

Abstract
Gram-negative bacteria are implicated in the causation of life-threatening hospital-acquired infections. They acquire rapid resistance to multiple drugs and available antibiotics. Hence, there is the need to discover new antibacterial agents with novel scaffolds. For the first time, this study explores the 1,3,5-triazine-2,4-diamine and 1,2,4-triazine-2,4-diamine group of compounds as potential inhibitors of E. coli DHFR, a pivotal enzyme in the thymidine and purine synthesis pathway. Using differential scanning fluorimetry, DSF, fifteen compounds with various substitutions on either the 3rd or 4th positions on the benzene group of 6,6-dimethyl-1-(benzene)-1,3,5-triazine-2,4-diamine were shown to bind to the enzyme with varying affinities. Then, the dose dependence of inhibition by these compounds was determined. Preliminary quantitative structure-activity relationship analysis and docking studies implicate the alkyl linker group and the sulfonyl fluoride group in increasing the potency of inhibition. 4-[4-[3-(4,6-diamino-2,2-dimethyl-1,3,5-triazin-1-yl)phenyl]butyl]benzenesulfonyl fluoride (NSC120927), the best hit from the study and a molecule with no reported inhibition of E. coli DHFR, potently inhibits the enzyme with a Ki value of 42.50 ± 5.34 nM, followed by 4-[6-[4-(4,6-diamino-2,2-dimethyl-1,3,5-triazin-1-yl)phenyl]hexyl]benzenesulfonyl fluoride(NSC132279), with a Ki value of 100.9 ± 12.7 nM. Detailed kinetic characterization of the inhibition brought about by five small-molecule hits shows that these inhibitors bind to the dihydrofolate binding site with preferential binding to the NADPH-bound binary form of the enzyme. Furthermore, in search of novel diaminotriazine scaffolds, it is shown that lamotrigine, a 1,2,4-triazine-3,5-diamine and a sodium-ion channel blocker class of antiepileptic drug, also inhibits E. coli DHFR. This is the first comprehensive study on the binding and inhibition brought about by diaminotriazines of a gram-negative prokaryotic enzyme and provides valuable insights into the SAR as an aid to the discovery of novel antibiotics.
Keywords: Mechanistic characterization; Inhibition kinetics; Escherichia coli dihydrofolate reductase; QSAR; drug discovery; 2,4-diamino-1,3,5-triazine; 2,4-diamino 1,2,4-triazine
Introduction
The emergence and rapid dissemination of drug resistance in disease-causing gram-negative bacteria presents a challenge to the treatment of life-threatening hospital-acquired infections. This also poses a general threat to prevalent healthcare management practices by creating species resistant to all currently available antibacterial agents. There are several mechanisms by which gram negative bacteria acquire resistance to known drugs in the shortest possible time span. Most prominent among these are drug efflux pumps, acquisition of plasmids encoding antibiotic-resistance genes and acquisition of mutations in a biological target making it refractory to the action of the drug1, 2. In fact, a survey of reported antibiotics of natural origin showed that among those compounds that showed activity against gram-positive bacteria, more than 90% lacked activity at a useful level against E. coli3. Given the alarming rise in instances of hospital-acquired infections by drug-resistant gram-negative bacteria4–11, it becomes imperative to search for novel antibiotic agents against these organisms.
Dihydrofolate reductase, DHFR, is an important enzyme in the de novo pathway of purine and thymidine synthesis. Small-molecules targeting this enzyme have demonstrated utility as potential antibiotics12. However, this enzyme acquires rapid resistance to available anti-folates. Several classes of compounds have been explored for their potential anti-folate activity. Predominant classes include diaminoquinazoline13–18, diaminopyrimidine19–22, diaminopteridine23 and diaminotriazines24.
Triazines are organic nitrogen containing heterocycles. They are classified into three different types based on the separation of the nitrogen atoms on the ring: 1,2,3-triazines, 1,2,4-triazines and 1,3,5-triazines. Among the three isomers, 1,3,5-triazine compounds are the best studied and are also known as symmetric triazines or s-triazines while 1,2,3-triazines are the least studied because of their poor stability25. 1,2,4-triazines have better solubility than 1,2,3-triazines and are comparatively better known, with the most prominent example being lamotrigine, a sodium-channel blocker class of antiepileptic drug26. 1,2-dihydro-1,3,5-triazine (baker triazines) compounds are by far the best known with several studies exploring the potential of 2,4-diamino-1,3,5-triazines as inhibitors of eukaryotic DHFRs24. The most prominent example of the diaminotriazine group of compounds as DHFR inhibitors is the inhibition of Plasmodium falciparum DHFR by cycloguanil, a derivative of proguanil27, 28. Detailed QSAR analysis of diaminotriazine derivatives has been carried out on DHFRs from several different organisms29–39. Recently, hybrids of triazines have also been demonstrated to show inhibitory activity on DHFRs40, 41.
However, most studies have focused on understanding the inhibitory effect of diaminotriazines on DHFRs from eukaryotic sources. Moreover, there is a total lack of kinetic characterization to understand their mechanism of DHFR inhibition. Knowledge of detailed kinetic mechanism is useful for designing effective drugs. One possible reason for the non-exploration of diaminotriazines as inhibitors of gram-negative bacterial DHFRs in general, and EcDHFR in particular, might stem from the consistently low affinities and poor inhibition by this class of molecules of prokaryotic DHFRs.
This study is the first attempt at detailed mechanistic characterization of diaminotriazine family of compounds by taking recourse to inhibition kinetics to assess their effect on EcDHFR. It also sheds valuable insights into the QSAR of 2,4-diamino-1,3,5-triazine inhibition of E. coli DHFR. Further, macromolecular docking studies provides valuable insights into the physical interactions that are likely responsible for conferring potency and specificity of the small-molecule/protein interaction. For the first time, we report two novel nanomolar inhibitors (NSC120927 and NSC132279) of a gram-negative prokaryotic enzyme from the 1,3,5-triazine-2,4-diamine class of molecules. This, combined with studies on the use of 1,2,4-triazine-3,5-diamine as novel scaffolds for E. coli DHFR inhibitors, opens up the possibility of exploring a new class of molecules that could potentially yield novel antibiotic candidates.
Results
Binding of 1,3,5-triazine-2,4-diamine analogues to EcDHFR
Binding of a small molecule ligand to the protein target of interest is a prerequisite for specific inhibition. Fifteen derivatives of 1,3,5-triazine-2,4-diamine were assessed for their ability to bind to the apo form of EcDHFR (Fig. 1). Differential scanning fluorimetry (DSF), a methodology relying on the increased protein stability conferred by small molecule binding as assessed by the environmentally-sensitive extrinsic fluorophore dye Sypro-Orange, was used to assess binding.
Fig. 1.

Structures of diaminotriazines employed in this study. The structures were downloaded from PubChem with following PubChem CIDs: 3929273, 280860, 272645, 409219, 9049, 88868, 271921, 309795, 308877, 54606350, 280527, 274731, 419313, 3878, 1986. The molecules are binned into 5 different clusters based on a Tanimoto coefficient cutoff of 0.4.
Fig 2 and Table 1 summarize the binding assay results. For ease of comprehension of results, molecules were classified into four different classes based on the chemical nature of substituents on the 1-phenyl-6,6-dimethyl-1,3,5-triazine-2,4-diamine group. Halide (NSC3074), methyl (NSC212229), methoxy (NSC19732) and ethoxy (NSC19745) substituents gave ΔTm values ranging from ∼10°C-13°C (Fig 2A and Table 1). Nitrile (NSC115928) and dimethylamino (CHEMBL597262) substituents at R1 position and aminomethyl substituents (NSC168184) at the R2 position of 1-phenyl-6,6-dimethyl-1,3,5-triazine-2,4-diamine gave comparable ΔTm values ranging from ∼9°C-13°C (Fig 2B and Table 1). However substitution of the aminomethyl group at the R1 position (NSC211137) drastically reduced binding as assessed by the magnitude of the thermal shift (∼4 °C) (Fig 2B and Table 1). This might be because of unfavorable electrostatic interactions of this ligand with the apo form of the enzyme. Molecules having alkyl benzenesulfonyl fluoride (NSC120927 and NSC1332279), phenoxypropoxyphenyl (NSC133071), phenylbutyl (NSC132277) and fluorosulfonylphenylaminocarbonyl (NSC113909) substituents at either R2 or R1 position of 1-phenyl-6,6-dimethyl-1,3,5-triazine-2,4-diamine gave ΔTm values ranging from ∼11°C- 23°C (Fig. 2C and Table 1). Molecules in this class were by far the best binders as ranked by the apparent dissociation constants for all the ligands assessed in this study. It should be pointed out here that NSC132279 and NSC132277 were assessed at 10 µM concentrations rather than 500 µM, as was done for the other small-molecules. However, 2,4-dichlorophenylbutyl (NSC104129) substituent at R1 position gave a poor ΔTm value of 5.8 °C (Fig 2C and Table 1). NSC117268, a compound containing 4-chlorophenyl and sulfonamide substitution at the R3 and R1 positions of 1,3,5-triazine-4,6-diamine group, gave a reasonable ΔTm value of 7.6 °C (Fig 2D and Table 1). The results from binding analysis indicates that all 15 diaminotriazine small-molecules showed binding to E. coli DHFR to variable extents depending on the nature of the substituents.
