

Abstract
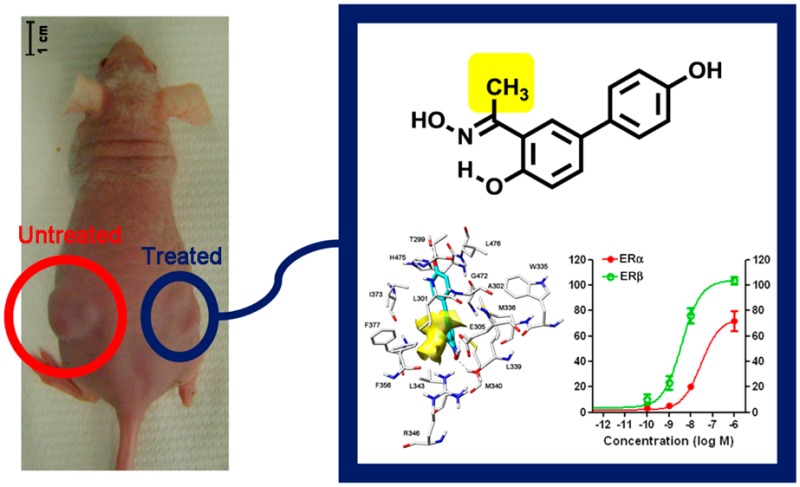
Estrogen receptor β (ERβ) selective agonists are considered potential therapeutic agents for a variety of pathological conditions, including several types of cancer. Their development is particularly challenging, since differences in the ligand binding cavities of the two ER subtypes α and β are minimal. We have carried out a rational design of new salicylketoxime derivatives which display unprecedentedly high levels of ERβ selectivity for this class of compounds, both in binding affinity and in cell-based functional assays. An endogenous gene expression assay was used to further characterize the pharmacological action of these compounds. Finally, these ERβ-selective agonists were found to inhibit proliferation of a glioma cell line in vitro. Most importantly, one of these compounds also proved to be active in an in vivo xenograft model of human glioma, thus demonstrating the high potential of this type of compounds against this devastating disease.
Introduction
Estrogen receptors (ERs) are nuclear transcription factors that mediate the physiological functions of estrogenic compounds. These receptors exert many of their actions in the nucleus, where they bind to associated DNA regulatory sequences and modulate the transcription of specific target genes. Two ER subtypes, α (ERα) and β (ERβ), are known,1 and subsequent studies have indicated the presence of up to five different ERβ isoforms (ERβ1–5) that arise from alternative splicing of the last exon coding for ERβ.2 Nevertheless, the only fully functional ERβ isoform appears to be the originally cloned 59 kDa ERβ1 isoform; hence, this is the isoform referred to simply as ERβ.
Both ERα and ERβ are widely distributed throughout the human body, where they modulate biological functions in several organ systems. In addition to their obvious control of the female reproductive system, they also play key roles in skeletal, cardiovascular, and central nervous systems. ERα plays a more prominent role in the mammary gland and uterus, on the preservation of bone homeostasis, and on the regulation of metabolism. ERβ has more pronounced effects on the central nervous system (CNS) and immune system. Moreover, the β-subtype generally counteracts the ERα-promoted cell hyperproliferation in tissues such as breast and uterus and is generally considered a tumor suppressor in these organs. This antiproliferative effect exerted by ERβ was also observed in several cancer tissues, such as, for example, breast,3 prostate,4 colon,5 renal,6 pleural mesothelioma,7 and glioma.8 In particular, the protective role of ERβ in gliomas is also supported by the fact that the incidence of developing this type of cancer is smaller in women than in men,9 and the use of exogenous estrogens further reduces this incidence.10 All this evidence suggests that selective activation of this receptor subtype may be exploited in order to obtain an antitumor effect.
Several efforts have been dedicated so far to the development of ERα- or ERβ-selective ligands.11 In particular, a great deal of attention has been focused on ERβ-selective agonists,12 which have the potential to be used as antitumor agents because they predominantly activate the β-subtype, thus being free from the undesired ERα-promoted proliferative effects on breast and uterus. However, this endeavor is particularly difficult since, in spite of a limited overall sequence identity (59%) in the ligand binding domains (LBD) of the two subtypes, the differences within the ligand binding cavities are at only two amino acid positions and consist of minor changes between hydrophobic residues. Thus, Leu384 and Met421 in ERα are replaced by Met336 and Ile373, respectively, in ERβ. A more important difference arises from the smaller volume of ERβ binding pocket when compared to that of ERα, which may be exploited in the design of ERβ-selective ligands.
We have been involved in the optimization of selective ERβ agonists that were developed by structural refinements of a monoaryl-substituted salicylaldoxime scaffold.13 In this article we describe how molecular modeling has indicated a simple way to introduce molecular variations that produced some salicylketoxime derivatives displaying significant improvements in binding affinity, transactivation activity, and subtype selectivity over their aldoxime counterparts. Furthermore, for the first time further pharmacological evaluations were conducted on our oxime-based ERβ-agonists, both in vitro, on a glioma U87 cell line, and in vivo on a murine xenograft model of the same tumor.
Results and Discussion
Molecular Modeling and Design
Some of the most potent and selective salicylaldoxime-based ERβ-selective agonists were obtained by interchanging the respective positions of the hydroxyl and oxime groups of the Salaldox A class, to produce compounds belonging to the Salaldox B class (Figure 1).13c We then decided to further analyze the complex derived by a docking procedure of the simplest member of the Salaldox B class, compound 1 (Figure 1), into ERβ-binding cavity, in order to search for additional productive interactions that might enhance ERβ binding affinity or selectivity. From this modeling analysis we realized that there is an empty hydrophobic cavity that abuts the aldoxime hydrogen atom and is delimited by Phe356 and Leu301 (Figure 1 and Figure 2A). Therefore, we decided to fill this cavity with a suitable small lipophilic group, such as a methyl, an ethyl, or a trifluoromethyl substituent so that the binding affinity of the resulting compounds could be improved. A similar docking analysis of a methyl-ketoxime analogue of 1, such as compound 2a (Figure 1), showed that this compound nicely fits into the receptor binding site and neatly places its methyl group in the lipophilic cavity (Figure 2B). It should be noted that the presence of the ketoxime portion slightly distorts the pseudocycle, which is formed because of the intramolecular H-bond of these oxime derivatives. This is due to the larger steric interaction of the methyl group compared to a hydrogen atom with the adjacent arene C–H bond. Nevertheless, the most important interactions that are typical of these types of ERβ ligands13c are maintained: in detail, both 1 and 2a have their oxime OH group participating in a highly energetic H-bond network with residues Glu305 and Arg346, and their antipodal phenolic OH group forms another H-bond with Thr299 (Figure 2A and Figure 2B).
Figure 1.
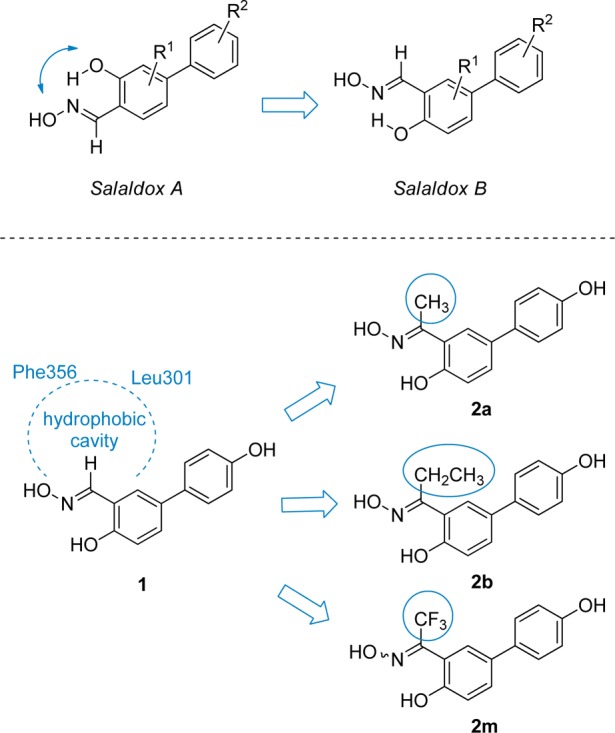
General structures of Salaldox A and Salaldox B compounds; position of the hydrophobic cavity in the complex of aldoxime 1 into ERβ; design of new ketoxime derivatives containing a methyl (2a), an ethyl (2b), or a trifluoromethyl (2m) group.
Figure 2.

Docking analysis of salicylaldoxime 1 (A) and salicylketoxime 2a (B) into ERβ binding site. The volume of the small hydrophobic cavity is represented in yellow (PDB code of the starting ERβ crystal structure is 2I0G).
In light of these theoretical results, we planned the synthesis of a selected series of methyl-, ethyl-, and trifluoromethyl-ketoximes (2a–m, Chart 1), where we could also investigate the effect due to the variation of the substitution pattern in the aryl substituent (R2, Chart 1). In particular, we wanted to verify whether the addition of an extra substituent (F, Cl, or CH3) in the 3-position or the replacement of the phenol 4-OH with a group that is exclusively able to act as a H-bond acceptor (OCH3, F, Cl) could lead to any further improvement of ERβ affinity and selectivity. In fact, that portion of the molecules binds to the OH group of a threonine residue (Thr299) of the receptor, which may act as a H-bond donor or acceptor. In fact, compounds possessing a p-OH group (2a–d, 2i, 2k, 2m, R2 = OH; Chart 1) are likely to mainly donate a H-bond to Thr299, whereas the others (2e–h, 2j, 2l, R2 = OCH3, F, Cl; Chart 1) can only function as H-bond acceptors in their interaction with the same residue. It should be noted that oximes 2a–l were obtained as single (E)-diastereoisomers, whereas trifluoromethyl-substituted oxime 2m could only be obtained as a (E/Z)-diastereoisomeric mixture (see discussion below).
