

Abstract
Human mast cells (huMCs) are involved in both innate and adaptive immune responses where they release mediators including amines, reactive oxygen species (ROS), eicosanoids and cytokines. We have reported that interferon-γ (IFN-γ) enhances FcγR-dependent ROS production. The aim of this study was to extend these observations by investigating the effect of IFN-γ on the biological responses of huMCs to Staphylococcus aureus. We found that exposure of huMCs to S. aureus generated intracellular and extracellular ROS, which were enhanced in the presence of IFN-γ. IFN-γ also promoted bacteria killing, β-hexosaminidase release and eicosanoid production. Interferon-γ similarly increased expression of mRNAs encoding CCL1 to CCL4, granulocyte–macrophage colony-stimulating factor (GM-CSF), tumour necrosis factor-α and CXCL8 in S. aureus-stimulated huMCs. The ability of IFN-γ to increase CXCL8 and GM-CSF protein levels was confirmed by ELISA. Fibronectin or a β1 integrin blocking antibody completely abrogated IFN-γ-dependent S. aureus binding and reduced S. aureus-dependent CXCL8 secretion. These data demonstrate that IFN-γ primes huMCs for enhanced anti-bacterial and pro-inflammatory responses to S. aureus, partially mediated by β1 integrin.
Keywords: bacteria, innate immunity, β1 integrin, interferon-γ, mast cells
Introduction
Mast cells (MCs) are tissue-resident cells that, although classically associated with allergic inflammation,1 have been reported to participate in the innate immune response to infectious organisms including bacteria.2 In vitro challenge of mouse MCs with Staphylococcus aureus or its cell wall component peptidoglycan generates cytokines including interleukin-6 (IL-6) and tumour necrosis factor-α (TNF-α)3,4 and induces MC degranulation.5 Furthermore, studies in mice lacking MCs due to defective c-Kit signalling (WBB6F1-KitW/W-v and C57BL/6-Kitw-sh/w-sh) reveal a protective role for MCs against S. aureus infection and bacterial cell wall components,6–8 although when MC deficiency was independent of KIT signalling (Mcpt5-Cre+ × R-DTA), MCs were not essential in protection.3
There is less known concerning the response of human MCs (huMCs) to bacteria. What has been reported is that huMCs phagocytose and kill FimH+ Escherichia coli and S. aureus, and release TNF-α and chemokines,9–12 in part via a Toll-like receptor 2 (TLR2) and CD48-dependent mechanism.13,14 Human MCs are also reported to release extracellular traps for bacteria.15,16 Following the observation that mouse MCs generate reactive oxygen species (ROS) that contribute to bacterial killing,17 we reported that MCs generate ROS from the enzymes 5-lipoxygenase (5LO) and cyclooxygenase (COX) following FcεRI and FcγR aggregation.18–20 This is of interest because it is known that huMCs respond to interferon-γ (IFN-γ) by up-regulating the surface expression of FcγRI21–24 and TLR4.25
Based both on the observations that huMCs are able to engulf bacteria and that IFN-γ up-regulates the expression of some surface receptors on huMCs, we were interested in determining whether IFN-γ, as would be produced by infiltrating inflammatory cells, would further enhance the response of huMCs to bacteria (S. aureus). As will be shown, IFN-γ substantially increases the ability of huMCs to respond to S. aureus by enhancing ROS production, MC degranulation and eicosanoid, cytokine and chemokine production. We also found that these responses are mediated, in part, through a β1 integrin receptor-mediated process. These observations are consistent with the conclusion that the involvement of huMCs in innate defence mechanisms against bacteria is further enhanced by IFN-γ, providing a mechanism to increase the efficiency of these tissue-resident effector cells in control of infection as inflammation progresses.
Materials and methods
Mice and materials
Wild-type C57BL6 mice (< 6 months old, 20 g), Tlr2-deficient and Scarb1-deficient mice were obtained from Jackson Laboratories (Bar Harbor, ME). Aged-matched wild-type and Msr1-deficient mice were a kind gift from Dr Andrij Holian (Department of Biomedical and Pharmaceutical Sciences, University of Montana, MT). Mice were maintained and killed in accordance with the National Institutes of Health guidelines on animal care and use, which were reviewed and approved by the NIAID Animal Use Committee.
The materials used were supplied as follows: BSA, diphenyleneiodonium (DPI), saponin, superoxide dismutase and trypan blue (Sigma Aldrich, St Louis, MI); dichlorofluorescein diacetate (DCF-DA; EMD Biosciences, La Jolla, CA); FR122047 and zileuton (Cayman Chemicals, Ann Arbor, MI); diogenes (National Diagnostics, Atlanta, GA); cell culture media and supplements (Invitrogen, Carlsbad, CA and Biosource International, Camarillo, CA), human recombinant granulocyte–macrophage colony-stimulating factor (GM-CSF), IFN-γ, IL-3, IL-4, IL-6, stem cell factor (SCF) and TNF-α (Peprotech, Rocky Hill, NJ); dextran (T-500) and Ficoll-Hypaque (Amersham Biosciences, Piscataway, NJ); and Luria–Bertani broth and agar (KD Medical, Columbia, MD).
Cell cultures
Human mast cells were derived from CD34+ peripheral blood mononuclear cells obtained following informed consent from normal volunteers on a protocol approved by the NIAID Institutional Review Board. CD34+ cells were cultured in IL-3 (week 1 only, 30 ng/ml), SCF (100 ng/ml) and IL-6 (100 ng/ml) as described.19 The HuMCs were > 99% pure by toluidine blue staining of cytospin preparations and were used after 7–10 weeks of culture.
Polymorphonuclear leucocytes (PMNs) were isolated from peripheral blood of normal human volunteers20 using a protocol approved by the NIAID Institutional Review Board. The PMNs were kept on ice in PBS containing glucose (10 mm) until studied.
Mouse bone-marrow-derived MCs (mBMMCs) were cultured from femoral marrow cells in RPMI-1640 medium supplemented with fetal bovine serum (10%), penicillin (100 U/ml), streptomycin (100 μg/ml), HEPES (25 mm), sodium pyruvate (1 mm), non-essential amino acids (1 mm), 2-mercaptoethanol (0·0035%) and mouse IL-3 (30 ng/ml) as described.20 Murine BMMCs were > 99% pure by toluidine blue staining of cytospin preparations and were used after 4–6 weeks of culture.
Staphylococcus aureus culture
A 10 μl frozen stock of S. aureus (ATCC 27217; strain 502A) was added to Luria–Bertani broth (6 ml) and cultured overnight at 37° with shaking (200 rpm). Then, a 100-fold dilution of the culture was incubated for a further 2·5 hr with shaking (200 rpm) to obtain bacteria in late logarithmic stage growth [with optical density at 600 nm (OD600) < 1]. Viable bacteria/ml was determined by measuring OD600 and comparing this value to the known OD600 values for colony-forming units (CFU)/ml from a pre-determined standard curve. Bacteria were then centrifuged at 10 000 g for 4 min and washed three times with appropriate media before use.
Intracellular ROS detection by microfluorimetry
Intracellular ROS production was measured in a 96-well microplate assay employing DCF (the intracellular product of DCF-DA that fluoresces in the presence of ROS). The MCs or PMNs (1 × 106/ml) were incubated with DCF-DA (20 μm, 20 min) at 4° with rotation. Cells were then washed (HEPES buffer) and added (2 × 105/well) to a black opaque 96-well microplate containing enzyme inhibitors and bacteria on ice. The activation of huMCs by S. aureus was then synchronized by centrifugation of the microplate at 170 g for 8 min at 4°. DCF fluorescence was monitored at 30-second intervals for 1 hr at 37° using a GENios fluorescent plate reader (ReTirSoft Inc., Toronto, ON, Canada) with excitation and emission wavelengths of 492 nm and 535 nm, respectively. Fluorescence was expressed as relative fluorescent units and the kinetic data were collected using an XFlour4 macro within Microsoft Excel.
Extracellular ROS detection by microflourimetry
Extracellular ROS production was measured in a 96-well plate assay employing the chemiluminescent probe Diogenes which is used to detect extracellular superoxide. Briefly, huMCs or PMNs were washed (HEPES buffer) and added (4 × 105/well) to a white opaque 96-well microplate containing enzyme inhibitors, stimulants (bacteria) and Diogenes reagent. The assay was then synchronized by centrifugation (170 g, 8 min at 4°) before monitoring chemiluminescence at 120-second intervals for 1 hr at 37° using a GENios fluorescent plate reader (ReTirSoft Inc.) and expressed as relative luminescent units. The kinetic data were collected using an XFlour4 macro within Microsoft Excel.