Fig. 2.

Differential scanning fluorimetry, DSF, curves and their first derivatives for E. coli DHFR in the presence of 15 analogues of 1,3,5-triazine-2,4-diamine. (A) DSF curves for halide, methyl, methoxy and ethoxy substituents at the R1 position of 1-phenyl-6,6-dimethyl-1,3,5-triazine-2,4-diamine. (B) DSF curves for nitrile, dimethylamino and aminomethyl substituents at either R1 or R2 position of 1-phenyl-6,6-dimethyl-1,3,5-triazine-2,4-diamine. (C) DSF curves for alkyl benzenesulfonyl fluoride, phenoxypropoxyphenyl, phenylbutyl, 2,4-dichlorophenylbutyl and fluorosulfonylphenylaminocarbonyl substituents at either R1 or R2 position of 1-phenyl-6,6-dimethyl-1,3,5-triazine-2,4-diamine. Note that this class of molecules showed the largest thermal shifts. (D) DSF curve for 4-chlorophenyl and sulfonamide substitution at the R1 and R3 position of 1-phenyl-1,3,5-triazine-4,6-diamine. The y-axis in primary unfolding curves represents normalized fluorescence and the x-axis shows the temperature in degrees Celsius. The inhibitors were kept fixed at 500 µM except NSC132277 and NSC132299, which were done at 10 µM concentration. Right panel in each plot shows the Gaussian fit of first-derivative for curves from left panel. Note that the molecules NSC117268, NSC133071, NSC168184, NSC104129, CHEMBL597262 and NSC333873 were also independently picked by the virtual ligand screening algorithm, PoLi.
Table 1.
Summary of binding and inhibition parameters for diaminotriazine derivatives employed in this study.
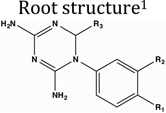 | |||||||
|---|---|---|---|---|---|---|---|
| Chem ID | Position | Substitution | Binding and inhibition potency | ||||
| ΔTm (°C)2 | KDapp( µM)3 | ClogP6 | IC-50 (µM) | Ki app*(µM) | |||
| NSC120927 | R2, R3 | 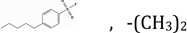 |
14.05 | 6.93 | 3.8490 | 1.15 ± 0.37 | 0.0497 ± 0.004 |
| NSC1322795 | R1, R3 | 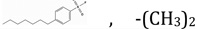 |
23.13 | 0.01 | 4.9070 | 1.84 ± 0.13 | 0.0926 ± 0.010 |
| NSC1322775 | R2, R3 | 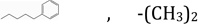 |
11.04 | 0.34 | 3.6860 | 7.59 ± 0.24 | 0.3591 ± 0.032 |
| NSC133071 | R1, R2, R3 | 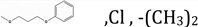 |
14.45 | 6.16 | 3.1958 | 18.45 ± 0.33 | 0.8802 ± 0.075 |
| NSC1041294 | R1, R3 | 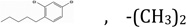 |
5.8 | 82.55 | 5.1120 | ND | ND |
| NSC115928 | R2, R3 | N ≡ C—, −(CH3)2 | 13.26 | 8.74 | 0.0115 | 20.19 ± 0.56 | 0.958 ± 0.06 |
| NSC3074 | R1, R3 | −Cl, −(CH3)2 | 12.25 | 11.77 | 1.0658 | 20.91 ± 1.01 | 1.02 ± 0.12 |
| NSC212229 | R1, R3 | −CH3, −(CH3)2 | 13.17 | 9.32 | 0.6810 | 32.27 ± 0.83 | 1.55 ± 0.11 |
| NSC19732 | R1, R3 | −O−CH3, −(CH3)2 | 10.97 | 17.23 | 0.4547 | 89.10 ± 2.75 | 4.02 ± 0.46 |
| CHEMBL5972624 | R1, R3 | −N(CH3)2, −(CH3)2 | 10.73 | 18.51 | 0.6520 | ND | ND |
| NSC19745 | R1, R3 | −O−CH2−CH3, −(CH3)2 | 9.79 | 24.53 | 0.9837 | 262.7 ± 6.68 | 11.16 ± 1.14 |
| NSC168184 | R2, R3 | −CH2−NH2, −(CH3)2 | 8.79 | 33.16 | −0.8660 | 478.4 ± 7.28 | 20.22 ± 3.10 |
| NSC211137 | R1, R3 | CH2−NH2, −(CH3)2 | 3.99 | 144.54 | −0.866 | 721.6 ± 6.65 | 39.30 ± 4.05 |
| NSC113909 | R2, R3 |  |
21.92 | 0.73 | 1.8274 | 62.83 ± 2.13 | 2.89 ± 0.35 |
| NSC117268 | R1, R3 |  |
7.57 | 48.01 | 0.4315 | 1548 ± 40 | 105.0 ± 13.51 |
| Lamotrigine1 | NA | ND | ND | 2.5343 | 348.9 ± 6.7 | 15.24 ± 2.78 | |
The Kiapp values are computed by fitting the dose-dependence inhibition curves to Morrison equation.
All structures except lamotrigine.
Thermal shift reported are at 500 µM ligand concentration in the absence of NADPH. The Tm for protein alone curve is 51.9 °C.
Approximate dissociation constant computed from the magnitude of thermal shift.
No inhibition done.
No signal seen at 500 µM and hence values reported for 10 µM.
Values computed by ChemBioDraw 14.0. ND, not determined. NA, not applicable.
Inhibition of EcDHFR by 1,3,5-triazine-2,4-diamine analogues
To further understand whether binding by these small molecules translates into inhibition, fourteen small molecules were tested for the dose-dependence of inhibition. NSC104129 and CHEMBL597262 were excluded from this analysis due to lack of sufficient quantities for inhibition studies. Fig 3A shows the experimental curves for dose-dependence of inhibition, and Table 1 summarizes the IC50 values for the 14 derivatives of 1,3,5-triazine-2,4-diamine tested. The apparent inhibition constants (Kiapp) were computed by fitting the curves to Morrison’s quadratic equation (Fig 3B and Table 1). Once again, the values in Table 1 clearly show that derivatives with alkyl benzenesulfonyl fluoride (NSC120927 and NSC132279), phenoxypropoxyphenyl (NSC133071) and phenylbutyl (NSC132277) at either the R2 or R1 position of 1-phenyl-6,6-dimethyl-1,3,5-triazine-2,4-diamine are better inhibitors than the other small molecules employed in the study. The best hit was NSC120927 with a butyl benzenesulfonyl fluoride substitution at R2 position on 1-phenyl-6,6-dimethyl-1,3,5-triazine-2,4-diamine; it inhibited the enzyme with an IC50 and Kiapp value of ∼1 µM and ∼50 nM, respectively. The second best hit was NSC132279, a hexylbenzenesulfonyl fluoride substituted at R1 position, which showed an IC50 and Kiapp value of ∼2 µM and ∼93 nM, respectively.
Fig. 3.

Dose-dependent inhibition of E. coli DHFR by analogues of 1,3,5-triazine-2,4-diamine. (A) IC50 determination of 14 compounds. (B) Fit of the experimental dose-response curves to Morrison’s equation to compute the Kiapp for various inhibitors of E. coli DHFR. Curves showing inhibition by derivatives having halide, methyl, methoxy, ethoxy, nitrile, dimethylamino and aminomethyl substituents at either the R1 or R2 position of 1-phenyl-6,6-dimethyl-1,3,5-triazine-2,4-diamine are shown in blue; derivatives having alkyl benzenesulfonyl fluoride, phenoxypropoxyphenyl and phenylbutyl at either R1 or R2 position of 1-phenyl-6,6-dimethyl-1,3,5-triazine-2,4-diamine are shown in green; derivative having fluorosulfonylphenylaminocarbonyl at either R2 position of 1-phenyl-6,6-dimethyl-1,3,5-triazine-2,4-diamine is shown in gold; derivative having a 4-chlorophenyl and benzenesulfonamide substitution at the R1 and R3 position of 1-phenyl-1,3,5-triazine-4,6-diamine is shown in pink; Lamotrigine, a 6-(2,3-dichlorophenyl)-1,2,4-triazine-3,5-diamine is shown in red. All activities are expressed as percentage activity. The numbered notations for the various inhibitor molecules represent NSC numbers.
