPH binding activity, were analyzed. HP-I, and HP-II, are largely identical except for the presence of NADPH tightly bound to HP-I. Using recombinant enzymes, we determined that HP-1 but not HP-2 produced ROS in response to UVB light, suggesting a role for NADPH in oxidant generation (Fig. 1). In additional studies, spectrophotometric analysis revealed that proteins lacking NADPH absorbed significantly less UVB light, further supporting the theory that the nucleotide is important in UVB-mediated oxidant generation.14
Figure 1.
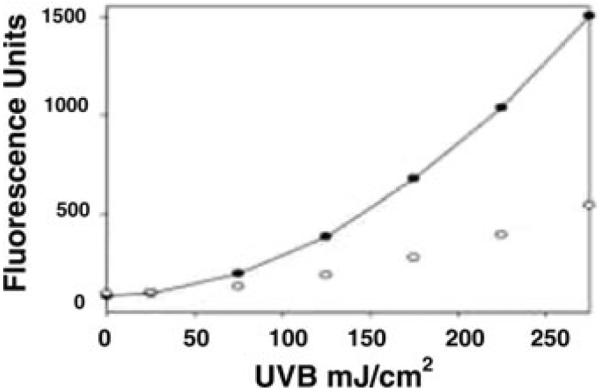
Differential effects of UVB on NADPH binding (HP I) and NADPH-independent (HP II) E-coli catalases. Recombinant catalases [(catalase (2.2 μM)] were incubated with 50 μM 2′,7′-dichlorofluorescein diacetate (Molecular Probes, Eugene, OR) and the reaction mixtures were irradiated in uncovered 96-well tissue culture plates (Costar, Corning, NY) with UVB light emitted from two Westinghouse FS20 light tubes. The UVB lights were calibrated with an IL 442A Phototherapy Radiometer (International Light, Newburyport, MA). Fluorescence was quantified using an HTS 7000 plus bio-assay reader (PerkinElmer Life Sciences, Beaconsfield Buckinghamshire, UK) with 495 nm excitation and 520 nm emission filters. E. coli hydroperoxidases were generously provided by Peter Loewen, U. Manotoba).
Further insight into potential mechanisms mediating ROS generation was provided by inhibitor studies using mouse and human catalases. As previously mentioned, it is well established that catalase possesses an activity responsible for degrading hydrogen peroxide via the reaction: 2H2O2 → 2H2O + O2.19–21 In addition, in the presence of low con-centrations of hydrogen peroxide, less than 10 μM, catalase exhibits significant levels of peroxidatic activity oxidizing ethanol and low molecular weight phenolic compounds.20,21 Biochemical mechanisms by which catalase functions in both catalatic and peroxidatic reactions have been the subject of numerous investigations focusing on the role of the heme-coordinated iron. It has become well recognized that in phase I of the catalatic reaction, a single molecule of H2O2 undergoes transient reduction resulting in an oxyferryl heme group with a p-cationic porphyrin protein radical, termed compound I.21,26–31 In the subsequent phase II of the reaction, a second molecule of H2O2 is oxidized by compound I resulting in return of the heme iron to it's resting +3 state, and the generation of water and molecular oxygen. In this phase of the reaction, iron-bound heme uncoordinated by water is required for enzymatic activity.32,33 More recently, a 1.5-Å resolution structural analysis of catalase demonstrated that the inhibitor cyanide blocks substrate access to the heme iron at the active site.33 Structural analysis revealed that a critical component required for both the catalatic and peroxidatic activities of the enzyme is a highly confined hydrogen peroxide binding site with strictly limited hydration. This site restricts both peroxide and water access to the heme iron and functions to optimally position peroxide molecules for cleavage by catalase.33–35 The enzyme inhibitor 3-amino-1,2,4-triazole (3-AT) has been reported to inactivate catalase by disrupting this binding site.33,36 It is important to note that analysis of the 2-Å-resolution structure of inhibitor-bound enzyme indicates the potential presence of water molecules within the substrate channel. It has been suggested that this alteration modifies both substrate access and subsequent peroxide hydrolysis.33 Interestingly, the oxidation of NADPH also results in similar structural alterations, inhibits peroxide degradation and promotes the interaction of water with the compound I intermediate and subsequent oxidant generation.