Chart 1. Structures of the Newly Synthesized Salicylketoximes 2a–m.

Chemistry
The synthesis of methyl- and ethyl-ketoximes 2a–l followed a common, straightforward reaction sequence, starting from commercially available 5-bromo-2-hydroxyacetophenone (3) or 5-bromo-2-hydroxypropiophenone (4), respectively (Scheme 1). The first step involved a Pd-catalyzed cross-coupling reaction under classical Suzuki conditions.14 In detail, compounds 3 and 4 were treated with 1.2 equiv of the appropriate arylboronic acid in the presence of aqueous sodium carbonate and a solvent mixture composed of toluene/ethanol (1:1). Catalyst Pd(PPh3)4 was formed in situ by reaction of palladium acetate with a 5-fold excess of triphenylphosphine. Conventional heating at 100 °C overnight produced the desired aryl-substituted acetophenone (5e–h, 5j, 5l) and propiophenone (6, 7) derivatives in good yields. Ketoxime containing methoxy- or halogen-substituted aryl rings (2e–h, 2j, 2l) were then obtained by a direct condensation with hydroxylamine hydrochloride. On the other hand, an intermediary BBr3-promoted deprotection of compounds 5e, 5f, 5j, 5l, 6, and 7 was needed to produce hydroxyaryl-substituted ketoximes (2a–d, 2i, 2k), via the initial formation of ketones (8a–d, 8i, 8k) followed by final condensation with hydroxylamine hydrochloride.
Scheme 1. Synthesis of Salicylketoximes 2a–l.

Reagents and conditions: (a) ArB(OH)2 (1.2 equiv), Pd(OAc)2 (0.04 equiv), PPh3 (0.2 equiv), aqueous 2 M Na2CO3, 1:1 toluene/EtOH, 100 °C, 16 h; (b) NH2OH·HCl, EtOH–H2O, 50 °C, 16 h; (c) BBr3, CH2Cl2, −78 to 0 °C, 1 h.
As previously observed for structurally related salicylaldoxime derivatives,13 all methyl- and ethyl-ketoximes obtained in this manner possess the E-configuration in their oxime portion, which is also consistent with the high degree of stabilization induced by the energetic intramolecular H-bond between the oxime nitrogen atom and the adjacent OH group. These configurations were demonstrated by 1H and 13C NMR analysis of the final compounds 2a–l. In the case of the methyl-ketoximes (2a, 2c, 2e–l), the 1H NMR chemical shift values (δ) of the methyl protons (2.44 ≤ δ ≤ 2.46 ppm) closely correspond to the values reported in the literature for the E-isomer of analogous aromatic methyl-ketoximes.15 Moreover, the most significant results are given by the 13C NMR chemical shift values of the methyl carbon atom (10.80 ≤ δ ≤ 10.87 ppm), which nicely overlaps with the values observed for the E-isomer of previously reported aromatic methyl-ketoximes (11.5 ≤ δ ≤ 12.4 ppm) and which differs substantially from the values reported for the Z-isomers (21 ≤ δ ≤ 21.4 ppm).16 By analogy, an E-configuration was assigned to ethyl-ketoximes 2b,d.
The synthesis of trifluoromethyl-ketoxime 2m required a different reaction sequence starting from commercially available 5-bromo-2-methoxybenzaldehyde (9), as shown in Scheme 2. We followed a trifluoromethylation/oxidation protocol,17 which had already been applied to the synthesis of trifluoromethylketone 11, via the isolation of intermediate trifluoromethylcarbinol 10.18 Subsequent Pd-catalyzed cross-coupling reaction of bromoaryl 11 with 4-methoxyphenylboronic acid produced biphenyl derivative 12, which was deprotected with BBr3. The resulting dihydroxylated trifluoromethylketone 13 was then condensed with hydroxylamine hydrochloride, thus affording the final product 2m.
Scheme 2. Synthesis of Trifluoromethylketoxime 2m.

Reagents and conditions: (a) TMS–CF3, TBAF, THF, rt, 10 h; then HCl (4.4 M), rt, 1 h; (b) TEMPO, PhI(OAc)2 CH2Cl2, rt, 13 h; (c) 4-methoxyphenylboronic acid (1.2 equiv), Pd(OAc)2 (0.04 equiv), PPh3 (0.2 equiv), aqueous 2 M Na2CO3, 1:1 toluene/EtOH, 100 °C, 16 h; (d) BBr3, CH2Cl2, −78 to 0 °C, 1 h; (e) NH2OH·HCl, EtOH–H2O, 50 °C, 16 h.
In distinction with the ketoximes 2a–l, which were obtained as single (E)-diastereoisomers, the trifluoromethylketoxime 2m was obtained as an 8:2 E/Z-diastereoisomeric mixture (note that in 2m there is a nominal inversion of the E/Z-diastereoisomers). Assignment of the (E)-geometry to the most abundant isomer is based on a comparison of the NMR signals with a 2-hydroxyaryl-substituted trifluoromethylketoxime, which was characterized by crystallographic X-ray analysis.19 In fact, 13C NMR spectra of 2m display peaks of the major isomer corresponding to the CF3 at 122.23 (quartet, 1JC–F = 273.0 Hz) and to the oxime carbon atom at 146.23 (quartet, 2JC–F = 33.0 Hz), which compare favorably to the corresponding peaks reported in the literature for the (E)-isomer of 1-(2,4-dihydroxy-3,5-dipropylphenyl)-2,2,2-trifluoroethanone oxime: 123.1 (quartet, 1JC–F = 274.5 Hz), 148.3 (quartet, 2JC–F = 27.9 Hz).19 This peculiar outcome of the condensation reaction of trifluoromethyl-ketone 13 with hydroxylamine is probably dictated by the substantial stereoelectronic repulsion that takes place when the OH and CF3 groups are placed on the same side, as in the (Z)-isomer (Figure 3).
Figure 3.
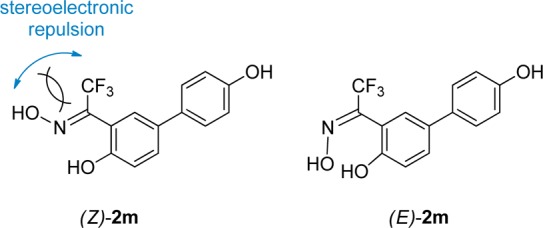
Stereoelectronic repulsion occurring in the (Z)-diastereoisomer of 2m, which favors the formation of (E)-2m.
Unfortunately, we were not able to separate the two isomers, and therefore, compound 2m was submitted as a 8:2 (E/Z)-diastereoisomeric mixture to the receptor binding assays reported below.
Biological Evaluation
The binding affinity of ketoximes 2a–l for ERα and ERβ was measured by a radiometric competitive binding assay by using previously reported methods.20,21 The relative binding affinity (RBA) values for the newly reported compounds, together with that previously obtained for reference aldoxime 1,13b are summarized in Table 1. RBA values are reported as percentage (%) of that of estradiol, which is set at 100%.
Table 1. Relative Binding Affinitiesa of Aldoxime 1 and Ketoximes 2a–l for the Estrogen Receptors α and β.
| ligand | hERα (%) | hERβ (%) | β/α ratio |
|---|---|---|---|
| estradiol | (100) | (100) | 1 |
| 1b | 0.064 ± 0.016 | 2.6 ± 0.6 | 41 |
| 2a | 0.54 ± 0.03 | 46 ± 14 | 85 |
| 2b | 3.0 ± 0.6 | 46 ± 9 | 15 |
| 2c | 0.16 ± 0.00 | 12 ± 3 | 75 |
| 2d | 1.9 ± 0.5 | 49 ± 5 | 26 |
| 2e | <0.005 | 0.011 ± 0.000 | >2 |
| 2f | <0.005 | <0.005 | |
| 2g | <0.005 | 0.015 ± 0.003 | >3 |
| 2h | <0.005 | 0.099 ± 0.030 | >20 |
| 2i | 0.10 ± 0.02 | 1.6 ± 0.1 | 16 |
| 2j | <0.005 | <0.005 | |
| 2k | 0.033 ± 0.008 | 1.1 ± 0.1 | 33 |
| 2l | <0.005 | <0.005 | |
| 2m | 0.036 ± 0.006 | 0.76 ± 0.10 | 21 |
Determined by a competitive radiometric binding assay with [3H]estradiol. Preparations of purified, full-length human ERα and ERβ (PanVera) were used; see Experimental Section. Values are reported as the mean ± the range or SD of two or more independent experiments. The Kd of estradiol for ERα is 0.2 nM and for ERβ is 0.5 nM. Ki values for the new compounds can be readily calculated by using the formula Ki = (Kd[estradiol]/RBA) × 100.
See ref (13b).
The most important finding from these binding measurements is the confirmation of our initial hypothesis derived from the computer-aided drug design. In fact, when a methyl group is inserted onto the oxime portion of aldoxime 1, a general improvement of the binding affinity of the resulting ketoxime 2a is observed, which is particularly evident for ERβ. In fact, 2a displays a 17-fold improvement in ERβ-binding affinity and a 2-fold improvement in ERβ-selectivity over its nonmethylated counterpart 1. It should be noted that the ERβ-RBA value of 45.7% observed for 2a corresponds to a Ki of 1.1 nM, thus confirming the remarkably high affinity of this compound for the β-subtype. An enlargement of the oxime alkyl substituent, from a methyl to an ethyl group, produces a compound (2b), which preserves an excellent affinity for ERβ but also gains some affinity for ERα, thus resulting in an ERβ selectivity that is less than that of 2a. The introduction of a m-fluorine atom into the 4-hydroxyphenyl group of compounds 2a and 2b, respectively, produced compounds 2c and 2d, which did not display any significant improvements over their nonfluorinated counterparts. Rather, a marked loss of affinity for ERβ was observed for methyl-ketoxime 2c. Furthermore, when a m-methyl group (2i) or a m-chlorine atom (2k) was analogously inserted in the structure of 2a, an even more dramatic reduction of the binding to ERβ was observed. As previously observed for other oxime derivatives,13 the presence of a p-methoxy group into the aryl substituent of these ketoxime derivatives always compromises the receptor binding affinities of the resulting compounds (2e, 2f, 2j, and 2l). On the other hand, the replacement of the p-hydroxy group with a fluorine (2g) or chlorine atom (2h) restores a certain, though minimal, affinity for ERβ. Overall, these binding assays confirm that the ERβ ligand cavity may profitably host methyl- (2a) and an ethyl- (2b) ketoxime portion, although the highest β-selectivity is obtained with the former, and that the 4-hydroxyphenyl substituent still constituted an ideal moiety for an efficient binding to ERβ.
