

González-Redondo et al. use PET to compare the extent of atrophy versus hypometabolism in Parkinson's disease with cognitive deficits. In patients with mild cognitive impairment, hypometabolism exceeds atrophy in several cortical regions. In patients with dementia, hypometabolism in these areas is replaced by atrophy, surrounded by zones of hypometabolism.
Keywords: Parkinson’s disease, dementia, mild cognitive impairment, FDG-PET, MRI
Abstract
The pathophysiological process underlying cognitive decline in Parkinson’s disease is not well understood. Cerebral atrophy and hypometabolism have been described in patients with Parkinson’s disease and dementia or mild cognitive impairment with respect to control subjects. However, the exact relationships between atrophy and hypometabolism are still unclear. To determine the extension and topographical distribution of hypometabolism and atrophy in the different cognitive states of Parkinson’s disease, we examined 46 patients with Parkinson’s disease (19 female, 27 male; 71.7 ± 5.9 years old; 14.6 ± 4.2 years of disease evolution; modified Hoehn and Yahr mean stage 3.1 ± 0.7). Cognitive status was diagnosed as normal in 14 patients, as mild cognitive impairment in 17 and as dementia in 15 patients. Nineteen normal subjects (eight female, 11 male; 68.1 ± 3.2 years old) were included as controls. 18F-fluorodeoxyglucose positron emission tomography and magnetic resonance imaging scans were obtained, co-registered, corrected for partial volume effect and spatially normalized to the Montreal Neurological Institute space in each subject. Smoothing was applied to the positron emission tomography and magnetic resonance imaging scans to equalize their effective smoothness and resolution (10 mm and 12 mm full-width at half-maximum and Gaussian kernel, respectively). Z-score maps for atrophy and for hypometabolism were obtained by comparing individual images to the data set of control subjects. For each group of patients, a paired Student’s t-test was performed to statistically compare the two Z-map modalities (P < 0.05 false discovery rate corrected) using the direct voxel-based comparison technique. In patients with mild cognitive impairment, hypometabolism exceeded atrophy in the angular gyrus, occipital, orbital and anterior frontal lobes. In patients with dementia, the hypometabolic areas observed in the group with mild cognitive impairment were replaced by areas of atrophy, which were surrounded by extensive zones of hypometabolism. Areas where atrophy was more extended than hypometabolism were found in the precentral and supplementary motor areas in both patients with mild cognitive impairment and with dementia, and in the hippocampus and temporal lobe in patients with dementia. These findings suggest that there is a gradient of severity in cortical changes associated with the development of cognitive impairment in Parkinson’s disease in which hypometabolism and atrophy represent consecutive stages of the same process in most of the cortical regions affected.
Introduction
Cognitive impairment is common in Parkinson’s disease, which is associated with a long-term prevalence of dementia of up to 80% (Aarsland et al., 2005b; Hely et al., 2008). Mild cognitive impairment (MCI) is a cognitive decline that is not normal for age, but where normal functional activities can be maintained (Foltynie et al., 2004; Caviness et al., 2007; Williams-Gray et al., 2007; Litvan et al., 2011). MCI and age represent the two major risk factors for the development of dementia in Parkinson’s disease (Janvin et al., 2006; Broeders et al., 2013; Pedersen et al., 2013). Studies using MRI analysed with voxel-based morphometry and PET with 18F-FDG (fluorodeoxyglucose) have shown that patients with dementia in Parkinson’s disease have extensive areas of cerebral atrophy (i.e. reduction of grey matter volume) (Burton et al., 2004; Nagano-Saito et al., 2005; Summerfield et al., 2005; Beyer et al., 2007a, b; Ibarretxe-Bilbao et al., 2008; Song et al., 2011; Melzer et al., 2012) and hypometabolism (i.e. reduced uptake of FDG) in comparison with control subjects (Peppard et al., 1992; Huang et al., 2007a, 2008; Yong et al., 2007; Hosokai et al., 2009; Liepelt et al., 2009; Garcia-Garcia et al., 2012). In patients with Parkinson’s disease with MCI, more localized atrophy in the temporal, parietal and frontal cortices and hypometabolism in the occipito-temporo-parietal junction and the frontal cortex are observed (Huang et al., 2008; Hosokai et al., 2009; Lyoo et al., 2010; Pappata et al., 2011; Garcia-Garcia et al., 2012). Simple comparisons of the results obtained in these studies point to consider that hypometabolism is more extended than atrophy both in patients with Parkinson’s disease with MCI and those with dementia in Parkinson’s disease. Interestingly, while there are no significant differences in the extent of cerebral atrophy between patients with Parkinson’s disease with MCI and cognitively normal patients with Parkinson’s disease (Apostolova et al., 2010; Dalaker et al., 2010; Hattori et al., 2012), patients with Parkinson’s disease with MCI exhibit a reduced FDG uptake in the frontal and parietal regions (Huang et al., 2008; Hosokai et al., 2009; Lyoo et al., 2010; Garcia-Garcia et al., 2012). Based on these data, we hypothesized that in the neurodegenerative process leading to dementia, the reduction of FDG uptake precedes and exceeds the loss of grey matter volume. To test this hypothesis, we directly compared the cerebral regions where atrophy and hypometabolism are located in patients with Parkinson’s disease with MCI and Parkinson’s disease with dementia . For this purpose, we generated MRI PET Z-score maps for each patient relative to normative data obtained from a sample of control subjects; this was done by following the method developed by Chételat et al. (2008) to study patients with Alzheimer’s disease. This approach permits one to integrate MRI grey matter volume and 18F-FDG-PET metabolism data from the same subject for direct comparison (Chételat et al., 2008). We show here that there is a gradient of severity in the cortical changes associated with the development of cognitive impairment in Parkinson’s disease whereby hypometabolism and atrophy could be consecutive stages of the same neurodegenerative process.
Materials and methods
Subjects
A cross-sectional study was conducted in patients with Parkinson’s disease, diagnosed according to the UK Brain Bank criteria (Hughes et al., 1992) aged over 60 years and with a disease duration of at least 10 years, as this profile represents the Parkinson’s disease population at highest risk of developing cognitive impairment (Hobson and Meara, 2004). Exclusion criteria were other neurological or major psychiatric illness (including major depression), abnormal findings in the cerebral MRI (i.e. tumour, hydrocephalus or severe vascular lesions), previous cerebral surgery or metabolic co-morbidities that could contribute to cognitive impairment. Age and sex-matched healthy control subjects were recruited from members of the Association of Blood Donors of Navarra (Spain). Cases with any history of neurological, psychiatric or major medical illness, memory complaints, scores below normal for age and education-appropriate test norms in the neuropsychological assessment, having MRI abnormalities or taking drugs with CNS effects were ruled out. The Ethics Committee for Medical Research of the University of Navarra approved the study, and all patients or legal representatives and controls provided informed consent to participate in accordance to the Declaration of Helsinki. All evaluations were performed for each subject within a 1-week period.