However, the derivative with fluorosulfonylphenylaminocarbonyl substitution (NSC113909), in spite of showing very tight binding as evident in a ΔTm value of ∼22°C, poorly inhibited the enzyme with an IC50 and Kiapp value of ∼63 µM and ∼3 µM, respectively (Table 1). This might be because of the planarity of the peptide bond and partial charges introduced by the peptide group as a linker.
It should be noted that lamotrigine, a 1,2,4-triazine-3,5-diamine and a sodium-ion channel blocker class of antiepileptic drug, is also an inhibitor of E. coli DHFR inhibiting the enzyme with an IC50 and Kiapp of 348.9 ± 6.7 µM and 15.24 ± 2.78 µM, respectively (Table 1 and Fig 3). Weak inhibition by this compound could be ascribed to the lack of dimethyl hydrophobic substituents at the 6th position. Lamotrigine is substantially different structurally from the other 6,6-dimethyl-1,3,5-triazine-2,4-diamine derivatives employed in this study (Fig 1) and represents a novel scaffold that could be modified for design of new inhibitor molecules with activity against E. coli DHFR.
An important aspect to note is that the inhibition by this family of compounds does not conform to the slow-onset tight binding mechanism as evident in the linear time-course curves. It should be noted here that most other reported inhibitors of EcDHFR display slow-onset tight binding inhibition.
To further understand the relationship between the binding of these small-molecules to the apo form of the enzyme and inhibition assessed on the holo-enzyme, a scatter plot of 1/IC50 vs. ΔTm was generated (Fig S1A). A Pearson correlation computed between the two parameters gave an R-value of 0.5774 and an R2 of 0.33 indicating poor correlation. Lack of strong linear correlation between the magnitude of the thermal shift (hence binding) and inhibition indicates that there might be substantial differences between the mode of binding and inhibition. Moreover, since binding was assessed in the absence of NADPH, it is possible that occupancy of the NADPH binding pocket may change the charge distribution in the diaminotriazine small-molecule binding pocket.
Quantitative structure-activity relationship analysis and macromolecular docking
To quantitatively understand the inhibition brought about the diaminotriazine series of compounds studied and to increase the predictive value of the current study in its ability to find more potent inhibitors, a QSAR analysis was carried out. Various physiochemical parameters (like hydrogen bond donors, hydrogen bond acceptors, total polar surface area, geometrical shape coefficient etc) of the molecules were computed employing OpenBabel, Joelib and ChemmineR descriptors. Most of the parameters, when individually assessed, showed no correlation with inhibition potency and a representative scatter plot for tPSA depicting this lack of correlation is shown in Fig S1B. However, inhibition by these molecules showed good correlation to both atomistic (LogP) and fragment-based partition coefficient (CLogP) (Fig S1C and S1D), whereby increasing the hydrophobicity of a compound led to increased inhibition. The equations defining the relationship between the partition coefficients of the small-molecules and the respective inhibition constants are shown in the figure along with the regression measure. This result is in broad agreement with already reported QSAR analysis for DHFRs from eukaryotic sources which had indicated that hydrophobic triazines are particularly effective against P. carinii DHFR34 and mammalian DHFRs31. This similarity in QSAR behavior is in spite of the low sequence similarity shared by the prokaryotic and eukaryotic DHFRs.
To perform a more definitive QSAR, a genetic algorithm (GA)-based heuristic methodology was employed for selection of significant variables that influences inhibition42. Genetic algorithms are a subclass of evolutionary algorithms (EA) that are employed for generating solutions to optimization problems involving multiple variables employing the principles of natural evolution like inheritance, mutation, crossover and selection. The best solution with good statistics of fit is given below
Log 1/Kiapp = 0.90(±0.79) −0.61(±0.22) bonds + 0.80(±0.40) nF −1.2(±0.26) HBD + 0.6 (±0.22) atoms; R=0.922
“bonds” indicate the number of single bonds, “nF” indicates the number of fluorine atoms and “HBD” indicates hydrogen-bonding donors.
This result, along with the correlation that LogP score showed, indicates that the number of fluorine atoms, hydrogen bonding donors and LogPs contribute to better fits. Thus, the presence of an alkyl benzene group in general, and the alkyl benzenesulfonyl fluoride group in particular, has a major influence in determining inhibition potency. This provides vital clues about the type of modifications that might be desirable on the small-molecules to increase their potency of inhibition.
To understand the physical basis for inhibitor discrimination, template-based macromolecular rigid body docking was carried out. The conformation of folate in complex with DHFR 43 was used as reference for docking the small molecules inside DHFR’s binding site. To understand the procedure, it is helpful to describe the interaction of folate with DHFR in more detail, as shown in Figure 4A. The folate molecule can be broken into three fragments based on the interaction each fragment has with DHFR’s active-site pocket and the pocket’s neighboring residues; (a) The 2-amino-4-oxo-1,4-dihydropteridin group interacts with the active-site pocket residues of DHFR, (b) The methylaminobenzoyl group in the middle of folate is in contact with four hydrophobic amino acids (Leu28, Phe31, Ile50 and Leu54) and finally (c) The polar and negatively charged glutamic acid tail fragment of the folate interacts with the positive triad of Lys32, Arg52 and Arg57 located at the entrance of the binding site.
Fig. 4.

Template-based docking of the best hits from the study (A) Folate (B) NSC120927 (C) NSC132279 (D) NSC132277 (E) NSC133071 (F) NSC113909. The positive charged ring located at the entrance of the pocket (Lys 32, Arg52 and Arg57) is shown in green van der Waals representation. DHFR is shown in a white cartoon representation. The hydrophobic ring (Leu28, Phe31, Ile50 and Leu54) is shown in a pink van der Waals representation. Ligands are presented in licorice format showing the heavy atoms: carbon (cyan), nitrogen (blue), oxygen (red), sulfur (yellow), fluorine (pink), chlorine (ochre). The figures were rendered using Tachyon 64 and VMD 65 was used for visualization.
After aligning different conformations of the various small molecules to folate (see Methods section), the best alignment for each case was chosen based on the highest combined shape and chemical similarity scores (Table 2) given by LIGSIFT44. All five small-molecules share the diaminotriazine group, which is aligned to the 2-amino-4-oxo-1,4-dihydropteridin group of folate and ends up interacting with the binding site amino acids (Figure 4 B–F). Therefore, the differential affinity of these molecules could be due to the dissimilarity in the other regions that we next explain in more detail. The molecule that shows good binding and most potent inhibition, NSC120927 (Figure 4B), shows the best scores for shape and chemical similarity when compared to the rest of the molecules, with significant p-values as shown in Table 2. In this case, the sulfonyl fluoride group aligns to the tail fragment of folate allowing it to preserve the favorable interaction with the triad of positive residues located at the entrance of the binding site. Also, the central hydrophobic fragment of NSC120927 (two aromatic rings linked by four carbons) aligns to the middle fragment of folate and can interact strongly with the hydrophobic patch made of Leu28, Phe31, Ile50 and Leu54.
Table 2.
The best alignment scores and the corresponding P-value for each of the predicted molecules.
| Name | ShapeSim1 | P-value | ChemSim2 | P-value |
|---|---|---|---|---|
| NSC120927 | 0.617 | 3.96×10−2 | 0.460 | 6.78×10−2 |
| NSC132279 | 0.599 | 7.58×10−2 | 0.421 | 1.85×10−1 |
| NSC132277 | 0.634 | 2.37×10−2 | 0.447 | 1.05×10−1 |
| NSC1330713 | 0.564 | 7.87×10−2 | 0.329 | 7.03×10−1 |
| NSC113909 | 0.596 | 8.34×10−2 | 0.461 | 6.98×10−2 |
Shape-similarity score calculated by LIGSIFT.
Chemical-similarity score calculated by LIGSIFT.
This case was the only one with no significant score when considering both shape and chemistry mode of LIGSIFT for the alignment, therefore only shape mode was used for this case. For the other cases shape+chemistry mode of LIGSIFT was used to generate the alignments.
The second best molecule, NSC132279 (Figure 4C), has almost the same structure as the first with a minor difference. Now, the two aromatic rings are connected with a longer linker (six carbons) making it slightly harder for it to fit within the pocket (reflected in the lower shape and chemical similarity scores in Table 2) and preserve the interaction with the hydrophobic patch. Therefore, the strong binding and inhibition by NSC120927 and NSC132279 can be ascribed to possible charged group interactions formed by the negative groups on the ligands with the apo protein target as well as their interaction with the hydrophobic patch. Hydrogen bonding of the charged moiety is possible; as evident in Fig S2, which shows that the distances and angles of the charged portion of NSC120927 are compatible with formation of moderate to strong hydrogen bonds45, 46. As depicted in the figure, the angles of the predicted hydrogen bonds are between 145–160 degrees and the distances are between 1.56–2.80 Å. This observation is in agreement with obtained QSAR results that selected “hydrogen-bonding donor” as a possible variable that determines efficacy of inhibition.
