

Abstract
High-throughput screening (HTS) was employed to discover APOBEC3G inhibitors and multiple 2-furylquinolines (e.g., 1) were found. Dose-response assays with 1 from the HTS sample, as well as commercial material, yielded similar confirmatory results. Interestingly, freshly synthesized and DMSO-solubilized 1 was inactive. Repeated screening of the DMSO aliquot of synthesized 1 revealed increasing APOBEC3G inhibitory activity with age, suggesting 1 decomposes into an active inhibitor. Laboratory aging of 1 followed by analysis revealed that 1 undergoes oxidative decomposition in air, resulting from a [4+2] cycloaddition between the furan of 1 and 1O2. The resulting endoperoxide then undergoes additional transformations, highlighted by Baeyer-Villager rearrangements, to deliver lactam, carboxylic acid, and aldehyde products. The endoperoxide also undergoes hydrolytic opening followed by further transformations to a bis-enone. Eight structurally related analogues from HTS libraries were similarly reactive. This study constitutes a cautionary to validate 2-furylquinolines for structure and stability prior to chemical optimization campaigns.
1. Introduction
The translation of high-throughput screening (HTS) hits to mechanistic probes and lead molecules for drug discovery is often plagued by false positives: compounds that exhibit the desired assay outcome though not as a result of specific interaction with the intended biological target.1 In 2007, Inglese, Shamu & Guy recommended guidelines for reporting HTS-identified small molecules after recognizing the absence of standards for publishing such discoveries.2 Included in this commentary is caution for scientists employing HTS to structurally validate their identified small molecules prior to publication, as false positives can not only result in inaccurate data reporting, but their characterization and elimination can burden a drug discovery campaign.2,3
Annotations of chemical libraries to identify scaffolds prone to hit in unrelated assays have been performed in recent years. These offending compounds, termed Pan Assay Interference Scaffolds or PAINS, result as a consequence of the physical and chemical properties of the small molecule and/or its interactions with certain components of the HTS assay.4-7 False positives can arise through various mechanisms including colloidal aggregation, compromised chemical integrity, or inherent reactivity of the small molecule; in addition to organic and/or inorganic impurities in the compound stock solution.8-14 Maintaining the chemical integrity of small molecule libraries as dimethylsulfoxide (DMSO) stock solutions upon long-term storage and freeze/thaw cycles remains a fundamental concern.15-17 It is known that a percentage of HTS library members decompose when stored in DMSO, which is the most common solvent employed.9 A number of decomposition mechanisms are possible, such as hydrolysis, oxidation, isomerization, and rearrangement reactions. Moreover, compounding factors such as organic (e.g., residual solvent) and inorganic (e.g., salt) impurities may promote decomposition.9 Herein, we report the decomposition of a seemingly stable HTS scaffold whose substructure is present in 133 of the 329510 NIH MLPCN library members.
Our laboratories have performed HTS to identify small molecule inhibitors of the DNA cytosine deaminases APOBEC3A (A3A) and APOBEC3G (A3G).18 The APOBEC3 (apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3) family functions in vivo to deaminate single-stranded DNA (ssDNA) cytosines to uracils (C-to-U), initiating the protective mechanism of foreign DNA degradation.19 APOBEC3s, in most cases, play a protective role as an innate immune defense mechanism in cells; however the mutative capacity of A3 enzymes may also contribute to human disease. A3G is predicted to drive human immunodeficiency virus-1 (HIV-1) evolution by enabling a consistent source of sublethal mutation in the HIV-1 genome.20-22 Moreover, the expression of nuclear-localized APOBEC3B (A3B) is up-regulated in over half of primary breast carcinomas, accounting for a large proportion of the mutational load in these tumors.23 A3B over-expression correlates to a higher overall mutational frequency, and poorer clinical outcomes, such as disease-free and overall survival, in estrogen receptor positive (ER+) breast cancer patients.23,24 Additionally, A3B over-expression has been implicated in the mutagenesis of bladder, cervix, head and neck, and lung cancers, both adenocarcinoma and squamous, suggesting that A3B-contributed mutation is a common paradigm in cancer mutagenesis.25,26 Consequently, the identification of small molecule inhibitors of the APOBEC3 enzymes may provide leads that can be further developed into clinical candidates for suppressing mutation and evolution in HIV-1 and cancer.
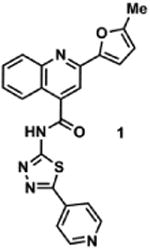
Using a previously reported fluorescence-based ssDNA C-to-U deaminase assay, we identified 62 structurally unique dual inhibitors of A3A (92% identical to A3B) and A3G from an HTS of 21126 small molecules at the University of Minnesota, which included compounds from the following commercial libraries: Sigma LOPAC (LO1280), Tocriscreen, Prestwick Chemical, NIH Clinical Collection and MicroSource Discovery.18 A detailed description of the screening assay can be found in the SI (Figure S1).18 Compound 1 was particularly intriguing because of its low micromolar potency against A3A (IC50 = 2.8 μM) and A3G (IC50 = 10.8 μM), and its low toxicity against 293T and HeLa cells (>85% cell viability when treated with 50 μM compound after 5 days). We confirmed this observed activity by purchasing 1 (Figure 1), commercially available from ChemBridge Corporation (#7922691), and subjecting the commercial material to dose response experiments against recombinant A3A and A3G. Purchased 1 substantiated our HTS observations demonstrating inhibition of both A3A (IC50 = 9.8 μM) and A3G (IC50 = 50.3 μM), albeit to a lesser extent.
Figure 1.