Neuropsychological evaluation
Global cognitive function, five cognitive domains (memory, attention, language, executive and visuospatial function), and functional state were evaluated by using an extensive battery of neuropsychological tests (Garcia-Garcia et al., 2012; González-Redondo et al., 2012) (Supplementary Table 1). All tests were administered by an experienced neuropsychologist to control subjects and patients in the ON medication state. The Movement Disorder Society criteria were used to diagnose patients with Parkinson’s disease as having dementia (Emre et al., 2007) or MCI (Litvan et al., 2012). With respect to MCI, the level II category guidelines of the Movement Disorder Society were applied (Litvan et al., 2012). This features that there was a cognitive decline reported by either the patient or informant, or observed by the neurologist, but this decline did not interfere significantly with the functional independence of the patient as evaluated with the Interview for Daily Living Activities (Teunisse and Derix, 1991) and that the patient scored >1.5 standard deviations (SD) below control values in at least two tests in the neuropsychological battery, either within a single cognitive domain or across different cognitive domains. An insidious onset of the cognitive decline from premorbid level, slow progression, impairment in at least two cognitive domains with deficits severe enough to impair activities of daily living were required for the diagnosis of dementia (Emre et al., 2007). Patients not fulfilling criteria for MCI or dementia were considered as cognitively normal Parkinson’s disease.
Image data acquisition
Magnetic resonance imaging
For each subject, a 3D T1-weighted gradient-echo sequence was acquired with a 1.5 T Siemens Symphony scan using the following parameters: 144 coronal slices, repetition time/echo time/inversion time 1900/3.36/1100, flip angle 15°, field of view 187 × 250, matrix 192 × 256 and voxel size 0.98 × 1.5 × 0.98 mm.
Positron emission tomography
Patients were studied under the effect of their usual dopaminergic regime. Other CNS drugs such as benzodiazepines, antipsychotic, antidepressant or anti-acetyl-cholinesterase treatments were withdrawn according to their pharmacological kinetics. Quetiapine was used in four patients with Parkinson’s disease with MCI and in eight patients with Parkinson’s disease with dementia because of mild visual hallucinations. Five patients with dementia were on treatment with rivastigmine. Selective serotonin reuptake inhibitors were used in three patients with normal cognition, in five patients with Parkinson’s disease with MCI and in seven patients with Parkinson’s disease with dementia because of sleep disorders, anxiety or mild depression. Benzodiazepines were given to two patients with normal cognition, three patients with Parkinson’s disease with MCI and four patients with Parkinson’s disease with dementia because of anxiety or sleep disorders. Additionally, subjects fasted overnight before PET scanning. Before injection of the radiopharmaceutical agent, blood glucose was checked and confirmed to be <120 mg/dl in all cases. After a few minutes of rest in silence and with dimmed lighting, 18F-FDG 3 (370 MBq) was injected intravenously and subjects were required to rest for 40 min in the supine position in the PET scanner bed with their eyes closed. Then 74 planes (128 × 128 matrix) were acquired with a voxel size of 2.06 × 2.06 × 2.06 mm during a 20-min scan using a Siemens ECAT EXAT HR+ scanner. A transmission scan in 3D mode for attenuation correction was performed at the end of the acquisition period. Images were reconstructed by means of a filtered back-projection method using ECAT software (version 7.4; Siemens).
Image handling and transformations
Magnetic resonance imaging
Images were segmented using the SPM8 new segmentation tool (Wellcome Department of Neurology, London, UK) (Ashburner and Friston, 2005) in Matlab 7.4 (Mathworks Inc.). Grey and white matter templates were generated from the entire image data set using the DARTEL technique (Ashburner, 2007). After an initial affine registration of the grey matter DARTEL templates to the tissue probability maps in Montreal Neurological Institute (MNI) space (Collins et al., 1994), non-linear warping was performed to the grey matter images to normalize them onto the MNI space. Finally, the normalized grey matter partitions were masked and smoothed as described in Fig. 1.
Figure 1.
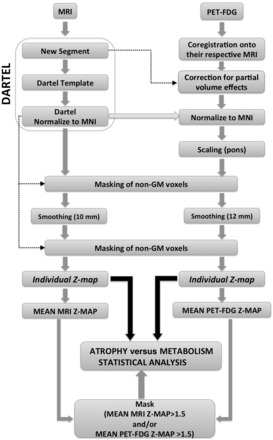
Schematic representation of the procedures for MRI and PET data handling and transformation steps. GM = grey matter.
Positron emission tomography
Images were processed using SPM8 software implemented in Matlab 7.4. FDG-PET images for each patient were co-registered with their corresponding MRI images. All images were corrected for partial volume effect using the voxel-by-voxel method proposed by Müller-Gärtner et al. (1992) combined with the modification proposed by Rousset et al. (1998) as previously used by others (Chételat et al., 2003, 2008; Villain et al., 2010). Correction was undertaken using ‘PVE-lab’ software (Quarantelli et al., 2004) including as inputs the co-registered FDG-PET and the segmented MRI images obtained from the segmentation process (Fig. 1). The spatial normalization parameters of each MRI were then applied to each partial volume corrected FDG-PET image. For every spatially normalized PET image, voxel values were normalized to pons activity (Bq/cm3) using the pons volume of interest (Nifti format) from WFU PickAtlas v3.0 (Minoshima et al., 1995; Maldjian et al., 2003, 2004; Bohnen et al., 2011; Garcia-Garcia et al., 2012). The resulting PET scans were masked and smoothed as described in Fig. 1.
Differential smoothing
A smoothing procedure was used to blur individual variations in the anatomy of gyri and to increase the signal-to-noise ratio. Different smoothing was applied to the masked spatially-normalized grey matter and PET-FDG images to equalize their effective smoothness and resolution (12 mm and 10 mm full-width at half-maximum Gaussian kernel, respectively) (Richardson et al., 1997; Van Laere and Dierckx, 2001).
Masking
All images were masked to include only grey matter voxels of interest and to prevent contamination by misclassified voxels. The mask was obtained by thresholding the grey matter DARTEL template above a value of 0.2 and then spatially normalizing this to the MNI space (Chételat et al., 2008). This binary mask was applied to both MRI and PET data sets twice (before and after smoothing), to avoid contamination of misclassified voxels by smoothing.
Z-score maps
Z-score maps for each patient and each image (MRI and PET) were respectively obtained by comparing individual images to the data set of control subjects [(−1) x (patient individual value − control mean) / control SD], for each patient and each modality (Kawachi et al., 2006; Chételat et al., 2008). Individual Z-score maps were then averaged across patients to provide the whole-brain profile of grey matter atrophy and hypometabolism, both expressed as mean Z-scores, for each particular group (cognitively normal patients with Parkinson’s disease; patients with Parkinson’s disease with MCI; patients with Parkinson’s disease with dementia).
Comparison between atrophy and hypometabolism Z-scores
For each group of patients (above) a paired Student’s t-test was performed to statistically compare the two Z-map modalities using SPM8 software (P < 0.05, false discovery rate corrected) to contrast the extent of grey matter reduction and FDG hypometabolism. A mean PET-hypometabolism map and a mean MRI-atrophy Z-score map were obtained separately for the cognitively normal patients with Parkinson’s disease, patients with Parkinson’s disease with MCI and patients with Parkinson’s disease with dementia groups. For each group, a mask image was created from the mean Z-map to consider exclusively those voxels with a mean MRI Z-map and/or a mean FDG-PET Z-map > 1.5. The obtained mask was then applied to the individual Z-maps (Fig. 1). The regional mean Z-scores for PET and MRI were calculated from masked images (mask including mean MRI and/or mean FDG-PET Z-map > 1.5), using an Automated Anatomical Labelling tool (Tzourio-Mazoyer et al., 2002) to provide quantitative insight into the regional hypometabolism and atrophy. This analysis was implemented for each group of patients with a custom-designed software running under Matlab 7.4. Gender is implicitly modelled as a confounding factor in this type of statistical design.
