

Abstract
Hypoxia is often found in solid tumors and is associated with tumor progression and poor clinical outcomes. The exact mechanisms related to hypoxia-induced invasion and metastasis remain unclear. We elucidated the mechanism by which the nuclear damage associated molecular pattern molecule, high mobility group box 1 (HMGB1), released under hypoxic stress can induce an inflammatory response to promote invasion and metastasis in hepatocellular carcinoma (HCC) cells. Caspase-1 activation was found to occur in hypoxic HCC cells in a process that was dependent on the extracellular release of HMGB1 and subsequent activation of both TLR4 and RAGE signaling pathways. Downstream from hypoxia induced caspase-1 activation, cleavage and release of proinflammatory cytokines, IL-1β and IL-18 occurred. We further demonstrate that overexpression of HMGB1 or treatment with recombinant HMGB1 enhanced invasiveness of HCC cells while stable knockdown of HMGB1 remarkably reduced HCC invasion. Moreover, in a murine model of HCC pulmonary metastasis, stable knockdown of HMGB1 suppressed HCC invasion and metastasis. Conclusion: These results suggest that in hypoxic HCC cells, HMGB1 activates TLR4 and RAGE signaling pathways to induce caspase-1 activation with subsequent production of multiple inflammatory mediators which in turn promotes cancer invasion and metastasis.
Keywords: Hypoxia, Inflammasome, Inflammation, Tumor, Damage Associated Molecular Pattern molecules
Introduction
A worldwide increase in mortality associated with hepatocellular carcinoma (HCC) has recently been reported (1). Clinical treatment of HCC remains challenging due to a lack of effective chemotherapy and clearly defined endpoints for clinical protocols (2, 3). The advanced nature of disease at presentation, often in the background of steatosis (4) and chronic hepatitis C (5), is a major change in the etiology from that observed in the past (6). The main cause of death in HCC patients, irrespective of etiology, is cancer metastasis within or outside the liver. The underlying mechanisms responsible for invasiveness and metastatic spread of HCC are still not fully understood.
Hypoxia is found in a wide range of human malignancies including liver, breast, prostate, and pancreatic cancers as well as brain tumors and melanoma (7). The extent of hypoxia is associated with tumor progression and poor clinical outcomes. Hypoxia has a dual role: insufficient oxygen limits tumor cell division while at the same time selects for more malignant cells and induces cell adaptations which allow for more invasive behavior (8). Cancer cells may promote tumor metastasis through the release of paracrine or endocrine signals that enhance invasiveness and promote the tumor pre-metastatic niche (9-11). Tumor-derived mediators not only inhibit apoptosis of tumor cells, but also promote cell invasiveness and metastasis.
High mobility group box1 (HMGB1) is an evolutionarily conserved chromatin-binding protein that has been implicated in several disease states, including sepsis, arthritis, ischemiareperfusion injury, and cancer (12, 13). Cancer cells that have undergone necrotic cell death can release HMGB1 into the local microenvironment. HMGB1 is also actively secreted by inflammatory cells, acting as an endogenous danger signal and binding with high affinity to several receptors including the receptor for advanced glycation end products (RAGE), Toll-like receptors (TLR)-2, TLR-4 and TLR-9 (12). Extracellular HMGB1 can lead to chronic inflammatory-reparative responses that, in the setting of cancer, may lead to tumor cell survival, expansion and metastases. Overexpression of HMGB1 is associated with all of the central hallmarks of cancer (13). Interestingly, numerous studies suggest that HMGB-1 plays a role in metastasis development, and thus link it to poor prognosis in a variety of cancers including prostate, breast, liver, and colon (13).
Caspase-1, a prototypical member of the inflammatory caspases, is responsible for the maturation of pro- interleukin(IL)-1β and pro-IL-18 (14). Once activated by cellular infection or stress, including hypoxia, caspase-1 cleaves IL-1β and IL-18 in inflammasomes. Endogenous damage associated molecular pattern (DAMP) molecules, such as HMGB1, are either normal cell constituents or derived from the extracellular matrix and can be released into the extracellular milieu during states of cellular stress or damage. DAMPs subsequently activate inflammasomes and promote pro-inflammatory responses. Cytokines downstream of caspase-1 can attract and activate immune cells to induce inflammation and affect tumor progression, including cell survival and metastasis (15, 16).
Therefore, we evaluated whether HMGB1 can induce caspase-1 activation and subsequently contribute to tumor invasiveness in hypoxic HCC cells. We show that HMGB1 is overly expressed in human HCC tumors compared to noncancerous liver tissue and higher in the serum of patients with HCC. HMGB1 is also released from hypoxic HCC cells and then subsequently activates caspase-1. In addition, both HMGB1 receptors, TLR4 and RAGE, play important roles in hypoxia-induced caspase-1 activation. Inhibition of HMGB1 or caspase-1 can decrease cell invasiveness in the setting of hypoxia; and knockdown of HMGB1 in HCC cells suppresses lung metastasis in a murine model of liver cancer
Materials and methods
Patient Samples, Cell lines and reagents
Patient serum, human HCC and their corresponding nontumorous liver samples were collected at the time of surgical resection at the University of Pittsburgh (IRB-approved serum and tumor registry). Cell lines and reagents information are described in the Supporting Materials and Methods.
Animals
Male wild-type (C57BL/6) mice (8-week-old) were purchased from Jackson ImmunoResearch Laboratories. Animal protocols were approved by the Animal Care and Use Committee of the University of Pittsburgh and the experiments were performed in adherence to the National Institutes of Health Guidelines for the Use of Laboratory Animals.
Establishment of Stable HMGB1 Expressing Cells and HMGB1 Knockdown Cells
The stable transfection is described in the Supporting Methods.