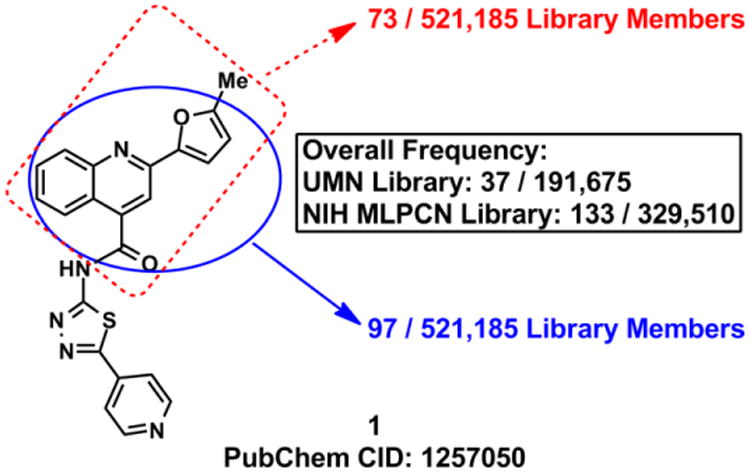
Chemical structure of 1 and the frequency with which the 2-(5-methylfuran-2-yl)quinoline-4-carbonyl (red) and the 2-(furan-2-yl)quinoline-4-carbonyl (blue) substructures occur in the University of Minnesota and the NIH MLPCN libraries (521,185 total compounds when combined). The boxed text represents the overall frequency of these chemotypes in each individual library. Overlap between the University of Minnesota and NIH MLPCN libraries is <10%.
Quinoline 1 is a privileged scaffold as related analogues bearing the 2-furan moiety have been investigated as leads for neurokinin-3 receptor inhibition, and in antimicrobial, antiviral, contraceptive, antitrypanosomal, and anticancer applications, though to the best of our knowledge, none of these molecules have advanced to clinical studies.27-34 Consistent with this observation, the 2-furylquinoline-4-carbonyl substructure is present in 170 HTS library members available in the University of Minnesota and the NIH MLPCN screening libraries and in many of our own HTS hits (Figure 1). As a result of marked A3 inhibition in HTS screening, a low toxicity profile against human cells, and the lack of PAINS warnings, we initiated a structural optimization campaign to refine 1 for potency and selectivity.
2. Results & Discussion
2.1 Synthesis and Enzymatic Assays with 1
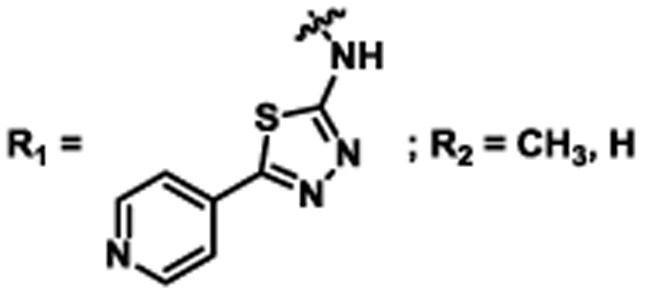
In an effort to investigate this scaffold as a potential lead molecule for structure-activity relationship (SAR) studies, we synthesized 1 (Scheme 1). A short two-step sequence delivered 1 in 17% overall yield, which began with the preparation of 3. Pfitzinger-Borsche chemistry was employed by reacting isatin (2) with 5-acetyl-2-methylfuran under basic conditions to deliver 3 in 79% yield.34 Commercially available 4 and carboxylic acid 3 were coupled under EDCI·HCl/HOBt conditions to achieve 1 in 22% yield. Amine 4 can also be synthesized as previously reported.35
Scheme 1.
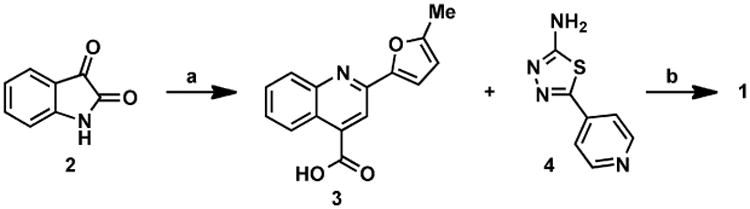
Reagents and conditions: (a) 5-acetyl-2-methylfuran, KOH, EtOH, 65 °C to reflux, 79%; (b) EDCI·HCl, HOBt, NMM, DMF, 22%.
Synthetic 1 was subsequently evaluated for A3A and A3G inhibition using our previously described fluorescence-based deamination assay.18,36 However, we were surprised to find that 1 exhibited no inhibition of either A3 enzyme when tested in dose response assays up to 200 μM concentrations (Figure 2; day 0). To verify our results, the same DMSO stock of 1 was re-tested on multiple occasions in the days that followed, and curiously, A3G inhibitory activity was noted, with the magnitude of A3G inhibition paralleling the age of the DMSO stock solution. A testing schedule that assayed for A3G inhibition at days 3, 21, 38, and 72 demonstrated that the DMSO stock solution of 1, aged in a closed microcentrifuge tube at 20-22 °C (on lab bench), increasingly and incrementally gained A3G inhibitory activity over a period of approximately two months, and as early as three days. Specifically, at 200 μM treatment, the aged stock of 1 reduced A3G deamination efficiency to 82% (3 days), 55% (21 days), 29% (38 days), and 29% (74 days) (Figure 2). It is worth noting that although this phenomenon with A3G was consistently reproduced, inhibitory activity against A3A, as seen in the HTS, was never recovered.
Figure 2.
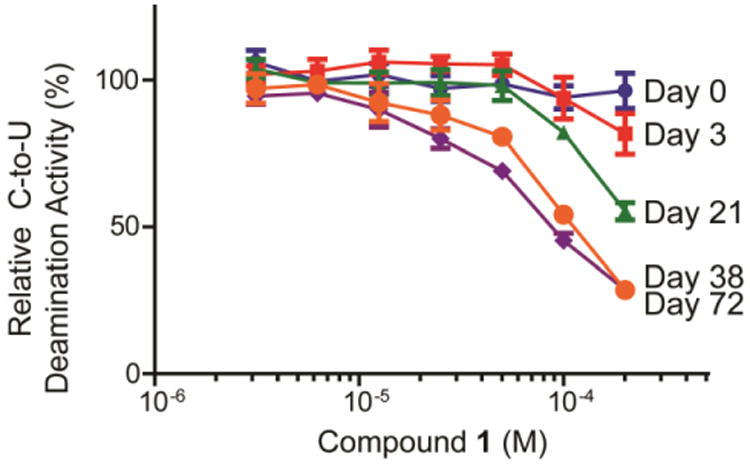
Dose response assays for A3G inhibition by freshly synthesized and solubilized 1 at 0, 3, 21, 38 and 72 days. Data indicates that the stock solution of 1 gains A3G inhibitory activity over time. Assays were performed in triplicate and deaminase activity was quantified as previously reported.10 Means ± standard deviations are indicated.