Statistics
Differences in the demographic and clinical characteristics between the distinct Parkinson’s disease groups and controls were analysed using Fisher’s exact test in cases of categorical variables, ANOVA with post hoc Bonferroni multiple comparison test in cases of continuous normally distributed variables, and the Kruskal-Wallis and Mann-Whitney U-tests for continuous non-parametric variables. The normal distribution of the variables was assessed using the Kolmogorov-Smirnov test. A value of P < 0.05 was considered to indicate statistical significance. Statistical analyses were performed using SPSS 15.0 software.
Results
Demographic and clinical features
Forty-six patients with Parkinson’s disease, classified as cognitively normal patients with Parkinson’s disease (n = 14), patients with Parkinson’s disease with MCI (n = 17) and patients with Parkinson’s disease with dementia (n = 15), and 19 healthy control subjects were studied. The general demographic and clinical features of the groups are summarized in Table 1. Patients with Parkinson’s disease with dementia were older on average than cognitively normal patients and controls. They also had more severe parkinsonism (significant higher Unified Parkinson’s Disease Rating Scale-III score and Hoehn and Yahr scale) than the cognitively normal patients with Parkinson’s disease and patients with Parkinson’s disease with MCI. The educational level was higher in control subjects and cognitively normal patients with Parkinson’s disease than in the patients with Parkinson’s disease with MCI and patients with Parkinson’s disease with dementia groups. The neuropsychological data of each group are summarized in Supplementary Table 1.
Table 1.
General features of patients with Parkinson’s disease and controls
| Controls n = 19 | PD-NC n = 14 | PD-MCI n = 17 | PDD n = 15 | |
|---|---|---|---|---|
| Age | 68.1 (3.2) | 69.3 (7.6) | 70.6 (4.2) | 75.4 (5.9)*,# |
| Parkinson’s disease evolution (years) | - | 14.1 (3.8) | 15.0 (4.3) | 14.8 (4.5) |
| Gender (% male) | 57.9 | 64.3 | 70.6 | 40 |
| UPDRS-III ‘OFF’ | - | 33.2 (8.7) | 32.5 (10.4) | 51.4 (10.8)*,† |
| UPDRS-III ‘ON’ | - | 13.5 (6.9) | 16.3 (7.6) | 31.2 (12.2)**,†† |
| Hoehn and Yahr | - | 2.5 (0.6) | 3 (0.7) | 3.7 (0.8)*,† |
| LEDD (mg) | - | 1031 (372) | 1158 (430) | 1039 (650) |
| Education (years) | 10.8 (4.7) | 12.8 (3.8) | 9.9 (3.2)*,# | 9.1 (2.3)*,# |
Values are mean (SD).
*P < 0.05 versus cognitively normal patients with Parkinson’s disease (PD-NC).
**P < 0.001 versus cognitively normal patients with Parkinson’s disease.
†P < 0.05 versus patients with Parkinson’s disease with MCI (PD-MCI).
††P < 0.001 versus patients with Parkinson’s disease with MCI.
#P < 0.05 versus control subjects.
LEDD = Levodopa equivalent daily dose was calculated for each patient (Grosset et al., 2004).
PDD = patients with Parkinson’s disease with dementia; UPDRS-III = Unified Parkinson’s Disease Rating Scale motor section.
Atrophy
Patients with Parkinson’s disease with MCI had bilateral (predominantly on the left) areas of atrophy in angular and middle occipital gyri, left middle frontal and precentral gyri, the left supplementary motor area and right inferior frontal gyri (Fig. 2 and Supplementary Table 2). Patients with dementia exhibited bilateral grey matter loss in the angular, middle occipital, medial-superior frontal and middle temporal gyri, and in the right inferior frontal gyrus and left hippocampus. The olfactory cortices and the left precentral and supplementary motor areas were also reduced in those with Parkinson’s disease with dementia (Fig. 3 and Supplementary Table 3). In cognitively normal patients with Parkinson’s disease, small areas of reduced grey matter volume were observed bilaterally in the angular gyri, right occipital and left frontal lobes (Supplementary Fig. 1).
Figure 2.
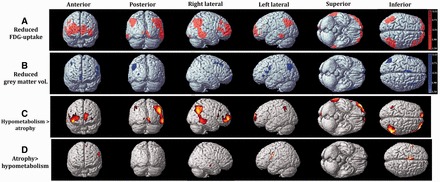
Regions with reduced FDG-uptake (A) and grey matter volume (B) in patients with Parkinson’s disease with mild cognitive impairment in comparison with control subjects using the MRI and PET Z-score maps, respectively. Cortical areas where hypometabolism exceeded atrophy (C) and where atrophy exceeded hypometabolism (D) in the comparison between MRI and PET Z-score maps.
Figure 3.

Regions with reduced FDG-uptake (A) and grey matter volume (B) in patients with Parkinson’s disease with dementia in comparison with control subjects using MRI and PET Z-score maps, respectively. Cortical areas where hypometabolism exceeded atrophy (C) and where atrophy exceeded hypometabolism (D) in the comparison between MRI and PET Z-score maps.
Hypometabolism
Patients with Parkinson’s disease with MCI exhibited bilateral reduction of FDG uptake in the parietal (angular > inferior > supramarginal > superior gyri), occipital (calcarine > middle > lingual > superior gyri) and frontal (middle-orbital > superior-medial > superior-orbital > superior > middle > inferior-orbital >inferior-opercular gyri) lobes. Small hypometabolic regions were also observed in the temporal lobe (middle and inferior gyri) (Fig. 2 and Supplementary Table 2). Patients with Parkinson’s disease with dementia exhibited hypometabolism in those areas affected in patients with Parkinson’s disease with MCI, but with a greater extension in the parietal, occipital, frontal, and temporal lobes (Fig. 3 and Supplementary Table 3) and also in the caudate and right thalamus. In cognitively normal patients with Parkinson’s disease, hypometabolism was restricted to very small areas of the angular gyrus and occipital and frontal lobes (Supplementary Fig. 1).
Comparison between atrophy and hypometabolism
Hypometabolism > atrophy
The comparison between the MRI and PET Z-scores revealed that hypometabolism was more extended than atrophy in several brain regions in patients with Parkinson’s disease with MCI and patients with dementia in Parkinson’s disease. In patients with Parkinson’s disease with MCI these areas were bilaterally distributed in the angular, inferior parietal, calcarine and superior frontal gyri, and in the right middle orbito frontal, middle frontal, middle temporal, supramarginal and lingual gyri (Fig. 2; Supplementary Table 2). In patients with dementia, these areas were more widespread in the occipital and parietal lobes and in a small region of the frontal lobe (maximal for the middle frontal gyrus) (Fig. 3; Supplementary Table 3).
Atrophy > hypometabolism
Small areas where atrophy exceeded hypometabolism were found in the precentral and supplementary motor area in patients with Parkinson’s disease with MCI and those with Parkinson’s disease with dementia, and in the right temporal lobe, left hippocampus, bilateral putamen, pallidum and left thalamus in the Parkinson’s disease with dementia group (Figs 2–4 and Supplementary Tables 2 and 3). In the Parkinson’s disease with dementia group, atrophy and hypometabolism in the caudate nucleus were overall of similar degree. Patients with normal cognition did not exhibit areas of hypometabolism > atrophy or atrophy > hypometabolism.
Figure 4.