Immunoblot Analysis
Whole cell protein was extracted with Cell Lysis Buffer (Sigma-Aldrich). Cytoplasmic protein extraction and Western blot analysis were performed following a standard protocol as described previously (17).
RNA Interference (RNAi) by siRNA
The RNAi experiment protocol is described in the Supporting Methods.
ELISA
HMGB1 level in the serums from human and mouse was detected by ELISA (IBL) according to the manufacturer's instructions.
Confocal Microscopy
Cultures were fixed, stained and examined under a confocal microscope (Olympus) as described in the Supporting Methods (18).
Caspase-1 Colorimetric Assay
Caspase-1 Colorimetric Assay kit (RD Systems) was used according to the manufacturer's protocol.
Cell Migration and Invasion Assays
We determined migration and invasion as previously described (18).
Experimental Metastasis Model
To examine the metastatic potential of stable HMGB1 knockdown clones, 2×106 Hepa1-6 in 0.3 mL of phosphate-buffered saline, were injected into the tail vain of C57BL/6 mice. For in vivo tracking, the Hepa1-6 cells were stably transfected with firefly luciferase. One hundred milligrams per kilogram D-luciferin (Caliper) was injected into the peritoneal cavities of mice, and bioluminescence detected with the IVIS 100 Imaging System.
Statistical Analysis
Results are expressed as the mean ± standard error of the mean (SEM). Statistical analysis was performed using Student's t test or one-way ANOVA test. All statistical analyses were performed using Sigma Stat v.3.5 (Systat Software, Inc., Chicago, USA). Graphs were generated using Sigma Plot v.10 (Systat Software, Inc., Chicago, USA). P <0.05 was denoted as statistically significant.
Results
HMGB1 is over expressed in Hepatocellular Carcinoma
Overexpression of HMGB1 is associated with tumor progression(13). To study the role of HMGB1 in HCC, we first examined the amount of HMGB1 in twenty HCC tissue samples and their corresponding nontumor liver by immunoblot analysis. The detailed clinicopathological information of 20 cases was shown in Supplemental Table 1. We found that expression of HMGB1 was higher in all HCC tissues (Fig. 1A). Compared to normal primary hepatocytes, expression of HMGB1 was also much stronger in five HCC cell lines (Fig. 1B). We then examined the level of nuclear and cytoplasmic HMGB1 by the fractionation of nuclear and cytoplasmic proteins in HCC tissues and nontumor liver tissues. The amount of nuclear HMGB1 in HCC tissues and nontumor tissues was not significantly different (data not shown). However, cytoplasmic HMGB1 was absent or was present at low levels in nontumor tissues, whereas cytoplasmic HMGB1 was found at high levels in HCC tissues (Fig. 1C). High cytoplasmic levels of HMGB1 usually occur in the context of active HMGB1 release. Indeed, compared to serum obtained from normal volunteers, HMGB1 levels were significantly increased in patients with HCC (Fig. 1D). These findings suggest that HMGB1 may in fact be actively released into the circulation of patients with HCC.
Fig. 1. Overexpression and nuclear and cytoplasmic localization of HMGB1 in HCC cells.

(A) HMGB1 protein levels were measured with western blot in twenty paired HCC samples and their nontumor counterparts. Protein expression results were normalized to internal control β-actin. ***P<0.001. N, nontumorous liver; T, tumor. (B) Expressions of HMGB1 in two primary hepatocytes (mouse and human hepatocytes) and five HCC cell lines were detected by western blot analysis. (C) The expression of HMGB1 in cytoplasm was measured with in HCC samples and their nontumor counterparts. (D) The expression of HMGB1 in normal and HCC serums were detected by ELISA. **P <0.01. (E) The nontumor and HCC tissues were stained for HMGB1. Red, HMGB1; blue, nuclei. (F) Hepa1-6 and Huh7 cells were analyzed in normoxia or hypoxia by immunostaining. Green, HMGB1; blue, nuclei; red, F-actin. Imaging shown is representative of three experiments with similar results.
Hypoxia induces HMGB1 release from hepatocellular carcinoma cells
Hypoxia is a hallmark of solid tumors, including HCC(8). Accordingly, we found HIF-1α levels to be substantially increased in liver homogenates of HCC specimens compared to nontumor tissue (Supplementary Fig. S1). Immuno-fluorescence showed that HMGB1 mainly localized in the nucleus of hepatocytes in nontumor liver. However, in addition to nuclear HMGB1, cytoplasmic HMGB1 was also present at high levels in HCC cells (Fig. 1E). To determine whether hypoxia plays a role in inducing expression or translocation of HMGB1 in HCC cells, Hepa1-6 cells and Huh7 cells were cultured under normoxic (21% O2) or hypoxic (1% O2) conditions. Under normoxia in both cell lines, HMGB1 remained located predominantly in the nucleus. After exposure to hypoxia, the expression of HMGB1 increased in the cytoplasm (Fig. 1F).These findings indicate that hypoxia leads to the nuclear to cytoplasmic translocation of HMGB1 in HCC cells.
To determine whether hypoxia induces expression or translocation of HMGB1 in HCC, Western blot analysis was performed. The expression of HMGB1 in whole cell lysates was not significantly changed during hypoxia at either the protein or mRNA level (Fig. 2A, Supplementary Fig. S2). In contrast, hypoxia led to a time-dependent increase of HMGB1 translocation to the cytoplasm and HMGB1 release into the media in both cell lines (Fig. 2B, 2C). In addition, cell viability was not substantially different when comparing HCC cells exposed to hypoxia and normoxia as assessed in a crystal violet viability assay (Fig. 2D), These findings demonstrate that hypoxia leads to the translocation and release of HMGB1 in HCC cells.
Fig. 2. Hypoxia induces translocation and release of HMGB1.