In order to evaluate the binding disposition of these derivatives in ERα, the most interesting compounds 2a and 2b were also analyzed for their interaction with this receptor subtype (Figure 4). As already reported,13b the interaction of Thr299 with the 4′-hydroxyl of 2a and 2b is only possible in ERβ because only in this subtype is there enough space for the phenol group to reach Thr299, because of its proximity to Met336, one of the two nonconserved residues that in ERα is replaced by bulkier Leu384. As shown in Figure 3, the docking results showed that for both compounds 2a and 2b the phenolic OH interacts in the ERα with the Glu305–Arg346–water hydrogen bonding system, the pseudocycle/oxime system does not show any important interactions, and the methyl (2a)/ethyl (2b) substituents show lipophilic interactions with Phe404, Phe425, and Leu428. This binding disposition is very similar to that hypothesized for compound 1,13b and the lipophilic interactions of the methyl/ethyl group may explain the increase in ERα affinity associated with ketoximes 2a and 2b, when compared to that of aldoxime 1. Nevertheless, the ERβ-binding affinities of 2a increase to a larger extent (17-fold) when compared to its improvement in binding to ERα (8-fold), and therefore, the newly synthesized ketoxime 2a display a substantial enhancement of both affinity and selectivity for ERβ.
Figure 4.
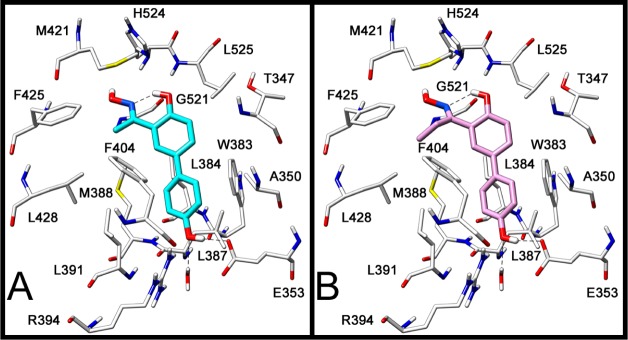
Docking analysis of 2a (A) and 2b (B) into ERα (PDB code of the starting ERα crystal structure is 2I0J).
The compounds showing the highest levels of β-selectivity in the receptor binding assays (2a and 2c) were assayed for transcriptional activity through ERα and ERβ by two different methods, and estradiol was always used as the reference receptor activator. The first method was a reporter gene transfection assay, which was conducted in human endometrial (HEC-1) cells, using expression plasmids for either full-length human ERα or ERβ and an estrogen-responsive luciferase reporter gene system.22,23 These assays (Figure 5, Table 2) showed that estradiol (E2) has a 2.4-fold preference in favor of ERα in terms of transcriptional potency (EC50[ERα] = 0.16 nM vs EC50[ERβ] = 0.38 nM), as has been widely noted. We had previously reported13b that aldoxime 1 is also a potent ERβ full agonist, with an EC50 of 10 nM, but it also stimulated ERα with an EC50 of 17 nM. New ketoxime 2a displayed significant improvements over aldoxime 1, in terms of both potency on ERβ (EC50[ERβ] = 3.97 nM) and subtype-selectivity (EC50[ERα] = 29.3 nM). Ketoxime 2c proved to suffer from a ∼4-fold reduction in its potency on ERβ (EC50[ERβ] = 14.7 nM) when compared to 2a, although an even more pronounced loss of activity on ERα (EC50[ERα] = 194 nM) elevates its β-selectivity.
Figure 5.
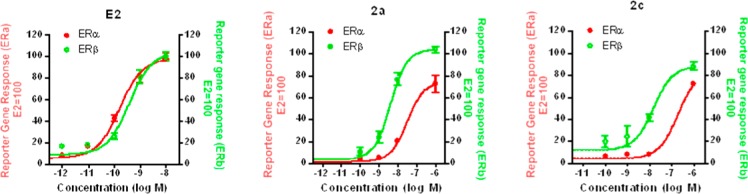
Dose–response curves for transcriptional activation by estradiol (E2), 2a, and 2c through ERα (red line) and ERβ (green line) in the reporter gene transfection assay. Human endometrial cancer (HEC-1) cells were transfected with expression vectors for ERα or ERβ and an (ERE)2-pS2-luc reporter gene and were treated for 24 h with estradiol, 2a, or 2c at the concentrations indicated. Luciferase activity was expressed relative to β-galactosidase activity from an internal control plasmid. The maximal activity with 1 nM E2 was set at 100%. Values are the mean and SD of triplicate determinations.
Table 2. Reporter Gene Transfection Assay in Human Endometrial (HEC-1) Cells: Transcriptional Activities of Estradiol and Compounds 2a and 2ca.
| hERα |
hERβ |
|||||
|---|---|---|---|---|---|---|
| ligand | EC50 (nM) | RTP (%) | EC50 (nM) | RTP (%) | EC50(β/α) ratio | RTP(β/α) ratio |
| estradiol | 0.16 ± 0.03 | 100 | 0.38 ± 0.09 | 100 | 0.42 | 1 |
| 2a | 29.3 ± 7.6 | 0.546 | 3.97 ± 0.76 | 9.62 | 7.4 | 18 |
| 2c | 194 ± 98.6 | 0.083 | 14.7 ± 5.2 | 2.59 | 13 | 31 |
To compare the ER subtype transcriptional potencies of these compounds with their subtype binding affinities in a more proper way, we calculated their relative transcriptional potency (RTP) values from their EC50 values according to the formula RTP = (EC50(estradiol)/EC50(ligand)) × 100 (RTP of estradiol = 100). These RTP values provide an estimate of transcriptional potency relative to that of estradiol and, therefore, are more appropriate to compare with their binding affinities, which are also measured relative to estradiol as RBA. By these metrics (Tables 1 and 2), compound 2a has an RBA(β/α) ratio of 84 and an RTP(β/α) ratio of 18, and compound 2c has an RBA(β/α) ratio of 72 and an RTP(β/α) ratio of 31. It is worth noting that differences in ERβ-selectivity in terms of transcriptional potency vs binding affinity may be ascribed to changes in the manner in which the ERα- and ERβ-ligand complexes interact with numerous cellular coregulators, which act as modulators of ligand potency. Therefore, when measured relative to estradiol, most of the ERβ affinity preference of these compounds is actually maintained in their ERβ transcriptional potency preference.
In addition to the reporter gene assays, we also examined the regulation of two endogenous genes, the progesterone receptor (PR), which is mostly activated through ERα, and otubain 2 (OTUB2), which is stimulated largely through ERβ.24−26 We studied the activation of these genes in MCF-7 breast cancer cells containing either ERα only, or ERα and ERβ (Figure 5 and Table 3).
Table 3. Activation of Endogenous Genes in Human Breast Cancer (MCF-7) Cells Containing Only ERα or Both ERα and ERβ: Transcriptional Activities of Estradiol and Compounds 2a and 2ca.
| hERα |
hERα+β |
|||||
|---|---|---|---|---|---|---|
| ligand | EC50 (nM) | RTP (%) | EC50 (nM) | RTP (%) | EC50((α + β)/α) ratio | RTP((α + β)/α) ratio |
| estradiol | 0.0084 ± 0.0060 | 100 | 0.30 ± 0.20 | 100 | 0.028 | 1 |
| 2a | 83.8 ± 17.0 | 0.0095 | 12.7 ± 5.0 | 2.35 | 6.58 | 247 |
| 2c | 2090 ± 1781 | 0.0004 | 35.0 ± 20.0 | 0.857 | 59.7 | 2140 |
As we have observed before,24,25 the PR gene (Figure 6, red curves) is effectively stimulated by estradiol (E2) in cells with only ERα (solid red curve), with this stimulation being somewhat less in cells containing ERα and ERβ (dashed red curve). By contrast, the OBUT2 gene (green curves) requires the presence of ERβ (ERα plus ERβ cells, dashed green curve) to be stimulated effectively, the response being very minimal in cells with only ERα (solid green curve). The OTUB2 gene is also stimulated in ERα + ERβ cells with the two ERβ-selective ketoximes (2a and 2c; green dashed curves), though at a somewhat lowered potency, and as expected, no stimulation of OTUB2 with these compounds was seen in cells with only ERα (solid green curves). While there was some stimulation of PR by these compounds, this required high concentrations (the red curves were right shifted compared to that for E2; see also Table 3).
Figure 6.
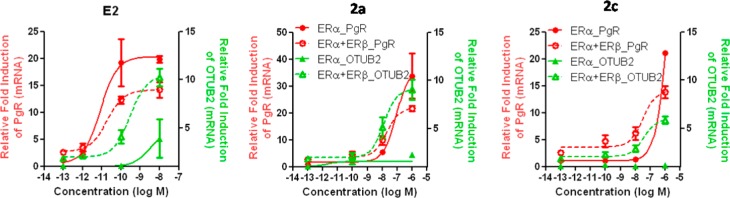
Dose–response curves for transcriptional activation of endogenous genes by estradiol (E2), 2a, and 2c. The response of progesterone receptor (PR, red curves), a gene activated predominantly through ERα, and otubain 2 (OTUB2, green curves), a gene activated predominantly through ERβ, was measured by qPCR at 24 h after exposure of human breast cancer (MCF-7) cells containing only ERα (solid curves) or both ERα and ERβ (dashed curves) to compounds at the indicated concentrations. Values are the mean ± SD of triplicate determinations. For details, see ref (24).