Bacterial killing assay
Human MCs or PMNs (4 × 105/well) were added to a 48-well plate containing bacteria, activation by S. aureus was synchronized by centrifugation (170 g, 8 min at 4°), and assay plates were then incubated at 37° for 0·5–3 hr. Bacteria in the absence of huMCs or PMNs were used as controls for bacterial viability. At each time-point, samples were placed on ice and lysed (0·1% saponin, 15 min); serial dilutions were prepared before 50-μl aliquots were spread on Luria–Bertani agar (1·5%) plates, inverted and incubated overnight at 37°. The number of CFU/plate was counted and the percentage of bacteria killed by huMCs or PMNs was determined using the following equation; [CFU (samples containing host cells)/CFU (samples without host cells)] × 100. In experiments where enzyme inhibitors were used, these were added 20 min before the start of the assay.
Cell degranulation assay
Immunoglobulin E-sensitized MCs (1 × 104/well) in HEPES buffer were added to a 96-well microplate containing bacteria (ratios of 2 : 1, 20 : 1, 100 : 1 MC) or antigen (streptavidin; 100 ng/ml) on ice. Activation of huMCs by S. aureus was then synchronized by centrifugation (170 g, 8 min at 4°) before incubation at 37° for 1 hr. Degranulation was measured as percent release of β-hexosaminidase.20
Leukotriene C4 and prostaglandin D2 measurements
Immunoglobulin E-sensitized MCs (2 × 105/well) in HEPES buffer were added to a 96-well microplate containing bacteria (ratio of 2 : 1, 20 : 1, 100 : 1 MC) or antigen (streptavidin; 100 ng/ml) on ice and assays were synchronized by centrifugation (170 g, 8 min at 4°). Following a 1 hr incubation at 37°, cell-free supernatants were removed and analysed for leukotriene C4 (LTC4) and prostaglandin D2 (PGD2) by enzyme immunoassay (Cayman Chemicals) according to the manufacturer's instructions. Supernatants were diluted 1/1000 before analysis of PGD2 release.
Chemokine and cytokine focused SuperArrays
Total RNA was isolated (Superarray, Frederick, MD), 6 μg was treated for genomic DNA contamination and then reverse transcribed (Superarray). For each quantitative PCR assay, cDNA (50 ng/25 μl of PCR mastermix) was added to a Superarray plate containing primer sets for chemokines (CCL1-19, CXCL1-13; a total of 84 genes), cytokines (IL1-25, CSF1-2, IFNs, TGFs, TNF, TNFSF10–14, BMPs, GDFs; a total of 84 genes) and housekeeping genes (B2M, HPRT1, RPL13A, GAPDH, ACTB) as designed by the manufacturer (Superarray). As a control, DNA and RNA preparations were run on a quality control plate before running samples on the pathway-focused Superarrays. All reactions were performed on single samples for 40 cycles as per the manufacturer's instructions and gene expression was analysed using the Real-Time PCR cycler ABI PRISM 7700 (Applied Biosystems, Foster City, CA).
Real-time PCR analysis
Total RNA was isolated (RNeasy; Qiagen Inc., Valencia, CA); 2 μg was treated for genomic DNA contamination and reverse transcribed (Qiagen Inc.). For each quantitative PCR assay, cDNA (50 ng) was mixed with PCR mastermix containing primer sets for CD14, CD36, TLR2, TLR6, MSR1, SCARB1 and SCARB2 or the housekeeping gene ACTB as designed by the manufacturer (Qiagen Inc.). As a control, RNA that had not been reverse transcribed to cDNA was used in some PCRs. All reactions were performed in triplicate for 40 cycles and gene expression was analysed using the Real-Time PCR cycler ABI PRISM 7700 (Applied Biosystems).
Cytokine measurements
Human MCs (4 × 105/well) were added to a 48-well plate containing bacteria at a ratio of 20 : 1 huMCs and cultures were synchronized by centrifugation (170 g, 8 min at 4°). Cultures were then placed at 37° for 1–24 hr and cell-free supernatants were analysed for CXCL8 and GM-CSF by ELISA according to the manufacturer's instructions (R&D Systems, Minneapolis, MN).
Receptor expression by flow cytometric analysis
Human mast cells were washed three times (PBS + 0·1% BSA) and 100-μl (1 × 106/ml) aliquots were incubated with the following antibodies for 20 min at 20°: allophycocyanin-conjugated mouse anti-human CD14 (clone 61D3, subclass IgG1к), phycoerythrin (PE) -conjugated mouse anti-human TLR2 (clone TL2.1, subclass IgG2a), FITC-conjugated mouse anti-human CD36 (clone NL07, subclass IgM), rat anti-human TLR6 (clone hPer6, subclass IgG2aк) (eBiosciences, San Diego, CA) followed by PE-conjugated goat anti-rat IgGγ (Southern Biotech, Birmingham, AL), mouse anti-human SR-AI/MSRI (clone 351615, subtype IgG2b) then allophycocyanin-conjugated goat anti-mouse IgG2b (Southern Biotech) or rabbit polyclonal anti-human SCARB1 (Lifespan Biosciences, Seattle, WA) followed by PE-conjugated goat anti-rabbit IgG (Southern Biotech), PE-conjugated mouse anti-human CR3 (CD11b) (subclass IgG1к), PE-conjugated mouse anti-human CR4 (CD11c) (clone 3.9, subclass IgG1к), PE-conjugated anti-human or PE-labelled mouse anti-human CD49d (α4 integrin) (clone 9F10, subclass IgG1к) PE-conjugated mouse anti-human CD49e (α5 integrin) (clone SAM-1, subclass IgG2b), FITC-conjugated mouse anti-human CD51/61 integrin (αVβ3) (clone 23C6, subclass IgG1к) or PE-conjugated mouse anti-human CD29 (β1) (clone MEM-101A, subclass IgG1) (eBioscience), PE-labelled mouse anti-human dectin-1 (clone 259931, subclass IgG2b) (R&D Systems) or PE-conjugated mouse anti-human CD205 (clone MG38, subclass IgG2b) or allophycocyanin-labelled mouse anti-human CD206 (clone 19·2, subclass IgG1к) [Becton Dickinson (BD), San Jose, CA]. Cells were then washed, resuspended in PBS + 0·1% BSA and analysed using a FACScalibur and CellQuest software (BD) of 10 000 events in a CD117/FcεRI double-positive gated area.
Binding of S. aureus to huMCs by flow cytometric analysis
Staphylococcus aureus was grown to mid-log phase (OD600 < 1), heat killed [60° for 0·5 hr (1 × 109/ml in PBS)], washed twice in PBS, resuspended in an FITC [0·1 mg/ml in NaHCO3 (0·1 m)] solution, and incubated for 40 min with rotation. Following three washes, the FITC-labelled bacteria were resuspended in appropriate media and stored on ice until required. The binding of S. aureus to huMCs was determined by incubating huMCs with increasing ratios of FITC-labelled bacteria (20 : 1, 100 : 1; bacteria : huMCs) for 1 hr at 4°. After washing twice (PBS + 0·1% BSA), huMCs were resuspended in PBS + 0·1% BSA (100 μl) and analysed for FL1 using a FACScalibur (BD) and CellQuest software (BD).
Data presentation and statistical analyses
Real-time kinetic readings and grouped data are from a minimum of three separate experiments performed in duplicate using one to three donors to obtain CD34+ derived huMCs. The area under the curve data for total ROS production were determined after baseline subtraction by Prism (GraphPad Software, San Diego, CA). Differences between groups were tested for statistical significance by SigmaPlot. Where data were normally distributed, two-way repeated measures analysis of variance with Holm–Sidak correction for multiple comparisons was used. For non-parametric data, a Kruskal–Wallis one-way analysis of variance on ranks with Bonferroni correction for multiple comparisons or Wilcoxon signed rank test was used.