Recent studies indicate that catalatic and peroxidatic enzymatic activity may also require a charge-relay network that stabilizes reaction intermediates and facilitates the heterolytic cleavage of peroxide. This network has been hypothesized to regulate catalase activity at the metal-binding site by minimizing the charge of a porphyrin-cation radical and an electron deficient oxyferryl moiety at Tyr358.33 In cyanide-inhibited enzymes, increased electron density formed by the presence of cyanide at the heme iron may serve to alter this charge relay network. Interactions between amino acid residues forming this network and azide or 3-amino triazole, moieties that, in turn, disrupt channel activity, are likely to account for the competitive inhibition of the enzyme.29,30,33,34
Our observations that the effects of UVB light on catalase were highly pH-sensitive and oxygen-dependent, but did not require additional substrates, suggest that the transfer of water-derived protons and subsequent interactions with molecular oxygen may form the resulting ROS. To investigate this possibility, inhibitors of the catalytic and peroxidatic activity of catalase were used. We found that sodium cyanide induced a biphasic response, enhancing catalase activity at low concentrations but inhibiting the generation of ROS at higher concentrations equal molar with heme moieties, suggesting a role for the heme iron in ROS generation (Fig. 3). However, emerging studies suggesting interaction of ferrocyanide with enzyme-bound NADPH and resultant disruption of NADPH-heme electronic linkage offer an alternative explanation for this effect.36 In our studies, both sodium azide and 3-AT, which disrupt peroxide access, enhanced UVB light-mediated ROS production.5 The actions of these inhibitors on the activities of catalase suggest a role for altering substrate access in the mechanism mediating UVB light-induced responses. However, the possibility that increases in production of ROS at low concentrations of sodium cyanide, or in the presence of 3-AT or sodium azide, result from inhibition of a competing hydrogen peroxide degrading activity of catalase can not be ruled out. In the presence of higher concentrations of cyanide, interaction of the anion with catalase heme is likely to block substrate access to heme iron as well as limiting the activity of the charge relay network, rendering both structures unable to participate in the UVB-induced production of ROS. The increased generation of ROS that is observed in the presence of 3-AT and sodium azide suggests that the presence of reactants at the peroxide binding site, and the activity of the charge relay network implicated in peroxide degradation,35–38 are both important in UVB light-mediated oxidant production. This is not in agreement with the contention that ROS generation results from light-induced heme degradation.39,40 Analysis of these findings prompted us to hypothesize that, similar to 3AT, UVB light-induced effects alter the peroxide binding site allowing water molecules to access the heme iron. In our scheme, proton extraction from water molecules is also facilitated by azide-induced disruption of the charge relay network. Therefore, acting as substrates, water molecules provide a source for the generation of protons, which subsequently interact with molecular oxygen to generate ROS.
Figure 3.
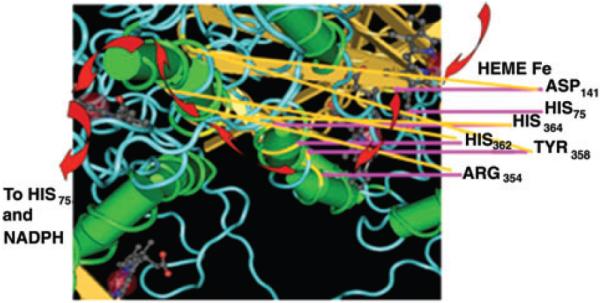
Detail of potential charge relay network in one catalase tertromer. The red arrow identifies the proposed path of charge relay. Mutations to residues noted have inhibited the rate of peroxide degradation in mammalian and bacterial catalases and enhanced ROS generation in response to UVB light (data not shown).13,22,26,33,36
If disruption of the heme preserving substrate access channel and charge relay network through chemical inhibitors or NADPH absorption of UVB light is central to disrupting hydrogen peroxide degradation and facilitating ROS generation, it becomes critical to better understand the fundamental role(s) of NADPH in catalase function. To address this challenge, we have developed a potential scheme for the reaction that involves the formation of functional dimers. In this scheme, two catalase monomers are linked via a network of residues capable of supporting charge relay. Through this virtual electronic circuit, an enzymatic reduction in one heme is coupled to the corresponding oxidation at the linked heme, driving the overall reaction and facilitating high substrate turnover. A critical aspect of this process is the existence of paired Tyr370 residues (Fig. 2). We speculate that amino acid residues Asp141, His75, His364, His362, Tyr358, and Arg354, residues we and others have found critical to full enzyme activity, are important in this relay network. Special alterations which further separate the dimer's paired residues in inhibited and irradiated catalase mediate reduced catalatic activity and facilitate ROS formation (Fig. 3).11,36–38 In this scheme, a complete catalase structure, comprised of four monomers, requires the intertwined protein arms results in a holoenzyme comprised of two functional dimers. This scheme provides a role for the requirement for the complex linkage that is needed for full enzyme activity.