Again, as was the case with the reporter gene assays (Figure 5, Table 2), comparisons of potencies in transcription assays with binding affinities require that the EC50 values from the transcription assay be referenced to the values of E2, expressed as RTP values (Table 3). Clearly, the RTP values of compounds 2a and 2c are far greater for the ERβ-mediated response (OTUB2 activation in ERα + ERβ cells) than the ERα-mediated response (PR activation in ERα only cells). The RTP (α + β)/α ratios, in particular, highlight the very high ERβ selectivity of these compounds.
We then wanted to evaluate a possible application of our most promising ERβ agonists as antitumor agents. Among the beneficial effects of estrogens, a possible protective role in the progression of gliomas has been reported.27 In fact, this deadly disease has a significantly lower incidence in reproductive-aged females than in males. Recently, a naturally occurring ERβ agonist, liquiritigenin, was shown to be active in vitro against U87 glioma cells and also in a murine model of the same disease.8 Therefore, we decided to test the ability of our best ketoximes 2a and 2c, together with aldoxime 1 and liquiritigenin, a major component in licorice root extracts, to block proliferation of glioma (U87), colon (LoVo, HCT), and breast (MDA-MB-231, MCF7, SKBR3) cancer cells. The results are reported in Table 4.
Table 4. Effects on Cell Growth (IC50, μM) of Human Glioma (U87), Colon (LoVo, HCT), and Breast (MDA-MB-231, MCF7, SkBr3) Cancer Cells by Compounds 1, 2a, and 2c and Liquiritigenina.
| IC50 (μM) |
||||||
|---|---|---|---|---|---|---|
| compd | U87 | LoVo | HCT | MDA231 | MCF7 | SkBr3 |
| liquiritigenin | 88.3 ± 7.8 | 77.7 ± 2.9 | 51.4 ± 2.9 | 167 ± 9 | >250 | 175.6 ± 8.9 |
| 1 | 45.8 ± 4.2 | 36.7 ± 2.7 | 27.4 ± 4.1 | 12.4 ± 1.4 | 16.4 ± 2.6 | 96.2 ± 3.3 |
| 2a | 35.0 ± 1.7 | 38.1 ± 1.6 | 17.2 ± 2.9 | 9.3 ± 1.2 | 8.7 ± 1.4 | 86.3 ± 5.6 |
| 2c | 68.8 ± 6.5 | 27.1 ± 2.8 | 15.2 ± 2.0 | 10.0 ± 3.1 | 2.7 ± 0.7 | 92.4 ± 4.8 |
IC50: inhibitory concentration causing a 50% reduction in cell growth, in μM. Mean values ± SD calculated from at least two triplicate cytotoxicity experiments (see Experimental Section).
As for the U87 glioma cells, in our hands, liquiritigenin displayed an IC50 value of 88.3 μM in this assay, whereas all our oxime-based ERβ-agonists proved to be more potent than this. In particular, ketoxime 2a, which was also found to be the most potent ERβ agonist in both the reporter (Table 2) and the endogenous (Table 3) gene assays, demonstrated the highest potency as an antiproliferative agent from this series, with an IC50 value of 35.0 μM. In addition, since activation of ERβ is also known to exert an antiproliferative effect in colorectal and breast tumors,10 we extended our in vitro screening to colon (LoVo, HCT) and breast (MDA231, MCT7) cancer cell lines. In all these cell lines, oxime derivatives 1, 2a, and 2c still displayed the most potent inhibition of proliferation when compared to liquiritigenin. Finally, we included in our study the ERβ-negative breast cancer cell line SkBr328 in order to verify the contribution of ERβ activation to the antiproliferative effect of these compounds. Notably, the activities of the oxime-based ERβ agonists 1, 2a, and 2c against SkBr3 cells were significantly lower than their activities against all the other ERβ-positive cancer cells. For a more significant evaluation of the involvement of ERβ in the antiproliferative effect of these compounds, we can restrict our comparison to only breast cancer cells. Then the differences in the IC50 values found in ERβ-positive MDA231 and MCF7 cells (2.7–16.4 μM range) and those in ERβ-negative SkBr3 cells (86.3–96.2 μM) are even more evident, with 6- to 34-fold reductions of antiproliferative activities in the ERβ-negative cancer cells. Of course, we cannot exclude the involvement of other mechanisms in the inhibition of proliferation by compounds 1, 2a, and 2c, since some activity is also noted in SkBr3 cells. Nevertheless, these results confirm a highly significant involvement of the activation of ERβ in the antiproliferative effects of these compounds.
To complete the evaluation of our compounds 1, 2a, and 2c against glioma, we carried out an in vivo study using U87 glioma cells grown as xenografts in nude mice, an in vivo model that has already been used to evaluate the antiglioma effect of liquiritigenin.8 After tumors reached a measurable size, the indicated compounds (10 mg/kg/mouse/day) were administered for 14 days, and the tumor volume was measured with a caliper. After 14 days of treatment no signs of animal weight loss were observed (see Table S1, Supporting Information). As shown in Figure 7, ketoxime 2a produced a statistically significant reduction of the tumor volume when compared to control vehicle, whereas the effects of 1 and 2c were negligible. This is not surprising since compound 2a, when compared to the other two compounds, displayed the highest ERβ-binding affinity and ERβ-transcriptional activation, as well as the most potent antiproliferative activity against U87 cells. Therefore, these in vivo results indicate that our most potent ERβ-agonist, 2a, is effective in reducing the progression of tumor growth in both an in vitro and in vivo model of this glioma.
Figure 7.

Tumor volume (normalized to control, which was set at 1) of subcutaneous implanted U87 cells in nude mice after 14 days of treatment with 10 mg/kg compounds. The values are derived from ≥5 mice, and bars show SE.
Conclusion
In summary, molecular modeling studies drove us to the rational design of some new ketoxime derivatives by introducing small alkyl groups on the oxime carbon atom of previously developed salicylaldoxime-based ERβ agonists. Some of the newly synthesized compounds displayed remarkably high subtype-selective binding affinities for ERβ, which is unprecedented for this chemical class of ligands. In particular, these compounds proved to behave as full agonists on ERβ and to activate transcription of reporter genes and endogenous genes, highlighting their very high ERβ potency selectivity. Finally, we could then demonstrate that one of these ketoxime derivatives efficiently inhibited tumor progression of ERβ-expressing human glioma cells, both in vitro and in vivo. These results further extend the therapeutic potential of ERβ-selective agonists.
Experimental Section
General Procedures and Materials
All solvents and chemicals were used as purchased without further purification. Chromatographic separations were performed on silica gel columns by flash (Kieselgel 40, 0.040–0.063 mm; Merck) or gravity column (Kieselgel 60, 0.063–0.200 mm; Merck) chromatography. Reactions were followed by thin-layer chromatography (TLC) on Merck aluminum silica gel (60 F254) sheets that were visualized under a UV lamp. Evaporation was performed in vacuo (rotating evaporator). Sodium sulfate was always used as the drying agent. Proton (1H) and carbon (13C) NMR spectra were obtained with a Varian Gemini 200 MHz spectrometer using the indicated deuterated solvents. Chemical shifts are given in parts per million (ppm) (δ relative to residual solvent peak for 1H and 13C). Electron impact (EI, 70 eV) mass spectra were obtained on a HP-5988A mass spectrometer. High-resolution mass spectrometry (HRMS) analysis was performed using a Waters Quattro II quadrupole–hexapole–quadrupole liquid chromatography/mass spectrometry apparatus (Waters, Milford, MA) equipped with an electrospray ionization source. Purity of the final compounds 2a–m was determined by high performance liquid chromatography (HPLC) on a Waters SunFire RP 18 (3.0 mm × 150 mm, 5 μm) column (Waters, Milford, MA, www.waters.com) using a Beckmann SystemGold instrument consisting of chromatography 125 solvent module and a 166 UV detector set at 254 and 300 nm, and injection volume was 30 μL (see below for details about mobile phase and flow rate.The purity was always ≥95%, unless otherwise specified. Yields refer to isolated and purified products derived from nonoptimized procedures.
Preparation of Ketones 5e–h, 5j, 5l, 6, 7, and 12. General Procedure
A solution of Pd(OAc)2 (50 mg, 0.22 mmol) and triphenylphosphine (294 mg, 1.12 mmol) in ethanol (12 mL) and toluene (12 mL) was stirred at rt under nitrogen for 10 min. After that period, 5.8 mmol of commercially available 5-bromo-2-hydroxyacetophenone (3) or 5-bromo-2-hydroxypropiophenone (4), a 2 M aqueous solution of Na2CO3 (13 mL), and the appropriate arylboronic acid (1.2 equiv) were sequentially added. The resulting mixture was heated at 100 °C in a sealed vial under nitrogen overnight. After being cooled to rt, the mixture was diluted with water and extracted with EtOAc. The combined organic phase was dried and concentrated. The crude product was purified by flash chromatography over silica gel. Elution with n-hexane/EtOAc (95:5 to 8:2) afforded the desired ketone intermediates. The same procedure was applied to 1-(5-bromo-2-methoxyphenyl)-2,2,2-trifluoroethanone (11), which was synthesized as previously reported.18
Preparation of O-Deprotected Ketones 8a–d, 8i, 8k, and 13. General Procedure
A solution of pure ketones 5e, 5f, 5j, 5l, 6, 7, and 12 (0.90 mmol) in anhydrous CH2Cl2 (11 mL) was cooled to −78 °C and treated dropwise with a 1.0 M solution of BBr3 CH2Cl2 (3 mL) under nitrogen. The mixture was left under stirring at the same temperature for 5 min and then at 0 °C for 1 h. The mixture was then diluted with water and extracted with ethyl acetate. The organic phase was dried and concentrated. The crude product was purified by flash chromatography over silica gel. Elution with n-hexane/EtOAc (8:2 to 7:3) afforded the desired O-deprotected ketones.