The third molecule, NSC132277 (Figure 4D), has the same structure as NSC120927 but lacks the sulfonyl fluoride group. Hence, it loses favorable interactions with the positively charged ring, and thus, has lower affinity (Table 1). The next molecule, NSC133071 (Figure 4E), also lacks the sulfonyl fluoride group of NSC120927. Additionally, chlorine is introduced in one of the aromatic rings, and the chlorophenoxypropoxy linker between the two rings has two oxygen atoms rather than a phenyl alkyl linker (Fig 1). The docking pose of NSC133071 shows that the chlorine and one of the two oxygen atoms are in the vicinity of the hydrophobic patch, therefore resulting in unfavorable interactions and lower binding affinity.
NSC113909 is a unique molecule that shows good binding, but poor inhibition of E. coli DHFR. NSC113909 (Figure 4F) shows higher shape similarity than NSC133071 to folate and the chemical similarity is same as that of the best molecule (NSC120927). The disparity between binding and inhibition could be explained by the fact that the charged interactions made by the sulfonyl fluoride group is preserved while the peptide bond as linker between two aromatic rings is unfavorable for bringing about effective inhibition. We suggest that the peptide bond locks the molecule in fewer conformations and reduces its degrees of freedom, thereby making it harder for it to access the binding site due to steric effects in the NADPH-bound holo enzyme. Moreover, burying the charges on the peptide moiety of NSC113909 in the NADPH-bound holo protein might impose an energetic cost making it a weaker inhibitor.
In summary, preliminary QSAR and macromolecular rigid-body docking studies indicates that the length of the alkyl linker and the sulfonyl fluoride group play pivotal roles in determining the potency of inhibition.
1,3,5-triazine-2,4-diamine analogues as competitive inhibitors of H2F binding
To further understand the inhibition shown by derivatives of 1,3,5-triazine-2,4-diamines, we resorted to detailed inhibition kinetics. The top four inhibitors showing the lowest Kiapp values, indicating highest affinities, were selected to understand the mechanism of inhibition. To better understand the mechanism, the analysis was split into inhibition brought about by alkyl benzenesulfonyl fluoride substituents at either R2 (NSC120927) or R1 (NSC132279) position of 1-phenyl-6,6-dimethyl-1,3,5-triazine-2,4-diamine ring and those shown by phenoxypropoxyphenyl (NSC133071) and phenylbutyl (NSC132277) substituents.
Substrate dihydrofolate (H2F) was titrated at several fixed concentrations of NSC120927 and NSC132279, and the resulting curves from the experiment were globally fit to models for the various types of inhibition. A sum-of-square F-test was performed to validate the non-linear fits. Both the curves for NSC120927 and NSC132279 showed the best fit to the model for competitive inhibition (Fig 5A and 4B) yielding a Ki, the equilibrium dissociation constant for the competitive inhibitor, of 42.50 ± 5.34 nM and 100.9 ± 12.7 nM, respectively (Table 3). Further, for visual understanding, the data were transformed and plotted as the double-reciprocal Lineweaver-Burk plot, LB. The right hand panels in Fig 5A and 5B show the lines of the LB-plot intersecting on the ordinate. This is further indicative of competitive displacement of substrate dihydrofolate at saturating NADPH by NSC120927 and NSC132279, whereby increasing concentration of the inhibitors decreases the affinity for H2F without unduly affecting the maximal velocity of the reaction. It should be pointed out that for NSC120927 and NSC132279, the Ki values are ∼27-fold and ∼18-fold lower than the obtained IC50 values for the respective ligands. The above data is conclusive about the inhibitors binding to the same site as the substrate H2F, competing with the substrate for high-affinity interactions with the enzyme. This competitive displacement can be ascribed to the diaminotriazine group that shares substantial structural similarity with the 2-amino-4-oxo-1,4,7,8-tetrahydropteridin group on the substrate and may form the common motif for binding.
Fig. 5.

Competition experiments of NSC120927, NSC132279, NSC133071 and NSC132277 against substrate H2F for E. coli DHFR. The figure shows the fit of the primary data to the competitive inhibition model and double reciprocal Lineweaver-Burk plot for H2F titration at several fixed concentrations of (A) NSC120927 (B) NSC132279. (C) NSC133071 and (D) NSC132277. The y-axis shows the kcat and kcat−1 value and the x-axis shows the [H2F] and 1/[H2F] values for the primary and LB plot, respectively. The experimental data points were fit to the respective models using the non-linear curve-fitting algorithm of GraphPad Prism v 6.0e.
Table 2.
Parameters from inhibition kinetics of E. coli DHFR.
| Inhibitors | Substrate | Inhibition | Ki/αKi (nM)* |
|---|---|---|---|
| NSC120927 | H2F | Competitive | 42.50 ± 5.34 |
| NADPH | Uncompetitive | 946.4 ± 62.3 | |
| NSC132279 | H2F | Competitive | 100.9 ± 12.7 |
| NADPH | Uncompetitive | 2019 ± 155 | |
| NSC132277 | H2F | Competitive | 394.3 ± 58.4 |
| NADPH | Uncompetitive | 7432 ± 375 | |
| NSC133071 | H2F | Competitive | 430 ± 53.8 |
| NADPH | Uncompetitive | 12300 ± 793 | |
| NSC113909 | H2F | Competitive | 3816.0 ± 434.0 |
| NADPH | Uncompetitive | 62240 ± 3557 |
The Ki is reported for competitive inhibition while αKi is reported for uncompetitive inhibition;
Further, substrate dihydrofolate was titrated at several fixed concentrations of NSC133071 and NSC132277. When the resulting experimental curves were fit to the models for various types of inhibition, they showed equally good global fits to both competitive and linear mixed-type inhibition models (Fig S3A–B). However, based on visual assessment of the double-reciprocal Lineweaver-Burk plots, lines in which were seen intersecting on, or proximal to, the ordinate, the global fits shown are for the competitive displacement model (Fig 5C and 5D). Further, Dixon analysis also showed that the lines intersect in the second quadrant, reinforcing the suggestion that the inhibition is competitive (data not shown). However, it should be noted that the long phenoxyalkyl and phenylalkyl substituents on the small molecules might give rise to some interaction with the protein that might lead to slight perturbation in the maximum velocity observed, apart from its role in competitively displacing the substrate. This may lead to slight perturbation of the Vmax, giving rise to the observed ambiguity in the nonlinear fits. However, conclusive refutation of this model would require further experimentation. Moreover, an exact physical basis for this behavior might require detailed structural studies.
1,3,5-triazine-2,4-diamine analogues as uncompetitive inhibitors of NADPH binding
To understand the effect of 1,3,5-triazine-2,4-diamine derivatives on the cofactor NADPH binding, the latter was titrated at several fixed concentrations of the inhibitors. As before, the top four small molecules with the lowest Kiapp values, i.e., NSC120927, NSC132279, NSC133071 and NSC132277, were selected for this analysis. The resulting curves from the primary plot for all four small molecules, when globally fit to various types of inhibition models, showed the best fit to the model for uncompetitive inhibition (Fig 6A–D). This yielded αKi values, the equilibrium dissociation constant for the uncompetitive inhibitor, as specified in Table 3. These higher αKi values, compared to the Ki values obtained from competition with substrate H2F, show that the inhibitor binding site is fully formed only when the enzyme is bound to NADPH. It is worthwhile to point out that most reported inhibitors of E. coli DHFR show synergy with respect to NADPH binding47. However, it is evident from Fig 6A–B that the fit of the primary curves to the global uncompetitive model deviates substantially from the experimental data points obtained at high inhibitor concentrations. This can be ascribed to the underestimation of inhibition at really high inhibitor concentrations by the model. To overcome this ambiguity by appropriate visual assessment, the resulting data were transformed and plotted as double-reciprocal LB plots. The right hand panels in Fig 6A–D show parallel lines on the LB-plot. These confirm the fit of primary data to model for uncompetitive inhibition. The data on competition of the diaminotriazine derivatives with NADPH is strongly indicative of an ordered binding event whereby NADPH binding facilitates inhibitor binding.
Fig. 6.

Competition experiments of NSC120927, NSC132279, NSC133071 and NSC132277 against substrate NADPH for E. coli DHFR. The plots show the fit of the primary data to the uncompetitive inhibition model and double reciprocal Lineweaver-Burk plot for NADPH titration at several fixed concentrations of (A) NSC120927, (B) NSC132279, (C) NSC133071 and (D) NSC132277. The y-axis shows the kcat and kcat−1 value and the x-axis shows the [NADPH] and 1/[NADPH] values for the primary and LB plot, respectively. The experimental data points were fit to the respective models using the non-linear curve-fitting algorithm of GraphPad Prism v 6.0e.