Results
IFN-γ enhances ROS generation and killing of S. aureus by huMCs
We initially investigated the ability of huMCs to generate ROS and kill S. aureus in response to T helper type 1 (Th1) and Th2 cytokines as might be present as inflammation progresses. As shown in Fig.1(a), pre-incubation of huMCs with IFN-γ, TNF-α or GM-CSF, but not IL-4, resulted in S. aureus-dependent ROS generation. ROS production in huMCs exposed to S. aureus and IFN-γ was greater than that in cells exposed to S. aureus combined with GM-CSF or TNF-α (Fig.1a). Killing by untreated huMCs was 2% compared to 25% in cells treated with IFN-γ (P < 0·001). By comparison, TNF-α, GM-CSF or IL-4 had a minimal effect on the ability of huMCs to kill S. aureus (Fig.1b). These data demonstrate that of the cytokines tested, IFN-γ was by far the most effective at enhancing ROS production and promoting killing of S. aureus by huMCs.
Figure 1.

Interferon-γ (IFN-γ) and not interleukin-4 (IL-4), tumour necrosis factor-α (TNF-α) or granulocyte–macrophage colony-stimulating factor (GM-CSF) primes human mast cells (huMCs) for enhanced production of reactive oxygen species (ROS) and bacterial killing of Staphylococcus aureus. Human MCs were pre-treated with IFN-γ (20 ng/ml), IL-4 (20 ng/ml), TNF-α (20 ng/ml) or GM-CSF (20 ng/ml) for 48 hr before the addition of S. aureus (20 bacteria : one huMC). Extracellular ROS production was determined by Diogenes chemiluminescence for 1 hr at 30-second intervals at 37° (a) and bacterial killing determined after 3 hr incubation by counting CFUs of cell lysates on agar plates after a 24-hr culture at 37° (b). Results are shown as either kinetic data of single experiments of cells in the absence (thin lines) or presence (bold lines) of bacteria (a) or means ± SE (b) performed in triplicate and n = 3 independent experiments for one huMC donor. Differences between individual groups was tested for statistical significance by Kruskal–Wallis one-way analysis of variance on ranks with Bonferroni correction for multiple comparisons (***P ≤ 0·001 for comparison with control cells not treated with cytokines).
As we have demonstrated that FcεRI- and FcγR-dependent intracellular ROS production in huMCs are 5LO- and COX-dependent,20 we next examined the mechanisms by which IFN-γ alters the production of intracellular and extracellular ROS by huMCs exposed to S. aureus. As shown in Fig.2(a), there was little or no production of ROS by huMCs exposed to S. aureus alone. However, huMCs pre-exposed to IFN-γ produced ROS in response to S. aureus, and the rate of ROS production was maximal 10–20 min after stimulation (Fig.2a). This S. aureus-dependent ROS generation was ratio dependent, and similar ROS levels were achieved with heat-killed bacteria. In contrast, coating of bacteria by serum opsonization or with S. aureus-specific IgG elicited lower levels of ROS from huMCs (see Supplementary material, Fig. S1a–c). Also, S. aureus-derived peptidoglycan (PGN) or lipoteichoic acid (LTA) did not induce ROS production (see Supplementary material, Fig. S1d). The ability of S. aureus to trigger extracellular production of ROS in huMCs following treatment with IFN-γ was verified by a Diogenes chemiluminescence assay (Fig.2b).
Figure 2.

Interferon-γ (IFN-γ) primes human mast cells (huMCs) for enhanced reactive oxygen species (ROS) generation following Staphylococcus aureus exposure. Human MCs were incubated with or without IFN-γ (20 ng/ml) for 48 hr then exposed to dichlorodifluorescein diacetate (DCF-DA; 20 μm) or medium for 20 min. After washing huMCs were incubated in the absence (a, b) or presence of FR122047 (300 nm) (c, d), zileuton (20 μm) (c, d), FR122047 (300 nm) + zileuton (20 μm) (c, d), DPI (5 μm) (e, f) or l-NMMA (100 μm) (e) for 10 min before the addition of S. aureus (20 : 1) or HEPES buffer (negative control). Intracellular ROS production was determined by DCF fluorescence (a, c, e, g) whereas extracellular ROS production was determined by Diogenes chemiluminescence (b, d, f, h) for 1 hr at 30-second intervals at 37°. Area under the curve (AUC) data were calculated for each kinetic curve and averaged (g, h). Results are shown as either kinetic data of single experiments performed in duplicate, n = 4 (a, c, e) or n = 2 (b, d, f) independent experiments from two donors (a–f) or means ± SE for AUC data calculated for each kinetic curve, n = 4 independent experiments from two donors in duplicate (g) or n = 2, two donors in duplicate (h). Results are for huMCs treated with or without IFN-γ (a, b) or IFN-γ only (c–h). AUC data were calculated for inhibitor curves (g, h) and statistical significance was determined by Kruskal–Wallis one-way analysis of variance on ranks with Bonferroni correction for multiple comparisons (+P < 0.05 and +++P ≤ 0·001 for comparison with control cells and *P ≤ 0·05, **P < 0.01 and ***P ≤ 0·001 for comparison with S. aureus-treated cells).
We next explored the enzymes responsible for ROS production. Incubation of huMCs with the COX inhibitor FR122047, the 5LO inhibitor zileuton (ZT) or a combination of both caused a significant reduction in S. aureus-dependent DCF fluorescence (Fig.2c,g) whereas the flavoenzyme inhibitor DPI and the NOS inhibitor l-NMMA had no inhibitory effect (Fig.2e,g). ZT or FR122047 alone or in combination also had no effect (Fig.2d,h) whereas DPI completely abrogated S. aureus-dependent Diogenes chemiluminescence in huMCs (Fig.2f,h). Hence, 5LO and COX were the enzymes responsible for S. aureus-dependent intracellular ROS production, while extracellular ROS production was primarily NADPH oxidase-dependent in IFN-γ-treated huMCs.
We then compared IFN-γ-primed huMCs with PMNs for their ability to generate ROS and kill S. aureus. As shown in Fig.3(a), PMNs, as expected, generated high levels of ROS following exposure to S. aureus. Generation of ROS by PMNs was approximately 50-fold greater than that of huMCs (Fig.3c). Killing of S. aureus by PMNs was more rapid than that by huMCs (1·5 hr for PMNs versus 3 hr for huMCs) (see Supplementary material, Fig. S2a,b) – where, by 1·5 hr, there was a significant killing of S. aureus by PMNs, but little killing of the organism by huMCs (Fig.3b,d). These data are consistent with the conclusion that the reduced ability of huMCs to generate ROS (compared with PMNs) limits the capacity to kill S. aureus.
Figure 3.

Interferon-γ (IFN-γ) -primed human mast cells (huMCs) generate less reactive oxygen species (ROS) and are less efficient at killing Staphylococcus aureus than polymorphonuclear cells (PMNs). Human MCs were pre-treated with IFN-γ (20 ng/ml) for 48 hr (c, d, e) or PMNs (a, b, f) were stimulated with S. aureus (20 : 1) or HEPES buffer (negative control) in the absence or presence of zileuton (20 μm), FR122047 (300 nm) or a combination of both (e, f). Diogenes chemiluminescence was then monitored for 60 min at 30-second intervals at 37° (a, c) or bacterial killing was determined after 1·5 hr (c, d) or 3 hr (e, f) by counting CFUs of cell lysates on agar plates after 24 hr culture at 37° (c–f). Results are shown as either kinetic data of single experiments performed in duplicate, n = 3 separate huMC donors (a, b) or means ± SE of independent experiments performed in duplicate for 2 (e) or 4 (c) separate PMN donors or three independent experiments performed in duplicate on a single huMC donor (d, f). Differences between individual groups was tested for statistical significance by Wilcoxon signed rank test (*P ≤ 0·05 and **P ≤ 0·01 for comparison with S. aureus alone)
We next investigated whether 5LO and COX inhibition would affect the killing of S. aureus by huMCs. Interferon-γ-primed huMCs were incubated with ZT, FR122047 or a combination of both inhibitors before S. aureus exposure for 3 hr. As shown in Fig.3(f), pre-incubation of huMCs with 5LO or COX inhibitors had no effect on the killing of S. aureus. These findings were confirmed with PMNs (Fig.3e). Assessment of the role of NADPH oxidase in the killing of S. aureus by huMCs and PMNs was hindered by the fact that DPI was toxic to S. aureus. These data demonstrate that IFN-γ-primed huMCs are capable of killing S. aureus, albeit less efficiently than PMNs, and that intracellular ROS derived from 5LO and COX are not involved. These data are consistent with the conclusion that extracellular ROS-derived from NADPH oxidase is involved in killing of bacteria by IFN-γ-primed huMCs.