Preparation of Final Products 2a–m. General Procedure
A solution of pure ketones 5e–h, 5j, 5l, 8a–d, 8i, 8k, 13 (1.2 mmol) in ethanol (20 mL) was treated with a solution of hydroxylamine hydrochloride (257 mg, 3.72 mmol) in water (4 mL), and the mixture was heated to 50 °C for 16 h. After being cooled to rt, part of the solvent was removed under vacuum, and the mixture was diluted with water and extracted with EtOAc. The organic phase was dried and evaporated to afford a crude residue that was purified by column chromatography (n-hexane/ethyl acetate 7:3) to afford the desired ketoxime derivatives.
(E)-1-(6-Hydroxy-3-(4-hydroxyphenyl)phenyl)ethanone Oxime (2a)
White solid; yield 50% from 8a; mp 188 °C. 1H NMR (acetone-d6) δ (ppm): 2.44 (s, 3H), 6.88–6.94 (m, 3H), 7.44–7.52 (m, 3H), 7.70 (d, 1H, J = 2.2 Hz), 8.36 (exchangeable s, 1H), 10.70 (exchangeable s, 1H), 11.45 (exchangeable s, 1H). 13C NMR (acetone-d6) δ (ppm): 10.80, 116.43 (2C), 117.98, 119.91, 126.32, 128.32 [2C], 129.06, 132.74, 132.83, 157.45, 157.69, 159.18. MS m/z 243 (M+, 71), 242 (M+ – H, 100), 227 (M+ – H2O, 31). HPLC analysis: retention time = 1.550 min; peak area, 98.97% (254 nm), 99.17% (300 nm); eluent A, NH4OAc solution (10 mM); eluent B, CH3CN; a gradient was formed from 50% to 95% of B in 10 min and held at 95% for 15 min; flow rate was 1.0 mL/min. HRMS (ESI): m/z calculated for C14H13NO3 + H+ [M + H+]: 244.0974. Found: 244.0971.
(E)-1-(6-Hydroxy-3-(4-hydroxyphenyl)phenyl)propan-1-one Oxime (2b)
White solid; yield 37% from 8b; mp 171 °C. 1H NMR (acetone-d6) δ (ppm): 1.24 (t, 3H, J = 7.6 Hz), 3.02 (q, 2H, J = 7.6 Hz), 6.87–6.96 (m, 3H), 7.43–7.50 (m, 3H), 7.69 (d, 1H, J = 2.2 Hz), 11.52 (exchangeable s, 1H). 13C NMR (acetone-d6) δ (ppm): 11.60, 18.39, 116.42 [2C], 118.28, 118.48, 126.00, 128.36 [2C], 129.20, 132.82, 132.90, 157.41, 158.24, 164.03. MS m/z 257 (M+, 100), 239 (M+ – H2O, 39). HPLC analysis: retention time = 0.983 min; peak area, 99.01% (254 nm), >99.95% (300 nm); eluent A, NH4OAc solution (10 mM); eluent B, CH3CN; a gradient was formed from 70% to 95% of B in 10 min and held at 95% for 10 min; flow rate was 1.0 mL/min. HRMS (ESI): m/z calculated for C15H15NO3 + H+ [M + H+]: 258.1130. Found: 258.1129.
(E)-1-(6-Hydroxy-3-(3-fluoro-4-hydroxyphenyl)phenyl)ethanone Oxime (2c)
White solid; yield 91% from 8c; mp 193 °C. 1H NMR (acetone-d6) δ (ppm): 2.46 (s, 3H), 6.94 (d, 1H, J = 8.6 Hz), 7.05 (dd, 1H, J = 9.0, 8.6 Hz 1H), 7.30 (ddd, 1H, J = 8.4, 2.2, 0.9 Hz), 7.40 (dd, 1H, J = 12.6, 2.2 Hz), 7.50 (dd, 1H, J = 8.4, 2.4 Hz), 7.74 (d, 1H, J = 2.4 Hz), 11.52 (exchangeable s, 1H). 13C NMR (acetone-d6) δ (ppm): 10.87, 114.74 (d, J = 19.2 Hz), 118.13, 118.93, 120.01, 123.26 (d, J = 2.7 Hz), 126.56, 129.18, 131.51, 133.89 (d, J = 6.4 Hz), 144.61 (d, J = 12.8 Hz), 152.54 (d, J = 239.0 Hz), 158.71, 159.26. MS m/z 261 (M+, 100), 243 (M+ – H2O, 51), 228 (M+ – H2O – CH3, 57). HPLC analysis: retention time = 10.100 min; peak area, >99.95% (254 nm), 99.91% (300 nm); eluent A, NH4OAc solution (10 mM); eluent B, CH3CN; a gradient was formed from 5% to 80% of B in 10 min and held at 95% for 10 min; flow rate was 0.7 mL/min. HRMS (ESI): m/z calculated for C14H12FNO3 + H+ [M + H+]: 262.0879. Found: 262.0880.
(E)-1-(6-Hydroxy-3-(3-fluoro-4-hydroxyphenyl)phenyl)propan-1-one Oxime (2d)
White solid; yield 45% from 8d; mp 172 °C. 1H NMR (acetone-d6) δ (ppm): 1.24 (t, 3H, J = 7.6 Hz), 3.04 (q, 2H, J = 7.6 Hz), 6.95 (d, 1H, J = 8.4 Hz), 7.05 (dd, 1H, J = 9.0, 8.4 Hz 1H), 7.30 (ddd, 1H, J = 8.4, 2.2, 0.9 Hz), 7.39 (dd, 1H, J = 12.5, 2.2 Hz), 7.50 (dd, 1H, J = 8.4, 2.2 Hz), 7.73 (d, 1H, J = 2.2 Hz), 8.70 (exchangeable bs, 1H), 10.61 (exchangeable bs, 1H), 11.58 (exchangeable s, 1H). 13C NMR (acetone-d6) δ (ppm): 11.60, 18.41, 114.76 (d, J = 18.3 Hz), 118.42, 118.62, 118.97, 123.30 (J = 2.7 Hz), 126.22, 129.31, 131.60, 133.96 (d, J = 6.4 Hz), 144.62 (d, J = 12.8 Hz), 152.50 (d, J = 239.9 Hz), 158.74, 164.11. MS m/z 275 (M+, 100), 259 (M+ – O, 18), 257 (M+ – H2O, 12). HPLC analysis: retention time = 10.533 min; peak area, 98.25% (254 nm), 98.99% (300 nm); eluent A, NH4OAc solution (10 mM); eluent B, CH3CN; a gradient was formed from 5% to 80% of B in 10 min and held at 95% for 10 min; flow rate was 0.7 mL/min. HRMS (ESI): m/z calculated for C15H14FNO3 + H+ [M + H+]: 276.1036. Found: 276.1034.
(E)-1-(6-Hydroxy-3-(4-methoxyphenyl)phenyl)ethanone Oxime (2e)
White solid; yield 74% from 5e; mp 164 °C. 1H NMR (acetone-d6) δ (ppm): 2.45 (s, 3H), 3.83 (s, 3H), 6.94 (d, 1H, J = 8.6 Hz), 6.99 (AA′XX′, 2H, JAX = 8.8 Hz, JAA′/XX′ = 2.6 Hz), 7.49 (dd, 1H, J = 8.4, 2.4 Hz), 7.57 (AA′XX′, 2H, JAX = 8.8 Hz, JAA′/XX′ = 2.6 Hz), 7.73 (d, 1H, J = 2.4 Hz), 10.71 (exchangeable s, 1H), 11.48 (exchangeable s, 1H). 13C NMR (acetone-d6) δ (ppm): 10.83, 55.59, 115.00 [2C], 118.06, 119.95, 126.45, 128.27 [2C], 129.18, 132.39, 133.94, 157.88, 159.15, 159.74. MS m/z 257 (M+, 90), 239 (M+ – H2O, 34), 224 (M+ – H2O – CH3, 100). HPLC analysis: retention time = 1.583 min; peak area, >99.95% (254 nm), 99.00% (300 nm); eluent A, NH4OAc solution (10 mM); eluent B, CH3CN; a gradient was formed from 70% to 95% of B in 10 min and held at 95% for 10 min; flow rate was 1.0 mL/min.
(E)-1-(6-Hydroxy-3-(3-fluoro-4-methoxyphenyl)phenyl)ethanone Oxime (2f)
White solid; yield 68% from 5f; mp 171 °C. 1H NMR (acetone-d6) δ (ppm): 2.46 (s, 3H), 3.91 (s, 3H), 6.95 (d, 1H, J = 8.4 Hz), 7.18 (t, 1H, J = 8.9 Hz), 7.37–7.48 (m, 2H), 7.52 (dd, 1H, J = 8.6, 2.4 Hz), 7.76 (d, 1H, J = 2.2 Hz), 10.76 (exchangeable s, 1H), 11.60 (exchangeable s, 1H). 13C NMR (acetone-d6) δ (ppm): 10.87, 56.61, 114.74 (d, J = 19.3 Hz), 114.87, 118.19, 120.03, 123.05 (d, J = 3.7 Hz), 126.69, 129.29, 131.19, 134.71 (d, J = 6.4 Hz), 147.49 (d, J = 10.1 Hz), 153.29 (d, J = 243.6 Hz), 158.33, 159.32. MS m/z 275 (M+, 100), 257 (M+ – H2O, 53). HPLC analysis: retention time = 1.517 min; peak area, >99.95% (254 nm), >99.95% (300 nm); eluent A, NH4OAc solution (10 mM); eluent B, CH3CN; a gradient was formed from 70% to 95% of B in 10 min and held at 95% for 10 min; flow rate was 1.0 mL/min. HRMS (ESI): m/z calculated for C15H14FNO3 + H+ [M + H+]: 276.1036. Found: 276.1037.