Kinetics of NSC113909, a tight binder with poor inhibition
NSC113909, containing a fluorosulfonylphenylaminocarbonyl substitution at R2 on 1-phenyl-6,6-dimethyl-1,3,5-triazine-2,4-diamine ring, showed the unique behavior of tight-binding and average inhibition. To understand whether this molecule shows differential kinetics in its inhibition mechanism and the order of addition to the enzyme, detailed inhibition kinetics was carried out with this molecule too. Fig 7A shows the primary curves for H2F titration at several different concentrations of NSC113909, fit to the model for competitive inhibition. Further, Fig 7B shows the double-reciprocal LB plot with lines intersecting on the ordinate. Likewise, Fig 7C shows the primary curves for NADPH titrations at several different concentrations of NSC113909 fit to the model for uncompetitive inhibition, and Fig 7D shows the double reciprocal LB plot, giving parallel lines. These patterns show that NSC113909 occupies the H2F binding site and preferentially binds to the NADPH-bound form of the enzyme, mirroring the behavior shown by the best hits assessed in this study. However, the Ki value of 3.82 ± 0.43 µM for NSC113909 is much higher, indicative of poor inhibition of the enzyme (Table 3). As speculated earlier, this poorer inhibition might be because of two reasons: Partial charges from the carbonyl and amino group introduced as part of the linker might be incompatible with the NADPH-bound holo form of the enzyme or the planarity of the peptide bond due to electron pair delocalization might restrain the available degrees of freedom leading to unfavorable energy of interaction.
Fig. 7.

Inhibition kinetics of NSC113909 for E. coli DHFR (A) Fit of the primary data to the competitive inhibition model for H2F titration at several fixed concentrations of NSC113909. (B) Double reciprocal Lineweaver-Burk plot of H2F titration at several fixed concentrations of NSC113909. (C) Fit of the primary data to the uncompetitive inhibition model for NADPH titration at several fixed concentrations of NSC113909. (D) Double reciprocal Lineweaver-Burk plot of NADPH titration at several fixed concentrations of NSC113909.
Discussion
E. coli DHFR follows a sequential mechanism, whereby product release is conditional upon binding of both substrates. Further, the substrates NADPH and H2F can add to the enzyme randomly to form the Enzyme-NADPH-ternary complex. Understanding the mechanism of inhibition in a two-substrate reaction is a must for the success of a medicinal chemistry optimization exercise. Lack of understanding of the mechanism of a reaction may render a QSAR based drug-discovery program unproductive. Understanding the relationship between steady-state rate parameters and the specific form of the enzyme to which the inhibitor binds goes a long way towards ensuring the success of a drug-discovery program by narrowing the optimization of the SAR to achieve enrichment in binding/inhibition of the drug to that particular form of enzyme. Detailed mechanistic characterization of the top hits obtained in this study shows that the inhibitor molecules preferentially bind to the NADPH-bound binary form of the enzyme and are mostly competitive with the H2F substrate. As pointed out by us in our previous study17, it is desirable for an inhibitor to bind to the NADPH-bound form of E. coli DHFR given that E. coli cytoplasm has equal concentrations of both NADPH and NADP48 and inhibition of the NADPH-bound, catalytically competent form of the enzyme would have maximum effect in enhancing the antibacterial activity of a compound.
The diaminotriazine family of compounds have been predominantly explored as inhibitors of eukaryotic DHFRs, with the most notable application being inhibitors of P. falciparum DHFR49, 50. Several studies have reported the application of diaminotriazine derivatives as drugs for wild type and drug-resistant mutants in P. falciparum51–53. However, to the best of our knowledge, there is a paucity of studies in which diaminotriazine compounds have been explored for their antibacterial activity, especially against gram-negative bacteria. This might be because of the poor inhibition of prokaryotic DHFRs by known diaminotriazine inhibitors. Structural analysis of various DHFR structures suggests that for a small-molecule to be a DHFRs potent inhibitor, it must have a protonated N in the heterocyclic ring to enable the formation of a charge-mediated hydrogen bond with the enzyme. Furthermore, partial delocalization of the positive charge on the amino group adjacent to the protonated N contributes to the formation of an additional hydrogen bond, effectively increasing the binding affinity. An additional amino group at the para position relative to the protonated N group leads to additional hydrogen bonding. This in turn acts to increase the strength of protein-ligand interactions. In addition, the presence of hydrophobic substituents perpendicular to the heterocyclic ring enables optimal polar and hydrophobic interactions with the enzyme54. The nanomolar inhibition constants of NSC132279 and NSC120927, the best inhibitors from our study against E. coli DHFR are encouraging from the perspective of utilizing diaminotriazines, a chemical scaffold that remains relatively unexplored for antibiotic discovery. It shows that, apart from the above-mentioned interactions pivotal for a small-molecule to function as an effective inhibitor, NSC120927 and NSC132279 make additional favorable interactions with the prokaryotic enzyme. Docking studies have indicated that the hydrophobic linker region and the terminal sulfonyl fluoride group makes several favorable interactions with the E. coli enzyme that can enable the development of this class of molecules as potent antibacterial agents. It should be pointed out here that nosocomial infections caused by multi-drug resistant E. coli are on the rise55. This clearly necessitates the discovery of newer scaffolds in antibiotic drug-discovery.
It should also be noted that none of the diaminotriazine compounds studied in this work displayed slow-onset of tight binding behavior. This is irrespective of the fact that two of the most potent hits had low nanomolar inhibition constants comparable to that shown by some of the well-known DHFR inhibitors. This is a unique observation, since it has been reported extensively in the literature that most, if not all, inhibitors of E. coli DHFR display slow-onset of tight binding56. In a previous study from our lab, we had shown that NSC309401, a 7-[(4-aminophenyl) methyl]-7H-pyrrolo [3,2-f] quinazoline-1,3-diamine, also shows slow-onset of tight binding inhibition of E. coli DHFR17. Further, we showed that the parent compound NSC339578, a 7H–pyrrolo [3,2-f] quinazoline-1,3-diamine, does not show slow-onset of inhibition in spite of being a tight binding inhibitor. This caused us to hypothesize that the nature of substitution on inhibitors of E. coli DHFR leads to the slow-onset behavior. In this study, none of the molecules showed slow-onset of inhibition irrespective of the nature of substituents. This suggests that the slow-onset of inhibition behavior might be a cooperative outcome of interactions made by the complete inhibitor molecule, rather than being an additive property of individual fragments.
Lamotrigine is a drug of the phenyltriazine class with inhibitory effects on voltage-sensitive sodium channels57 that has also been shown to weakly inhibit mammalian dihydrofolate reductases57, 58. It should be pointed out that lamotrigine is a 1,2,4-triazine-3,5-diamine, whereas the other compounds employed in this study are 1,3,5-triazine-2,4-diamine derivatives. This small-molecule was never explored for its potential as an inhibiting agent of prokaryotic DHFRs. In this study, we show for the first time that lamotrigine binds and inhibits E. coli DHFR with IC50 and Kiapp values of ∼350 µM and ∼15 µM, respectively. This finding opens up a whole new scaffold for antibacterial drug discovery and paves the way for next generation antibiotics to tackle the menace of increasing drug resistance in gram-negative bacteria.
Conclusions
In summary, this study is the first comprehensive binding and kinetic investigation of various derivatives of 1-phenyl-6,6-dimethyl-1,3,5-triazine-2,4-diamine to understand the effect of substitutions in their binding to and inhibition of E. coli DHFR. Further, this study throws light on the mechanism of inhibition by this family of compounds and shows that NSC120927 and NSC132279 are novel compounds with low nM inhibition constants against the E. coli DHFR. To the best of our knowledge, these molecules are the first ever nanomolar inhibitors of E. coli DHFR from this family of compounds and constitute potential antibiotic candidates.
Methods
Reagents
All reagents and chemicals, unless mentioned otherwise, were of high quality and were procured from Sigma-Aldrich Co., USA, Amresco, or Fisher Scientific. E. coli dihydrofolate reductase was provided by Prof. Eugene Shakhnovich, Harvard University. The small molecule NSC120927, NSC132279, NSC132277, NSC133071, NSC104129, NSC115928, NSC3074, NSC212229, NSC19732, NSC19745, NSC168184, NSC211137, NSC113909 and NSC117268 were provided by the Developmental Therapeutics Program (DTP) of the National Cancer Institute (NCI), National Institutes of Health (NIH). The small molecule CHEMBL597262 was kindly provided by Medicines for Malaria Venture (MMV) (http://www.mmv.org/malariabox). Dihydrofolate reductase assay kit (CS0340) was obtained from Sigma (Sigma-Aldrich, St. Louis, MO). Lamotrigine (L3791) was purchased from Sigma.
