

Abstract

To date, examples of α-arylation of carboxylic acids remain scarce. Using a deprotonative cross-coupling process (DCCP), a method for palladium-catalyzed γ-arylation of aryl acetic acids with aryl halides has been developed. This protocol is applicable to a wide range of aryl bromides and chlorides. A procedure for the palladium-catalyzed α-arylation of styryl acetic acids is also described.
Transition-metal-catalyzed α-arylation of enolates and their derivatives is of great importance due to the diversity of compounds that can be accessed.1 In 1997 pioneering studies on palladium-catalyzed α-arylation of ketones with aryl bromides was independently reported by the Miura,2 Buchwald,3 and Hartwig4 groups. Since these seminal works, intensive studies on α-arylation of carbonyl compounds have been reported.5,6 To date, however, examples of α-arylation of simple carboxylic acids remain scarce.7−9
In 2007, Daugulis and co-workers studied the palladium-catalyzed ortho arylation of benzoic acids. They reported a byproduct derived from the α-arylation of acetic acid with 3,5-dimethyl iodobenzene (Scheme 1A).7 The scope and mechanism of this reaction were recently investigated by Han and Zhao.8 In both cases, the reaction required use of excess acetic acid (3.5 to >50 equiv) and supra-stoichiometric silver acetate and gave poor to moderate yields (18–68%).
Scheme 1. Previous Examples of α-Arylation of Carboxylic Acid Derivatives.

In 2000, Wolfe and co-workers reported the reaction of phenyl acetic acid dienolate with aryl bromides and iodides in liquid ammonia under near-UV irradiation to afford alpha and para arylation products via a proposed SRN1 process (Scheme 1B).10 The inconvenient reaction conditions (NH3(l), hv) preclude large-scale applications of this chemistry.
During the preparation of this manuscript, Mori and co-workers described α-arylation of carboxylic acids in a two-step process (Scheme 1C). The substrates are first irreversibly deprotonated using EtMgCl to generate dienolates followed by palladium-catalyzed α-arylation with aryl bromides and iodides.9
Our approach to the α-arylation of carboxylic acids differs from that of Mori and co-workers. We envisioned reversible deprotonation of the substrate in the presence of the catalyst, allowing the use of less reactive bases. Under these conditions, the carboxylate would be in equilibrium with the dienolate, as shown in Scheme 1D. To develop this approach, a few issues must be considered. Prior studies on the generation of dienolates by double deprotonation of carboxylic acids11−14 indicate strong bases (e.g., nBuLi, LDA, Grignard reagents) are needed to achieve the second deprotonation.
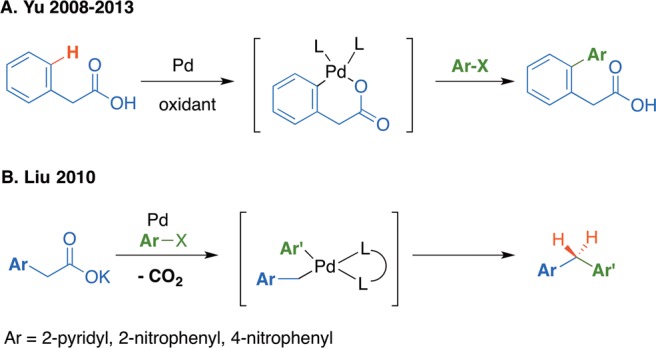
Other challenges include control of chemoselectivity, undesired reactivity of the aryl acetic acid starting materials, and decomposition/further reactions of the diaryl acetic acid product. As noted above, aryl acetic acid derivatives are known to undergo palladium-catalyzed ortho arylation via a C–H activation pathway.15,16 In this process the carboxylate moiety serves as a directing group to facilitate the C–H bond cleavage. For example, Yu and co-workers demonstrated that ortho-C–H activation of phenyl acetic acid derivatives by palladium could be followed by cross-coupling (Scheme 2A). Furthermore, aryl acetates have been reported to be unstable in the presence of palladium catalysts and undergo decarboxylative cross-coupling.17−23 This reaction pathway is exemplified by the report of Liu and co-workers, who demonstrated diarylmethanes can be formed using 2-pyridyl or nitroaryl acetates with aryl halides via palladium-catalyzed decarboxylative coupling (Scheme 2B).22,23 These studies indicate a potential side reaction involving intermediates such as those in Scheme 1D that may compete with the desired α-arylation of dienolates.
Scheme 2. Selectivity in Arylation of Aryl Acetic Acid Derivatives.