(E)-1-(6-Hydroxy-3-(4-fluorophenyl)phenyl)ethanone Oxime (2g)
White solid; yield 53% from 5g; mp 203 °C. 1H NMR (acetone-d6) δ (ppm): 2.45 (s, 3H), 6.96 (d, 1H, J = 8.4 Hz), 7.19 (double AA′XX′, 2H, 3JHF-o = 9.0 Hz, JAX = 8.8 Hz, JAA′/XX′ = 2.6 Hz), 7.52 (dd, 1H, J = 8.4, 2.2 Hz), 7.67 (double AA′XX′, 2H, 4JHF-m = 5.3 Hz, JAX = 8.8 Hz, JAA′/XX′ = 2.6 Hz), 7.77 (d, 1H, J = 2.2 Hz), 10.76 (exchangeable s, 1H), 11.57 (exchangeable s, 1H). 13C NMR (acetone-d6) δ (ppm): 10.85, 116.18 (d, 2C, J = 22.0 Hz), 118.20, 120.03, 126.98, 129.11 (d, 2C, J = 7.3 Hz), 129.57, 131.58, 137.92, 158.43, 159.28, 162.83 (d, J = 243.5 Hz). MS m/z 245 (M+, 100), 227 (M+ – H2O, 40). HPLC analysis: retention time = 1.817 min; peak area, 99.50% (254 nm), 97.90% (300 nm); eluent A, NH4OAc solution (10 mM); eluent B, CH3CN; a gradient was formed from 70% to 95% of B in 10 min and held at 95% for 10 min; flow rate was 1.0 mL/min. HRMS (ESI): m/z calculated for C14H12FNO2 + H+ [M + H+]: 246.0930. Found: 246.0931.
(E)-1-(6-Hydroxy-3-(4-chlorophenyl)phenyl)ethanone Oxime (2h)
White solid; yield 60% from 5h; mp 207 °C. 1H NMR (acetone-d6) δ (ppm): 2.46 (s, 3H), 6.98 (d, 1H, J = 8.4 Hz), 7.45 (AA′XX′, 2H, JAX = 8.6 Hz, JAA′/XX′ = 2.3 Hz), 7.55 (dd, 1H, J = 8.6, 2.3 Hz), 7.69 (AA′XX′, 2H, JAX = 8.6 Hz, JAA′/XX′ = 2.3 Hz), 7.80 (d, 1H, J = 2.2 Hz), 10.76 (exchangeable s, 1H), 11.61 (exchangeable s, 1H). 13C NMR (acetone-d6) δ (ppm): 10.87, 118.33 [2C], 120.15, 127.04, 128.89 [2C], 129.58 [2C], 131.24, 132.99, 140.27, 158.772, 159.28. MS m/z 261 (M+, 100), 243 (M+ – H2O, 45). HPLC analysis: retention time = 2.483 min; peak area, 98.79% (254 nm), 98.05% (300 nm); eluent A, NH4OAc solution (10 mM); eluent B, CH3CN; a gradient was formed from 70% to 95% of B in 10 min and held at 95% for 10 min; flow rate was 1.0 mL/min. HRMS (ESI): m/z calculated for C14H12ClNO2 + H+ [M + H+]: 262.0635. Found: 262.0634.
(E)-1-(6-Hydroxy-3-(4-hydroxy-3-methylphenyl)phenyl)ethanone Oxime (2i)
White solid; yield 60% from 8i; mp 209 °C. 1H NMR (acetone-d6) δ (ppm): 2.26 (s, 3H), 2.44 (s, 3H), 6.88 (d, 1H, J = 8.1 Hz), 6.91 (d, 1H, J = 8.4 Hz), 7.28 (dd, 1H, J = 8.2, 2.4 Hz), 7.38 (d, 1H, J = 2.4 Hz), 7.46 (dd, 1H, J = 8.4, 2.2 Hz), 7.69 (d, 1H, J = 2.2 Hz), 8.19 (exchangeable s, 1H), 10.67 (exchangeable s, 1H), 11.42 (exchangeable s, 1H). 13C NMR (acetone-d6) δ (ppm): 10.85, 16.35, 115.82, 117.95, 119.84, 125.30, 125.58, 126.31, 129.13, 129.73, 132.88, 132.95, 155.42, 157.62, 159.28. MS m/z 257 (M+, 100), 239 (M+ – H2O, 33). HPLC analysis: retention time = 1.000 min; peak area, >99.95% (254 nm), >99.95% (300 nm); eluent A, NH4OAc solution (10 mM); eluent B, CH3CN; a gradient was formed from 70% to 95% of B in 10 min and held at 95% for 10 min; flow rate was 1.0 mL/min. HRMS (ESI): m/z calculated for C15H15NO3 + H+ [M + H+]: 258.1130. Found: 258.1133.
(E)-1-(6-Hydroxy-3-(4-methoxy-3-methylphenyl)phenyl)ethanone Oxime (2j)
White solid; yield 50% from 5j; mp 154 °C. 1H NMR (acetone-d6) δ (ppm): 2.23 (s, 3H), 2.44 (s, 3H), 3.86 (s, 3H), 6.90–6.99 (m, 2H), 7.39–7.43 (m, 2H), 7.48 (dd, 1H, J = 8.4, 2.4 Hz), 7.72 (d, 1H, J = 2.4 Hz), 10.69 (exchangeable s, 1H), 11.46 (exchangeable s, 1H). 13C NMR (acetone-d6) δ (ppm): 10.87, 16.46, 55.77, 111.21, 118.02, 119.92, 125.67, 126.47, 127.20, 129.26, 129.49, 132.66, 133.63, 157.83, 157.86, 159.28. MS m/z 271 (M+, 100), 253 (M+ – H2O, 21), 238 (M+ – H2O – CH3, 54). HPLC analysis: retention time = 2.133 min; peak area, 99.17% (254 nm), 98.93% (300 nm); eluent A, NH4OAc solution (10 mM); eluent B, CH3CN; a gradient was formed from 70% to 95% of B in 10 min and held at 95% for 10 min; flow rate was 1.0 mL/min. HRMS (ESI): m/z calculated for C16H17NO3 + H+ [M + H+]: 272.1287. Found: 272.1284.
(E)-1-(6-Hydroxy-3-(3-chloro-4-hydroxyphenyl)phenyl)ethanone Oxime (2k)
White solid; yield 50% from 8k; mp 208 °C. 1H NMR (acetone-d6) δ (ppm): 2.46 (s, 3H), 6.94 (d, 1H, J = 8.6 Hz), 7.08 (d, 1H, J = 8.4 Hz), 7.44 (dd, 1H, J = 8.6, 2.2 Hz), 7.49 (dd, 1H, J = 8.4, 2.2 Hz), 7.62 (d, 1H, J = 2.2 Hz), 7.74 (d, 1H, J = 2.2 Hz), 8.80 (exchangeable bs, 1H), 10.60 (exchangeable bs, 1H), 11.52 (exchangeable s, 1H). 13C NMR (acetone-d6) δ (ppm): 10.87, 117.88, 118.17, 120.03, 121.48, 126.58, 126.93, 128.49, 129.24, 131.30, 134.45, 152.76, 158.23, 159.30. MS m/z 277 (M+, 90), 259 (M+ – H2O, 100). HPLC analysis: retention time = 1.050 min; peak area, >99.95% (254 nm), >99.95% (300 nm); eluent A, NH4OAc solution (10 mM); eluent B, CH3CN; a gradient was formed from 70% to 95% of B in 10 min and held at 95% for 10 min; flow rate was 1.0 mL/min. HRMS (ESI): m/z calculated for C14H12ClNO3 + H+ [M + H+]: 278.0584. Found: 278.0582.
(E)-1-(6-Hydroxy-3-(3-chloro-4-methoxyphenyl)phenyl)ethanone Oxime (2l)
White solid; yield 59% from 5l; mp 178 °C. 1H NMR (acetone-d6) δ (ppm): 2.46 (s, 3H), 3.94 (s, 3H), 6.95 (d, 1H, J = 8.6 Hz), 7.18 (d, 1H, J = 8.4 Hz), 7.49–7.59 (m, 2H), 7.67 (d, 1H, J = 1.8 Hz), 7.77 (d, 1H, J = 2.0 Hz), 10.73 (exchangeable s, 1H), 11.55 (exchangeable s, 1H). 13C NMR (acetone-d6) δ (ppm): 10.87, 56.61, 113.67, 118.20, 120.10, 123.14, 126.69, 126.84, 128.69, 129.29, 131.02, 135.05, 154.93, 158.35, 159.23. MS m/z 291 (M+, 100), 273 (M+ – H2O, 36), 258 (M+ – H2O – CH3, 91). HPLC analysis: retention time = 1.883 min; peak area, >99.95% (254 nm), 99.83% (300 nm); eluent A, NH4OAc solution (10 mM); eluent B, CH3CN; a gradient was formed from 70% to 95% of B in 10 min and held at 95% for 10 min; flow rate was 1.0 mL/min. HRMS (ESI): m/z calculated for C15H14ClNO3 + H+ [M + H+]: 292.0740. Found: 292.0737.