2.2 Identification of the Decomposition Products of 1
Based on the observed increase in inhibitory activity over time, we proposed that 1 decomposes into one or more chemically distinct A3G inhibitors. We first HPLC analyzed a 10 mM stock solution of synthesized 1 that was fortuitously dissolved in DMSO and stored in the laboratory for 16 months. This discrete sample primarily existed as a single product (SI, Figure S2). The major peak was isolated by HPLC, and the decomposition product was identified as 5 by 1H and 13C NMR, IR, and LC-MS. Spectroscopic data suggested that 5 exists as the more energetically stable lactam,37-39 which is supported by an IR spectrum that shows a strong C=O vibrational frequency (1771 cm-1) and the notable absence of a strong, broad –OH signal. To confirm that 5 is a decomposition product of 1 and that it inhibits A3G, we synthesized 5 by coupling amine 4 and commercially available 2-hydroxyquinoline-4-carboxylic acid with PyBOP in 59% yield (SI, Figure S3A).40 Following the isolation and characterization of 5, the purified aliquot was evaluated for A3A and A3G inhibitory activity. We found that isolated 5 reduced A3G-deamination efficiency to 83% at 200 μM, accounting for some of the inhibitory activity associated with the aged stock solution of 1 (SI, Figure S3B). Biochemical evaluation of synthesized 5 revealed a weak A3G inhibitor, reducing deamination efficiency by 50% (±1.5) at 200 μM (SI, Figure S3B). LC-MS/MS traces of isolated 5 and synthesized 5 were identical, thereby confirming the identity of the decomposition product (SI, Figures S3C,D). The slight discrepancy between the observed biochemical activities can be accounted for by the small amount of 5 (<1 mg) isolated from the aged DMSO stock solution. Inherently, the determined mass of purified 5 could reflect a higher probability of error, skewing the concentration of the stock solution, and thus, the calculated percent inhibition.
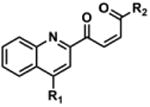
Next, analytical HPLC was employed to monitor the decomposition kinetics of 1 over the time frame that we observed increasing A3G inhibitory activity (t = 72 days, Figure 2). As expected at time 0, 1 exists in solution as a pure compound (Figure 3A). It was observed that by 21 days, however, notable decomposition of 1 had occurred, resulting in a mixture of decomposition products (Figure 3B). LC-MS analysis of the aged stock identified the masses of the three prominent peaks as 378.1, 464.1, and 430.1, respectively. The structures corresponding to these three masses were predicted to be oxidative decomposition products 6, 7a-7b, and 8 (Figure 3C), which were confirmed by HPLC isolation and 1H NMR and HRMS driven structural elucidation. To verify that the isolated samples were, in fact, decomposition products of 1, aliquots of the isolated samples (as 10 mM DMSO stock solutions) were spiked into a stock solution of 1 aged to 21 days. The anticipated enrichment of the intensity of decomposition peaks for 6, 7a-7b, and 8 (Figure 3B) was observed (SI, Figure S4). Decomposition products 6 and 8 were also synthesized by RuO2·H2O/NaIO4-catalyzed oxidation of 1 to give a 2:1:2 mixture of 6:1:8. These compounds were isolated by HPLC and characterized. LC-MS/MS traces of isolated and synthesized 6 and isolated and synthesized 8 were respectively identical, confirming the identities of these decomposition products (SI, Figures S5, S6). Moreover, aliquots of the synthesized compounds (as 10 mM DMSO stock solutions) were spiked into an aged stock of 1 at 21 days. Enrichment of the intensities of the appropriate peaks was observed (SI, Figure S4).
Figure 3.
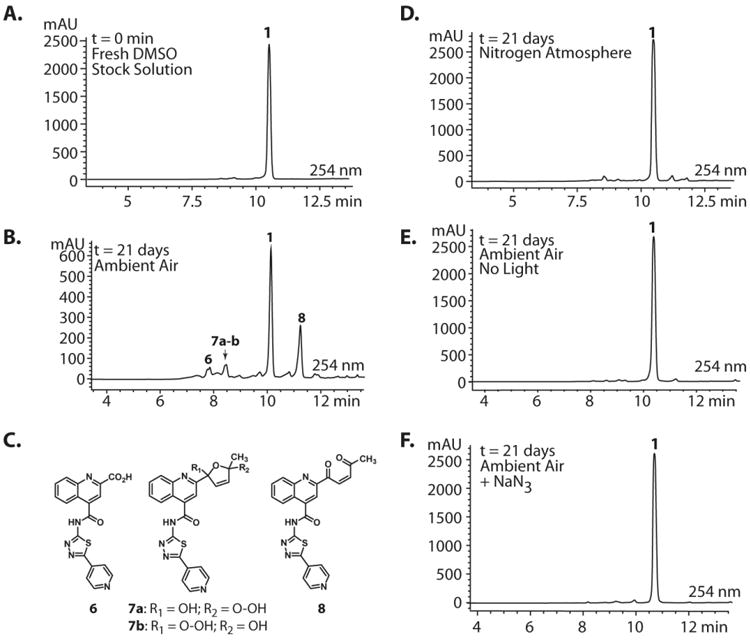
Analytical HPLC analyses of 10 mM DMSO stocks of 1. (A) Fresh stock solutions of 1 are >99% pure as determined by HPLC (See SI: Section III). (B) After 21 days of gentle shaking in ambient atmosphere at 25 °C, HPLC analysis shows evidence of decomposition through the appearance of multiple new peaks. (C) Compounds 6, 7a-b, and 8 can be assigned to the three prominent peaks in the decomposition mixutre. These compounds were isolated by HPLC and characterized by LC-MS, 1H NMR, HRMS, and co-injection of the isolated standards with aged samples of 1 (t = 21 d) (See SI: Sections II, V-VII, and IX for characterization data). (D) DMSO stocks of 1 aged under inert conditions (N2) exhibit drastically reduced decomposition. (E, F) DMSO stocks of 1 aged in the dark or in the presence of NaN3 exhibit no decomposition.