Binding studies using differential scanning fluorimetry, DSF
Binding of 16 small molecule compounds to E. coli DHFR was tested by differential scanning fluorimetry, DSF. The experiments were carried out following protocols reported in previous communications from our lab13, 17. Briefly, the reactions were carried out in 20 µl reaction volumes in 96 well plates on the RealPlex quantitative PCR instrument (Eppendorf, NY, USA). The reaction mixture consisted of 100 mM HEPES pH 7.3 and 150 mM NaCl with 5X concentration of the reporter dye Sypro orange. Various compounds were tested for binding at a final concentration of 500 µM with 5 µM of E. coli DHFR. Compounds NSC132277 and NSC132279 failed to give signal at 500 µM and hence, the experiments were done at 10 µM.
The first derivatives of the thermal melt curves were fit to a Gaussian equation (Eq. 1) for estimating the Tm (melting temperature) from the observed intensity of fluorescence, I.
| (1) |
Here, µ is the mean of the Gaussian distribution, σ is the standard deviation and σ2 is the variance. Thermodynamic parameters and apparent dissociation constants (KDapp) were estimated as specified in the previous literature13.
Dihydrofolate reductase assay
DHFR was assayed as previously reported17. Briefly, the formation of NADP was monitored by the decrease in absorbance at 340 nm for 100 seconds. The amount of product formed was computed from the slopes of time-course measurements using a molar extinction coefficient (ε) of 6.2 × 10 M−1 cm−1 for P-NADPH at 340 nm59. The non-enzymatic hydrolysis of NADPH was normalized. Assays were initiated with the addition of enzyme to the sample cuvette after zeroing the absorbance reading with respect to the reference cuvette. The initial velocities, where product formation was less than 5%, were measured for reaction mixtures containing 100 mM HEPES pH 7.3 at room temperature (∼ 22 ° C).
All the measurements were performed in duplicate, and the error values indicated are standard errors (S.E.). The concentration of E. coli DHFR used was 16.7 nM and was estimated by the method of velocity-titration curves as previously reported17 . Unless mentioned otherwise, all the data were fit using non-linear curve fitting subroutines of GraphPad Prism, version 4.0 (GraphPad Software, Inc., San Diego, CA).
Inhibition kinetics
14 compounds with varied substitutions of diaminotriazine were assessed for their inhibitory effect on the NADPH oxidizing ability of E. coli DHFR. Both the dose-dependence of inhibition and affinity of the inhibitor for the enzyme were computed by experimental IC50 determination and competition assays to determine its Ki. IC50 determination assays were carried out in 100 mM HEPES pH 7.3, 60 µM NADPH, 50 µM H2F and variable concentration of each inhibitor. The enzyme concentration was as specified above. The curves were fit to equation (2),
| (2) |
Where, I is the inhibitor concentration, and y is the percentage of activity.
Furthermore, Kiapp values were computed from the IC50 curves by fitting them to the quadratic Morrison equation (3) assuming competitive inhibition.
| (3) |
Where, vi represents velocity in the presence of inhibitor, v0 represents velocity in the absence of inhibitor, [E]T represents total enzyme, [I]T represents total inhibitor and Kiapp represents apparent Ki.
Experimental Ki value determinations were carried out by titrating the substrates H2F and NADPH, around their respective Km values at various fixed concentrations of the inhibitors around their Kiapp values. The substrate or cofactor that is not varied is kept at a fixed saturating concentration greater than 10 times their respective Km values. The resulting [substrate] vs. velocity curves were fit to models of competitive inhibition (equation 4), non-competitive inhibition (equation 5), uncompetitive inhibition (equation 6) and linear mixed-type inhibition (equation 7) in order to discriminate between the different types of inhibition and to estimate the various inhibition constants (Ki). Competitive:
| (4) |
Non-competitive:
| (5) |
Uncompetitive:
| (6) |
Linear Mixed-type:
| (7) |
| (8) |
| (9) |
Where, v is the velocity of the reaction, Vmax is the maximum velocity, [S] is the substrate concentration, and [I] is the inhibitor concentration. Km is the Michaelis-Menten constant, and Ki is the inhibition constant. Visual assessment of the type of inhibition was undertaken by plotting the double reciprocal Lineweaver-Burk plot from experimental data points constituting the primary plot.
QSAR analysis
The various physicochemical properties of the small molecules employed in this study were computed from ChemMine tools employing OpenBabel and Joelib descriptors60 and ChemBioDraw 14.0. Pearson’s correlation analysis was carried out individually on each parameter against the obtained inhibition constants and the equation was derived by linear regression with GraphPad Prism, version 4.0. Genetic algorithm analysis was carried out using the opensource QSAR model development algorithm provided by Nanobridges.
Template based docking of small molecules
In order to find the interacting pose of the predicted small molecules with DHFR, template based docking was performed using the structure of DHFR in complex with folate (PDB_ID: 1RX7) 43 as the template. This was achieved by using the known interacting pose of folate with DHFR as the template and aligning the small molecules to it. To do so, initially we generated 200 different conformers for each of the predicted ligands using the open source chemoinformatics software RDKit 61. Next, LIGSIFT 44 was used to perform a structural alignment of each of the conformers to folate’s structure obtained in its crystal structure with DHFR. Finally, the best conformer for each case with the most significant P-value was chosen and superposed on folate. Both shape and chemical similarities between the two molecules were used to produce the alignments, and in cases where there were no significant alignments, only shape similarity was used to align the two molecules. The initial 3D conformation of each of the predicted ligands was obtained from PubChem62, and Open Babel 63 was used for converting different file formats (e.g. SDF to MOL2 format).
Supplementary Material
Highlights.
Exploring Diaminotriazine compounds as potential inhibitors of E. coli DHFR.
Demonstrated binding by 15 diaminotriazines using DSF methodology.
Demonstrated inhibition and showed inhibition mechanism of diaminotriazines.
Showed site of binding of the inhibitors on E. coli DHFR.
QSAR & Docking to understand the functional groups responsible for inhibition.
Acknowledgements
This project was funded by GM-37408 and GM-48835 of the Division of General Medical Sciences of the NIH. The authors wish to thank Prof. Eugene Shakhnovich, Harvard University for providing purified E. coli DHFR protein. We would also like to thank the Developmental Therapeutics Program of the National Cancer Institute for providing the small molecules used in this study.
Abbreviations
- EcDHFR
Escherichia coli dihydrofolate reductase
- NADPH
nicotinamide adenine dinucleotide phosphate, reduced
- H2F
dihydrofolate
- H4F
tetrahydrofolate
- QSAR
Quantitative Structure-Activity Relationship
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
BS conceived of the study, participated in its design, carried out the experiments, analyzed and interpreted the results, and drafted the manuscript. ST-N performed the docking studies, and was involved in drafting and critically reviewing the manuscript. JS conceived of the study, participated in its design and coordination, provided appropriate resources, helped analyze the data, and was involved in drafting and critically reviewing the manuscript. All authors read and approved the final manuscript.
Supporting information
Supporting information contains Fig S1. Preliminary Structure-activity relationship analysis for the various diaminotriazine derivatives studied. Fig. S2. Hydrogen bond formation by the sulfonyl fluoride group on NSC120927 with three positively charged residues on E. coli DHFR. Fig S3. Non-linear curve fitting of experimental curves to models for both competitive and linear mixed-type inhibition for NSC133071 and NSC132277.