Figure 2.
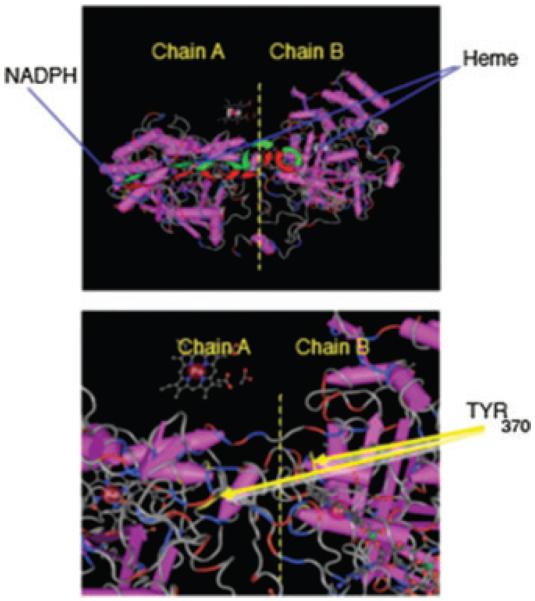
Model of proposed heme-heme and heme-NADPH linkage in human catalase. (Upper panel) Model illustrating functional dimmer in 1.5A structure of human catalase.14 (Lower panel) Position of migrating potential tyrosinate radical in adjacent protein domains. We speculate that the migration of this radical facilitates the formation of an electronic charge relay linkage associating two heme moieties in each functional dimer.13,22,26,33,36
In summary, we have devised a novel mechanism that encompasses the numerous unresolved observations stemming from years of analysis of the activity of the enzyme catalase. We speculate that this peculiar tetromeric enzyme functions as conjoined identical dimers, degrading hydrogen peroxide at an exceptional rate for an enzymatic reaction, through in an electrochemically driven process.
Acknowledgments
The study was supported by CA093798 and AR055073.
Footnotes
Conflicts of interest The authors declare no conflicts of interest.