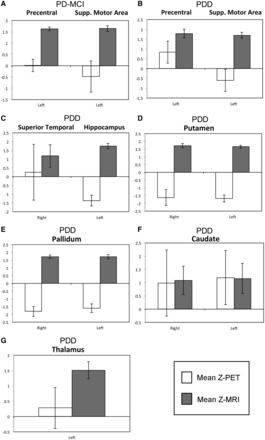
Representation of the extracted mean regional MRI and PET Z-scores in cerebral regions where atrophy exceeded hypometabolism in patients with Parkinson’s disease with mild cognitive impairment (PD-MCI) and patients with Parkinson’s disease with dementia (PDD). On the y- axis, negative values corresponds to higher FDG uptake (white) in patients than in controls and positive values denote reduced FDG uptake. For grey matter volume (grey), positive values indicate reduced grey matter volume and negative values indicate increased grey matter volume. Error bars represent 95% confidence intervals.
PET and MRI images of each group of patients were superimposed to further assess whether atrophic regions were contained in hypometabolic zones. In patients with Parkinson’s disease with dementia, regions where atrophy coincided with hypometabolism were displayed at the core of the hypometabolic areas of the occipito-parietal junction and frontal regions. In patients with Parkinson’s disease with MCI, a similar small region was found in the left occipito-parieto-temporal junction (Fig. 5).
Figure 5.
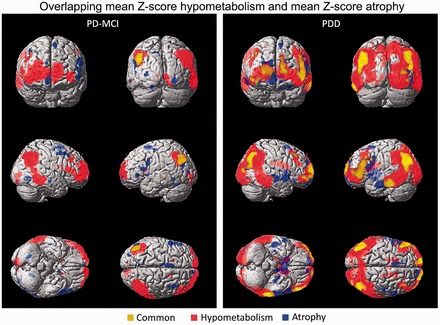
Overlapping areas of hypometabolism and atrophy in patients with Parkinson’s disease with MCI (left, PD-MCI) and dementia (right, PDD).
Discussion
We have performed a direct voxel-by-voxel comparison of MRI and PET in a cohort of patients with Parkinson’s disease from normal to dementia. This allowed us to compare the extension of grey matter volume loss and FDG uptake reduction at different cognitive stages compared with controls. Patients with Parkinson’s disease with cognitive decline have (i) regions showing only hypometabolism; (ii) regions showing an overlap of atrophy and hypometabolism; and (iii) small regions showing atrophy only. Patients with Parkinson’s disease with dementia showed large areas (occipito-parietal > frontal) of coincidental hypometabolism and atrophy which were surrounded by areas of reduced FDG uptake. In patients with Parkinson’s disease with MCI, hypometabolism was the main feature. Nevertheless, small atrophic areas were also identified in regions with hypometabolism in the left occipito-parieto-temporal junction. In addition, small areas of atrophy exceeding that of hypometabolism were observed in the precentral and supplementary motor areas of the frontal lobe in patients with Parkinson’s disease with MCI and patients with Parkinson’s disease with dementia, and in the temporal lobe, hippocampus, putamen, pallidum and thalamus exclusively in the Parkinson’s disease with dementia group. These results suggest that hypometabolism in Parkinson’s disease predates and is replaced by atrophy in a progressive and expanding manner as the cognitive state worsens. However, the relationship between changes in metabolism and atrophy is neither uniform nor complete, given that there are some regions in which atrophy seems to be independent of metabolic changes.
Strengths and limitations
We have directly compared, for the first time in patients with Parkinson’s disease with different cognitive states, the relative amounts of atrophy and hypometabolism by applying step-by-step the methodology previously used in patients with Alzheimer’s disease by Chételat et al. (2008). Patients were evaluated with a comprehensive neuropsychological battery and the cognitive diagnosis was performed using the Movement Disorder Society consensus criteria for MCI (Litvan et al., 2012) and dementia in Parkinson’s disease (Emre et al., 2007). Although the methodology here applied (Z-map scores) has not been generally applied to ascertain cerebral defects in Parkinson’s disease, our results are supported by the fact that the patterns of atrophy and hypometabolism here identified are consistent with those previously reported using standard methodology (Burton et al., 2004; Summerfield et al., 2005; Huang et al., 2008; Hosokai et al., 2009; Song et al., 2011; Weintraub et al., 2011; Garcia-Garcia et al., 2012; Melzer et al., 2012).
On the other hand, there are obvious limitations for MRI and PET techniques mostly regarding brain shape transformations in the normalization processes, which could cause to some degree a mismatch between the images obtained with MRI and PET. This is especially relevant for the regions where atrophy exceeded hypometabolism, as it cannot be excluded that the use of the ‘double-masking’ procedure causes some slight increases in atrophy Z-scores, especially in deep grey structures, such as the caudate nucleus, putamen, pallidum, thalamus or the hippocampus, even though atrophy in these structures has been reported in patients with Parkinson’s disease (Laakso et al., 1996; Camicioli et al., 2003; Burton et al., 2004; Junque et al., 2005; Nagano-Saito et al., 2005; Summerfield et al., 2005; Beyer et al., 2007a, b; Ibarretxe-Bilbao et al., 2008, Melzer et al., 2012).
Atrophy as a core of hypometabolic areas and hypometabolism
A critical question to be addressed is whether hypometabolism and atrophy are independent processes in the same regions, or conversely if they are two stages of a single process evolving in a step-wise pattern. Thus, extensive regions of hypometabolism surrounding a central area of atrophy were located bilaterally in the occipito-temporo-parietal junction and some regions of the frontal lobe in patients with Parkinson’s disease with dementia. In contrast, in patients with Parkinson’s disease with MCI, a small region of atrophy encircled by a broad hypometabolic area was only present in the left occipito-temporo-parietal junction. Moreover, MCI patients exhibited only hypometabolism in the right occipito-temporo-parietal junction and frontal pole, regions that showed atrophy surrounded by hypometabolism in patients with Parkinson’s disease with dementia. Accordingly, our data suggest that hypometabolism and atrophy are two steps of the same process initiated with a reduction of cortical glucose uptake evolving towards a decrease in grey matter volume, which seems to expand in an exocentric pattern. This is compatible with an evolving neurodegenerative process within the cortex probably paralleling cognitive decline. Thus, it is tantalizing to equate this evolution pattern with the concept of ischaemic penumbra for stroke. Here, the ‘neurodegenerative penumbra’ would imply areas where cell loss is putatively reversible. This concept requires definitive evidence in a prospective and longitudinal assessment and studies to unravel its histopathological and biochemical basis. This in turn could lead to newer and relevant therapeutic approaches.
We can only speculate the possible basis for the sequential pattern of hypometabolism exceeding atrophy in some brain regions. Synapses are the physiologically most active compartments of neurons and a dysfunction at this level can account for reduced glucose uptake. Limbic and cortical Lewy bodies are associated with dementia in Parkinson’s disease (Hurtig et al., 2000; Apaydin et al., 2002; Aarsland et al., 2005a) although this association is not always present (Halliday et al., 2008; Kalaitzakis et al., 2008a). In dementia with Lewy bodies, the majority of alpha-synuclein aggregates are located at presynaptic terminals, thus causing a pathological impact on synaptic function (Kramer and Schulz-Schaeffer, 2007) and probably on metabolism. On the other hand, a lack of function of alpha-synuclein in genetically modified mice impairs the release of glutamate from the synaptic pool (Gureviciene et al., 2007), which is necessary for the maintenance of dendrites in the postsynaptic component and synaptic connectivity (Verhage et al., 2000). In this sense, significant synaptic pathology with almost complete loss of dendritic spines at the postsynaptic level is observed in the cortex of patients with dementia with Lewy bodies (Kramer and Schulz-Schaeffer, 2007) and in patients with Parkinson’s disease with dementia (Zhan et al., 1993). Thus, a putative neuronal damage would follow the maintenance of prolonged periods of synaptic dysfunction. In addition, astrocytes have a key role in regulating synaptic glucose use (Magistretti, 2006) and failure of metabolic coupling between neurons and glia might be another potential explanation for the reduction of FDG signal.