Despite these issues, our experience with deprotonative cross-coupling processes (DCCP) of weakly acidic substrates (pKa > 25) positioned us to pursue the α-arylation of acids.24−31 An important feature of the DCCP is an in situ reversible metalation of the substrate via C–H deprotonation under catalytic cross-coupling conditions. Herein, we describe a general protocol for the palladium-catalyzed α-arylation of carboxylic acids with aryl chlorides and bromides involving reversible deprotonation of the carboxylate.
The first step in our reaction development focused on deprotonation of phenyl acetic acid to form the dienolate intermediate. To evaluate bases to deprotonate the carboxylate substrate, we employed a benzylation reaction with benzyl chloride to trap the dienolate (eq 1).
 |
1 |
We chose to use KN(SiMe3)2 as the base, because it exhibits good compatibility with catalysts, reagents, and products in deprotonative cross-coupling processes.28 Employing KN(SiMe3)2 at 80 °C (eq 1), the benzylation product 1aa was isolated in 80% yield, indicating formation of the dienolate under these conditions. With this result in hand, we chose the NiXantphos/Pd(OAc)2-based catalyst due to its ability to arylate weakly acidic substrates, such as diphenylmethanes (pKa = 32.3,32 eq 2).28
 |
2 |
We examined the α-arylation of phenyl acetic acid 1a (1.2 equiv) with 1-bromo-4-tert-butylbenzene 2a (limiting reagent) in the presence of KN(SiMe3)2 (2.5 equiv) and catalytic Pd(OAc)2 (5 mol %) and NiXantphos (7.5 mol %). These studies were initiated with a solvent screen. The reaction in cyclopentyl methyl ether (CPME), an excellent solvent for DCCPs, failed to afford an arylation product (Scheme 3, entry 1). Dioxane initially appeared promising, with a 56% yield (entry 2). Attempts to increase the yield, however, were unsuccessful. THF, 2-methyl-THF, and DME (entries 3–5) were examined at 80 °C, with DME giving up to 80% yield. Likewise, toluene at 110 °C afforded the product in 80% yield (entry 6). Both THF and DME resulted in generation of 5–10% of an inseparable byproduct that was not observed in toluene, making toluene a better choice for further optimization. In the reaction in toluene (entry 6), some decomposition of phenyl acetic acid was observed, presumably resulting in lower yields. To address this issue, the equivalents of aryl acetic acid and base were increased to 1.5 and 3, respectively, resulting in an 83% yield (entry 7). Reducing the reaction concentration to 0.05 M resulted in a >95% yield by 1H NMR of the crude product (entry 8). Lowering the temperature (entry 9) or catalyst loading (entry 10) resulted in lower yields.
Scheme 3. Optimization of Phenyl Acetic Acid α-Arylation.
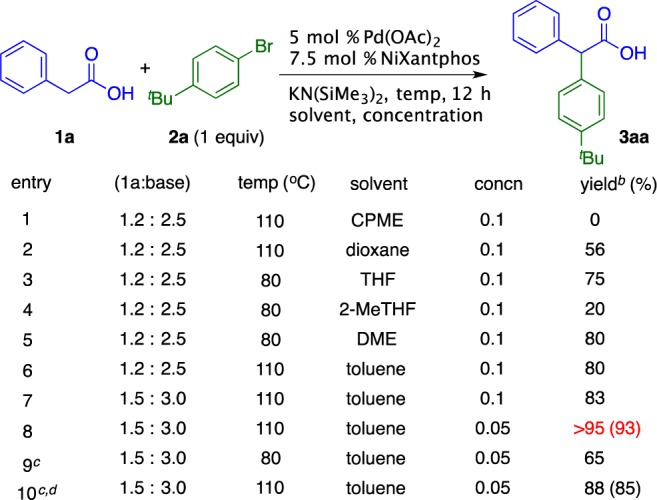
Reaction conducted on a 0.1 mmol scale. b Yield determined by 1H NMR spectroscopy of the crude reaction mixture. c Isolated yield after chromatographic purification. d 2.5 mol % Pd(OAc)2/3.75 mol % NiXantphos.