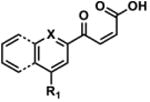
Biochemical evaluations of synthesized 6 and 8 identified 8 as the most potent A3G inhibitor, with an IC50 = 20 ± 4.2 μM (SI, Figure S7). Decomposition product 6 reduced deamination efficiency to 43% (± 2.8) at 200 μM, exhibiting activity similar to 5, which reduced deamination efficiency to 50% (±1.5) at 200 μM (SI, Figure S8). A freshly isolated sample of 7a-7b was also biochemically evaluated, but this intermediate exhibited no A3G inhibition. 2-Hydroxyquinonline-4-carboxylic acid, quinoline-2,4-carboxylic acid, and 5-(pyridin-4-yl)-1,3,4-thiadiazol-2-amine (4), which are substructures of the decomposition products, were also evaluated for A3G inhibitory activity and found to be inactive (data not shown). Taken together, decomposition product 8 (IC50 = 20 ± 4.2 μM for A3G) most closely mirrored the biochemical activity exhibited by the commercially-available ChemBridge material (IC50 = 50.3 μM for A3G) and in the HTS (IC50 = 10.8 μM for A3G).
2.3 Mechanism of Decomposition
To elucidate the mechanism by which 1 undergoes decomposition to 5, 6, 7a-7b, and 8, we first performed an equivalent aging experiment, except a saturated N2 atmosphere was used instead of laboratory air. Degradation of 1 was dramatically slowed under these conditions, supporting an oxidative mechanism of decomposition (Figure 3D). Moreover, decomposition was inhibited when DMSO stocks of 1 were maintained in the dark or in the presence of 1O2-quenching reagent NaN3 (Figures 3E,F). From these qualitative experiments, we proposed that the decomposition of 1 is enabled by the in situ generation of 1O2, which is (auto)photosensitized by 1. Additional experiments revealed that decomposition occurs at the same rate regardless of the temperature in which the DMSO stock is stored (-20 °C vs. rt vs. 37 °C; data not shown).
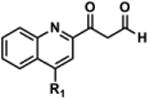
Singlet oxygen is 22 kcal/mol higher in energy than ground state oxygen and exhibits double bond character as a result of having paired electrons in the same LUMO.41 Schenck first proposed this [4+2] cycloaddition by hypothesizing that sensitized or auto-sensitized photooxygenations of furans produce endoperoxides.42 Subsequent work demonstrated that the resulting endoperoxides can yield dioxirane intermediates (via carbonyl oxides) that further rearrange to give “anomalous ozonolysis” products through an intramolecular Baeyer-Villiger mechanism.43-45 Evidence for the carbonyl oxide intermediate has been demonstrated through trapping experiments.46 A Baeyer-Villiger mechanism would explain the incorporation of the lactam carbonyl and the carboxylic acid at the 2-position of 1 (Figure 4, 5 & 6). Based on this previous literature, we propose that the furan of 1 undergoes [4+2] cycloaddition with 1O2 to give endoperoxide 9. The endoperoxide can collapse without directional bias to yield carbonyl oxide species that readily convert into dioxirane intermediates. This mechanism is unlikely to be concerted as previous research has demonstrated that carbonyl oxide intermediates are readily intercepted by olefins, sulfides, and ketones.47 Furthermore, cyclization to the dioxirane resonance is thought to be driven by destabilization of the carbonyl oxide by the enone.47 The resulting dioxiranes can then undergo Baeyer-Villiger-type rearrangements to produce four penultimate esters that are hydrolyzed in aqueous conditions. Our proposed mechanism could also yield α,β-unsaturated acid 10 and aldehyde 11, in addition to, lactam 5 and carboxylic acid 6. To account for observed decomposition products 7a, 7b, and 8 (Figure 3), endoperoxide 9 could alternatively be hydrolyzed by H2O present in the DMSO stock.48 We propose that the hydrolysis of 9 yields peroxides 7a and 7b, which are inherently unstable and can rearrange to enone 8 via the loss of -OOH and ring opening (Path E, Figure 4)48 or decomposition products 5, 6, 10 and 11 through the reversible loss of H2O and the aforementioned mechanisms (Figure 4). When isolated by HPLC, 10 mM DMSO stocks of 7a/7b exhibited a short shelf life of <48 h. After which, analysis of these stocks by analytical HPLC displayed primarily 6 and 8, which were confirmed by isolation and 1H NMR (data not shown).
Figure 4.
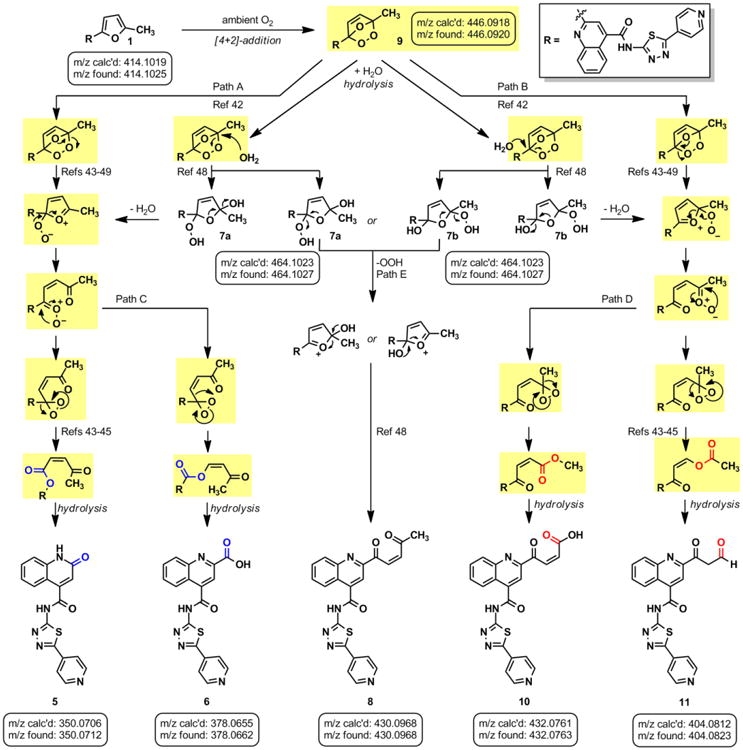
(A) Proposed mechanistic pathways for oxygen-mediated decomposition of 1. Intermediates highlighted in yellow exhibit m/z [M+H]+ = 446.1, which is observed in the LC-MS analysis.