(A) The expression of HMGB1 was determined by western blot in Hepa1-6 and Huh7 cells subjected to 24 h of hypoxia (1% O2). Cell supernatants and cytoplasmic extracts from Hepa1-6 cells (B) or Huh7 cells (C) that were subjected to a time course of hypoxia were collected. HMGB1 was determined by Western Blot. The blot shown is representative of three experiments with similar results. (D) Viability was determined by crystal violet assay on Hepa1-6 and Huh7 cells subjected to 24 h of hypoxia.
Hypoxia induces caspase-1 activation in HCC cells
Caspases play an important role in programmed cell death and inflammation (16, 19). Though apoptosis inducers caspase-3 and caspase-9 were activated in our hypoxic HCC cells (data not shown), we focused on caspase-1, which can induce inflammation and affect tumor progression. To characterize caspase-1 activation during hypoxia, Hepa1-6 and Huh7 cells were cultured under hypoxic conditions for various times. The 45-kDa pro-caspase-1 was cleaved into the active 20-kDa caspase-1 during hypoxia and increased in a time-dependent manner during hypoxia (Fig. 3A, 3B). Using a colorimetric assay to assess caspase-1 activity during hypoxia, we found that caspase-1 activity was also significantly increased in both Hepa1-6 and Huh7 cells subjected to hypoxia compared to normoxic controls (Fig. 3C). To confirm the localization of cleaved caspase-1, we performed immunofluorescent staining for cleaved caspase-1. After hypoxia, cleaved caspase-1 significantly increased in the cytoplasm diffusely in both cell lines (Supplementary Fig. S3). These findings demonstrate that hypoxia induces caspase-1 activation in HCC cells.
Fig. 3. Hypoxia induces caspase-1 activation in HCC cells.

Caspase-1 expression was determined by western blot in Hepa1-6 cells (A) or Huh7 cells (B) subjected to a time course of hypoxia. Blot shown is representative of three experiments with similar results. (C) Whole cell lysates were extracted from Hepa1-6 and Huh7 cells. Caspase-1 activity was determined by colorimetric assay.*P <0.05. Assay shown is representative of three experiments with similar results.
HMGB1 is necessary for hypoxia-induced activation of caspase-1 and also activates caspase-1 under normoxia
The previous results suggest that an association exists between HMGB1 release and caspase-1 activation in hypoxic HCC cells. Since HMGB1 can promote inflammation and inhibit apoptosis, we next sought to study whether HMGB1 participates in hypoxia-induced activation of the inflammation-related caspase-1. Hepa1-6 cells were treated with ethyl pyruvate or an anti-HMGB1 neutralizing antibody to inhibit HMGB1 release or block HMGB1, respectively. Either inhibiting HMGB1 release or blocking HMGB1 significantly decreased the production of cleaved caspase-1 in hypoxia (Fig. 4A). Treatment with ethyl pyruvate or anti-HMGB1 neutralizing antibody also resulted in a dramatic decrease in caspase-1 activity compared with hypoxic controls (Fig. 4B). These results suggest that HMGB1 released from hypoxic HCC cells is necessary for caspase-1 activation.
Fig. 4. HMGB1 is necessary for hypoxia-induced activation of caspase-1 and induces activation of caspase-1 in normoxia.

(A) Western blot analysis for caspase-1 from Hepa1-6 cells treated with 10 mM ethyl pyruvate (EP) or 1 μg/ml anti-HMGB1 neutralizing antibody (a-HMGB1) in hypoxia. The blot shown is representative of three experiments with similar results. (B) Caspase-1 activity was determined by colorimetric assay after various treatments. *P < 0.05 versus Hepa1-6 cells that were subjected to hypoxia. (C) Western blot analysis of caspase-1 in Hepa 1-6 cells treated with various dose rhHMGB1 for 24h. (D) Western blot analysis of caspase-1 in Hepa1-6 cells treated with 1 μg/ml rhHMGB1 for various times. (E) Ectopic overexpression of HMGB1-GFP fusion protein in Hepa1-6 stable clones. GFP and HMGB1-GFP fusion expression were confirmed via western blot analysis with GFP antibody. (F) Cleaved caspase-1 was significantly increased in HMGB1 stable transfectants as compared with vector controls via western blot analysis.
To further confirm that HMGB1 activates caspase-1, we treated Hepa1-6 cells with recombinant human HMGB1 (rhHMGB1) and studied these cells under normoxic cell culture conditions. rhHMGB1 treatment in normoxia induced a dose- and time-dependent significant increase in cleaved caspase-1 in Hepa1-6 cells (Fig. 4C, 4D). Constitutively active HMGB1 was also stably transfected into the Hepa1-6 cell line and the expression was confirmed via Western blot (Fig. 4E). HMGB1 stably expressing cells displayed a significant increase of cleaved caspase-1 compared with the vector control (Fig. 4F). These results indicate that HMGB1 is required for hypoxia-induced caspase-1 activation and HMGB1 overexpression independently induces caspase-1 activation in Hepa1-6 cells, even without exposure to hypoxia.
Signaling pathways downstream of HMGB1 are involved in hypoxia-induced activation of caspase-1
Several important receptors have been implicated in HMGB1 signaling, including RAGE, TLR2 and TLR4(12). To investigate whether these receptors were involved in hypoxia-induced caspase-1 activation, Western blot analysis was performed on whole cell protein from Hepa1-6 cells subjected to hypoxia. TLR4 and RAGE, but not TLR2, were detected in Hepa1-6 cells. The expression of TLR4 increased in a time-dependent manner in Hepa1-6 cells subjected to hypoxia (Fig. 5A). To determine whether hypoxia-induced caspase-1 activation is TLR4 dependent, Hepa1-6 cells were treated with TLR4 siRNA. After TLR4 siRNA treatment, the expression of TLR4 was significantly decreased (Supplementary Fig. S4A) and the hypoxia induced expression of cleaved caspase-1 was also significantly diminished (Fig. 5B). Anti-TLR4 neutralizing antibody also was used to confirm this result.