With the optimized conditions of Scheme 3 (entry 8), the scope of aryl halides in the α-arylation of phenyl acetic acid was examined (Scheme 4). 1-Bromo- and 1-chloro-4-tert-butylbenzene gave 3aa in 93% and 84% yield, respectively (entry 1). Substrates with electron-donating groups, such as 4-bromo- and 4-chloroanisole, exhibited good reactivity, generating 87% and 80% yields of the arylation products (entry 2). 4-Bromo-N,N-dimethylaniline furnished 3ac in 86% yield. Aryl bromides and chlorides with electron-withdrawing groups, such as 4-F, 4-Cl, and 3-CF3, were all tolerated. 1-Bromo- and 1-chloro-4-fluorobenzene furnished diaryl acetic acid in 75% and 62% yield, respectively, while 1-bromo-4-chlorobenzene underwent coupling to give 3ae in 65% yield. In the latter case, no byproducts derived from oxidative addition of the C–Cl bond were observed. Aryl bromide bearing a 3-CF3 substituent furnished the product in 77% yield. Sterically hindered 2-bromotoluene and 1-bromonaphthalene gave 3ag and 3ah in 71% and 80% yields, respectively. In the case of 1-bromo-3-(trifluoromethyl)benzene, both the aryl acetic acid and base equivalents were increased to 2 and 4, respectively. For the more challenging 2-chlorotoluene, 10% Pd(OAc)2 was required to render 3ag in 63% isolated yield. Heterocyclic N-methyl-5-bromoindole exhibited good reactivity with phenyl acetic acid to form 3ai in 82% yield. The TIPS silyl ether derived from 4-bromophenol was subjected to the reaction conditions and afforded 3ai in 53% yield (Scheme 4, entry 10). Unfortunately, aryl bromides containing ketone, ester, or amide groups failed to give desired products.
Scheme 4. Scope of Aryl Halides in the α-Arylation of Phenyl Acetic Acid,b.

Reaction conducted on a 0.1 mmol scale at 0.05 M. b Isolated yield after chromatographic purification. c 2 equiv of 1a and 4 equiv of KN(SiMe3)2. d 10 mol % Pd(OAc)2/15 mol % NiXantphos.
The scope of α-arylation with substituted aryl acetic acids was examined with 1-bromo-4-tert-butylbenzene 2a (Scheme 5). Electron-neutral 2-naphthyl acetic acid afforded the coupling product in 73% yield. Electron-donating 4-methoxy phenyl acetic acid underwent coupling in 86% yield. Aryl acetic acids with withdrawing 4-F, 4-Cl, and 3-CF3 groups furnished coupling products in 81%, 58%, and 65% yield, respectively. Sterically hindered 2-tolyl acetic acid proved to be a challenging substrate with a 53% yield obtained despite repeated attempts to optimize the reaction. We next examined 3-pyridyl acetic acid in the α-arylation. With our standard reaction conditions, only decomposition of the acid was observed. We then screened bases KN(SiMe3)2, NaN(SiMe3)2, LiN(SiMe3)2, KOtBu, NaOtBu, and LiOtBu and identified NaOtBu as a better option, generating a 70% isolated yield in the coupling (entry 7). In order to demonstrate the scalability of the protocol, a gram-scale reaction was performed with 4-methoxy phenyl acetic acid (1b) and 4-tert-butyl bromobenzene (2a). The arylation product 3ba (1.1 g) was isolated in 74% (Scheme 5, entry 1).
Scheme 5. Scope of α-Arylation with Aryl Acetic Acids,b.
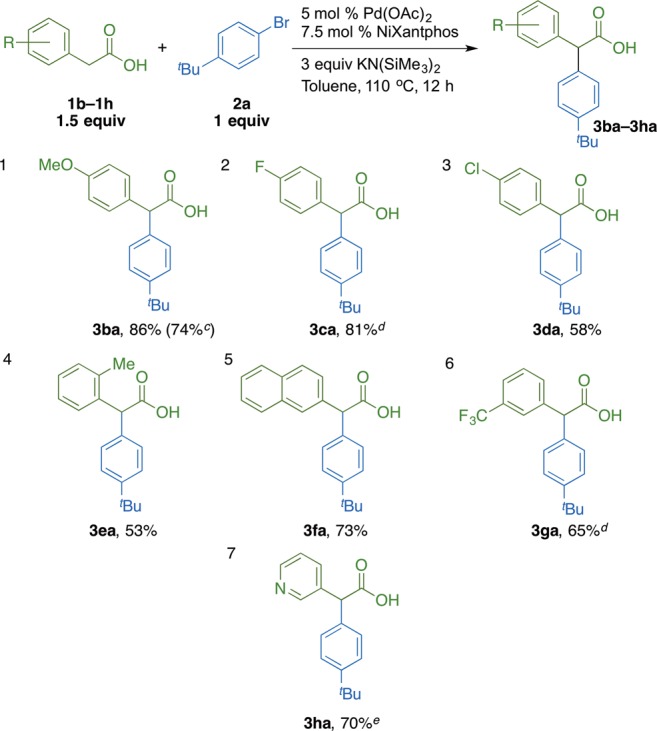
Reaction conducted on a 0.1 mmol scale at 0.05 M. b Isolated yield after chromatographic purification. c Reaction conducted on a 5 mmol scale at 0.05 M; 1.1 g of product was obtained. d 2 equiv of acid and 4 equiv of KN(SiMe3)2. e 2 equiv of acid and 6 equiv of NaOtBu.