On the other hand, cortical atrophy is associated with cell death but it may also be related to the reduced size of cell bodies and dendritic arborization or the loss of presynaptic terminals (Freeman et al., 2008). Thus, it can be hypothesized that an early step in the neurodegenerative process underlying cognitive dysfunction in Parkinson’s disease might be a synaptic dysfunction due to primary neuronal changes and/or neuron-glia decoupling detected by a reduced uptake of FDG in PET, which is the major finding in patients with Parkinson’s disease with MCI. This defect might in turn lead to a loss of dendritic arborization and presynaptic terminals, thus accounting for a reduction in grey matter volume, which is more evident in Parkinson’s disease with dementia. Moreover, other anomalies such as abnormal lipid metabolism that take place in the cortex and correlate with cognitive decline in Parkinson’s disease could also play a part in the metabolic and structural changes encountered in in vivo studies (Fabelo et al., 2011).
Atrophy exceeding hypometabolism
We identified a few regions in which atrophy does not coincide with nor is encircled by hypometabolic areas. This was the case for the precentral and supplementary motor areas of the frontal lobe, in patients with Parkinson’s disease with MCI and patients with Parkinson’s disease with dementia, and for the right temporal lobe, left hippocampus, bilateral putamen, pallidum and left thalamus in the Parkinson’s disease with dementia group. As discussed above, these findings could be related to methodological issues, especially in regions with small volume such as the pallidum, left putamen and left thalamus. Nevertheless, some alternative explanations need to be considered.
A previous pathological study described early neuronal loss in regions involved in motor control such as the presupplementary motor cortex in patients with Parkinson’s disease (MacDonald and Halliday, 2002). Accordingly, the extension of the atrophy in the precentral and supplementary motor area is similar in patients with Parkinson’s disease with MCI and those with Parkinson’s disease with dementia, suggesting that this finding is not related to cognition and therefore the causative process might be different. In contrast, only patients with Parkinson’s disease with dementia showed greater atrophy in the temporal lobe and hippocampus, putamen, pallidum and thalamus suggesting some relationship between these cortico-subcortical changes and progression of cognitive impairment. However, we do not have any direct evidence for the underlying putative mechanisms. Amyloid-β deposition in the allocortex/hippocampus and in the striatum strongly correlates with dementia in Parkinson’s disease (Jellinger and Attems, 2006; Kalaitzakis et al., 2008b) and it could be that the molecular interaction between different pathological proteins could lead to higher neuronal or synaptic loss (Kalaitzakis and Pearce, 2009). Theoretically, as shown in Alzheimer’s disease, disruption in hippocampus or temporal lobe circuitry could also lead to remote metabolic reduction in functionally connected areas but not in the hippocampus or temporal lobe themselves (Minoshima et al., 1999). However, this interpretation could be hardly applied to our subcortical findings as increased metabolism in the putamen, globus pallidus and thalamus is a feature of the parkinsonian state (Eidelberg et al., 1994; Ma et al., 2007; Teune et al., 2010) which increases linearly with progression of motor disability (Huang et al., 2007b), and atrophy of these nuclei have been reported in Parkinson’s disease with dementia (Burton et al., 2004; Nagano-Saito et al., 2005; Duncan et al., 2013). Nevertheless, it is noteworthy that the volume where atrophy exceeded hypometabolism was very small in these nuclei (<10%) and therefore its pathophysiological relevance is uncertain.
Conclusions and perspectives
Our findings suggest the existence of a severity gradient in the cortical changes associated with the development of cognitive impairment in Parkinson’s disease in which hypometabolism and atrophy are consecutive stages of the same process. In this hypothesis, hypometabolism would represent a synaptic or cellular dysfunction which, if maintained, evolves towards histological changes and neuronal death which manifests as a reduced grey matter volume. Thus, in Parkinson’s disease with dementia the greatest metabolic injury is focused on a central core of hypometabolism and atrophy surrounded by a broader area of non-atrophic hypometabolism. In MCI this pattern is less extensive, with predominance of hypometabolism, which is replaced by atrophy in the dementia stage. Thus, cognitive decline in Parkinson’s disease is heralded by an overlapping pattern of hypometabolism and atrophy, where non-atrophic hypometabolism might be considered the ‘metabolic penumbra’ that precedes the definitive cortical changes associated with atrophy and progression to dementia. We like to postulate that such ‘penumbral’ areas are putative pharmacological targets to halt cognitive decline in Parkinson’s disease. This supports the notion that recognition of the earliest cognitive changes in Parkinson’s disease may be critically important for recovering dysfunctional brain regions before the process evolves towards a definitive loss of grey matter volume and dementia. The data shown here could provide an approach to ascertain potential therapeutic interventions. Finally, regions of atrophy exceeding hypometabolism, such as the hippocampus and specially putamen, pallidum and thalamus observed only in patients with Parkinson’s disease with dementia should be interpreted cautiously.
Supplementary Material
Acknowledgements
We acknowledge Isabel Lamet for her assistance with the neuropsychological assessments and Dr Pablo Martinez-Lage for his contribution in the study of control subjects.
Glossary
Abbreviations
- FDG
fluorodeoxyglucose
- MCI
mild cognitive impairment
Funding
This study was funded in part by a grant from the Health department of Government of Navarra Government of Navarra (32/2007), by the University of Navarra-UTE agreement, Centro de Investigación Biomédica en Red sobre Enfermedades Neurodegenerativas (CIBERNED); Spain CIBERNED and by a grant from Spanish Health Institute Carlos III (FIS- PI08/1539) FIS (PI08/1539) in Spain.
Conflict of interest
Dr Rodriguez Oroz reports grants from Government of Navarra (32/2007) and FIS (ISCIII) PI08/1539, Spain, and from CIBERNED, Spain, during the conduct of the study; personal fees from UCB, Lundbeck and Medtronic and grants from Government of Navarra, Spain, Government of Basque Country, Spain, Spanish Health Institute and Eran-net Neuron. Europe, outside the submitted work. Dr Obeso reports personal fees from GSK, Lundbeck, UCB, TEVA (USA) and Boehringer Ingelheim (Mexico), grants from Spanish Science and Education Ministery and from European Union outside the submitted work; Dr Clavero reports personal fees and non-financial support from UCB, Lundbeck, and Novartis outside the submitted work. The rest of authors have no conflicts of interest concerning the research dealt with in this manuscript or outside the submitted work.
Supplementary material
Supplementary material is available at Brain online.