Fig. 5. Signaling pathways downstream of HMGB1 are involved in hypoxia-induced activation of caspase-1.

(A) Expression of TLR4 from Hepa1-6 cells subjected to hypoxia for various time points. (B) Western blot analysis for caspase-1 from hypoxic Hepa1-6 cells transfected with scrambled siRNA or TLR4 siRNA or treated with 5 μg/ml anti-TLR4 neutralizing antibody (a-TLR4). (C) Expression of RAGE from Hepa1-6 cells subjected to hypoxia for various time points. (D) Western blot analysis for caspase-1 from hypoxic Hepa1-6 cells transfected with scrambled siRNA or RAGE siRNA or treated with 5 μg/ml sRAGE. (E) Western blot analysis for caspase-1 from Hepa1-6 cells treated with a-TLR4 or/and sRAGE. The blot shown is representative of three experiments with similar results.
RAGE regulates metabolism, inflammation, and epithelial survival in the setting of stress (12). The expression of RAGE increased in a time-dependent manner in Hepa1-6 cells subjected to hypoxia (Fig. 5C). To further study whether hypoxia-induced caspase-1 activation is RAGE dependent, Hepa1-6 cells were treated with RAGE siRNA. Compared with scrambled siRNA, treatment with specific siRNA against RAGE resulted in a significant decrease of RAGE protein (Supplementary Fig. S4B). The expression of cleaved caspase-1 was also significantly decreased following RAGE siRNA transfection. The soluble isoform inhibitor of the receptor (sRAGE) also was used to confirm the result. As shown, cleaved caspase-1 was significantly inhibited after blocking RAGE with sRAGE treatment (Fig. 5D). To further confirm that both TLR4 and RAGE receptors were involved in hypoxia-induced caspase-1 activation, Hepa1-6 cells were simultaneously treated with anti-TLR4 neutralizing antibody and sRAGE. Cleaved caspase-1 was almost completely inhibited by blocking both TLR4 and RAGE (Fig. 5E). These results suggest that hypoxia-induced caspase-1 activation occurs through both TLR4 and RAGE signaling pathways.
NLRP3-dependent caspase-1 activation promotes maturation of IL-1β and IL-18 under hypoxia
To date, four cytoplasmic receptors have been described that form an inflammasome complex: NLRP1, NLRP3, IPAF, and AIM2(14). To investigate which one is involved in hypoxia-induced caspase-1 activation, Hepa1-6 cells were transfected with NLRP1 siRNA, NLRP3 siRNA, IPAF siRNA and AIM2 siRNA. All receptors were efficiently silenced by their respective siRNA (data not shown), but only NLRP3 siRNA was found to inhibit hypoxia induced caspase-1 activation (Fig. 6A, Supplementary Fig. S4C, S4D). These results suggest that hypoxia-induced caspase-1 activation in HCC cells is NLRP3 dependent.
Fig. 6. NLRP3-dependent caspase-1 activation promotes cleavage of IL-1β, IL-18 in hypoxia.

(A) Western blot analysis for caspase-1 from Hepa1-6 cells treated with NLRP3 siRNA in hypoxia. (B) Hepa1-6 cells were treated with a caspase-1 inhibitor Z-YVAD-FMK (10 μM) and then analyzed by western blot for caspase-1. (C) Western blot analysis for IL-1β after treatment with Z-YVAD-FMK. (D) Expression of IL-18 from Hepa1-6 cells was determined by western blot.
Next, we investigated signaling pathways downstream of caspase-1. The cytokines IL-1β and IL-18 begin as cytosolic precursors that require cleavage by the cysteine protease caspase-1 to generate biologically active molecules. Cleaved caspase-1, IL-1β, and IL-18 were all increased following hypoxia (Fig. 6B, 6C, 6D). In contrast, all three were decreased after treatment with the caspase-1 inhibitor Z-YVAD-FMK (Fig. 6B, 6C, 6D). As downstream effectors of caspase-1, both cytokines released from cancer cells can induce production and secretion of other cytokines and chemokines, recruit stromal cells, and induce angiogenesis, leading to tumor progression (15, 16).
HMGB1-induced caspase-1 activation is involved in hypoxia-induced cell migration and invasion
We next sought to determine the effects of hypoxia on HCC cell migration and invasion. Under hypoxia, Fig. 7A shows that Hepa1-6 cell migration was increased 2.04 ± 0.14-fold (P < 0.01; n = 6) in 1% O2. Similarly, invasion of Hepa1-6 cells through reconstituted 3D Matrigel matrices was increased 1.72 ± 0.12-fold (P < 0.01; n = 6) in 1% O2.
Fig. 7. HMGB1-induced caspase-1 activation is involved in hypoxia-induced cell invasion.

(A) Transwell chamber studies were performed for 24 h in 21% O2 or 1.0% O2. Migration and invasion are expressed relative to the levels observed with normoxic controls. (mean ± SEM; n = 6). **P < 0.01. (B) Hepa1-6 cells were treated with rhHMGB1 (1 μg/ml) or Z-YVAD-FMK (10 μM) in normoxia or a-HMGB1 (1 μg/ml) or Z-YVAD-FMK in hypoxia. Cells were allowed to invade matrigel for 24 h. (C) The number of invasive cells was quantified. Invasion is expressed relative to the levels observed with normoxic controls. *P < 0.05; **P < 0.01. (D) Huh7 cells were treated with a-HMGB1 or Z-YVAD-FMK in hypoxia. Cells were allowed to invade matrigel for 24 h. (E) The number of invasive cells was quantified. Invasion is expressed relative to the levels observed with normoxic controls. *P < 0.05.