After demonstrating the scope of aryl halides and aryl acetic acids, we turned our attention to styryl acetic acids (Scheme 6). trans-Styryl acetic acid 4a is an interesting substrate, because it has two sites that can potentially undergo arylation. After screening different bases with this substrate, we observed that, with NaN(SiMe3)2, only one regioisomeric arylation product was obtained. It appears that the initially formed product undergoes isomerization of the double bond to form the more conjugated product 4ap in 70% isolated yield. This product could also be envisioned to arise from a Heck reaction. We are unaware, however, of literature precedence involving trans-styryl acetic acids undergoing Heck-type reactions.
Scheme 6. Arylation of trans-Styryl Acetic Acid 4a.

It is noteworthy that the structure of 4ap is related to Amitriptyline, a medication that has been used in the treatment of migraines,33,34 diabetic neuropathy,35 postherpetic neuralgia,36 and chronic lower back pain.35,37 In addition, Brown and co-workers reported an analog of Amitriptyline, diphenylamine DHP-362, which exhibits higher potency as a sodium channel blocker.38 The synthesis of DHP-362 is only two steps away from 4ap. We next examined the arylation of trans-styryl acetic acid 4a with different aryl bromides. Both 4-tert-Bu and 4-F groups underwent reaction with excellent regioselectivity and afforded E/Z-mixtures in 71–78% yields.39
In summary, we have developed a general method for palladium-catalyzed α-arylation of aryl acetic acids with aryl bromides and chlorides. The reaction also affords product with trans-styryl acetic acid, which provides intermediates in route to biologically active compounds. We anticipate that this protocol will be an important complement to the existing arsenal of palladium-catalyzed α-arylation reactions.
Acknowledgments
We thank the National Science Foundation (CHE-1152488) and National Institutes of Health (NIGMS 104349) for financial support.
Supporting Information Available
Procedures, characterization data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Culkin D. A.; Hartwig J. F. Acc. Chem. Res. 2003, 36, 234–245. [DOI] [PubMed] [Google Scholar]
- Satoh T.; Kawamura Y.; Miura M.; Nomura M. Angew. Chem., Int. Ed. Engl. 1997, 36, 1740–1742. [Google Scholar]
- Palucki M.; Buchwald S. L. J. Am. Chem. Soc. 1997, 119, 11108–11109. [Google Scholar]
- Hamann B. C.; Hartwig J. F. J. Am. Chem. Soc. 1997, 119, 12382–12383. [Google Scholar]
- Johansson C. C. C.; Colacot T. J. Angew. Chem., Int. Ed. 2010, 49, 676–707. [DOI] [PubMed] [Google Scholar]
- Bellina F.; Rossi R. Chem. Rev. 2009, 110, 1082–1146. [DOI] [PubMed] [Google Scholar]
- Chiong H. A.; Pham Q.-N.; Daugulis O. J. Am. Chem. Soc. 2007, 129, 9879–9884. [DOI] [PubMed] [Google Scholar]
- Wu G.-J.; Guan J.; Han F.-S.; Zhao Y.-L. ChemCatChem 2014, 6, 1589–1593. [Google Scholar]
- Tanaka D.; Tanaka S.; Mori A. Eur. J. Org. Chem. 2014, 20, 4254–4257. [Google Scholar]
- Nwokogu G. C.; Wong J.-W.; Greenwood T. D.; Wolfe J. F. Org. Lett. 2000, 2, 2643–2646. [DOI] [PubMed] [Google Scholar]
- Hauser C. R.; Chambers W. J. J. Am. Chem. Soc. 1956, 78, 4942–4944. [Google Scholar]
- Creger P. L. J. Am. Chem. Soc. 1967, 89, 2500–2501. [Google Scholar]