LC-MS analysis of DMSO stock solutions of 1 aged 38 days under ambient air provided significant experimental support for our proposed mechanism of decomposition. Lactam 5 (m/z 350.1), carboxylic acid 6 (m/z 378.1), enone 8 (m/z 430.1), α,β-unsaturated acid 10 (m/z 432.1), and aldehyde 11 (m/z 404.1) were all found in our analysis (Figure 4; full mass chromatograms and extracted ion currents for each of the decomposition products and intermediates are shown in the SI, Section XVII). Furthermore, we also observed masses that correspond to either 7a or 7b (m/z 464.1), which would result from hydrolysis of endoperoxide 9. Hydrolysis of the carbonyl oxides or dioxiranes could also yield this same mass (m/z 464.1). Additionally, the mass corresponding to 1O2 addition to the furan (m/z 446.1) was observed in our sample, which could denote the endoperoxide 9, the carbonyl oxides, the dioxiranes, or the esters (Figure 4, possible m/z 446.1 intermediates are highlighted in yellow). Aging of a DMSO solution of 1 under an atmosphere of [18O]-O2 was performed to further support that 1 reacts with photosensitized molecular oxygen to yield the intermediates reported above. Accordingly, we found incorporation of heavy oxygen into 5* and 6* (m/z 352.1 and 380.1 for aging under [18O]-O2 versus 350.1 and 378.1 for 5 and 6, respectively), as well as isotopically-labeled 7a* or 7b* (+4 Daltons) in our analysis (SI, Section XIV).
Similar patterns of decomposition following laboratory aging in DMSO are observed regardless of the sample analysis method (e.g., 4.6 × 150 mm, 3.5 μm C18 column, trifluoroacetic acid modifier versus 0.5 × 150 mm, 5 μm C18 column, trifluoroacetic acid and formic acid eluent modifiers). The addition of formic acid to the eluent was required for mass spectrometry analysis (LC-MS). However, we do note some minor differences in the relative abundances of the decomposition products as visible in the UV chromatograms between these methods. Given that quantitative measurements of the decomposition products are outside the scope of this investigation, and that the masses of decomposition products are clearly observed (full mass chromatograms are provided in the SI), we employed these HPLC methods interchangeably for sample analysis in additional mechanistic queries.
To further demonstrate that 1 decomposes by the proposed cycloaddition-rearrangement sequences, we employed known photochemical conditions to an oxygenated solution of 1.49 A 10 mM DMSO solution of 1 was irradiated with visible light (300 W) under an oxygen-saturated environment (O2 balloon, 1 atm) in the presence of rose bengal (RB), a photosensitizer. LC-MS analysis of the crude reaction mixture exhibited m/z products corresponding to 5, 6, 7a-b, 8, the intermediates with the common m/z of 446.1, and related analogues 10 and 11 (Figure 5A; SI, Section XI). To demonstrate that 1 auto-sensitizes its photooxygenation, we also irradiated 1 in DMSO using visible light (300 W) in an oxygen-saturated atmosphere (O2 balloon, 1 atm) that was absent of sensitizer. Surprisingly, we observed more extensive decomposition without addition of photosensitizer (Figure 5B). These conditions permitted additional mechanistic work without the long incubation times used previously (e.g., weeks of time, Figure 3) To rule out the potential influence of heat generated by the lamp on the decomposition of 1, and to further provide evidence for oxidative decomposition, an identical experiment was performed under an N2 atmosphere (balloon, 1 atm). In the absence of oxygen, virtually no decomposition of 1 was observed over 9 h, although constantly irradiated (Figure 5C). Finally, we observed the incorporation of isotopically labeled [18O]-O2 by performing this photooxidation in a saturated 18O2 environment (balloon, 1 atm; Figure 5E). LC-MS analysis revealed incorporation of the isotope, yielding 5* (m/z = 352.1, + 2.0 Daltons), 6* (m/z 352.1, +2.0 Daltons), and hydrolysis products 7a* and/or 7b* (m/z 468.1, +4.0 Daltons). Conversely, irradiation of 1 in an O2 environment (balloon, 1 atm) in the presence of H218O yielded incorporation of the isotope only as a result of hydrolysis with [18O]-labeled water and no incorporation into any other intermediate (SI, Section XV).
Figure 5.
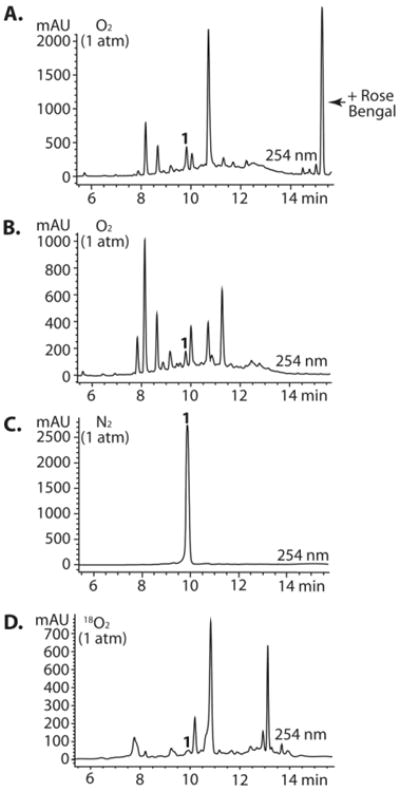
(A) The decomposition of 1 can be achieved in short reactions times (9h vs. 21d) by irradiating DMSO stocks of 1 with visible light (300W) in an oxygen saturated atmosphere. (B) Decomposition readily occurs in the absense of photosensitizer. (C) No decomposition occurs under an N2 atmosphere. (D) Decomposition in the presence of [18O]-O2. Rose bengal was not added to reactions B – D. LCMS traces can be found in the SI (Sections XI-XV).