To elucidate whether HMGB1-induced caspase-1 activation was responsible for the increase in cell invasion observed in hypoxia, Hepa1-6 cells were treated with anti-HMGB1 neutralizing antibodies. Blockade of HMGB1 inhibited the increase in cell invasion observed during hypoxia (Fig. 7B, 7C). We also examined caspase-1 inhibition in hypoxia-induced invasion. Hepa1-6 cells treated with the caspase-1 inhibitor Z-YVAD-FMK also exhibited significantly decreased hypoxia-induced invasion (Fig. 7B, 7C). In contrast, HCC cell invasiveness was increased after rhHMGB1 treatment, but this effect was abolished by caspase-1 inhibition (Fig. 7B, 7C). These findings were further confirmed using Huh7 HCC cells (Fig. 7D, 7E). Collectively, these results demonstrate that HMGB1-mediated caspase-1 activation is required for hypoxia-induced invasion in HCC cells.
HMGB1 promotes HCC cells invasion and metastasis in vitro and in vivo
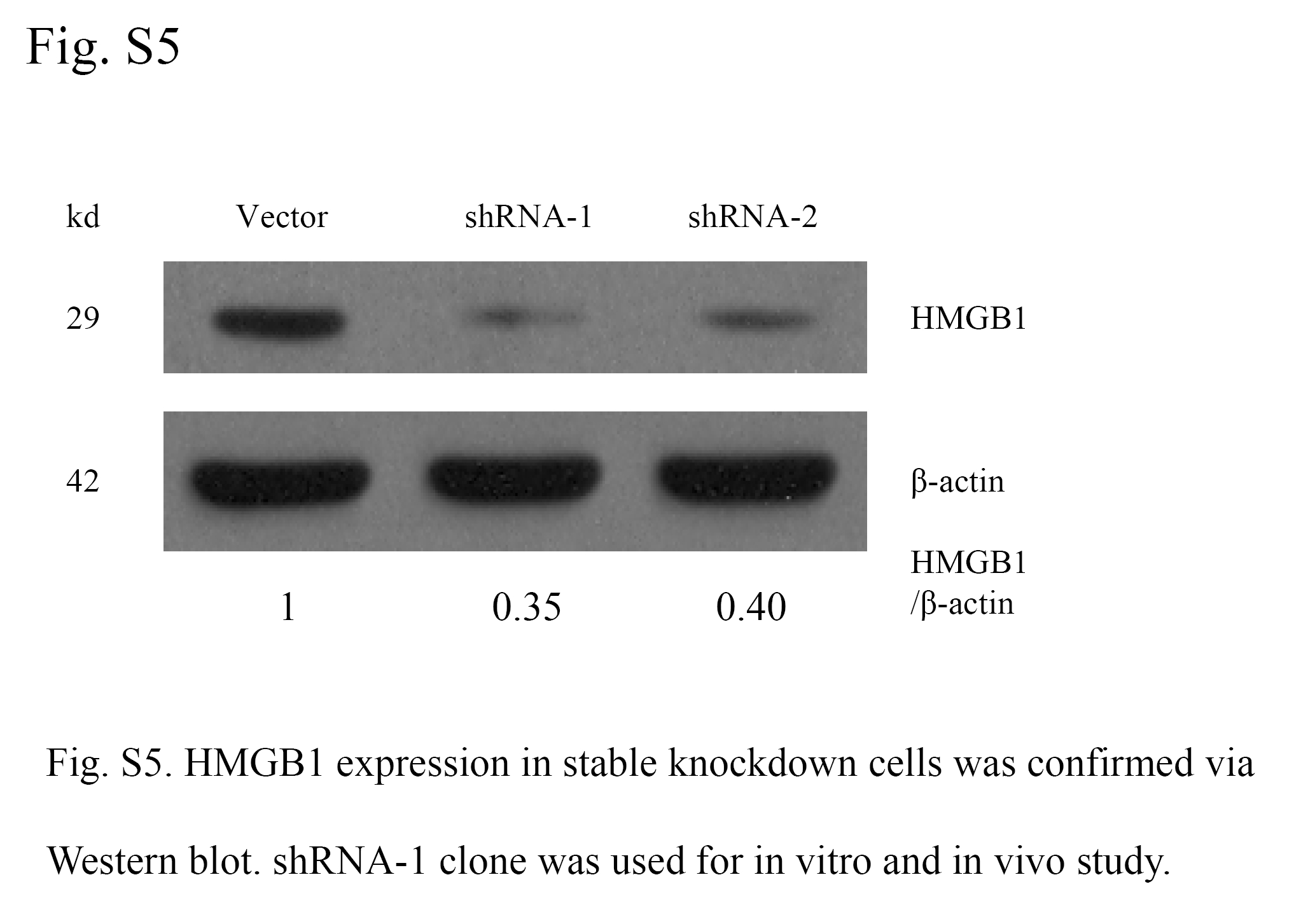
To further assess the effect of HMGB1 on HCC cell invasion, constitutively active HMGB1 was stably transfected into the Hepa1-6 cell line. HMGB1 stably expressing cells (pEGFPN1-HMGB1) displayed a significant increase in cell invasion ability compared with vector controls (Fig. 8A). In contrast to HMGB1 overexpression, stable knockdown of HMGB1 in Hepa1-6 cells using short hairpin RNA (Supplementary Fig. S5) considerably decreased the invasiveness of Hepa1-6 cells as evidenced by the transwell assay (Fig. 8B).
Fig. 8. HMGB1 promotes HCC cells invasion and metastasis in vitro and in vivo.

Stable HMGB1 overexpressing (A) or knockdown cells (B) were seeded onto the upper chamber of the transwell and allowed to invade matrigel for 24 h. The number of invasive cells was quantified. Invasion is expressed relative to the levels observed with vector controls. **P < 0.01. (C) A luciferase signal was generated from luciferase-labeled Hepa1-6 cells. The signal was observed in the lungs of the C57BL/6 mice. A stronger signal was observed in the vector control group. (D) The luciferase signal was determined once a week in both the vector control group and the shRNA group. Each group contained five mice. (E) The HMGB1 in serum was measured by ELISA in different groups. (F) Metastatic nodules on the surface of lung are shown (indicated by arrows). The numbers of tumor nodules were quantified.