- Pfeffer P. E.; Silbert L. S.; Chirinko J. M. Jr. J. Org. Chem. 1972, 37, 451–458. [Google Scholar]
- Streitwieser A.; Husemann M.; Kim Y.-J. J. Org. Chem. 2003, 68, 7937–7942. [DOI] [PubMed] [Google Scholar]
- Wang D.-H.; Mei T.-S.; Yu J.-Q. J. Am. Chem. Soc. 2008, 130, 17676–17677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X.-F.; Li Y.; Su Y.-M.; Yin F.; Wang J.-Y.; Sheng J.; Vora H. U.; Wang X.-S.; Yu J.-Q. J. Am. Chem. Soc. 2013, 135, 1236–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goossen L. J.; Rodríguez N.; Goossen K. Angew. Chem., Int. Ed. 2008, 47, 3100–3120. [DOI] [PubMed] [Google Scholar]
- Rodríguez N.; Goossen L. J. Chem. Soc. Rev. 2011, 40, 5030. [DOI] [PubMed] [Google Scholar]
- Fromm A.; van Wüllen C.; Hackenberger D.; Goossen L. J. J. Am. Chem. Soc. 2014, 136, 10007–10023. [DOI] [PubMed] [Google Scholar]
- Weaver J. D.; Recio A. III; Grenning A. J.; Tunge J. A. Chem. Rev. 2011, 111, 1846–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F.; Wang T.; Jiao N. Chem. Rev. 2014, 114, 8613–8661. [DOI] [PubMed] [Google Scholar]
- Shang R.; Yang Z.-W.; Wang Y.; Zhang S.-L.; Liu L. J. Am. Chem. Soc. 2010, 132, 14391–14393. [DOI] [PubMed] [Google Scholar]
- Shang R.; Huang Z.; Chu L.; Fu Y.; Liu L. Org. Lett. 2011, 13, 4240–4243. [DOI] [PubMed] [Google Scholar]
- Zheng B.; Jia T.; Walsh P. J. Adv. Synth. Catal. 2014, 356, 165–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia T.; Bellomo A.; Montel S.; Zhang M.; El Baina K.; Zheng B.; Walsh P. J. Angew. Chem., Int. Ed. 2013, 53, 260–264. [DOI] [PubMed] [Google Scholar]
- Montel S.; Jia T.; Walsh P. J. Org. Lett. 2014, 16, 130–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montel S.; Raffier L.; He Y.; Walsh P. J. Org. Lett. 2014, 16, 1446–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Bellomo A.; Creamer A. D.; Dreher S. D.; Walsh P. J. J. Am. Chem. Soc. 2012, 134, 13765–13772. [DOI] [PubMed] [Google Scholar]
- Zheng B.; Jia T.; Walsh P. J. Org. Lett. 2013, 15, 1690–1693. [DOI] [PubMed] [Google Scholar]
- Jia T.; Bellomo A.; Baina K. E.; Dreher S. D.; Walsh P. J. J. Am. Chem. Soc. 2013, 135, 3740–3743. [DOI] [PubMed] [Google Scholar]
- Zheng B.; Jia T.; Walsh P. J. Org. Lett. 2013, 15, 4190–4193. [DOI] [PubMed] [Google Scholar]
- Bordwell F. G.; Matthews W. S.; Vanier N. R. J. Am. Chem. Soc. 1975, 97, 442–443. [Google Scholar]
- Cerbo R.; Barbanti P.; Fabbrini G.; Pascali M. P.; Catarci T. Headache 1998, 38, 453–457. [DOI] [PubMed] [Google Scholar]
- Ashina S.; Bendtsen L.; Jensen R. Pain 2004, 108, 108–114. [DOI] [PubMed] [Google Scholar]
- Collins S. L.; Moore R. A.; McQuay H. J.; Wiffen P. J. Pain Symptom. Manage. 2000, 20, 449–458. [DOI] [PubMed] [Google Scholar]
- Bowsher D. Eur. J. Pain 2003, 7, 1–7. [DOI] [PubMed] [Google Scholar]
- Atkinson J. H.; Slater M. A.; Williams R. A.; Zisook S.; Patterson T. L.; Grant I.; Wahlgren D. R.; Abramson I.; Garfin S. R. Pain 1998, 76, 287–296. [DOI] [PubMed] [Google Scholar]
- Hudgens D. P.; Taylor C.; Batts T. W.; Patel M. K.; Brown M. L. Bioorg. Med. Chem. 2006, 14, 8366–8378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbert C. A.; Koe B. K.; Kraska A. R.; Welch W. M. Jr., Antidepressant derivatives of cis-4-phenyl-1,2,3,4-tetrahydro-1-naphthalenamine, U.S. Patent 4,536,518, August 20, 1985.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.