IFN-γ enhances the immediate secretory responses of huMCs to S. aureus
Because IFN-γ is known to enhance mediator release following FcγRI aggregation of IFN-γ-treated huMCs,22 we next examined mediator release from huMCs treated with IFN-γ and exposed to S. aureus. Human MCs exposed to S. aureus in the absence of IFN-γ released negligible β-hexosaminidase (Fig.4a). However, incubation of huMC with IFN-γ before S. aureus stimulation elicited a significant increase in percent β-hexosaminidase release [from 4·36 ± 0·78% to 14·40 ± 2·14% at 20 : 1 (P ≤ 0·001); from 5·78 ± 1·17% to 26·90 ± 2·52% at 100 : 1 (P ≤ 0·001)], which was multiplicity of infection-dependent (Fig.4a). Similar results were obtained with histamine release (data not shown). Moreover, IFN-γ induced LTC4 secretion by huMCs following exposure to S. aureus (at a ratio of 100 bacteria per huMC, LTC4 secretion by huMCs increased from 2·35 ± 1·88 ng/ml to 13·68 ± 6·89 ng/ml, P ≤ 0·05) (Fig.4b). Similarly, IFN-γ caused a significant increase in S. aureus-dependent PGD2 secretion (Fig.4c). These data demonstrate that IFN-γ induces an S. aureus-dependent release of β-hexosaminidase, LTC4 and PGD2 by huMCs.
Figure 4.
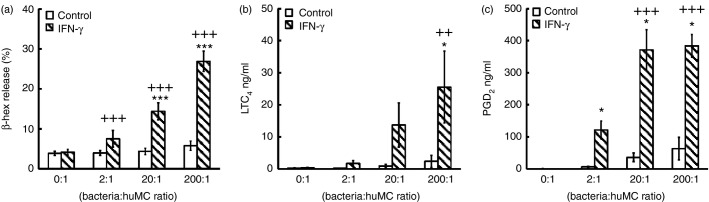
Interferon-γ (IFN-γ) enhances the immediate secretory responses of human mast cells (huMCs) to Staphylococcus aureus. Human MCs were pre-treated with or without IFN-γ (20 ng/ml) for 48 hr before the addition of S. aureus at a ratio of 2 : 1, 20 : 1 or 200 : 1 bacteria : huMC or HEPES buffer (negative control). Cell-free supernatants were collected after 1 hr and quantified for net β-hexosaminidase (a) or leukotriene C4 (LTC4) (b) and prostaglandin D2 (PGD2) (c) by competitive enzyme immunoassay. Results are means ± SE performed in duplicate, n = 4 independent experiments for two separate donors. Differences between individual groups were tested for statistical significance by two-way repeated measures analysis of variance with Holm–Sidak correction for multiple comparisons (++P < 0·01; +++P ≤ 0·001 for comparison of IFN-treated cells exposed to increasing ratios of S. aureus and *P > 0·05; **P > 0·01; ***P > 0·001 for comparison between untreated and IFN-γ-treated cells).
IFN-γ enhances production of chemokines and cytokines by huMCs exposed to S. aureus
Since IFN-γ enhanced the immediate secretory responses of huMCs to S. aureus, we next determined whether IFN-γ altered late secretory events. Human MCs were incubated with IFN-γ for 48 hr before exposure to S. aureus (20 : 1) for 2 hr, and changes in gene expression were analysed using cytokine and chemokine superarrays. The cytokine array revealed that IFN-γ-treated huMCs had a decrease in expression of CSF2 (0·18 ± 0·06 fold change), IL18 (0·16 ± 0·08 fold change) and IL1B (0·19 ± 0·07 fold change) whereas there were no alterations in baseline chemokine expression (data not shown). In response to S. aureus, huMCs showed a trend for increased expression of the chemokines CCL1-4, CXCL8, and the cytokines CSF1-2, IL5, IL13, INHBA, TNF and TNFSF14, which were further enhanced by IFN-γ (Fig.5a). The enhancing effect of IFN-γ on S. aureus-dependent CXCL8 mRNA expression was confirmed by quantitative real-time PCR (Fig.5b). To determine whether increases in mRNA correlated with increased levels of protein, we measured CXCL8 and GM-CSF secretion in cell-free supernatants of S. aureus-stimulated huMCs cultured with IFN-γ. As shown in Fig.5(c,d), exposure of huMCs to S. aureus induced CXCL8 and GM-CSF protein release as early as 2 hr post stimulation and IFN-γ pre-treatment significantly enhanced CXCL8 secretion at 2–4 hr and GM-CSF secretion at 2 hr post S. aureus stimulation. These data are consistent with the conclusion that IFN-γ enhances the ability of huMCs to release cytokines and chemokines following S. aureus exposure.
Figure 5.

Interferon-γ (IFN-γ) enhances Staphylococcus aureus-dependent CXCL8 and granulocyte–macrophage colony-stimulating factor (GM-CSF) secretion by human mast cells (huMCs). Human MCs were pre-treated with IFN-γ (20 ng/ml) for 48 hr before the addition of S. aureus (20 : 1). After 2 hr, mRNA was isolated, cDNA was transcribed and quantitative PCR was performed for analysis of cytokine and chemokine induction by S. aureus using a SuperArray platform (a) or real-time PCR (b). For quantification of CXCL8 and GM-CSF protein release, cell-free supernatants were collected after 1–8 hr and quantified for CXCL8 (c) and GM-CSF (d) by ELISA. Results are average fold change in mRNA levels relative to huMCs without bacterial exposure (a, b) or means ± SE performed in duplicate (c, d), n = 3–4 separate huMC donors. Differences between individual groups were tested for statistical significance by two-way repeated measures analysis of variance with Holm–Sidak correction for multiple comparisons (*P ≤ 0·05; **P > 0·01; ***P > 0·001 for comparison between control and IFN-γ-stimulated cells exposed to S. aureus).
IFN-γ-treated huMCs have increased surface binding of S. aureus and have increased expression of receptors associated with bacterial recognition
As it has been demonstrated that IFN-γ increases FcγRI surface expression,21 we determined whether IFN-γ also up-regulates receptors involved in bacterial recognition. To assess this, we initially investigated the binding of the bacterium to IFN-γ-treated huMCs using FITC-labelled S. aureus (FITC-SA) and flow cytometry and demonstrated that IFN-γ significantly increased the binding of S. aureus to huMCs (Fig.6a). We next investigated the transcript levels of receptors associated with bacterial recognition of S. aureus on huMCs by real-time PCR and flow cytometry including TLR2, TLR6, CD14 and CD36 and additional receptors that could potentially interact with S. aureus, including the scavenger receptors (mRNA/protein) (MSR1/SR-AI, SCARB1/SR-BI, SCARB2/SR-BII),26 lectin receptors [LY75/CD205(DEC-205), MRC1/CD206(mannose), CLEC7A/dectin-1], complement receptors [ITAGM/CR3(CD11b), ITAGX/CR4(CD11c)] and integrin receptors [ITAG4/CD49d(α4), ITAG5/CD49e(α5), ITAV/CD51(αV), ITGB1/CD29(β1), ITGB3/CD61(β3)].27,28 Human MCs expressed mRNA for the majority of receptors tested with the exception of TLR4 and ITAG4, and IFN-γ upregulated (fold change) the expression levels of MSR1 (1·56 ± 0·15), SCARB2 (2·28 ± 0·49), TLR2 (2·59 ± 0·38), ITAG5 (2·56 ± 0·24) and LY75 (3·39 ± 0·57) (see Supplementary material, Fig. S3).
Figure 6.

Interferon-γ (IFN-γ) enhances Staphylococcus aureus binding and increases Toll-like receptor (TLR) and scavenger receptors on human mast cells (huMCs). Human MCs were pre-treated with IFN-γ (20 ng/ml) for 48 hr before addition of FITC-labelled S. aureus (20 : 1) (a) or antibodies to surface receptors (b) and analysed by flow cytometry. Results are representative flow cytometric traces or means ± SE performed in duplicate, n = 3 huMC donors. Differences between individual groups were tested for statistical significance by paired Student's t-test (*P ≤ 0·05, **P ≤ 0·01 and ***P ≤ 0·001 for comparison between control and IFN-γ-stimulated cells).