References
- 1.Nikaido H. Multidrug efflux pumps of gram-negative bacteria. Journal of bacteriology. 1996;178:5853–5859. doi: 10.1128/jb.178.20.5853-5859.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Paterson DL. Resistance in gram-negative bacteria: Enterobacteriaceae. American journal of infection control. 2006;34:S20–S28. doi: 10.1016/j.ajic.2006.05.238. discussion S64–73. [DOI] [PubMed] [Google Scholar]
- 3.Vaara M. Antibiotic-supersusceptible mutants of Escherichia coli and Salmonella typhimurium. Antimicrobial agents and chemotherapy. 1993;37:2255–2260. doi: 10.1128/aac.37.11.2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Daxboeck F, Budic T, Assadian O, Reich M, Koller W. Economic burden associated with multi-resistant Gram-negative organisms compared with that for methicillin-resistant Staphylococcus aureus in a university teaching hospital. The Journal of hospital infection. 2006;62:214–218. doi: 10.1016/j.jhin.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 5.Fair RJ, Tor Y. Antibiotics and bacterial resistance in the 21st century. Perspectives in medicinal chemistry. 2014;6:25–64. doi: 10.4137/PMC.S14459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vasoo S, Barreto JN, Tosh PK. Emerging issues in gram-negative bacterial resistance: an update for the practicing clinician. Mayo Clinic proceedings. 2015;90:395–403. doi: 10.1016/j.mayocp.2014.12.002. [DOI] [PubMed] [Google Scholar]
- 7.Finley RL, Collignon P, Larsson DG, McEwen SA, Li XZ, Gaze WH, Reid-Smith R, Timinouni M, Graham DW, Topp E. The scourge of antibiotic resistance: the important role of the environment. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2013;57:704–710. doi: 10.1093/cid/cit355. [DOI] [PubMed] [Google Scholar]
- 8.Hawkey PM, Jones AM. The changing epidemiology of resistance. The Journal of antimicrobial chemotherapy. 2009;64(Suppl 1):3–10. doi: 10.1093/jac/dkp256. [DOI] [PubMed] [Google Scholar]
- 9.Ibrahim ME, Bilal NE, Hamid ME. Increased multi-drug resistant Escherichia coli from hospitals in Khartoum state, Sudan. African health sciences. 2012;12:368–375. doi: 10.4314/ahs.v12i3.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schlackow I, Stoesser N, Walker AS, Crook DW, Peto TE, Wyllie DH and Infections in Oxfordshire Research Database, T. Increasing incidence of Escherichia coli bacteraemia is driven by an increase in antibiotic-resistant isolates: electronic database study in Oxfordshire 1999–2011. The Journal of antimicrobial chemotherapy. 2012;67:1514–1524. doi: 10.1093/jac/dks082. [DOI] [PubMed] [Google Scholar]
- 11.Swami SK, Liesinger JT, Shah N, Baddour LM, Banerjee R. Incidence of antibiotic-resistant Escherichia coli bacteriuria according to age and location of onset: a population-based study from Olmsted County, Minnesota. Mayo Clinic proceedings. 2012;87:753–759. doi: 10.1016/j.mayocp.2012.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schweitzer BI, Dicker AP, Bertino JR. Dihydrofolate reductase as a therapeutic target. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 1990;4:2441–2452. doi: 10.1096/fasebj.4.8.2185970. [DOI] [PubMed] [Google Scholar]
- 13.Srinivasan B, Zhou H, Kubanek J, Skolnick J. Experimental validation of FINDSITE(comb) virtual ligand screening results for eight proteins yields novel nanomolar and micromolar binders. Journal of cheminformatics. 2014;6:16. doi: 10.1186/1758-2946-6-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fukunaga JY, Hansch C, Steller EE. Inhibition of dihydrofolate reductase. Structure-activity correlations of quinazolines. Journal of medicinal chemistry. 1976;19:605–611. doi: 10.1021/jm00227a006. [DOI] [PubMed] [Google Scholar]
- 15.Kuyper LF, Baccanari DP, Jones ML, Hunter RN, Tansik RL, Joyner SS, Boytos CM, Rudolph SK, Knick V, Wilson HR, Caddell JM, Friedman HS, Comley JC, Stables JN. High-affinity inhibitors of dihydrofolate reductase: antimicrobial and anticancer activities of 7,8-dialkyl-1,3-diaminopyrrolo[3,2-f]quinazolines with small molecular size. Journal of medicinal chemistry. 1996;39:892–903. doi: 10.1021/jm9505122. [DOI] [PubMed] [Google Scholar]
- 16.Rosowsky A, Hynes JB, Queener SF. Structure-activity and structure-selectivity studies on diaminoquinazolines and other inhibitors of Pneumocystis carinii and Toxoplasma gondii dihydrofolate reductase. Antimicrobial agents and chemotherapy. 1995;39:79–86. doi: 10.1128/aac.39.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Srinivasan B, Skolnick J. Insights into the slow-onset tight-binding inhibition of Escherichia coli dihydrofolate reductase: detailed mechanistic characterization of pyrrolo [3,2-f] quinazoline-1,3-diamine and its derivatives as novel tight-binding inhibitors. The FEBS journal. 2015 doi: 10.1111/febs.13244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Srinivasan BA, GA, US, Skolnick Jeffrey, Atlanta, GA, US, Zhou Hongyi., Marietta, GA, US . Molecules With Potent Dhfr Binding Affinity And Antibacterial Activity. Atlanta, GA, US United States: Georgia Tech Research Corporation; 2014. [Google Scholar]
- 19.Li RL, Poe M. Quantitative structure-activity relationships for the inhibition of Escherichia coli dihydrofolate reductase by 5-(substituted benzyl)-2,4-diaminopyrimidines. Journal of medicinal chemistry. 1988;31:366–370. doi: 10.1021/jm00397a017. [DOI] [PubMed] [Google Scholar]
- 20.Baccanari DP, Daluge S, King RW. Inhibition of dihydrofolate reductase: effect of reduced nicotinamide adenine dinucleotide phosphate on the selectivity and affinity of diaminobenzylpyrimidines. Biochemistry. 1982;21:5068–5075. doi: 10.1021/bi00263a034. [DOI] [PubMed] [Google Scholar]
- 21.Chio LC, Queener SF. Identification of highly potent and selective inhibitors of Toxoplasma gondii dihydrofolate reductase. Antimicrobial agents and chemotherapy. 1993;37:1914–1923. doi: 10.1128/aac.37.9.1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hansch C, Li R, Blaney JM, Langridge R. Comparison of the inhibition of Escherichia coli and Lactobacillus casei dihydrofolate reductase by 2,4-diamino-5-(substituted-benzyl)pyrimidines: quantitative structure-activity relationships, X-ray crystallography, and computer graphics in structure-activity analysis. Journal of medicinal chemistry. 1982;25:777–784. doi: 10.1021/jm00349a003. [DOI] [PubMed] [Google Scholar]
- 23.Williams JW, Morrison JF, Duggleby RG. Methotrexate, a high-affinity pseudosubstrate of dihydrofolate reductase. Biochemistry. 1979;18:2567–2573. doi: 10.1021/bi00579a021. [DOI] [PubMed] [Google Scholar]
- 24.Blaney JMHC, Silipo C, Vittoria A. Structure-Activity relationships of dihydrofolate reductase inhibitors. Chemical Reviews. 1984;84:333–407. [Google Scholar]
- 25.Akio Ohsawa HA Hidefumi Ohnishi and Hiroshi Igeta. 1,2,3-Triazine. J Chem. Soc., Chem. Commun. 1981:1174–1174. [Google Scholar]
- 26.Leach MJ, Baxter MG, Critchley MA. Neurochemical and behavioral aspects of lamotrigine. Epilepsia. 1991;32(Suppl 2):S4–S8. doi: 10.1111/j.1528-1157.1991.tb05882.x. [DOI] [PubMed] [Google Scholar]
- 27.Fidock DA, Nomura T, Wellems TE. Cycloguanil and its parent compound proguanil demonstrate distinct activities against Plasmodium falciparum malaria parasites transformed with human dihydrofolate reductase. Molecular pharmacology. 1998;54:1140–1147. doi: 10.1124/mol.54.6.1140. [DOI] [PubMed] [Google Scholar]
- 28.Yuthavong Y, Tarnchompoo B, Vilaivan T, Chitnumsub P, Kamchonwongpaisan S, Charman SA, McLennan DN, White KL, Vivas L, Bongard E, Thongphanchang C, Taweechai S, Vanichtanankul J, Rattanajak R, Arwon U, Fantauzzi P, Yuvaniyama J, Charman WN, Matthews D. Malarial dihydrofolate reductase as a paradigm for drug development against a resistance-compromised target. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:16823–16828. doi: 10.1073/pnas.1204556109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Booth RG, Selassie CD, Hansch C, Santi DV. Quantitative structure-activity relationship of triazine-anti-folate inhibition of Leishmania dihydrofolate reductase and cell growth. Journal of medicinal chemistry. 1987;30:1218–1224. doi: 10.1021/jm00390a017. [DOI] [PubMed] [Google Scholar]
- 30.Coats EA, Genther CS, Dietrich SW, Guo ZR, Hansch C. Comparison of the inhibition of methotrexate-sensitive and -resistant Lactobacillus casei cell cultures with purified Lactobacillus casei dihydrofolate reductase by 4,6-diamino-1,2-dihydro-2,2-dimethyl-1-(3-substituted-phenyl)-s-triazines. Use of quantitative structure-activity relationships in making inferences about the mechanism of resistance and the structure of the enzyme is situ compared with the enzyme in vitro. Journal of medicinal chemistry. 1981;24:1422–1429. doi: 10.1021/jm00144a010. [DOI] [PubMed] [Google Scholar]