To determine whether HMGB1 participated in HCC metastasis in vivo, a murine lung metastasis model was utilized. Mice were injected via tail vein with luciferase expressing tumors derived from HMGB1 shRNA and vector control clones and monitored weekly for bioluminescent signals. Four weeks following injection, mice were sacrificed and their lungs were examined. Bioilluminescent signals in the lungs from the control group were much stronger than from the HMGB1 shRNA group (Fig. 8C, 8D). Furthermore, serum HMGB1 levels from the control group was 43.48 ±10.91 ng/ml, much higher than that from HMGB1 shRNA treated group 17.12 ± 4.56 ng/ml (Fig. 8E). Tumor nodules were also more numerous in the control group than the shRNA group (Fig. 8F) and were confirmed with histology (data not shown), demonstrating that HMGB1 can promote metastasis.
Discussion
Hepatocellular cancer remains a leading cause of cancer related death worldwide. This is despite the fact that a number of advances in both surgical (transplantation, resection) and ablative (transcatheter arterial chemoembolization, radiofrequency ablation) techniques have developed in the past several decades (20) and is reflective of the advanced nature of disease with which many patients present as well as the lack of effective chemotherapeutic agents aimed at the treatment of widely metastatic disease. While loss of tumor suppressor gene function, oncogene activation, direct viral effects and angiogenesis all appear to be involved in the development of HCC (21), the lack of effective chemotherapy speaks to a gap in knowledge as to the precise molecular events and pathways involved in tumor development and progression. Therefore, further elucidating such mechanism is an important goal in developing novel strategies to both prevent and treat HCC.
Hypoxia is a hallmark of diverse human solid tumors and is associated with tumor progression(8). The extent of hypoxia in a tumor may represent an independent indicator of poor prognosis (22); however, the mechanism by which hypoxia affects cancer progression is still unclear. Therefore, understanding the effects of hypoxia on cancer cell biology is an important goal; however, it is also important to identify individual gene products that may be targeted to counteract the adverse effects of hypoxia in cancer. Our study sought to determine how HMGB1, a pro-inflammatory cytokine, affects tumor invasion and metastasis induced by hypoxia. We find that hypoxia induces casapse-1 activation in HCC cells. For the first time, we report that HMGB1 is essential for hypoxia-induced caspase-1 activation in HCC cells. HMGB1 translocates from the nucleus to the cytoplasm in hypoxic HCC cells, and inhibiting its release prevents hypoxia-induced caspase-1 activation. Furthermore, treatment with rhHMGB1 or overexpression of HMGB1 in HCC cells induces caspase-1 activation even in normoxic cell culture.
HMGB1 is recognized as the prototypical DAMP (23). Initially identified as a chromatin binding protein, HMGB1 can be released by both passive (24) and active (25) pathways. The passive pathway requires loss of cell membrane integrity, as seen in necrosis. The active pathway, appears to involve HMGB1 hyperacetylation and packaging into secretory vesicles. Our previous studies showed HMGB1 release from cultured hepatocytes is an active process regulated by reactive oxygen species (ROS) in the setting of hypoxia (17). In cancer cells, HMGB1 is actively released following anticancer agent treatment, and promotes cell survival (26). We postulated that HMGB1 release in HCC cells would be an active process under hypoxic conditions. Indeed, hypoxia did not promote the expression of HMGB1, but induced the nuclear to cytoplasmic translocation of HMGB1 and release of HMGB1 into the extracellular space in two HCC cell lines indicating that HMGB1 release in this setting may affect the biologic behavior of tumors.
Tumor development and progression is associated with caspase activation, which regulates apoptosis and inflammation (27). One hallmark of tumor cells is the intrinsic or acquired resistance to apoptosis. Surprisingly, recent studies demonstrate that apoptosis promotes early tumorigenesis (19). Apoptosis-related caspase, caspase-3 and caspase-9, were activated in hypoxic HCC cells (data not shown). In our study, cell invasiveness was increased after inhibiting caspase-3 in hypoxic HCC cells which suggests that apoptosis seems to inhibit hypoxia-induced invasion (data not shown).
Studies have implicated caspase-1, an inflammation-related caspase, in a variety of responses including the host response to microbial pathogens, inflammatory diseases, and metabolic and autoimmune disorders (28). Recent studies have also shown caspase-1 is involved in tumorigenesis and tumor progression (29). Caspase-1 activation, regulated by the inflammasome, promotes the maturation of pro-inflammatory cytokines such IL-1β and IL-18 and induces inflammatory responses. Caspase-1 is activated not only in immune but also in epithelial and mesenchymal cells in conditions such as tissue repair (30). Recently, Okamoto (16) found that fresh melanoma biopsies constitutively express activated caspase-1 and secrete IL-1β. After caspase-1 activation, IL-1β -secreting human melanoma cells recruit stromal cells and induce angiogenesis, leading to tumor progression. However, the mechanisms by which caspase-1 affects tumor cancer progression remain incompletely understood. We speculate that hypoxia promotes caspase-1 activation and maturation of IL-1β and IL-18, thus contributing to invasion and metastasis.