2.4 Mechanistic Determinants
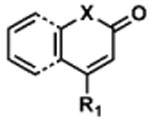
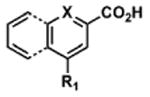
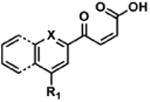
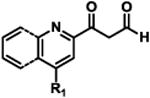
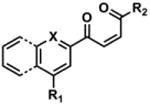
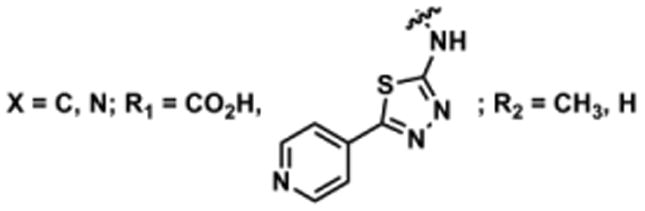
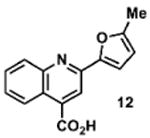
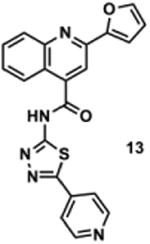
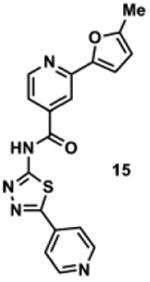
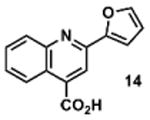
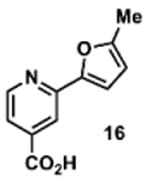
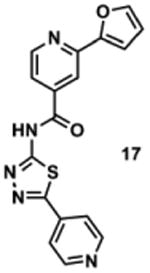
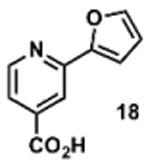
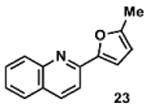
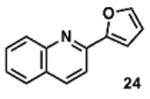
To determine the minimal sub-structure of 1 susceptible to this mechanism of decomposition, a series of mechanistic probes was synthesized and subjected to laboratory aging. The designed probes gauged the necessity for a 4-position amide, a methyl furan, a quinoline ring, and a 4-position carbonyl. Accordingly, analogues were studied which contained a free carboxylic acid as opposed to an amide linkage (12,14,16,18,20,22), a furan as opposed to a methyl furan (13,14,17,18,21,22), a pyridyl (15-18) or naphthyl ring (19-22) in place of the quinoline, or the absence a 4-position substituent (23,24). Syntheses were performed according to standard organic transformations. Generally, ring system-furan connections were accomplished under Stille or Suzuki conditions from commercial reagents, and amide bond couplings were accomplished with EDCI·HCl, HOBt, and DIPEA. DMSO stock solutions (10 mM) for the 13 probes were made identically to those prepared for 1 and solutions were exposed to laboratory air at 25 °C for 38 days. Aliquots were taken at 0, 2, 7, 21 and 38 days and analyzed by analytical HPLC. Percent decomposition was measured by dividing the ratio of the area under the curve (AUC) of compound over internal standard at day 38 by the ratio at time 0 (Table 1). The stock solutions of each compound were also subjected to LC-MS analysis at day 38. The extent of decomposition varied widely across the mechanistic probes. The following general trends were observed: (1) a higher degree of decomposition occurred in probes that contained 4-position amide linkages as compared to their free carboxylic acid counterparts, (2) a higher degree of decomposition was observed in probes with methyl functionalized furans in comparison to the non-substituted analogues, (3) probes lacking a 4-position carbonyl exhibited the least decomposition, and (4) the identity of the ring system had little effect on the extent of decomposition. Notable exceptions to these trends were observed, and thus, they must be accepted as only suggestions. For example, quinoline-based probe 14, which is absent of the 4-position amide linkage and methyl-substituted furan, showed more decomposition than probes 12 (methyl furan) and 13 (amide linkage). Additionally, naphthyl-based probe 21 showed extensive decomposition, although it lacked a furan methyl substituent. The percentage parent remaining for probes exhibiting a 4-position carboxylic acid ranged from 69-92%, for probes exhibiting a 4-position amide linkage from 0-99%, and >99% for probes lacking a 4-position carbonyl. Decomposition was observed for probes based on all three ring systems, with 6-99% parent remaining for the quinoline-based series, 34-99% remaining for the pyridine-based series, and 0-89% for the naphthylene-based series. The most common decomposition products observed by LC-MS were the putative endoperoxide intermediates, the carboxylic acid products (analogous to 6), and the enone products (analogous to 8); however, definitive proof for the formation of such decomposition products requires isolation and characterization of the individual compounds and/or co-injection with authentic standards, as performed for 1 (vide supra). Nonetheless, given the structural similarities of these probes with 1, a high degree of confidence can be attributed to our LC-MS-based approach to decomposition product characterization. Full mass chromatograms and extracted ion currents for each of the decomposition products and intermediates are shown in the SI, Sections XIX-XXXI. It is important to note that the reported percentages of probe remaining are in the context of 38 d, and that DMSO stocks analyzed at later times points (t = months) exhibited decomposition in every probe (data not shown). Taken together, each of the furan-substituted aromatic systems investigated in this study are prone to oxidative decomposition, albeit to varying rates.
Table 1. Decomposition Study of Sub-Structure Probes of 1 in DMSO Solution.