References
- Aarsland D, Perry R, Brown A, Larsen JP, Ballard C. Neuropathology of dementia in Parkinson's disease: a prospective, community-based study. Ann Neurol. 2005a;58:773–6. doi: 10.1002/ana.20635. [DOI] [PubMed] [Google Scholar]
- Aarsland D, Zaccai J, Brayne C. A systematic review of prevalence studies of dementia in Parkinson's disease. Mov Disord. 2005b;20:1255–63. doi: 10.1002/mds.20527. [DOI] [PubMed] [Google Scholar]
- Apaydin H, Ahlskog JE, Parisi JE, Boeve BF, Dickson DW. Parkinson disease neuropathology: later-developing dementia and loss of the levodopa response. Arch Neurol. 2002;59:102–12. doi: 10.1001/archneur.59.1.102. [DOI] [PubMed] [Google Scholar]
- Apostolova LG, Beyer M, Green AE, Hwang KS, Morra JH, Chou YY, et al. Hippocampal, caudate, and ventricular changes in Parkinson's disease with and without dementia. Mov Disord. 2010;25:687–95. doi: 10.1002/mds.22799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner J. A fast diffeomorphic image registration algorithm. Neuroimage. 2007;38:95–113. doi: 10.1016/j.neuroimage.2007.07.007. [DOI] [PubMed] [Google Scholar]
- Ashburner J, Friston KJ. Unified segmentation. Neuroimage. 2005;26:839–51. doi: 10.1016/j.neuroimage.2005.02.018. [DOI] [PubMed] [Google Scholar]
- Beyer MK, Janvin CC, Larsen JP, Aarsland D. A magnetic resonance imaging study of patients with Parkinson's disease with mild cognitive impairment and dementia using voxel-based morphometry. J Neurol Neurosurg Psychiatry. 2007a;78:254–9. doi: 10.1136/jnnp.2006.093849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer MK, Larsen JP, Aarsland D. Gray matter atrophy in Parkinson disease with dementia and dementia with Lewy bodies. Neurology. 2007b;69:747–54. doi: 10.1212/01.wnl.0000269666.62598.1c. [DOI] [PubMed] [Google Scholar]
- Bohnen NI, Koeppe RA, Minoshima S, Giordani B, Albin RL, Frey KA, et al. Cerebral glucose metabolic features of Parkinson disease and incident dementia: longitudinal study. J Nucl Med. 2011;52:848–55. doi: 10.2967/jnumed.111.089946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broeders M, de Bie RM, Velseboer DC, Speelman JD, Muslimovic D, Schmand B. Evolution of mild cognitive impairment in Parkinson disease. Neurology. 2013;81:346–52. doi: 10.1212/WNL.0b013e31829c5c86. [DOI] [PubMed] [Google Scholar]
- Burton EJ, McKeith IG, Burn DJ, Williams ED, O'Brien JT. Cerebral atrophy in Parkinson's disease with and without dementia: a comparison with Alzheimer's disease, dementia with Lewy bodies and controls. Brain. 2004;127:791–800. doi: 10.1093/brain/awh088. [DOI] [PubMed] [Google Scholar]
- Camicioli R, Moore MM, Kinney A, Corbridge E, Glassberg K, Kaye JA. Parkinson's disease is associated with hippocampal atrophy. Mov Disord. 2003;18:784–90. doi: 10.1002/mds.10444. [DOI] [PubMed] [Google Scholar]
- Caviness JN, Driver-Dunckley E, Connor DJ, Sabbagh MN, Hentz JG, Noble B, et al. Defining mild cognitive impairment in Parkinson's disease. Mov Disord. 2007;22:1272–7. doi: 10.1002/mds.21453. [DOI] [PubMed] [Google Scholar]
- Collins DL, Neelin P, Peters TM, Evans AC. Automatic 3D intersubject registration of MR volumetric data in standardized Talairach space. J Comput Assist Tomogr. 1994;18:192–205. [PubMed] [Google Scholar]
- Chételat G, Desgranges B, Landeau B, Mezenge F, Poline JB, de la Sayette V, et al. Direct voxel-based comparison between grey matter hypometabolism and atrophy in Alzheimer's disease. Brain. 2008;131:60–71. doi: 10.1093/brain/awm288. [DOI] [PubMed] [Google Scholar]
- Chételat G, Desgranges B, de la Sayette V, Viader F, Eustache F, Baron JC. Mild cognitive impairment: can FDG-PET predict who is to rapidly convert to Alzheimer’s disease? Neurology. 2003;60:1374–7. doi: 10.1212/01.wnl.0000055847.17752.e6. [DOI] [PubMed] [Google Scholar]
- Dalaker TO, Zivadinov R, Larsen JP, Beyer MK, Cox JL, Alves G, et al. Gray matter correlations of cognition in incident Parkinson's disease. Mov Disord. 2010;25:629–33. doi: 10.1002/mds.22867. [DOI] [PubMed] [Google Scholar]
- Duncan GW, Firbank MJ, O'Brien JT, Burn DJ. Magnetic resonance imaging: a biomarker for cognitive impairment in Parkinson's disease? Mov Disord. 2013;28:425–38. doi: 10.1002/mds.25352. [DOI] [PubMed] [Google Scholar]
- Eidelberg D, Moeller JR, Dhawan V, Spetsieris P, Takikawa S, Ishikawa T, et al. The metabolic topography of parkinsonism. J Cereb Blood Flow Metab. 1994;14:783–801. doi: 10.1038/jcbfm.1994.99. [DOI] [PubMed] [Google Scholar]
- Emre M, Aarsland D, Brown R, Burn DJ, Duyckaerts C, Mizuno Y, et al. Clinical diagnostic criteria for dementia associated with Parkinson's disease. Mov Disord. 2007;22:1689–707. doi: 10.1002/mds.21507. [DOI] [PubMed] [Google Scholar]
- Fabelo N, Martin V, Santpere G, Marin R, Torrent L, Ferrer I, et al. Severe alterations in lipid composition of frontal cortex lipid rafts from Parkinson's disease and incidental Parkinson's disease. Mol Med. 2011;17:1107–18. doi: 10.2119/molmed.2011.00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foltynie T, Brayne CE, Robbins TW, Barker RA. The cognitive ability of an incident cohort of Parkinson's patients in the UK. The CamPaIGN study. Brain. 2004;127:550–60. doi: 10.1093/brain/awh067. [DOI] [PubMed] [Google Scholar]
- Freeman SH, Kandel R, Cruz L, Rozkalne A, Newell K, Frosch MP, et al. Preservation of neuronal number despite age-related cortical brain atrophy in elderly subjects without Alzheimer disease. J Neuropathol Exp Neurol. 2008;67:1205–12. doi: 10.1097/NEN.0b013e31818fc72f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Garcia D, Clavero P, Gasca Salas C, Lamet I, Arbizu J, Gonzalez-Redondo R, et al. Posterior parietooccipital hypometabolism may differentiate mild cognitive impairment from dementia in Parkinson's disease. Eur J Nucl Med Mol Imaging. 2012;39:1767–77. doi: 10.1007/s00259-012-2198-5. [DOI] [PubMed] [Google Scholar]
- González-Redondo R, Toledo J, Clavero P, Lamet I, Garcia-Garcia D, Garcia-Eulate R, et al. The impact of silent vascular brain burden in cognitive impairment in Parkinson's disease. Eur J Neurol. 2012;19:1100–7. doi: 10.1111/j.1468-1331.2012.03682.x. [DOI] [PubMed] [Google Scholar]
- Grosset K, Needleman F, Macphee G, et al. Switching from ergot to nonergot dopamine agonists in Parkinson's disease: a clinical series and five-drug dose conversion table. Mov Disord. 2004;19:1370–4. doi: 10.1002/mds.20210. [DOI] [PubMed] [Google Scholar]
- Gureviciene I, Gurevicius K, Tanila H. Role of alpha-synuclein in synaptic glutamate release. Neurobiol Dis. 2007;28:83–9. doi: 10.1016/j.nbd.2007.06.016. [DOI] [PubMed] [Google Scholar]