Inflammation can promote tumorigenesis. Robust epidemiological data support the role of inflammation induced by chronic hepatitis B or C viral infections and alcohol abuse as key players in HCC development. Lymphotoxin, the proinflammatory and homeostatic cytokine, is induced by HBV or HCV infection and can promote HCC development (31). Similarly, HMGB1 can be secreted in response to HBC or HCV infection and can contribute to the pathogenesis of these infections (32, 33). Additionally, HMGB1 may be involved in the initial phases of tumorigenesis associated with these viral infections. Indeed, activation of the HMGB1 receptor RAGE significantly impacts tumorigenesis and hepatic tumor growth (34). Studies have shown that, when HMGB1 is over expressed, the oncoproteins CyclinD/E, which regulate cell proliferation, are overexpressed, while tumor suppressor protein p53 is repressed (35).
Overexpression of HMGB1 is also associated with tumor progression, including invasion and metastasis (13); however, the mechanism is still not fully understood. We hypothesized that during hypoxia, the release of HMGB1 may promote caspase-1 activation and thus contribute to invasion and metastasis of liver cancer. Our results demonstrate that inhibiting HMGB1 release or blocking its effects inhibits the activation of caspase-1 in hypoxia. In normoxia, rhHMGB1 can induce caspase-1 activation. This confirms that released HMGB1 from HCC cells can induce caspase-1 activation in both normoxia and hypoxia.
Using transwell experiments, we confirmed that hypoxia promotes liver cancer cell migration and invasion in vitro. Therefore, we wished to study what role HMGB1 plays in hypoxia-induced invasion. We found that HMGB1 induced caspase-1 activation promoted invasion in hypoxia. Conversely, knockdown of endogenous HMGB1 specifically using shRNA significantly reduced the invasiveness of HCC cells, indicating that HMGB1 is closely involved in HCC invasion. Furthermore, an in vivo murine model of HCC lung metastases also confirmed that HMGB1 is associated with tumor invasion and metastasis. Taken together, our data demonstrate that HMGB1 plays a pivotal role in HCC invasion and metastasis by way of enhancing invasiveness and activating caspase-1 with subsequent production of multiple mediators. These findings support the notion that HMGB1 may serve as a suitable target for the development of novel anticancer agents.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDMENTS
Expert technical assistance from X. Liao, L. Shao and J. Chen is appreciated. We thank J. Evankovich, L. Zhang, G. Nace and J. Klune for valuable advice and discussion.
Support: This work was supported by Howard Hughes Medical Institute Physician-Scientist Award (A.T.), American College of Surgeons Research Fellowship (A.T.).
Abbreviations
- DAMP
damage associated molecular pattern
- HCC
Hepatocellular Carcinoma
- HMGB1
High Mobility Group Box 1
- RAGE
receptor for advanced glycation end products
- TLR
Toll-Like Receptor
Footnotes
Authorship and contributions:
W.Y. performed the experiments and assisted with planning the experiments, data analysis and writing the manuscript; Y.C., X.L. and H.H. performed experiments; S.T. assisted with labeling the cells and animal works; J.C, S.M, D.G. and M.L. assisted with planning the experiments and revising the manuscript; A. T.coordinated the project, assisted with planning the experiments, data analysis and revising the manuscript.
References
- 1.Rampone B, Schiavone B, Confuorto G. Current management of hepatocellular cancer. Curr Oncol Rep. 2010;12:186–192. doi: 10.1007/s11912-010-0094-3. [DOI] [PubMed] [Google Scholar]
- 2.Llovet JM, Di Bisceglie AM, Bruix J, Kramer BS, Lencioni R, Zhu AX, Sherman M, et al. Design and endpoints of clinical trials in hepatocellular carcinoma. J Natl Cancer Inst. 2008;100:698–711. doi: 10.1093/jnci/djn134. [DOI] [PubMed] [Google Scholar]
- 3.Talwalkar JA, Gores GJ. Diagnosis and staging of hepatocellular carcinoma. Gastroenterology. 2004;127:S126–132. doi: 10.1053/j.gastro.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 4.Lu SC, Mato JM. Role of methionine adenosyltransferase and S-adenosylmethionine in alcohol-associated liver cancer. Alcohol. 2005;35:227–234. doi: 10.1016/j.alcohol.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 5.Di Bisceglie AM, Simpson LH, Lotze MT, Hoofnagle JH. Development of hepatocellular carcinoma among patients with chronic liver disease due to hepatitis C viral infection. J Clin Gastroenterol. 1994;19:222–226. doi: 10.1097/00004836-199410000-00011. [DOI] [PubMed] [Google Scholar]
- 6.Di Bisceglie AM, Rustgi VK, Hoofnagle JH, Dusheiko GM, Lotze MT. NIH conference. Hepatocellular carcinoma. Ann Intern Med. 1988;108:390–401. doi: 10.7326/0003-4819-108-3-390. [DOI] [PubMed] [Google Scholar]
- 7.Harrison L, Blackwell K. Hypoxia and anemia: factors in decreased sensitivity to radiation therapy and chemotherapy? Oncologist. 2004;9(Suppl 5):31–40. doi: 10.1634/theoncologist.9-90005-31. [DOI] [PubMed] [Google Scholar]
- 8.Finger EC, Giaccia AJ. Hypoxia, inflammation, and the tumor microenvironment in metastatic disease. Cancer Metastasis Rev. 2010;29:285–293. doi: 10.1007/s10555-010-9224-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ding W, You H, Dang H, LeBlanc F, Galicia V, Lu SC, Stiles B, et al. Epithelial-to-mesenchymal transition of murine liver tumor cells promotes invasion. Hepatology. 2010;52:945–953. doi: 10.1002/hep.23748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Erler JT, Bennewith KL, Cox TR, Lang G, Bird D, Koong A, Le QT, et al. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell. 2009;15:35–44. doi: 10.1016/j.ccr.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peinado H, Lavotshkin S, Lyden D. The secreted factors responsible for pre-metastatic niche formation: old sayings and new thoughts. Semin Cancer Biol. 2011;21:139–146. doi: 10.1016/j.semcancer.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 12.Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol. 2010;28:367–388. doi: 10.1146/annurev.immunol.021908.132603. [DOI] [PubMed] [Google Scholar]