To confirm whether changes in mRNA correlated with increased protein expression, we incubated huMCs with antibodies specific for these receptors. Human MCs did not express CD14, CD36, TLR6, the α subunits of the complement receptors (CR3, CR4), lectin receptors (CD205, CD206, dectin-1) or CD49e, the integrin α chain. Further, IFN-γ pre-treatment did not induce their expression (see Supplementary material, Fig. S4). In contrast, treatment of huMCs with IFN-γ caused a significant up-regulation of TLR2 and scavenger receptors (SR-AI, SR-BI/II), but not integrin receptors (CD49d, CD51/61 or CD29) (Fig.6b,c). Hence, huMCs treated with IFN-γ have increased gene expression and protein for TLR2 and scavenger receptors.
Integrin receptors and not TLR2 or SR-AI mediate binding of S. aureus to huMCs and the subsequent pro-inflammatory response
The contribution of bacterial recognition receptors to binding of S. aureus was next investigated by incubating huMCs with blocking antibodies to TLR2 or SR-AI. However, the ability of MCs to bind S. aureus was unaltered by pre-incubation of huMCs with blocking antibodies to TLR2 or SR-AI. Furthermore, binding of FITC-SA to tlr2-, msr1- or scarb1-deficient mBMMCs was also unchanged (Fig.7a–c). These data indicate that TLR2, SR-AI and SR-BI are not involved in the recognition of S. aureus by MCs. However, binding of FITC-SA to huMCs was blocked completely by pre-incubation with either the isotype (rabbit serum) or blocking antibody for SR-BI/II (Fig.7c, second panel). To further investigate the contribution of scavenger receptors, we pre-incubated huMCs with increasing concentrations of Poly I, Poly C or fucoidin (5–500 μg/ml) before FITC-SA, but no reduction in FITC-SA binding was observed (data not shown).
Figure 7.

The binding of Staphylococcus aureus to mast cells (MCs) is not mediated via Toll-like receptor 2 (TLR2) or scavenger receptors. Human (hu) MCs were pre-treated with interferon-γ (IFN-γ) for 48 hr and incubated with blocking antibodies to TLR2, SR-AI or SR-BI/II (a–c) for 1 hr before either huMCs or mouse bone-marrow-derived MCs (mBMMCs) from tlr2-deficient mice (a), msr1-deficient mice (b) or scarb1-deficient mice (c) were incubated with FITC-SA for a further 1 hr. After which MCs were analysed by flow cytometry for FL1 fluorescence. Results are representative flow cytometric traces or means ± SE performed in duplicate, n = 3 separate huMC or mBMMC donor.
Since the scarb1-deficient mBMMC lacked altered FITC-SA binding, we speculated that a component of rabbit serum was inhibiting FITC-SA binding to huMC. We therefore tested the effect of vitronectin, fibronectin and BSA (as a control) on SA-binding to huMCs in the presence or absence of IFN-γ. As shown in Fig.8(a), fibronectin reduced both the IFN-γ-enhanced binding of FITC-SA to huMCs as well as in untreated huMCs, whereas BSA had no effect. Vitronectin had no effect on FITC-SA binding in control or IFN-γ-treated huMCs (data not shown). Furthermore, blocking β1 integrin but not α4 integrin resulted in complete blockade of FITC-SA binding to huMCs (Fig.8b). These data are consistent with the involvement of integrin receptors, specifically β1 integrin, in the binding of S. aureus to huMCs in the presence and absence of IFN-γ.
Figure 8.

Binding of Staphylococcus aureus to human mast cells (huMCs) is mediated by integrin receptors. Human MCs were incubated with interferon-γ (IFN-γ) for 48 hr before a 1-hr exposure to fibronectin (100 μg/ml) or BSA (100 μg/ml) (a) or blocking antibodies to β1 integrin or α4 integrin (b). After which huMCs were exposed to FITC-SA for a further 1 hr. The S. aureus binding to huMCs was then analysed by flow cytometry for FL1 fluorescence. Results are representative flow cytometric traces or means ± SE performed in duplicate, n = 3 or n = 4 separate donors of huMCs. Differences between individual groups was tested for statistical significance by two-way repeated measures analysis of variance with Holm–Sidak correction for multiple comparisons (**P < 0·01; ***P < 0·001 for comparison of Fibronectin or anti-CD29 antibody-treated cells with control or isotype antibody, respectively)
We next determined whether the recognition of S. aureus by huMCs was indeed mediated via integrin receptors and if such an interaction could influence subsequent secretory responses. Pre-incubation of both control and IFN-γ-treated huMCs with fibronectin resulted in a concentration-dependent inhibition of FITC-SA binding, with complete blockade at 100 μg/ml (Fig.9). Pre-incubation of huMCs with an β1 integrin blocking antibody did not alter FITC-SA binding to control cells but did result in > 80% reduction of such binding to IFN-γ-treated huMCs (Fig.9a). These data suggest that binding of S. aureus to huMCs is mediated via β1 integrin under certain conditions including exposure to IFN-γ.
Figure 9.

Blockade of integrin receptors on human mast cells (huMCs) reduces Staphylococcus aureus-dependent binding and CXCL8 secretion. Human MCs were incubated with IFN-γ for 48 hr before a 1-hr exposure to increasing concentrations of fibronectin (1–100 μg/ml) or β1 integrin blocking antibody. Binding of FITC-SA was analysed by flow cytometry after 1 hr at 4° (a) or S. aureus-induced CXCL8 secretion in cell-free supernatants after 2 hr at 37° (b) was then determined. Results are means ± SE performed in duplicate, n = 3 or n = 4 huMC donors. Differences between individual groups were tested for statistical significance by two-way repeated measures analysis of variance with Holm–Sidak correction for multiple comparisons (+P < 0·05; ++P ≤ 0·01; +++P ≤ 0·001 for comparison between control and IFN-γ-stimulated cells and *P < 0·05; **P ≤ 0·01 and ***P ≤ 0·001 for comparison with S. aureus-treated cells).
Next, we investigated the involvement of integrin receptors in S. aureus-dependent CXCL8 secretion by huMCs. Following pre-incubation with fibronectin, there was a concentration-dependent inhibition of CXCL8 secretion in control and IFN-γ-treated huMCs (Fig.9b). A blocking antibody specific for β1 integrin partially inhibited S. aureus-dependent CXCL8 secretion in IFN-γ-treated huMCs (Fig.9b). These data together suggest a major role for β1 integrin in the recognition of S. aureus by huMCs and a partial role for β1 integrin in the secretory responses to the pathogen.
Discussion
In this study we demonstrated that IFN-γ enhances the antibacterial response of huMCs to S. aureus and increased ROS production. Interferon-γ pre-treatment also induced huMC degranulation and enhanced eicosanoid and pro-inflammatory chemokine and cytokine secretion by S. aureus. Interferon-γ increased binding of S. aureus to huMCs and up-regulated receptors associated with bacterial recognition. Blockade of β1 integrin completely abrogated binding of S. aureus with huMCs and partially inhibited cytokine secretion by these cells.
We investigated the ability of Th1 and Th2 cytokines to alter/enhance generation of extracellular ROS by huMCs, and in turn, their ability to kill S. aureus. We determined that while the Th2 cytokine IL-4 was capable of eliciting ROS production by huMCs, there was no effective killing of S. aureus. Of the Th1 cytokines tested, only IFN-γ caused significant generation of extracellular ROS by huMCs, and promoted killing of S. aureus. These data are similar to what has been reported for PMNs, where GM-CSF, TNF-α and IFN-γ enhance fMLP-dependent ROS production.29,30 The small amount of ROS generated by pre-incubation of huMCs with IL-4 is in contrast to PMNs, which do not generate ROS in response to IL-4.31
We have reported that IFN-γ-treated huMCs generate enhanced intracellular ROS following stimulation with IgG-coated latex beads and that the enzymes responsible are 5LO and COX1. This is in contrast to neutrophils, which primarily use NADPH oxidase for ROS production.20 In the present study, we demonstrate that IFN-γ enhances intracellular ROS production induced by S. aureus and that this also was 5LO- and COX1-dependent. Extracellular ROS production by huMCs, in contrast, was mediated by NADPH oxidase, since the NADPH oxidase inhibitor DPI and the antioxidant superoxide dismutase completely abrogated the extracellular ROS production. These findings support the conclusion that different enzymes are responsible for the generation of intracellular versus extracellular ROS in S. aureus-stimulated huMCs. We next investigated the role of 5LO and COX1 in mediating killing of S. aureus by huMCs. Using inhibitors of these enzymes, we demonstrated that 5LO and COX1 were not involved, indicating that extracellular and not intracellular ROS is involved in the killing of the bacterium. The investigation of NADPH oxidase was hampered by the fact that the enzyme inhibitor DPI had direct antibacterial activity in the absence of host cells. We did not employ other ROS scavengers/inhibitors such as l-NAC or apocynin, as these may have non-specific effects.