- Halliday G, Hely M, Reid W, Morris J. The progression of pathology in longitudinally followed patients with Parkinson's disease. Acta Neuropathol. 2008;115:409–15. doi: 10.1007/s00401-008-0344-8. [DOI] [PubMed] [Google Scholar]
- Hattori T, Orimo S, Aoki S, Ito K, Abe O, Amano A, et al. Cognitive status correlates with white matter alteration in Parkinson's disease. Hum Brain Mapp. 2012;33:727–39. doi: 10.1002/hbm.21245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hely MA, Reid WG, Adena MA, Halliday GM, Morris JG. The Sydney multicenter study of Parkinson's disease: the inevitability of dementia at 20 years. Mov Disord. 2008;23:837–44. doi: 10.1002/mds.21956. [DOI] [PubMed] [Google Scholar]
- Hobson P, Meara J. Risk and incidence of dementia in a cohort of older subjects with Parkinson's disease in the United Kingdom. Mov Disord. 2004;19:1043–9. doi: 10.1002/mds.20216. [DOI] [PubMed] [Google Scholar]
- Hosokai Y, Nishio Y, Hirayama K, Takeda A, Ishioka T, Sawada Y, et al. Distinct patterns of regional cerebral glucose metabolism in Parkinson's disease with and without mild cognitive impairment. Mov Disord. 2009;24:854–62. doi: 10.1002/mds.22444. [DOI] [PubMed] [Google Scholar]
- Huang C, Mattis P, Perrine K, Brown N, Dhawan V, Eidelberg D. Metabolic abnormalities associated with mild cognitive impairment in Parkinson disease. Neurology. 2008;70:1470–7. doi: 10.1212/01.wnl.0000304050.05332.9c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Mattis P, Tang C, Perrine K, Carbon M, Eidelberg D. Metabolic brain networks associated with cognitive function in Parkinson's disease. Neuroimage. 2007a;34:714–23. doi: 10.1016/j.neuroimage.2006.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Tang C, Feigin A, Lesser M, Ma Y, Pourfar M, et al. Changes in network activity with the progression of Parkinson's disease. Brain. 2007b;130:1834–46. doi: 10.1093/brain/awm086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson's disease: a clinicopathological study of 100 cases. J Neurol Neurosurg Psychiatry. 1992;55:181–4. doi: 10.1136/jnnp.55.3.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurtig HI, Trojanowski JQ, Galvin J, Ewbank D, Schmidt ML, Lee VM, et al. Alpha-synuclein cortical Lewy bodies correlate with dementia in Parkinson's disease. Neurology. 2000;54:1916–21. doi: 10.1212/wnl.54.10.1916. [DOI] [PubMed] [Google Scholar]
- Ibarretxe-Bilbao N, Ramirez-Ruiz B, Tolosa E, Marti MJ, Valldeoriola F, Bargallo N, et al. Hippocampal head atrophy predominance in Parkinson's disease with hallucinations and with dementia. J Neurol. 2008;255:1324–31. doi: 10.1007/s00415-008-0885-8. [DOI] [PubMed] [Google Scholar]
- Janvin CC, Larsen JP, Aarsland D, Hugdahl K. Subtypes of mild cognitive impairment in Parkinson's disease: progression to dementia. Mov Disord. 2006;21:1343–9. doi: 10.1002/mds.20974. [DOI] [PubMed] [Google Scholar]
- Jellinger KA, Attems J. Does striatal pathology distinguish Parkinson disease with dementia and dementia with Lewy bodies? Acta Neuropathol. 2006;112:253–60. doi: 10.1007/s00401-006-0088-2. [DOI] [PubMed] [Google Scholar]
- Junque C, Ramirez-Ruiz B, Tolosa E, Summerfield C, Marti MJ, Pastor P, et al. Amygdalar and hippocampal MRI volumetric reductions in Parkinson's disease with dementia. Mov Disord. 2005;20:540–4. doi: 10.1002/mds.20371. [DOI] [PubMed] [Google Scholar]
- Kalaitzakis ME, Graeber MB, Gentleman SM, Pearce RK. Controversies over the staging of alpha-synuclein pathology in Parkinson's disease. Acta Neuropathol. 2008a;116:125–8. doi: 10.1007/s00401-008-0381-3. [DOI] [PubMed] [Google Scholar]
- Kalaitzakis ME, Graeber MB, Gentleman SM, Pearce RK. Striatal beta-amyloid deposition in Parkinson disease with dementia. J Neuropathol Exp Neurol. 2008b;67:155–61. doi: 10.1097/NEN.0b013e31816362aa. [DOI] [PubMed] [Google Scholar]
- Kalaitzakis ME, Pearce RK. The morbid anatomy of dementia in Parkinson's disease. Acta Neuropathol. 2009;118:587–98. doi: 10.1007/s00401-009-0597-x. [DOI] [PubMed] [Google Scholar]
- Kawachi T, Ishii K, Sakamoto S, Sasaki M, Mori T, Yamashita F, et al. Comparison of the diagnostic performance of FDG-PET and VBM-MRI in very mild Alzheimer's disease. Eur J Nucl Med Mol Imaging. 2006;33:801–9. doi: 10.1007/s00259-005-0050-x. [DOI] [PubMed] [Google Scholar]
- Kramer ML, Schulz-Schaeffer WJ. Presynaptic alpha-synuclein aggregates, not Lewy bodies, cause neurodegeneration in dementia with Lewy bodies. J Neurosci. 2007;27:1405–10. doi: 10.1523/JNEUROSCI.4564-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laakso MP, Partanen K, Riekkinen P, Lehtovirta M, Helkala EL, Hallikainen M, et al. Hippocampal volumes in Alzheimer's disease, Parkinson's disease with and without dementia, and in vascular dementia: an MRI study. Neurology. 1996;46:678–81. doi: 10.1212/wnl.46.3.678. [DOI] [PubMed] [Google Scholar]
- Liepelt I, Reimold M, Maetzler W, Godau J, Reischl G, Gaenslen A, et al. Cortical hypometabolism assessed by a metabolic ratio in Parkinson's disease primarily reflects cognitive deterioration-[18F]FDG-PET. Mov Disord. 2009;24:1504–11. doi: 10.1002/mds.22662. [DOI] [PubMed] [Google Scholar]
- Litvan I, Aarsland D, Adler CH, Goldman JG, Kulisevsky J, Mollenhauer B, et al. MDS task force on mild cognitive impairment in Parkinson's disease: critical review of PD-MCI. Mov Disord. 2011;26:1814–24. doi: 10.1002/mds.23823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litvan I, Goldman JG, Troster AI, Schmand BA, Weintraub D, Petersen RC, et al. Diagnostic criteria for mild cognitive impairment in Parkinson's disease: movement disorder society task force guidelines. Mov Disord. 2012;27:349–56. doi: 10.1002/mds.24893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyoo CH, Jeong Y, Ryu YH, Rinne JO, Lee MS. Cerebral glucose metabolism of Parkinson's disease patients with mild cognitive impairment. Eur Neurol. 2010;64:65–73. doi: 10.1159/000315036. [DOI] [PubMed] [Google Scholar]
- Ma Y, Tang C, Spetsieris PG, Dhawan V, Eidelberg D. Abnormal metabolic network activity in Parkinson's disease: test-retest reproducibility. J Cereb Blood Flow Metab. 2007;27:597–605. doi: 10.1038/sj.jcbfm.9600358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald V, Halliday GM. Selective loss of pyramidal neurons in the pre-supplementary motor cortex in Parkinson's disease. Mov Disord. 2002;17:1166–73. doi: 10.1002/mds.10258. [DOI] [PubMed] [Google Scholar]