- 31.Hansch C, Dietrich SW, Fukunaga JY. Inhibition of bovine and rat liver dihydrofolate reductase by 4,6-diamino-1,2-dihydro-2,2-dimethyl-1-(4-substituted-phenyl)-s-triazines. Journal of medicinal chemistry. 1981;24:544–549. doi: 10.1021/jm00137a013. [DOI] [PubMed] [Google Scholar]
- 32.Hansch C, Hathaway BA, Guo ZR, Selassie CD, Dietrich SW, Blaney JM, Langridge R, Volz KW, Kaufman BT. Crystallography, quantitative structure-activity relationships, and molecular graphics in a comparative analysis of the inhibition of dihydrofolate reductase from chicken liver and Lactobacillus casei by 4,6-diamino-1,2-dihydro-2,2-dimethyl-1-(substituted-phenyl)-s-triazine s. Journal of medicinal chemistry. 1984;27:129–143. doi: 10.1021/jm00368a006. [DOI] [PubMed] [Google Scholar]
- 33.Kim KH, Dietrich SW, Hansch C, Dolnick BJ, Bertino JR. Inhibition of dihydrofolate reductase. 3. 4.6-Diamino-1,2-dihydro-2,2-dimethyl-1-(2-substituted-phenyl)-s-triazine inhibition of bovine liver and mouse tumor enzymes. Journal of medicinal chemistry. 1980;23:1248–1251. doi: 10.1021/jm00185a022. [DOI] [PubMed] [Google Scholar]
- 34.Marlowe CK, Selassie CD, Santi DV. Quantitative structure-activity relationships of the inhibition of Pneumocystis carinii dihydrofolate reductase by 4,6-diamino-1,2-dihydro-2,2-dimethyl-1-(X-phenyl)-s-triazines. Journal of medicinal chemistry. 1995;38:967–972. doi: 10.1021/jm00006a016. [DOI] [PubMed] [Google Scholar]
- 35.da Cunha EFFRTC, Maia ER, de Alencastra RB. The search for new DHFR inhibitors: a review of patents, january 2001-february 2005. Expert Opin. Ther. Patents. 2005;15:1–20. [Google Scholar]
- 36.Hathaway BA, Guo ZR, Hansch C, Delcamp TJ, Susten SS, Freisheim JH. Inhibition of human dihydrofolate reductase by 4,6-diamino-1,2-dihydro-2,2-dimethyl-1-(substituted-phenyl)-s-triazine s. A quantitative structure-activity relationship analysis. Journal of medicinal chemistry. 1984;27:144–149. doi: 10.1021/jm00368a007. [DOI] [PubMed] [Google Scholar]
- 37.Lele ACRA, Ray MK, Rajan MGR, Degani MS. Design and synthesis of diaminotriazines as anti-tuberculosis DHFR inhibitors. Current Research in drug discovery. 2014;1:45–40. [Google Scholar]
- 38.Selassie CD, Strong CD, Hansch C, Delcamp TJ, Freisheim JH, Khwaja TA. Comparison of triazines as inhibitors of L1210 dihydrofolate reductase and of L1210 cells sensitive and resistant to methotrexate. Cancer research. 1986;46:744–756. [PubMed] [Google Scholar]
- 39.Jacobus DP, Schiehser GA, Shieh HM, Jensen NP, Terpinski J. Biguanide and dihydrotriazine derivatives, Google Patents. 2007 [Google Scholar]
- 40.Singla P, Luxami V, Paul K. Triazine-benzimidazole hybrids: anticancer activity, DNA interaction and dihydrofolate reductase inhibitors. Bioorganic & medicinal chemistry. 2015;23:1691–1700. doi: 10.1016/j.bmc.2015.03.012. [DOI] [PubMed] [Google Scholar]
- 41.Deepak Kumar SI, Prija Ponnan K, Rawat Diwan S. Triazine-pyrimidine based molecular hybrids: synthesis, docking studies and evaluation of antimalarial activity. New J. Chem. 2014;38:5087–5095. [Google Scholar]
- 42.Roy K, Mitra I. On various metrics used for validation of predictive QSAR models with applications in virtual screening and focused library design. Combinatorial chemistry & high throughput screening. 2011;14:450–474. doi: 10.2174/138620711795767893. [DOI] [PubMed] [Google Scholar]
- 43.Sawaya MR, Kraut J. Loop and subdomain movements in the mechanism of Escherichia coli dihydrofolate reductase: crystallographic evidence. Biochemistry. 1997;36:586–603. doi: 10.1021/bi962337c. [DOI] [PubMed] [Google Scholar]
- 44.Roy A, Skolnick J. LIGSIFT: an open-source tool for ligand structural alignment and virtual screening. Bioinformatics. 2015;31:539–544. doi: 10.1093/bioinformatics/btu692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J Comp Chem. 1983;4:187–217. [Google Scholar]
- 46.Taylor R, Kennard O. Crystallographic Evidence for the Existence of CHO, CHN and CHCl hydrogen bonds. J. Am. Chem. Soc. 1982;104:5063–5070. [Google Scholar]
- 47.Stone SR, Morrison JF. Mechanism of inhibition of dihydrofolate reductases from bacterial and vertebrate sources by various classes of folate analogues. Biochimica et biophysica acta. 1986;869:275–285. doi: 10.1016/0167-4838(86)90067-1. [DOI] [PubMed] [Google Scholar]
- 48.Appleman JR, Beard WA, Delcamp TJ, Prendergast NJ, Freisheim JH, Blakley RL. Unusual transient- and steady-state kinetic behavior is predicted by the kinetic scheme operational for recombinant human dihydrofolate reductase. The Journal of biological chemistry. 1990;265:2740–2748. [PubMed] [Google Scholar]
- 49.Jensen NP, Ager AL, Bliss RA, Canfield CJ, Kotecka BM, Rieckmann KH, Terpinski J, Jacobus DP. Phenoxypropoxybiguanides, prodrugs of DHFR-inhibiting diaminotriazine antimalarials. Journal of medicinal chemistry. 2001;44:3925–3931. doi: 10.1021/jm010089z. [DOI] [PubMed] [Google Scholar]
- 50.Nzila A. Inhibitors of de novo folate enzymes in Plasmodium falciparum. Drug discovery today. 2006;11:939–944. doi: 10.1016/j.drudis.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 51.Peterson DS, Milhous WK, Wellems TE. Molecular basis of differential resistance to cycloguanil and pyrimethamine in Plasmodium falciparum malaria. Proceedings of the National Academy of Sciences of the United States of America. 1990;87:3018–3022. doi: 10.1073/pnas.87.8.3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maitarad P, Kamchonwongpaisan S, Vanichtanankul J, Vilaivan T, Yuthavong Y, Hannongbua S. Interactions between cycloguanil derivatives and wild type and resistance-associated mutant Plasmodium falciparum dihydrofolate reductases. Journal of computer-aided molecular design. 2009;23:241–252. doi: 10.1007/s10822-008-9254-z. [DOI] [PubMed] [Google Scholar]
- 53.Basak SC, Mills D. Quantitative structure-activity relationships for cycloguanil analogs as PfDHFR inhibitors using mathematical molecular descriptors. SAR and QSAR in environmental research. 2010;21:215–229. doi: 10.1080/10629361003770951. [DOI] [PubMed] [Google Scholar]
- 54.Xiang M. Department of Pharmacy. Singapore: National University of Singapore; 2006. Synthesis and bioactivity study of 4,6-diamino-1,3,5-triazines; pp. 41–42. [Google Scholar]
- 55.Mathers AJ, Peirano G, Pitout JD. The role of epidemic resistance plasmids and international high-risk clones in the spread of multidrug-resistant Enterobacteriaceae. Clinical microbiology reviews. 2015;28:565–591. doi: 10.1128/CMR.00116-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stone SR, Montgomery JA, Morrison JF. Inhibition of dihydrofolate reductase from bacterial and vertebrate sources by folate, aminopterin, methotrexate and their 5-deaza analogues. Biochemical pharmacology. 1984;33:175–179. doi: 10.1016/0006-2952(84)90472-6. [DOI] [PubMed] [Google Scholar]
- 57.Ma X, Poon TY, Wong PT, Chui WK. Synthesis and in vitro evaluation of 2,4-diamino-1,3,5-triazine derivatives as neuronal voltage-gated sodium channel blockers. Bioorganic & medicinal chemistry letters. 2009;19:5644–5647. doi: 10.1016/j.bmcl.2009.08.052. [DOI] [PubMed] [Google Scholar]
- 58.Laxer KD. Guidelines for treating epilepsy in the age of felbamate, vigabatrin, lamotrigine, and gabapentin. The Western journal of medicine. 1994;161:309–314. [PMC free article] [PubMed] [Google Scholar]
- 59.Horecker BL, Kornberg A. The extinction coefficients of the reduced band of pyridine nucleotides. The Journal of biological chemistry. 1948;175:385–390. [PubMed] [Google Scholar]
- 60.Backman TW, Cao Y, Girke T. ChemMine tools: an online service for analyzing and clustering small molecules. Nucleic acids research. 2011;39:W486–W491. doi: 10.1093/nar/gkr320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Landrum G. RDKit: Open-source cheminformatics [Google Scholar]
- 62.Li Q, Cheng T, Wang Y, Bryant SH. PubChem as a public resource for drug discovery. Drug discovery today. 2010;15:1052–1057. doi: 10.1016/j.drudis.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: An open chemical toolbox. Journal of cheminformatics. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stone J. Computer Science. University of Missouri; 1998. An Efficient Library for Parallel Ray Tracing and Animation. [Google Scholar]
- 65.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. Journal of molecular graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.