We also investigated inflammatory mediator secretion by huMCs in response to S. aureus ± IFN-γ. Human MCs did not degranulate in response to S. aureus but generated low levels of PGD2. In the presence of IFN-γ, degranulation occurred and production of LTC4 and PGD2 release was enhanced. This is consistent with a report that rat peritoneal MCs degranulate in response to E. coli32 and that rats challenged with S. aureus have a reduction in granular MCs.33 Additionally, mice challenged with peptidoglycan show increased histamine levels.7 Degranulation of MCs has also been reported to occur in response to exotoxins from S. aureus including δ-toxin5 and in response to superantigens.34 Our data demonstrate that MC degranulation can occur in direct response to S. aureus following pre-treatment of huMCs with IFN-γ. This suggests that the ability of huMCs to degranulate in response to bacterial stimuli is enhanced in the context of inflammatory lesions with a Th1 profile such as occurs in chronic lesions of atopic dermatitis.35
IFN-γ also enhanced LTC4 and PGD2 production induced by S. aureus in huMCs. There is little previous evidence of eicosanoid generation induced by S. aureus. Rat peritoneal MCs show LTC4 generation but no degranulation in response to S. aureus-derived peptidoglycan,36 whereas in humans Protein A (enterotoxins from S. aureus) induced histamine release, leukotriene and prostaglandin production from nasal polyps.37
As IFN-γ is known to up-regulate FcγRI on huMCs,21 we hypothesized that it would also up-regulate receptors for bacterial recognition on huMCs. We used quantitative PCR and flow cytometry to investigate the expression of a range of receptors both at baseline and induced by IFN-γ. We determined that huMCs increased expression of TLR2 and scavenger receptors following IFN-γ exposure and had high baseline levels of integrin receptors. However, using blocking antibodies to these receptors or mBMMCs deficient in these receptors, there was no change in SA-FITC binding, suggesting that these receptors are not involved in the recognition and binding of S. aureus by huMCs. This is in agreement with a report demonstrating that S. aureus binding to cord blood MCs (CBMCs) is not dependent on TLR2.13 There has been no investigation to date regarding the role of scavenger receptors in the huMC response to S. aureus but these receptors are known to interact with S. aureus in certain cells types.26 We determined that the β1 integrin was critical for S. aureus binding to IFN-γ-treated huMCs using blocking antibodies to this receptor or by blocking integrin receptors via fibronectin. FN-γ did not increase the mRNA or protein levels of β1 integrin, but it is possible that IFN-γ promotes a conformational change of β1 integrin or increases clustering of β1 integrin receptors resulting in increased binding of bacteria or activation intracellularly. Staphylococcus aureus has been reported to be internalized in mammalian cells by a ‘sandwich mechanism’ whereby fibronectin-binding proteins on the bacterium bind to fibronectin and the fibronectin-coated bacteria are then recognized by fibronectin-binding integrins on the cell surface, allowing internalization.27 Mouse fibroblasts with mutations in the β1 integrin receptor have a reduced capacity to internalize S. aureus38 and the epithelial cell line A549 using blocking antibodies or small interfering RNA for β1-inhibited internalization of S. aureus.39 Furthermore, α5β1 integrin has also been demonstrated in epithelial cells to be the receptor involved in mediating cell death, and TNF-α release by S. aureus α-toxin and pre-incubation with fibronectin reversed these effects.40 These studies support our data demonstrating that in huMCs the β1 integrin receptor is critical for the recognition and internalization of S. aureus.
We also investigated the role of β1 integrin in S. aureus-mediated cytokine secretion in IFN-γ-treated huMCs and demonstrated a partial role in CXCL8 production. This is in contrast to the report by Rocha-de-Souza et al.13 who concluded that a combination of TLR2 and CD48 was involved in S. aureus-induced cytokine production. This difference may relate to the source of the huMC cultures where Rocha-de-Souza et al. used cord-blood-derived huMCs whereas we used peripheral blood-derived huMCs. We did use a TLR2 blocking antibody to investigate the potential role of TLR2 in CXCL8 production but the antibody was without effect (data not shown). We also used blocking antibodies to scavenger receptors [msr1 (SR-AI), scarb1 (SR-BI/II)] as well as inhibitors of SR-BI (Block lipid transport-1). However, tlr2-deficient mBMMCs did have a 50% reduction in S. aureus-dependent CCL3 production and scarb1-deficient mBMMCs had complete abrogation of S. aureus-dependent CCL3 production (data not shown). Again, we observed no altered binding of S. aureus in these mBMMCs. These data appear to demonstrate a difference between mouse and human MCs in the receptors used to recognize S. aureus and those involved in mounting a cytokine response. Furthermore, it may not be a single receptor that is involved in recognition and cytokine secretion by mBMMCs but a combination of multiple receptors. Scavenger receptors do not mediate intracellular signalling and have to rely on co-receptors to induce their signals. In mBMMCs, it may be that the combination of scavenger receptors with TLR2 and others are involved in the response to S. aureus. We were unable to investigate the response in β1 integrin-deficient mBMMCs as these mice are embryonically lethal.41 Overall, our data demonstrate that integrin receptors are involved in the binding of S. aureus and the subsequent cytokine response in huMCs.
In summary, this study demonstrates that huMCs exposed to IFN-γ have enhanced intracellular and extracellular ROS production in response to the bacterium S. aureus and this relates to bacterial killing. Interferon-γ increases the sensitivity of huMCs to S. aureus by inducing degranulation and up-regulating eicosanoid, cytokine and chemokine secretion. Binding of S. aureus is increased to IFN-γ-treated huMCs and is mediated by integrin receptors. The secretory response of huMCs to S. aureus is partially mediated by integrin receptors. These data show the complexity of the MC response in relationship to the cytokine environment and help explain the role of MCs in innate immunity and in inflammatory processes such as those involved in atopic dermatitis.
Acknowledgments
EJS designed and performed experiments, analysed data and composed the manuscript. JB and MR provided some of the mast cell cultures and helped with PCRs and ELISAs and edited the manuscript. FRD and DD critically reviewed data and assisted in writing the manuscript. This work was supported by the Division of Intramural Research, NIAID/NIH.
Glossary
- CFU
colony-forming units
- COX
cyclooxygenase
- DCF-DA
dichlorodifluorescein diacetate
- DPI
diphenyleneiodonium
- huMCs
human mast cells
- IFN
interferon
- IL
interleukin
- LTC4
leukotriene C4
- 5-LO
5-lipoxygenase
- mBMMCs
mouse bone-marrow-derived mast cells
- PE
phycoerythrin
- PGD2
prostaglandin D2
- PMNs
polymorphonuclear cells
- ROS
reactive oxygen species
- Th1
T helper type 1
- TLR
Toll-like receptor
- TNF
tumour necrosis factor
Disclosure
The authors declare no conflict of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. Human mast cells (huMCs) incubated with pre-coated bacteria or cell wall components of Staphylococcus aureus have reduced production of reactive oxygen species (ROS). Human MCs were incubated with or without interferon-γ (IFN-γ; 20 ng/ml) for 48 hr then exposed to DCF-DA (20 μm) or medium for 20 min. After washing huMCs were incubated with increasing ratios of 2 : 1, 20 : 1 or 100 : 1 (S. aureus : huMC) (a), heat-killed S. aureus (b), opsonized or IgG-coated S. aureus (c) or the S. aureus-derived cell wall components peptidoglycan (PGN; 100 ng/ml) or lipoteichoic acid (LTA; 100 μg/ml) (d) or HEPES buffer (negative control). Intracellular ROS production was determined by DCF fluorescence for 1 hr at 30-second intervals at 37°. Results are shown as kinetic data of single experiments performed in duplicate and are representative of n = 3 (a–c) or n = 1 (d) from a single huMC donor.