(E/Z)-1-(6-Hydroxy-3-(4-hydroxyphenyl)phenyl)-2,2,2-trifluoroethanone Oxime (2m)
White solid; yield 68% (unresolved 8:2 E/Z-mixture) from 13; mp 187 °C. 1H NMR (acetone-d6; E/Z mixture, asterisk denotes minor isomer peaks) δ (ppm): 6.91 (AA′XX′, 2H, JAX = 8.6 Hz, JAA′/XX′ = 2.5 Hz), 7.02* (d, 1H, J = 8.4 Hz), 7.05 (d, 1H, J = 8.5 Hz), 7.40 (d, 1H, J = 2.1 Hz), 7.44 (AA′XX′, 2H, JAX = 8.6 Hz, JAA′/XX′ = 2.6 Hz), 7.55 (dd, 1H, J = 8.6, 2.4 Hz), 8.36 (exchangeable bs, 1H). 1H NMR (CD3OD; E/Z mixture, asterisk denotes minor isomer peaks) δ (ppm): 6.83 (AA′XX′, 2H, JAX = 8.7 Hz, JAA′/XX′ = 2.5 Hz), 6.88* (d,1H, J = 8.5 Hz), 6.93 (d,1H, J = 8.5 Hz), 7.21 (d,1H, J = 2.2 Hz), 7.30* (d,1H, J = 2.4 Hz), 7.35 (AA′XX′, 2H, JAX = 8.7 Hz, JAA′/XX′ = 2.5 Hz), 7.46 (dd, 1H, J = 8.5, 2.4 Hz). 13C NMR (acetone-d6; E/Z mixture, asterisk denotes minor isomer peaks) δ (ppm): 116.55, 116.58 [2C], 117.29, 122.23 (q, 1JC–F = 273.0 Hz), 128.08, 128.40 [2C], 129.55*, 130.10*, 130.16, 132.33*, 132.39*, 133.59, 133.60, 146.23 (q, 2JC–F = 33.0 Hz), 154.95, 157.65. HRMS (ESI): m/z calculated for C14H10F3NO3 + H+ [M + H+]: 298.0691. Found: 298.0686.
Modeling
The crystal structure of ERα (PDB code 2I0J) and ERβ (PDB code 2I0G)29 was taken from the Protein Data Bank.30 After addition of hydrogen atoms, the two proteins complexed with their reference inhibitor were minimized using AMBER 9 software31 and parm03 force field at 300 K. The two complexes were placed in a rectangular parallelepiped water box. An explicit solvent model for water, TIP3P, was used, and the complexes were solvated with a 10 Å water cap. Sodium ions were added as counterions to neutralize the system. Two steps of minimization were then carried out; in the first stage, we kept the protein fixed with a position restraint of 500 kcal/(mol·Å2) and we solely minimized the positions of the water molecules. In the second stage, we minimized the entire system through 5000 steps of steepest descent followed by conjugate gradient (CG) until a convergence of 0.05 kcal/(Å·mol). The two ligands were built using Maestro32 and were minimized by means of Macromodel33 in a water environment using the CG method until a convergence value of 0.05 kcal/(Å·mol), using the MMFFs force field and a distance-dependent dielectric constant of 1.0. Automated docking was carried out by means of the AUTODOCK 4.0 program;34 Autodock Tools35 was used in order to identify the torsion angles in the ligands, add the solvent model, and assign the Kollman atomic charges to the protein. The ligand charge was calculated using the Gasteiger method. In order to prevent the loss of the intramolecular H-bond of the pseudocycle/oxime system, during the docking we blocked the torsions involved in this intramolecular bond. The regions of interest used by Autodock were defined by considering SERBA-129 into both receptors as the central group; in particular, a grid of 50, 40, and 46 points in the x, y, and z directions was constructed centered on the center of the mass of this compound. A grid spacing of 0.375 Å and a distance-dependent function of the dielectric constant were used for the energetic map calculations. By use of the Lamarckian genetic algorithm, the docked compounds were subjected to 100 runs of the Autodock search, using 500 000 steps of energy evaluation and the default values of the other parameters. Cluster analysis was performed on the results using an rms tolerance of 1.0 Å, and the best docked conformation was used for the analysis. For the docking of compound 2b into ERβ, F356 was considered as a flexible residue. The reported docking procedure has recently been indirectly validated by the deposition of the crystal structure of ERα complexed with 2-chloro-3′-fluoro-3-[(E)-(hydroxyimino)methyl]biphenyl-4,4′-diol (PDB code 4IWF).36 Before the deposition of this structure, in 2011 we reported a docking analysis of this compound into ERα;13c superimposing the docking with the experimental results, the proposed binding mode was correctly predicted, as the two compounds showed a root-mean-square deviation of their disposition of 1.0 Å.
Relative Binding Affinity Assay
Relative binding affinities were determined by competitive radiometric binding assays with 2 nM [3H]E2 as tracer, as a modification of methods previously described.20,21 The source of ER was purified full-length human ERα and ERβ purchased from Pan Vera/Invitrogen (Carlsbad, CA). Incubations were done at 0 °C for 18–24 h, and hydroxyapatite was used to absorb the purified receptor–ligand complexes (human ERs).21 The binding affinities are expressed as relative binding affinity (RBA) values, where the RBA of estradiol is 100%; under these conditions, the Kd of estradiol for ERα is ∼0.2 nM, and for ERβ it is 0.5 nM. The determination of these RBA values is reproducible in separate experiments with a CV of 0.3, and the values shown represent the average ± range or SD of two or more separate determinations.
Reporter Gene Assays
The procedures used for the ERα and ERβ-responsive reporter gene assays in HEC-1 cells have been fully described in prior publications.22,23
Endogenous Gene Assays
The procedures used for the assay of ERα and ERβ-responsive endogenous gene in MCF-7 cells containing ERα only, ERβ only, or ERα + ERβ have been fully described in prior publications.24−26
Cell Viability Assay
U87-MG cells were purchased from Sigma and maintained at 37 °C in a humidified atmosphere containing 5% CO2 accordingly to the supplier. Cells (103) were plated in 96-well culture plates. The day after seeding, vehicle or compounds were added at different concentrations to the medium at a concentration ranging from 1000 to 0.1 μM. Cell viability was measured after 96 h according to the supplier (Promega, G7571) with a Tecan F200 instrument. IC50 values were calculated from logistical dose response curves. Averages and standard errors were obtained from three different experiments.
Nude Mice
All animal experiments were approved by the Ethical Committee for Animal Experimentation (CESA) and performed in accordance with the institution guidelines. For xenograft tumor assays, 2 × 106 U87 cells were mixed with 30% of Matrigel and implanted subcutaneously into the flanks of 6-week-old female nude mice. Once tumors reached measurable size, mice were treated with specify drug (10 mg/kg) subcutaneously once every other day for 14 days (n ≥ 5). Tumor volume was measured with a caliper instrument and calculated by using the formula 1/2(length × width2). Body weight was measured at weekly intervals to monitor drug toxicity.
Acknowledgments
Intramural funding support from the University of Pisa (to F.M.) and support from the National Institutes of Health (Grants PHS R01CA025836 and R01DK015556 to J.A.K.) are gratefully acknowledged. We thank Dr. Giorgio Placanica of the University of Pisa and Dr. Sung Hoon Kim of the University of Illinois for technical assistance in the analysis of the products and Dr. Yan Jiang of the University of Illinois for conducting some of the cell-based assays.
Glossary
Abbreviations Used
- ER
estrogen receptor
- CNS
central nervous system
- LBD
ligand binding domain
- E2
estradiol
- RBA
relative binding affinity
- RTP
relative transcriptional potency
- ERE
estrogen response element
- PR
progesterone receptor
- OTUB2
otubain 2
- qPCR
quantitative real-time polymerase chain reaction
- SD
standard deviation
- ppm
parts per million
- NMR
nuclear magnetic resonance
- HPLC
high performance liquid chromatography
- TLC
thin layer chromatography
- rt
room temperature
- MS
mass spectrometry
- EI
electron impact
- HRMS
high-resolution mass spectrometry
- s
singlet
- d
doublet
- dd
double doublet
- ddd
double double doublet
- t
triplet
- q
quartet
- m
multiplet
- bs
broad signal
- CG
conjugate gradient
- MMFF
Merck molecular force field
Supporting Information Available
HPLC chromatograms, mass spectra, analytical data of intermediate compounds, docking of 2b into ERβ binding site, time-based body weight analysis of treated animals. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Dahlman-Wright K.; Cavailles V.; Fuqua S. A.; Jordan V. C.; Katzenellenbogen J. A.; Korach K. S.; Maggi A.; Muramatsu M.; Parker M. G.; Gustafsson J.-Å. International Union of Pharmacology. LXIV. Estrogen Receptors. Pharmacol. Rev. 2006, 58, 773–781. [DOI] [PubMed] [Google Scholar]
- Leung Y.-K.; Mak P.; Hassan S.; Ho S.-M. Estrogen receptor (ER)-β isoforms: a key to understanding ER-β signalling. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 13162–13167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang E. C.; Frasor J.; Komm B.; Katzenellenbogen B. S. Impact of estrogen receptor beta on gene networks regulated by estrogen receptor alpha in breast cancer cells. Endocrinology 2006, 147, 4831–4842. [DOI] [PubMed] [Google Scholar]