| Decomposition Products Observedc,d | |||||||
|---|---|---|---|---|---|---|---|
| Probea | Exact Mass | % Parent Remaining ± SD (38 days)a |
|
|
|
|
|
|
|||||||
|
413.1 | 6 ± 1 | + 350.1 (350.1) | + 378.1 (378.1) | + 432.1 (432.1) | + 404.1 (404.1) | + 430.1 (430.1) |
|
253.1 | 83 ± 18 | + 190.1 (190.1) | + 218.1 (218.1) | + 272.1 (272.1) | - | + 270.1 (270.1) |
|
399.1 | 101 ± 7 | + 350.2 (350.1) | + 378.2 (378.1) | +e 432.2 (432.1) | - | - |
|
239.1 | 69 ± 7 | + 190.2 (190.1) | - | +e 272.1 (272.1) | - | - |
|
363.1 | 34 ± 4 | - | + 328.2 (328.0) | - | - | + 380.2 (380.1) |
|
203.1 | 85 ± 8 | - | - | - | - | + 220.1 (220.1) |
|
349.4 | 90 ± 12 | - | - | +e 382.2 (382.1) | - | - |
|
189.0 | 92 ± 6 | - | - | - | - | - |
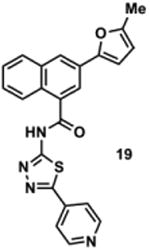
|
412.1 | 25 ± 1 | - | + 377.2 (377.1) | - | - | + 429.2 (429.1) |
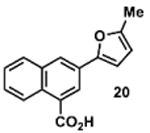
|
252.1 | 82 ± 31 | - | - | - | - | + 269.2 (269.1) |
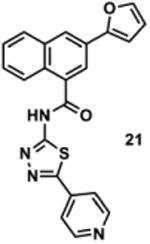
|
399.1 | 0 | - | + 377.2 (377.1) | +e 431.2 (431.1) | + 403.2 (403.1) | + 415.1 (415.1) |
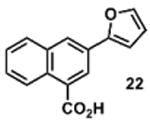
|
238.1 | 89 ± 8 | - | - | +e 271.1 (271.0) | + 243.1 (243.0) | + 254.1 (254.1) |
|
209.1 | 102 ± 12 | - | - | - | - | + 226.2 (226.2) |
|
195.1 | 117 ± 16 | - | - | +e 228.1 (228.1) | - | - |
10 mM solutions in DMSO were shaken gently at 25 °C open to the atmosphere. Percent remaining is the mean of three independent experiments.
DMSO stock solutions were analyzed by LC-MS on day 38. Chromatograms and spectra can be found in the SI (Sections XIX – XXXI).
Decomposition products positively identified are denoted by +, while products not detected are denoted by -. For observed decomposition products, the masses are presented as found mass, then calculated mass in parentheses. Masses were calculated for [M+H]+.
The aldehyde decomposition product could also be the enol tautomer.
For probes with a non-methylated furan, the carboxylic acid and [M+O2+H]+ intermediates (highlighted in yellow in Figure 4) have the same MW. The enone is represented in this table as identified; however, the observed m/z may correspond to an [M+O2+H]+ intermediate.
2.5 General Applicability of Oxidative Decomposition to Structurally Related HTS Library Scaffolds
Together, the University of Minnesota and the NIH MLPCN screening libraries contain 170 compounds based on this 2-furylquinoline-4-carbonyl substructure. To investigate if other 2-furylquinoline HTS members can decompose similarly to 1, we purchased eight representative compounds that were present in either the University of Minnesota library or the NIH MLPCN screening collection. DMSO stock solutions (10 mM) for the eight compounds were made identically to those prepared for 1 and solutions were exposed to laboratory air at 25 °C for 38 days. Aliquots were taken at 0, 2, 7, 21 and 38 days and analyzed by HPLC. Decomposition was observed in all of the compound stocks as early as 2 days post solvation, and at day 38, the stocks had decomposed between 16 – 81% (Table 2). Percent decomposition was calculated as described above. The stock solutions of each compound were subjected to LC-MS analysis, and in each case, peaks consistent with the formation of an oxygen-furan cycloaddition adduct [M+O2+H]+, the lactam decomposition product (equivalent to 5), and the carboxylic acid decomposition product (equivalent to 6) were observed. In the majority of experiments, our data supports the formation of the bis-enone (equivalent to 8), the α,β-unsaturated acid (equivalent to 10), and the aldehyde (equivalent to 11) decomposition products (SI, Sections XXXII - XXXIX). Our data suggests that HTS library compounds containing the 2-furylquinoline-4-carbonyl substructure are wholly subject to oxygen-mediated decomposition via the aforementioned mechanism and that this decomposition occurs within a timeframe relevant to HTS screening investigations.
Table 2. Decomposition of Furan-Functionalized Quinolines in DMSO Solution.
| Decomposition Products Observedc,d | ||||||||
|---|---|---|---|---|---|---|---|---|
| HTS Library Scaffolda | PubChem CID | Exact Mass | % Parent Remaining ± SD (38 days)b |
|
|
|
|
|
|
||||||||
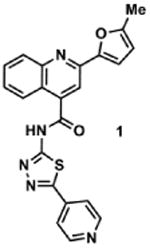
|
1257050 | 413.1 | 6 ± 1 | + 350.1 (350.1) | + 378.1 (378.1) | + 432.1 (432.1) | + 404.1 (404.1) | + 430.1 (430.1) |
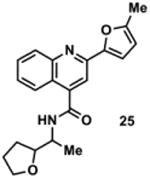
|
2965431 | 350.2 | 55 ± 4 | + 287.1 (287.1) | + 315.1 (315.1) | - | + 341.4 (341.1) | + 367.2 (367.2) |
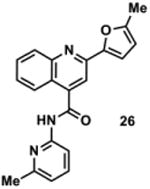
|
845057 | 343.1 | 73 ± 8 | + 280.1 (280.1) | + 308.1 (308.1) | + 362.1 (362.1) | + 334.2 (334.1) | + 360.2 (360.1) |
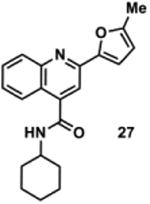
|
898877 | 334.2 | 54 ± 19 | + 271.1 (271.1) | + 299.1 (299.1) | + 353.2 (353.1) | + 325.2 (325.1) | + 351.2 (351.2) |
|
1290240 | 386.2 | 84 ± 32 | + 323.1 (323.1) | + 351.2 (351.1) | + 405.2 (405.1) | + 377.2 (377.1) | + 403.2 (403.2) |
|
1001397 | 386.1 | 56 ± 28 | + 323.1 (323.1) | + 351.2 (351.1) | + 405.1 (405.1) | + 377.1 (377.1) | + 403.1 (403.1) |
|
1295580 | 378.2 | 64 ± 19 | + 329.1 (329.1) | + 357.1 (357.1) | +e 411.1 (411.1) | + 383.1 (383.1) | - |
|
2220790 | 399.1 | 26 ± 4 | + 336.1 (336.1) | + 364.1 (364.1) | + 418.1 (418.1) | + 390.1 (390.1) | + 416.1 (416.1) |
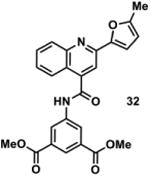
|
1000751 | 444.1 | 19 ± 10 | + 381.1 (381.1) | + 409.1 (409.1) | + 463.1 (463.1) | - | + 461.1 (461.1) |
Available in either the NIH MLPCN collection or the University of Minnesota small molecule collection.