References
- 1.Jamieson D, Chance B, Cadenas E, Boveris A. The relation of free radical production to hyperoxia. Annu. Rev. Physiol. 1986;48:703–719. doi: 10.1146/annurev.ph.48.030186.003415. [DOI] [PubMed] [Google Scholar]
- 2.Dröge W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 3.Heck DE, Kagan VE, Shvedova AA, Laskin JD. An epigrammatic (abridged) recounting of the myriad tales of astonishing deeds and dire consequences pertaining to nitric oxide and reactive oxygen species in mitochondria with an ancillary missive concerning the origins of apoptosis. Toxicology. 2005;208:259–271. doi: 10.1016/j.tox.2004.11.027. [DOI] [PubMed] [Google Scholar]
- 4.Kowaltowski AJ, de Souza-Pinto NC, Castilho RF, Vercesi AE. Mitochondria and reactive oxygen species. Free Radic. Biol. Med. 2009;47:333–343. doi: 10.1016/j.freeradbiomed.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 5.Heck DE, Vetrano AM, Mariano TM, Laskin JD. UVB light stimulates production of reactive oxygen species: unexpected role for catalase. J. Biol. Chem. 2003;278:22432–22436. doi: 10.1074/jbc.C300048200. [DOI] [PubMed] [Google Scholar]
- 6.Peus D, Meves A, Vasa RA, et al. H2O2 is required for UVB-induced EGF receptor and downstream signaling pathway activation. Free Radic. Biol. Med. 1999;27:1197–1202. doi: 10.1016/s0891-5849(99)00198-7. [DOI] [PubMed] [Google Scholar]
- 7.Rahman I, Adcock IM. Oxidative stress and redox regulation of lung inflammation in COPD. Eur. Respir. J. 2006;28:219–242. doi: 10.1183/09031936.06.00053805. [DOI] [PubMed] [Google Scholar]
- 8.Veal EA, Day AM, Morgan BA. Hydrogen peroxide sensing and signaling. Mol. Cell. 2007;26:1–14. doi: 10.1016/j.molcel.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 9.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2008;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sen CK, Roy S. Redox signals in wound healing. Biochim. Biophys. Acta. 2008;780:1348–1361. doi: 10.1016/j.bbagen.2008.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Veal EA, Day AM, Morgan BA. Hydrogen peroxide sensing and signaling. Mol. Cell. 2007;26:1–14. doi: 10.1016/j.molcel.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 12.Vetrano AM, Heck DE, Mariano TM, et al. Characterization of the oxidase activity in mammalian catalase. 2005. J. Biol. Chem. 280:35372–35381. doi: 10.1074/jbc.M503991200. [DOI] [PubMed] [Google Scholar]
- 13.Zamocky M, Furtmüller PG, Obinger C. Evolution of catalases from bacteria to humans. Antioxid Redox Signal. 2008;10:1527–1548. doi: 10.1089/ars.2008.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deisseroth A, Dounce AL. Catalase: physical and chemical properties, mechanism of catalysis, and physiological role. Physiol. Rev. 1970;50:319–375. doi: 10.1152/physrev.1970.50.3.319. [DOI] [PubMed] [Google Scholar]
- 15.Ivancich A, Jouve HM, Sartor B, Gaillard J. EPR investigation of compound I in Proteus mirabilis and bovine liver catalases: formation of porphyrin and tyrosyl radical intermediates. Biochemistry. 1997;36:9356–9364. doi: 10.1021/bi970886s. [DOI] [PubMed] [Google Scholar]
- 16.Jones P, Dunford HB. On the mechanism of compound I formation from peroxidases and catalases. J. Theor. Biol. 1977;69:457–470. doi: 10.1016/0022-5193(77)90152-7. [DOI] [PubMed] [Google Scholar]
- 17.George P. A comparison of the decomposition of hydrogen peroxide by catalase, ferrous and ferric ions, haemin and ferrous phthalocyanine. Chem. J. 1948;43:287–295. doi: 10.1042/bj0430287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kirkman HN, Gaetani GF. Mammalian catalase: a venerable enzyme with new mysteries. Trends Biochem. Sci. 2007;32:44–50. doi: 10.1016/j.tibs.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 19.Keilin D, Hartree EF. Catalase, peroxidase and metmyoglobin as catalysts of coupled peroxidatic reactions. Biochem. J. 1955;60:310–325. doi: 10.1042/bj0600310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kremer ML. Peroxidatic activity of catalase. Biochim. Biophys. Acta. 1970;198:199–209. doi: 10.1016/0005-2744(70)90052-5. [DOI] [PubMed] [Google Scholar]
- 21.Koller Zámocký M. Understanding the structure and function of catalases: clues from molecular evolution and in vitro mutagenesis. Prog. Biophys. Mol. Biol. 1999;72:19–66. doi: 10.1016/s0079-6107(98)00058-3. [DOI] [PubMed] [Google Scholar]