- 13.Tang D, Kang R, Zeh HJ, 3rd, Lotze MT. High-mobility group box 1 and cancer. Biochim Biophys Acta. 2010;1799:131–140. doi: 10.1016/j.bbagrm.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 15.Carrascal MT, Mendoza L, Valcarcel M, Salado C, Egilegor E, Telleria N, Vidal-Vanaclocha F, et al. Interleukin-18 binding protein reduces b16 melanoma hepatic metastasis by neutralizing adhesiveness and growth factors of sinusoidal endothelium. Cancer Res. 2003;63:491–497. [PubMed] [Google Scholar]
- 16.Okamoto M, Liu W, Luo Y, Tanaka A, Cai X, Norris DA, Dinarello CA, et al. Constitutively active inflammasome in human melanoma cells mediating autoinflammation via caspase-1 processing and secretion of interleukin-1beta. J Biol Chem. 2010;285:6477–6488. doi: 10.1074/jbc.M109.064907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsung A, Klune JR, Zhang X, Jeyabalan G, Cao Z, Peng X, Stolz DB, et al. HMGB1 release induced by liver ischemia involves Toll-like receptor 4 dependent reactive oxygen species production and calcium-mediated signaling. J Exp Med. 2007;204:2913–2923. doi: 10.1084/jem.20070247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yan W, Fu Y, Tian D, Liao J, Liu M, Wang B, Xia L, et al. PI3 kinase/Akt signaling mediates epithelial-mesenchymal transition in hypoxic hepatocellular carcinoma cells. Biochem Biophys Res Commun. 2009;382:631–636. doi: 10.1016/j.bbrc.2009.03.088. [DOI] [PubMed] [Google Scholar]
- 19.Tang D, Lotze MT, Kang R, Zeh HJ. Apoptosis promotes early tumorigenesis. Oncogene. 2010;30:1851–1854. doi: 10.1038/onc.2010.573. [DOI] [PubMed] [Google Scholar]
- 20.Jarnagin W, Chapman WC, Curley S, D'Angelica M, Rosen C, Dixon E, Nagorney D. Surgical treatment of hepatocellular carcinoma: expert consensus statement. HPB (Oxford) 2010;12:302–310. doi: 10.1111/j.1477-2574.2010.00182.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cha C, Dematteo RP. Molecular mechanisms in hepatocellular carcinoma development. Best Pract Res Clin Gastroenterol. 2005;19:25–37. doi: 10.1016/j.bpg.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 22.Chia SK, Wykoff CC, Watson PH, Han C, Leek RD, Pastorek J, Gatter KC, et al. Prognostic significance of a novel hypoxia-regulated marker, carbonic anhydrase IX, in invasive breast carcinoma. J Clin Oncol. 2001;19:3660–3668. doi: 10.1200/JCO.2001.19.16.3660. [DOI] [PubMed] [Google Scholar]
- 23.Yang D, Chen Q, Yang H, Tracey KJ, Bustin M, Oppenheim JJ. High mobility group box-1 protein induces the migration and activation of human dendritic cells and acts as an alarmin. J Leukoc Biol. 2007;81:59–66. doi: 10.1189/jlb.0306180. [DOI] [PubMed] [Google Scholar]
- 24.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 25.Youn JH, Shin JS. Nucleocytoplasmic shuttling of HMGB1 is regulated by phosphorylation that redirects it toward secretion. J Immunol. 2006;177:7889–7897. doi: 10.4049/jimmunol.177.11.7889. [DOI] [PubMed] [Google Scholar]
- 26.Tang D, Kang R, Cheh CW, Livesey KM, Liang X, Schapiro NE, Benschop R, et al. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene. 2010;29:5299–5310. doi: 10.1038/onc.2010.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 28.Davis BK, Wen H, Ting JP. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol. 2011;29:707–735. doi: 10.1146/annurev-immunol-031210-101405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu B, Elinav E, Huber S, Booth CJ, Strowig T, Jin C, Eisenbarth SC, et al. Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proc Natl Acad Sci U S A. 107:21635–21640. doi: 10.1073/pnas.1016814108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yazdi AS, Drexler SK, Tschopp J. The role of the inflammasome in nonmyeloid cells. J Clin Immunol. 2010;30:623–627. doi: 10.1007/s10875-010-9437-y. [DOI] [PubMed] [Google Scholar]
- 31.Haybaeck J, Zeller N, Wolf MJ, Weber A, Wagner U, Kurrer MO, Bremer J, et al. A lymphotoxin-driven pathway to hepatocellular carcinoma. Cancer Cell. 2009;16:295–308. doi: 10.1016/j.ccr.2009.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou RR, Zhao SS, Zou MX, Zhang P, Zhang BX, Dai XH, Li N, et al. HMGB1 cytoplasmic translocation in patients with acute liver failure. BMC Gastroenterol. 2011;11:21. doi: 10.1186/1471-230X-11-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jung JH, Park JH, Jee MH, Keum SJ, Cho MS, Yoon SK, Jang SK. Hepatitis C virus infection is blocked by HMGB1 released from virus-infected cells. J Virol. 2011;85:9359–9368. doi: 10.1128/JVI.00682-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DiNorcia J, Moroziewicz DN, Ippagunta N, Lee MK, Foster M, Rotterdam HZ, Bao F, et al. RAGE signaling significantly impacts tumorigenesis and hepatic tumor growth in murine models of colorectal carcinoma. J Gastrointest Surg. 2010;14:1680–1690. doi: 10.1007/s11605-010-1347-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gong H, Zuliani P, Komuravelli A, Faeder JR, Clarke EM. Analysis and verification of the HMGB1 signaling pathway. BMC Bioinformatics. 2010;11(Suppl 7):S10. doi: 10.1186/1471-2105-11-S7-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.