Figure S2. A comparison of the time-dependent killing of Staphylococcus aureus by polymorphonuclear cells (PMNs) or human mast cells (huMCs). PMNs (a) or huMCs pre-treated with interferon-γ (IFN-γ) (20 ng/ml) for 48 hr (b) were incubated with S. aureus (20 : 1) and bacterial killing was determined after 0–3 hr by counting CFUs of cell lysates on agar plates after 24 hr culture at 37°. Results are shown as means ± SE, n = 2 donors of PMNs or huMCs.
Figure S3. Interferon-γ (IFN-γ) enhances gene expression of receptors associated with cell recognition of Staphylococcus aureus. Human mast cells (huMCs) were pre-treated with IFN-γ (20 ng/ml) for 50 hr before mRNA isolation and transcription of cDNA. Quantitative PCR was then performed for detection of receptors associated with bacterial recognition including scavenger and Toll-like receptors (TLRs) (a) and integrin, complement and lectin receptors (b). Results are fold change compared with control cells and are means ± SE performed in triplicate on four donors of huMCs.
Figure S4. Interferon-γ (IFN-γ) does not induce expression of CD markers on human mast cells (huMCs) associated with Staphylococcus aureus binding. HuMCs were pre-treated with IFN-γ (20 ng/ml) for 48 hr before addition of antibodies to surface receptors and analysed by flow cytometry. Results are representative flow cytometric traces (a) or means ± SE. (b) performed in duplicate, n = 3 huMC separate donors.
References
- Metcalfe DD, Baram D, Mekori YA. Mast cells. Physiol Rev. 1997;77:1033–79. doi: 10.1152/physrev.1997.77.4.1033. [DOI] [PubMed] [Google Scholar]
- Chan CY, St John AL, Abraham SN. Plasticity in mast cell responses during bacterial infections. Curr Opin Microbiol. 2012;15:78–84. doi: 10.1016/j.mib.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronnberg E, Johnzon CF, Calounova G, Faroldi G, Grujic M, Hartmann K, et al. Mast cells are activated by Staphylococcus aureus in vitro but do not influence the outcome of intraperitoneal Staphylococcus aureus infection in vivo. Immunology. 2014;143:155–63. doi: 10.1111/imm.12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCurdy JD, Lin TJ, Marshall JS. Toll-like receptor 4-mediated activation of murine mast cells. J Leukoc Biol. 2001;70:977–84. [PubMed] [Google Scholar]
- Nakamura Y, Oscherwitz J, Cease KB, Chan Sm, Munoz-Planillo R, Hasegawa M, et al. Staphylococcus δ-toxin induces allergic skin disease by activating mast cells. Nature. 2013;503:397–401. doi: 10.1038/nature12655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullaly SC, Kubes P. The role of TLR2 in vivo following challenge with Staphylococcus aureus and prototypic ligands. J Immunol. 2006;177:8154–63. doi: 10.4049/jimmunol.177.11.8154. [DOI] [PubMed] [Google Scholar]
- Feng BS, He SH, Zheng PY, Wu L, Yang PC. Mast cells play a crucial role in Staphylococcus aureus peptidoglycan-induced diarrhea. Am J Pathol. 2007;171:537–47. doi: 10.2353/ajpath.2007.061274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jawdat DM, Rowden G, Marshall JS. Mast cells have a pivotal role in TNF-independent lymph node hypertrophy and the mobilization of Langerhans cells in response to bacterial peptidoglycan. J Immunol. 2006;177:1755–62. doi: 10.4049/jimmunol.177.3.1755. [DOI] [PubMed] [Google Scholar]
- Malaviya R, Gao Z, Thankavel K, van der Merwe PA, Abraham SN. The mast cell tumor necrosis factor α response to FimH-expressing Escherichia coli is mediated by the glycosylphosphatidylinositol-anchored molecule CD48. Proc Natl Acad Sci USA. 1999;96:8110–5. doi: 10.1073/pnas.96.14.8110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arock M, Ross E, Lai-Kuen R, Averlant G, Gao Z, Abraham SN. Phagocytic and tumor necrosis factor α response of human mast cells following exposure to gram-negative and gram-positive bacteria. Infect Immun. 1998;66:6030–4. doi: 10.1128/iai.66.12.6030-6034.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischoff SC, Lorentz A, Schwengberg S, Weier G, Raab R, Manns MP. Mast cells are an important cellular source of tumour necrosis factor α in human intestinal tissue. Gut. 1999;44:643–52. doi: 10.1136/gut.44.5.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulka M, Fukuishi N, Rottem M, Mekori YA, Metcalfe DD. Mast cells, which interact with Escherichia coli, up-regulate genes associated with innate immunity and become less responsive to FcεRI-mediated activation. J Leukoc Biol. 2006;79:339–50. doi: 10.1189/jlb.1004600. [DOI] [PubMed] [Google Scholar]
- Rocha-de-Souza CM, Berent-Maoz B, Mankuta D, Moses AE, Levi-Schaffer F. Human mast cell activation by Staphylococcus aureus: interleukin-8 and tumor necrosis factor α release and the role of Toll-like receptor 2 and CD48 molecules. Infect Immun. 2008;76:4489–97. doi: 10.1128/IAI.00270-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abel J, Goldmann O, Ziegler C, Holtje C, Smeltzer MS, Cheung AL, et al. Staphylococcus aureus evades the extracellular antimicrobial activity of mast cells by promoting its own uptake. J Innate Immun. 2011;3:495–507. doi: 10.1159/000327714. [DOI] [PubMed] [Google Scholar]
- von Kockritz-Blickwede M, Goldmann O, Thulin P, Heinemann K, Norrby-Teglund A, Rohde M, et al. Phagocytosis-independent antimicrobial activity of mast cells by means of extracellular trap formation. Blood. 2008;111:3070–80. doi: 10.1182/blood-2007-07-104018. [DOI] [PubMed] [Google Scholar]
- Branitzki-Heinemann K, Okumura CY, Vollger L, Kawakami Y, Kawakami T, Naim HY, et al. A novel role for the transcription factor HIF-1α in the formation of mast cell extracellular traps. Biochem J. 2012;446:159–63. doi: 10.1042/BJ20120658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaviya R, Ross EA, MacGregor JI, Ikeda T, Little JR, Jakschik BA, et al. Mast cell phagocytosis of FimH-expressing enterobacteria. J Immunol. 1994;152:1907–14. [PubMed] [Google Scholar]
- Swindle EJ, Hunt JA, Coleman JW. A comparison of reactive oxygen species generation by rat peritoneal macrophages and mast cells using the highly sensitive real-time chemiluminescent probe pholasin: inhibition of antigen-induced mast cell degranulation by macrophage-derived hydrogen peroxide. J Immunol. 2002;169:5866–73. doi: 10.4049/jimmunol.169.10.5866. [DOI] [PubMed] [Google Scholar]
- Swindle EJ, Metcalfe DD, Coleman JW. Rodent and human mast cells produce functionally significant intracellular reactive oxygen species but not nitric oxide. J Biol Chem. 2004;279:48751–9. doi: 10.1074/jbc.M409738200. [DOI] [PubMed] [Google Scholar]
- Swindle EJ, Coleman JW, DeLeo FR, Metcalfe DD. FcεRI- and Fcγ receptor-mediated production of reactive oxygen species by mast cells is lipoxygenase- and cyclooxygenase-dependent and NADPH oxidase-independent. J Immunol. 2007;179:7059–71. doi: 10.4049/jimmunol.179.10.7059. [DOI] [PubMed] [Google Scholar]
- Okayama Y, Kirshenbaum AS, Metcalfe DD. Expression of a functional high-affinity IgG receptor, FcγRI, on human mast cells: up-regulation by IFN-γ. J Immunol. 2000;164:4332–9. doi: 10.4049/jimmunol.164.8.4332. [DOI] [PubMed] [Google Scholar]
- Okayama Y, Hagaman DD, Metcalfe DD. A comparison of mediators released or generated by IFN-γ-treated human mast cells following aggregation of FcγRI or FcεRI. J Immunol. 2001;166:4705–12. doi: 10.4049/jimmunol.166.7.4705. [DOI] [PubMed] [Google Scholar]