- 22.Ko TP, Day J, Malkin AJ, McPherson AS. Structure of orthorhombic crystals of beef liver catalase. Acta Crystallogr. 1998;55:1383–1394. doi: 10.1107/s0907444999007052. [DOI] [PubMed] [Google Scholar]
- 23.Fita I, Rossmann MG. The NADPH binding site on beef liver catalase. Proc. Natl. Acad. Sci. USA. 1984;82:1604–1608. doi: 10.1073/pnas.82.6.1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kirkman HN, Gaetani GF. Mammalian catalase: a venerable enzyme with new mysteries. Trends Biochem. Sci. 2007;32:44–50. doi: 10.1016/j.tibs.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 25.Kalko SG, Gelpí JL, Fita I, Oroczo M. Theorethical study of the mechanisms of substrate recognition by catalase. J. Am. Chem. Soc. 2001;123:9665–9672. doi: 10.1021/ja010512t. [DOI] [PubMed] [Google Scholar]
- 26.George P. The effect of the peroxide concentration and other factors on the decomposition of hydrogen peroxide by catalase. Biochem. J. 1949;44:197–205. doi: 10.1042/bj0440197. 1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones P, Suggett A. The Catalase-Hydrogen Peroxide System: kinetics of catalatic action at high substrate concentrations. Biochem. J. 1968;110:617–620. doi: 10.1042/bj1100617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chance B. The primary and secondary compounds of catalase and methyl or ethyl hydrogen peroxide; kinetics and activity. J. Biol. Chem. 1949;179:1341–1369. [PubMed] [Google Scholar]
- 29.Chance B. The reactions of catalase in the presence of the notatin system. Biochem. J. 1950;46:387–402. doi: 10.1042/bj0460387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jones DP. Radical-free biology of oxidative stress. Am. J. Physiol. Cell Physiol. 2008;295:C849–C868. doi: 10.1152/ajpcell.00283.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schäfer M, Werner S. Oxidative stress in normal and impaired wound repair. Pharmacol. Res. 2008;58:165–171. doi: 10.1016/j.phrs.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 32.Deisseroth A, Dounce AL. Catalase: physical and chemical properties, mechanism of catalysis, and physiological role. Physiol. Rev. 1970;50:319–375. doi: 10.1152/physrev.1970.50.3.319. [DOI] [PubMed] [Google Scholar]
- 33.Chelikani P, Fita I, Loewen PC. Diversity of structures and properties among catalases. Cell. Mol. Life Sci. 2004;61:192–208. doi: 10.1007/s00018-003-3206-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Putnam CD, Arvai AS, Bourne Y, Tainer JA. Active and inhibited human catalase structures: ligand and NADPH binding and catalytic mechanism. J. Mol. Biol. 2000;296:295–309. doi: 10.1006/jmbi.1999.3458. [DOI] [PubMed] [Google Scholar]
- 35.Gibbons NC, Wood JM, Rokos H, Schallreuter KU. Computer simulation of native epidermal enzyme structures in the presence and absence of hydrogen peroxide (H2O2): potential and pitfalls. J. Invest. Dermatol. 2006;126:2576–2582. doi: 10.1038/sj.jid.5700612. [DOI] [PubMed] [Google Scholar]
- 36.Andreoletti P, Mouesca JM, Gouet P, et al. Verdoheme formation in Proteus mirabilis catalase. Biochim. Biophys. Acta. 2009;1790:741–753. doi: 10.1016/j.bbagen.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 37.Chelikani P, Carpena X, Fita I, Loewen PC. An electrical potential in the access channel of catalases enhances catalysis. J. Biol. Chem. 2003;278:31290–31296. doi: 10.1074/jbc.M304076200. [DOI] [PubMed] [Google Scholar]
- 38.Gao B, Boeglin WE, Brash AR. Role of the conserved distal heme asparagine of coral allene oxide synthase (Asn137) and human catalase (Asn148): mutations affect the rate but not the essential chemistry of the enzymatic transformations. Arch. Biochem. Biophys. 2008;477:285–290. doi: 10.1016/j.abb.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wood JM, Schallreuter KU. UVA-irradiated pheomelanin alters the structure of catalase and decreases its activity in human skin. J. Invest. Dermatol. 2006;26:13–14. doi: 10.1038/sj.jid.5700051. [DOI] [PubMed] [Google Scholar]
- 40.Aronoff S. Catalase: kinetics of photooxidation. 1965;150:72–73. doi: 10.1126/science.150.3692.72. [DOI] [PubMed] [Google Scholar]