- Magistretti PJ. Neuron-glia metabolic coupling and plasticity. J Exp Biol. 2006;209:2304–11. doi: 10.1242/jeb.02208. [DOI] [PubMed] [Google Scholar]
- Maldjian JA, Laurienti PJ, Burdette JH. Precentral gyrus discrepancy in electronic versions of the Talairach atlas. Neuroimage. 2004;21:450–5. doi: 10.1016/j.neuroimage.2003.09.032. [DOI] [PubMed] [Google Scholar]
- Maldjian JA, Laurienti PJ, Kraft RA, Burdette JH. An automated method for neuroanatomic and cytoarchitectonic atlas-based interrogation of fMRI data sets. Neuroimage. 2003;19:1233–9. doi: 10.1016/s1053-8119(03)00169-1. [DOI] [PubMed] [Google Scholar]
- Melzer TR, Watts R, MacAskill MR, Pitcher TL, Livingston L, Keenan RJ, et al. Grey matter atrophy in cognitively impaired Parkinson's disease. J Neurol Neurosurg Psychiatry. 2012;83:188–94. doi: 10.1136/jnnp-2011-300828. [DOI] [PubMed] [Google Scholar]
- Minoshima S, Frey KA, Foster NL, Kuhl DE. Preserved pontine glucose metabolism in Alzheimer disease: a reference region for functional brain image (PET) analysis. J Comput Assist Tomogr. 1995;19:541–7. doi: 10.1097/00004728-199507000-00006. [DOI] [PubMed] [Google Scholar]
- Minoshima S, Cross DJ, Foster LN, Henry TR, Kuhl DE. Discordance between traditional pathologic and energy metabolic changes in very early Alzheimer's disease. Pathophysiological implications. Ann N Y Acad Sci. 1999;893:350–2. doi: 10.1111/j.1749-6632.1999.tb07852.x. [DOI] [PubMed] [Google Scholar]
- Müller-Gärtner HW, Links JM, Prince JL, Bryan RN, McVeigh E, Leal JP. Measurement of radiotracer concentration in brain gray matter using positron emission tomography: MRI-based correction for partial volume effects. J Cereb Blood Flow Metab. 1992;12:571–83. doi: 10.1038/jcbfm.1992.81. [DOI] [PubMed] [Google Scholar]
- Nagano-Saito A, Washimi Y, Arahata Y, Kachi T, Lerch JP, Evans AC, et al. Cerebral atrophy and its relation to cognitive impairment in Parkinson disease. Neurology. 2005;64:224–9. doi: 10.1212/01.WNL.0000149510.41793.50. [DOI] [PubMed] [Google Scholar]
- Pappata S, Santangelo G, Aarsland D, Vicidomini C, Longo K, Bronnick K, et al. Mild cognitive impairment in drug-naive patients with PD is associated with cerebral hypometabolism. Neurology. 2011;77:1357–62. doi: 10.1212/WNL.0b013e3182315259. [DOI] [PubMed] [Google Scholar]
- Pedersen KF, Larsen JP, Tysnes OB, Alves G. Prognosis of mild cognitive impairment in early Parkinson disease: the Norwegian ParkWest study. JAMA Neurol. 2013;70:580–6. doi: 10.1001/jamaneurol.2013.2110. [DOI] [PubMed] [Google Scholar]
- Peppard RF, Martin WR, Carr GD, Grochowski E, Schulzer M, Guttman M, et al. Cerebral glucose metabolism in Parkinson's disease with and without dementia. Arch Neurol. 1992;49:1262–8. doi: 10.1001/archneur.1992.00530360060019. [DOI] [PubMed] [Google Scholar]
- Quarantelli M, Berkouk K, Prinster A, Landeau B, Svarer C, Balkay L. Integrated software for the analysis of brain PET/SPECT studies with partial-volume-effect correction. J Nucl Med. 2004;45:192–201. [PubMed] [Google Scholar]
- Richardson MP, Friston KJ, Sisodiya SM, Koepp MJ, Ashburner J, Free SL, et al. Cortical grey matter and benzodiazepine receptors in malformations of cortical development. A voxel-based comparison of structural and functional imaging data. Brain. 1997;120:1961–73. doi: 10.1093/brain/120.11.1961. [DOI] [PubMed] [Google Scholar]
- Rousset OG, Ma Y, Evans AC. Correction for partial volume effects in PET: principle and validation. J Nucl Med. 1998;39:904–11. [PubMed] [Google Scholar]
- Song SK, Lee JE, Park HJ, Sohn YH, Lee JD, Lee PH. The pattern of cortical atrophy in patients with Parkinson's disease according to cognitive status. Mov Disord. 2011;26:289–96. doi: 10.1002/mds.23477. [DOI] [PubMed] [Google Scholar]
- Summerfield C, Junque C, Tolosa E, Salgado-Pineda P, Gomez-Anson B, Marti MJ, et al. Structural brain changes in Parkinson disease with dementia: a voxel-based morphometry study. Arch Neurol. 2005;62:281–5. doi: 10.1001/archneur.62.2.281. [DOI] [PubMed] [Google Scholar]
- Teune LK, Bartels AL, de Jong BM, Willemsen AT, Eshuis SA, de Vries JJ, et al. Typical cerebral metabolic patterns in neurodegenerative brain diseases. Mov Disord. 2010;25:2395–404. doi: 10.1002/mds.23291. [DOI] [PubMed] [Google Scholar]
- Teunisse S, Derix MM. Measurement of activities of daily living in patients with dementia living at home: development of a questionnaire [in Dutch] Tijdschr Gerontol Geriatr. 1991;22:53–9. [PubMed] [Google Scholar]
- Tzourio-Mazoyer N, Landeau B, Papathanassiou D, Crivello F, Etard O, Delcroix N, et al. Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single-subject brain. Neuroimage. 2002;15:273–89. doi: 10.1006/nimg.2001.0978. [DOI] [PubMed] [Google Scholar]
- Van Laere KJ, Dierckx RA. Brain perfusion SPECT: age- and sex-related effects correlated with voxel-based morphometric findings in healthy adults. Radiology. 2001;221:810–7. doi: 10.1148/radiol.2213010295. [DOI] [PubMed] [Google Scholar]
- Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, et al. Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science. 2000;287:864–9. doi: 10.1126/science.287.5454.864. [DOI] [PubMed] [Google Scholar]
- Villain N, Fouquet M, Baron JC, Mézenge F, Landeau B, de La Sayette V, et al. Sequential relationships between grey matter and white matter atrophy and brain metabolic abnormalities in early Alzheimer's disease. Brain. 2010;133:3301–14. doi: 10.1093/brain/awq203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintraub D, Doshi J, Koka D, Davatzikos C, Siderowf AD, Duda JE, et al. Neurodegeneration across stages of cognitive decline in Parkinson disease. Arch Neurol. 2011;68:1562–8. doi: 10.1001/archneurol.2011.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams-Gray CH, Foltynie T, Brayne CE, Robbins TW, Barker RA. Evolution of cognitive dysfunction in an incident Parkinson's disease cohort. Brain. 2007;130:1787–98. doi: 10.1093/brain/awm111. [DOI] [PubMed] [Google Scholar]
- Yong SW, Yoon JK, An YS, Lee PH. A comparison of cerebral glucose metabolism in Parkinson's disease, Parkinson's disease dementia and dementia with Lewy bodies. Eur J Neurol. 2007;14:1357–62. doi: 10.1111/j.1468-1331.2007.01977.x. [DOI] [PubMed] [Google Scholar]
- Zhan SS, Beyreuther K, Schmitt HP. Quantitative assessment of the synaptophysin immuno-reactivity of the cortical neuropil in various neurodegenerative disorders with dementia. Dementia. 1993;4:66–74. doi: 10.1159/000107299. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.