- Woolhiser MR, Okayama Y, Gilfillan AM, Metcalfe DD. IgG-dependent activation of human mast cells following up-regulation of FcγRI by IFN-γ. Eur J Immunol. 2001;31:3298–307. doi: 10.1002/1521-4141(200111)31:11<3298::aid-immu3298>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Okayama Y, Tkaczyk C, Metcalfe DD, Gilfillan AM. Comparison of FcεRI- and FcγRI-mediated degranulation and TNF-α synthesis in human mast cells: selective utilization of phosphatidylinositol-3-kinase for FcγRI-induced degranulation. Eur J Immunol. 2003;33:1450–9. doi: 10.1002/eji.200323563. [DOI] [PubMed] [Google Scholar]
- Okumura S, Kashiwakura J, Tomita H, Matsumoto K, Nakajima T, Saito H, et al. Identification of specific gene expression profiles in human mast cells mediated by Toll-like receptor 4 and FcεRI. Blood. 2003;102:2547–54. doi: 10.1182/blood-2002-12-3929. [DOI] [PubMed] [Google Scholar]
- Peiser L, Mukhopadhyay S, Gordon S. Scavenger receptors in innate immunity. Curr Opin Immunol. 2002;14:123–8. doi: 10.1016/s0952-7915(01)00307-7. [DOI] [PubMed] [Google Scholar]
- Hauck CR, Ohlsen K. Sticky connections: extracellular matrix protein recognition and integrin-mediated cellular invasion by Staphylococcus aureus. Curr Opin Microbiol. 2006;9:5–11. doi: 10.1016/j.mib.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Takada Y, Ye X, Simon S. The integrins. Genome Biol. 2007;8:215. doi: 10.1186/gb-2007-8-5-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewas C, Dang PM, Gougerot-Pocidalo MA, El-Benna J. TNF-α induces phosphorylation of p47(phox) in human neutrophils: partial phosphorylation of p47phox is a common event of priming of human neutrophils by TNF-α and granulocyte-macrophage colony-stimulating factor. J Immunol. 2003;171:4392–8. doi: 10.4049/jimmunol.171.8.4392. [DOI] [PubMed] [Google Scholar]
- Tennenberg SD, Fey DE, Lieser MJ. Oxidative priming of neutrophils by interferon-γ. J Leukoc Biol. 1993;53:301–8. doi: 10.1002/jlb.53.3.301. [DOI] [PubMed] [Google Scholar]
- Reglier-Poupet H, Hakim J, Gougerot-Pocidalo MA, Elbim C. Absence of regulation of human polymorphonuclear oxidative burst by interleukin-10, interleukin-4, interleukin-13 and transforming growth factor-β in whole blood. Eur Cytokine Netw. 1998;9:633–8. [PubMed] [Google Scholar]
- Malaviya R, Ross E, Jakschik BA, Abraham SN. Mast cell degranulation induced by type 1 fimbriated Escherichia coli in mice. J Clin Invest. 1994;93:1645–53. doi: 10.1172/JCI117146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudo LS, Betti F, Hanada S, Sertie JA, Zelante F. Lymphatic mast cell response and effect of compound 48/80 on popliteal lymph node reaction in rats following intracutaneous injection of Staphylococcus aureus. Agents Actions. 1994;42:135–40. doi: 10.1007/BF01983479. [DOI] [PubMed] [Google Scholar]
- Zhang N, Holtappels G, Gevaert P, Patou J, Dhaliwal B, Gould H, et al. Mucosal tissue polyclonal IgE is functional in response to allergen and SEB. Allergy. 2011;66:141–8. doi: 10.1111/j.1398-9995.2010.02448.x. [DOI] [PubMed] [Google Scholar]
- Gittler JK, Shemer A, Suarez-Farinas M, Fuentes-Duculan J, Gulewicz KJ, Wang CQ, et al. Progressive activation of TH2/TH22 cytokines and selective epidermal proteins characterizes acute and chronic atopic dermatitis. J Allergy Clin Immunol. 2012;130:1344–54. doi: 10.1016/j.jaci.2012.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierzbicki M, Brzezinska-Blaszczyk E. Diverse effects of bacterial cell wall components on mast cell degranulation, cysteinyl leukotriene generation and migration. Microbiol Immunol. 2009;53:694–703. doi: 10.1111/j.1348-0421.2009.00174.x. [DOI] [PubMed] [Google Scholar]
- Patou J, Gevaert P, Van Zele T, Holtappels G, van Cauwenberge P, Bachert C. Staphylococcus aureus enterotoxin B, protein A, and lipoteichoic acid stimulations in nasal polyps. J Allergy Clin Immunol. 2008;121:110–5. doi: 10.1016/j.jaci.2007.08.059. [DOI] [PubMed] [Google Scholar]
- Fowler T, Johansson S, Wary KK, Hook M. Src kinase has a central role in in vitro cellular internalization of Staphylococcus aureus. Cell Microbiol. 2003;5:417–26. doi: 10.1046/j.1462-5822.2003.00290.x. [DOI] [PubMed] [Google Scholar]
- Wang JH, Zhang K, Wang N, Qiu XM, Wang YB, He P. Involvement of phosphatidylinositol 3-Kinase/Akt signaling pathway in β1 integrin-mediated internalization of Staphylococcus aureus by alveolar epithelial cells. J Microbiol. 2013;51:644–50. doi: 10.1007/s12275-013-3040-x. [DOI] [PubMed] [Google Scholar]
- Liang X, Ji Y. Involvement of α5β1-integrin and TNF-α in Staphylococcus aureus α-toxin-induced death of epithelial cells. Cell Microbiol. 2007;9:1809–21. doi: 10.1111/j.1462-5822.2007.00917.x. [DOI] [PubMed] [Google Scholar]
- Stephens LE, Sutherland AE, Klimanskaya IV, Andrieux A, Meneses J, Pedersen RA, et al. Deletion of β1 integrins in mice results in inner cell mass failure and peri-implantation lethality. Genes Dev. 1995;9:1883–95. doi: 10.1101/gad.9.15.1883. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Human mast cells (huMCs) incubated with pre-coated bacteria or cell wall components of Staphylococcus aureus have reduced production of reactive oxygen species (ROS). Human MCs were incubated with or without interferon-γ (IFN-γ; 20 ng/ml) for 48 hr then exposed to DCF-DA (20 μm) or medium for 20 min. After washing huMCs were incubated with increasing ratios of 2 : 1, 20 : 1 or 100 : 1 (S. aureus : huMC) (a), heat-killed S. aureus (b), opsonized or IgG-coated S. aureus (c) or the S. aureus-derived cell wall components peptidoglycan (PGN; 100 ng/ml) or lipoteichoic acid (LTA; 100 μg/ml) (d) or HEPES buffer (negative control). Intracellular ROS production was determined by DCF fluorescence for 1 hr at 30-second intervals at 37°. Results are shown as kinetic data of single experiments performed in duplicate and are representative of n = 3 (a–c) or n = 1 (d) from a single huMC donor.
Figure S2. A comparison of the time-dependent killing of Staphylococcus aureus by polymorphonuclear cells (PMNs) or human mast cells (huMCs). PMNs (a) or huMCs pre-treated with interferon-γ (IFN-γ) (20 ng/ml) for 48 hr (b) were incubated with S. aureus (20 : 1) and bacterial killing was determined after 0–3 hr by counting CFUs of cell lysates on agar plates after 24 hr culture at 37°. Results are shown as means ± SE, n = 2 donors of PMNs or huMCs.
Figure S3. Interferon-γ (IFN-γ) enhances gene expression of receptors associated with cell recognition of Staphylococcus aureus. Human mast cells (huMCs) were pre-treated with IFN-γ (20 ng/ml) for 50 hr before mRNA isolation and transcription of cDNA. Quantitative PCR was then performed for detection of receptors associated with bacterial recognition including scavenger and Toll-like receptors (TLRs) (a) and integrin, complement and lectin receptors (b). Results are fold change compared with control cells and are means ± SE performed in triplicate on four donors of huMCs.
Figure S4. Interferon-γ (IFN-γ) does not induce expression of CD markers on human mast cells (huMCs) associated with Staphylococcus aureus binding. HuMCs were pre-treated with IFN-γ (20 ng/ml) for 48 hr before addition of antibodies to surface receptors and analysed by flow cytometry. Results are representative flow cytometric traces (a) or means ± SE. (b) performed in duplicate, n = 3 huMC separate donors.