- McPherson S. J.; Hussain S.; Balanathan P.; Hedwards S. L.; Niranjan B.; Grant M.; Chandrasir U. P.; Toivanen R.; Wang Y.; Taylor R. A.; Risbridger G. P. Estrogen receptor-β activated apoptosis in benign hyperplasia and cancer of the prostate is androgen independent and TNFα mediated. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 3123–3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman J.; Edvardsson K.; Lindberg K.; Zhao C.; Williams C.; Ström A.; Gustafsson J.-Å. Tumor repressive functions of estrogen receptor β in SW480 colon cancer cells. Cancer Res. 2009, 69, 6100–6106. [DOI] [PubMed] [Google Scholar]
- Yu C. P.; Ho J. Y.; Huang Y. T.; Cha T. L.; Sun G. H.; Yu D. S.; Chang F. W.; Chen S. P.; Hsu R. J. Estrogen inhibits renal cell carcinoma cell progression through estrogen receptor-β activation. PLoS One 2013, 8, e56667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinton G.; Thomas W.; Bellini P.; Manente A. G.; Favoni R. E.; Harvey B. J.; Mutti L.; Moro L. Estrogen receptor β exerts tumor repressive functions in human malignant pleural mesothelioma via EGFR inactivation and affects response to gefitinib. PLoS One 2010, 5, e14110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sareddy G. R.; Nair B. C.; Gonugunta V. K.; Zhang Q.-G.; Brenner A.; Brann D. W.; Tekmal R. R.; Vadlamudi R. K. Therapeutic significance of estrogen receptor β agonists in gliomas. Mol. Cancer Ther. 2012, 11, 1174–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll R. S.; Zhang J.; Dashner K.; Sar M.; Black P. M. Steroid hormone receptors in astrocytic neoplasms. Neurosurgery 1995, 37, 496–503. [DOI] [PubMed] [Google Scholar]
- a Huang K.; Whelan E. A.; Ruder A. M.; Ward E. M.; Deddens J. A.; Davis-King K. E.; Carreón T.; Waters M. A.; Butler M. A.; Calvert G. M.; Schulte P. A.; Zivkovich Z.; Heineman E. F.; Mandel J. S.; Morton R. F.; Reding D. J.; Rosenman K. D.; Reproductive factors and risk of glioma in women. Cancer Epidemiol., Biomarkers Prev. 2004, 13, 1583–1588. [PubMed] [Google Scholar]; b Hatch E. E.; Linet M. S.; Zhang J.; Fine H. A.; Shapiro W. R.; Selker R. G.; Black P. M.; Inskip P. D. Reproductive and hormonal factors and risk of brain tumors in adult females. Int. J. Cancer 2005, 114, 797–805. [DOI] [PubMed] [Google Scholar]
- a Paterni I.; Granchi C.; Katzenellenbogen J. A.; Minutolo F. Estrogen receptors alpha (ERα) and beta (ERβ): subtype-selective ligands and clinical potential. Steroids 2014, 90, 13–29. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Nilsson S.; Gustafsson J.-Å. Estrogen receptors: therapies targeted to receptor subtypes. Clin. Pharmacol. Ther. 2011, 89, 44–55. [DOI] [PubMed] [Google Scholar]
- Minutolo F.; Macchia M.; Katzenellenbogen B. S.; Katzenellenbogen J. A. Estrogen receptor β ligands: recent advances and biomedical applications. Med. Res. Rev. 2011, 31, 364–442. [DOI] [PubMed] [Google Scholar]
- a Minutolo F.; Bellini R.; Bertini S.; Carboni I.; Lapucci A.; Pistolesi L.; Prota G.; Rapposelli S.; Solati F.; Tuccinardi T.; Martinelli A.; Stossi F.; Carlson K. E.; Katzenellenbogen B. S.; Katzenellenbogen J. A.; Macchia M. Monoaryl-substituted salicylaldoximes as ligands for estrogen receptor β. J. Med. Chem. 2008, 51, 1344–1351. [DOI] [PubMed] [Google Scholar]; b Minutolo F.; Bertini S.; Granchi C.; Marchitiello T.; Prota G.; Rapposelli S.; Tuccinardi T.; Martinelli A.; Gunther J. R.; Carlson K. E.; Katzenellenbogen J. A.; Macchia M. Structural evolutions of salicylaldoximes as selective agonists for estrogen receptor β. J. Med. Chem. 2009, 52, 858–867. [DOI] [PubMed] [Google Scholar]; c Bertini S.; De Cupertinis A.; Granchi C.; Bargagli B.; Tuccinardi T.; Martinelli A.; Macchia M.; Gunther J. R.; Carlson K. E.; Katzenellenbogen J. A.; Minutolo F. Selective and potent agonists for estrogen receptor beta derived from molecular refinements of salicylaldoximes. Eur. J. Med. Chem. 2011, 46, 2453–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyaura N.; Suzuki A. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar]
- Ngwerume S.; Camp J. E. Synthesis of highly substituted pyrroles via nucleophilic catalysis. J. Org. Chem. 2010, 75, 6271–6274. [DOI] [PubMed] [Google Scholar]
- Moehrle H.; Wehefritz B.; Steigel A. Stereochemistry of α-aminoacetophenone oximes, study of solvent effects. Tetrahedron 1987, 43, 2255–2260. [Google Scholar]
- Wayman K. A.; Sammakia T. O-Nucleophilic amino alcohol acyl-transfer catalysts: the effect of acidity of the hydroxyl group on the activity of the catalyst. Org. Lett. 2003, 5, 4105. [DOI] [PubMed] [Google Scholar]
- Litvinas N. D.; Brodsky B. H.; Du Bois J. C-H hydroxylation using a heterocyclic catalyst and aqueous H2O2. Angew. Chem., Int. Ed. 2009, 48, 4513–4516. [DOI] [PubMed] [Google Scholar]
- Cvetovich R. J.; Chung J. Y. L.; Kress M. H.; Amato J. S.; Matty L.; Weingarten M. D.; Tsay F.-R.; Li Z.; Zhou G. An efficient synthesis of a dual PPAR α/γ agonist and the formation of a sterically congested α-aryloxyisobutyric acid via a bargellini reaction. J. Org. Chem. 2005, 70, 8560–8563. [DOI] [PubMed] [Google Scholar]
- Katzenellenbogen J. A.; Johnson H. J. Jr.; Myers H. N. Photoaffinity labels for estrogen binding proteins of rat uterus. Biochemistry 1973, 12, 4085–4092. [DOI] [PubMed] [Google Scholar]
- Carlson K. E.; Choi I.; Gee A.; Katzenellenbogen B. S.; Katzenellenbogen J. A. Altered ligand binding properties and enhanced stability of a constitutively active estrogen receptor: evidence that an open-pocket conformation is required for ligand interaction. Biochemistry 1997, 36, 14897–14905. [DOI] [PubMed] [Google Scholar]
- Sun J.; Meyers M. J.; Fink B. E.; Rajendran R.; Katzenellenbogen J. A.; Katzenellenbogen B. S. Novel ligands that function as selective estrogens or antiestrogens for estrogen receptor-α or estrogen receptor-β. Endocrinology 1999, 140, 800–804. [DOI] [PubMed] [Google Scholar]
- Meyers M. J.; Sun J.; Carlson K. E.; Katzenellenbogen B. S.; Katzenellenbogen J. A. Estrogen receptor subtype-selective ligands: asymmetric synthesis and biological evaluation of cis- and trans-5,11-dialkyl-5,6,11,12-tetrahydrochrysenes. J. Med. Chem. 1999, 42, 2456–2468. [DOI] [PubMed] [Google Scholar]
- Chang E. C.; Charn T. H.; Park S. H.; Helferich W. G.; Komm B.; Katzenellenbogen J. A.; Katzenellenbogen B. S. Estrogen receptors alpha and beta as determinants of gene expression: influence of ligand, dose, and chromatin binding. Mol. Endocrinol. 2008, 22, 1032–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y.; Gong P.; Madak-Erdogan Z.; Martin T.; Jeyakumar M.; Carlson K. E.; Khan I.; Smillie T. J.; Chittiboyina A. G.; Rotte S. C.; Helferich W. G.; Katzenellenbogen J. A.; Katzenellenbogen B. S. Mechanisms enforcing the estrogen receptor β selectivity of botanical estrogens. FASEB J. 2013, 27, 4406–4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charn T. H.; Liu E. T.; Chang E. C.; Lee Y. K.; Katzenellenbogen J. A.; Katzenellenbogen B. S. Genome-wide dynamics of chromatin binding of estrogen receptors alpha and beta: mutual restriction and competitive site selection. Mol. Endocrinol. 2010, 24, 47–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabat G. C.; Etgen A. M.; Rohan T. E. Do steroid hormones play a role in the etiology of glioma?. Cancer Epidemiol., Biomarkers Prev. 2010, 19, 2421–2427. [DOI] [PubMed] [Google Scholar]
- Skliris G. P.; Munot K.; Bell S. M.; Carder P. J.; Lane S.; Horgan K.; Lansdown M. R. J.; Parkes A. T.; Hanby A. M.; Markham A. F.; Speirs V. Reduced expression of oestrogen receptor β in invasive breast cancer and its re-expression using DNA methyl transferase inhibitors in a cell line model. J. Pathol. 2003, 201, 213–220. [DOI] [PubMed] [Google Scholar]
- Norman B. H.; Dodge J. A.; Richardson T. I.; Borromeo P. S.; Lugar C. W.; Jones S. A.; Chen K.; Wang Y.; Durst G. L.; Barr R. J.; Montrose-Rafizadeh C.; Osborne H. E.; Amos R. M.; Guo S.; Boodhoo A.; Krishnan V. Benzopyrans are selective estrogen receptor beta agonists with novel activity in models of benign prostatic hyperplasia. J. Med. Chem. 2006, 49, 6155–6157. [DOI] [PubMed] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case D. A.; Darden T. A.; Cheatham T. E. III; Simmerling C. L.; Wang J.; Duke R. E.; Luo R.; Merz K. M.; Pearlman D. A.; Crowley M.; Walker R. C.; Zhang W.; Wang B.; Hayik S.; Roitberg A.; Seabra G.; Wong K. F.; Paesani F.; Wu X.; Brozell S.; Tsui V.; Gohlke H.; Yang L.; Tan C.; Mongan J.; Hornak V.; Cui G.; Beroza P.; Mathews D. H.; Schafmeister C.; Ross W. S.; Kollman P. A.. AMBER 9; University of California: San Francisco, CA, 2006.
- Maestro, version 9.0; Schrödinger Inc.: Portland, OR, 2009.
- Macromodel, version 9.7; Schrödinger Inc.: Portland, OR, 2009.
- Morris G. M.; Goodsell D. S.; Halliday R. S.; Huey R.; Hart W. E.; Belew R. K.; Olson A. J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan S.; Nwachukwu J. C.; Parent A. A.; Cavett V.; Nowak J.; Hughes T. S.; Kojetin D. J.; Katzenellenbogen J. A.; Nettles K. W. Ligand-binding dynamics rewire cellular signaling via estrogen receptor-alpha. Nat. Chem. Biol. 2013, 9, 326–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.