10 mM solutions in DMSO were shaken gently at 25 °C open to the atmosphere. Percent remaining is the mean of three independent experiments.
DMSO stock solutions were analyzed by LC-MS on day 38. Chromatograms and spectra can be found in the SI (Sections XXXII – XXXIX). Decomposition products positively identified are denoted by +, while products not detected are denoted by -. For observed decomposition products, the masses are presented as found mass and calculated mass in parentheses. Masses were calculated for [M+H]+.
The aldehyde decomposition product could also be the enol tautomer.
For probes with a non-methylated furan, the carboxylic acid and [M+O2+H]+ intermediates (highlighted in yellow in Figure 4) have the same MW. The enone is represented in this table as identified; however, the observed m/z may correspond to an [M+O2+H]+ intermediate.
3. Conclusions
In summary, it was found that the chemical integrity of 1 was compromised by air oxidation of the furan, yielding intermediates that rearrange through a Baeyer-Villiger mechanism to products 5, 6, 10, and 11, and via hydrolysis-promoted rearrangement to 8. Oxygen mediated decomposition of the 2-furylquinoline-4-carbonyl moiety appears to occur in chemically analogous scaffolds as demonstrated through the aging of eight randomly selected HTS library compounds. Our observations reinforce the phenomenon that small molecule instability in DMSO continues to plague HTS-based drug discovery efforts. Moreover, the 2-furylquinoline-4-amide substructure should be considered a PAINS scaffold and only prioritized as a drug lead with extensive structural validation. Finally, taking the steps to deconvolute this impure screening hit translated to the discovery of a modestly potent small molecule inhibitor of A3G.
4. Experimental Section
General Synthesis Information
Chemical reagents were purchased from commercial sources and used without additional purification. The eight representative 2-furylquinolines (25-32) were purchased from Chembridge Corporation, and determined to be >95% by two-wavelength HPLC (254 and 215 nm). Bulk solvents were from Fisher Scientific and anhydrous N,N′-dimethylformamide (DMF) was purchased from Sigma-Aldrich. Anhydrous solvents were obtained from an MBraun Solvent Purification system. Reactions were performed under an atmosphere of dry N2 where noted. Silica gel chromatography was performed on a Teledyne-Isco Combiflash Rf-200 instrument using Redisep Rf Gold High Performance silica gel columns (Teledyne-Isco) or self-packed columns with SiliaFlash 60Å silica gel (SiliCycle). HPLC analyses were performed on an Agilent 1200 series instrument equipped with a diode array detector and a Zorbax SB-C18 column (4.6 × 150 mm, 3.5 μm, Agilent Technologies). Compounds used in biological testing were no less than 98% pure as determined by two-wavelength HPLC analysis (254 and 215 nm). Nuclear magnetic resonance (NMR) spectroscopy was performed using a Bruker Avance instrument operating at 400 MHz or 500 MHz (for 1H) and 100 MHz or 125 MHz (for 13C) at ambient temperature. Chemical shifts are reported in parts per million and normalized to internal solvent peaks or tetramethylsilane (0 ppm). High-resolution mass spectrometry (HRMS) was recorded in positive-ion mode on a Bruker BioTOF II instrument.
2-(5-Methylfuran-2-yl)-N-(5-(pyridin-4-yl)-1,3,4-thiadiazol-2-yl)quinoline-4-carboxamide (1)
To a 0 °C solution of 2-(5-Methylfuran-2-yl)quinoline-4-carboxylic acid (3)34 (101 mg, 0.40 mmol) and pyridine thiadiazole 435 (79 mg, 0.44 mmol) in DMF (4 mL) were added EDCI·HCl (86 mg, 0.45 mmol) and HOBt (82 mg, 0.61 mmol), followed by NMM (240 μL). The reaction solution was slowly warmed to room temperature and stirred for 18 h. The resulting solution was poured into sat. aq. NH4Cl (30 mL) and extracted with EtOAc (3 × 30 mL). The combined organic layers were washed with brine (2 × 30 mL), dried over anhydrous Na2SO4, filtered and concentrated. The crude product was purified via SiO2 column chromatography using a gradient of MeOH (0% to 50%) in CH2Cl2 to afford 36 mg (22%) of 1 as a yellow solid. IR (neat) 2968 cm-1, 1774 cm-1, 1506 cm-1; 1H NMR (DMSO-d6) δ = 13.82 (s, 1H), 8.77 (dd, J = 4.5 Hz, 1.6 Hz, 2H), 8.27 (s, 1H), 8.15 (d, J = 8.0 Hz, 1H), 8.09 (d, J = 8.0 Hz, 1H), 8.01 (dd, J = 4.5 Hz, 1.6 Hz, 2H), 7.83 (ddd, J = 8.4 Hz, 7.0 Hz, 1.3Hz, 1H), 7.64 (ddd, J = 8.2 Hz, 7.0 Hz, 1.1 Hz, 1H), 7.39 (d, J = 3.3 Hz, 1H), 6.40 (dd, J = 3.3 Hz, 0.9 Hz, 1H), 2.45 (s, 3H); 13C (DMSO-d6) δ = 165.4, 160.3, 159.8, 154.9, 150.9, 150.8, 147.8, 147.8, 139.0, 137.0, 130.6, 129.1, 127.2, 124.9, 122.4, 120.9, 116.9, 113.1, 109.2, 13.6; HRMS-ESI+ m/z [M + H]+ calc'd for C22H15N5O2S: 414.1025, found: 414.1033.
2-oxo-N-(5-pyridin-4-yl)-1,3,4-thiadiazol-2-yl)-1,2-dihydroquinoline-4-carboxamide (5)
2-oxo-1,2-dihydroquinoline-4-carboxylic acid (6) (50 mg, 0.27 mmol) and PyBOP (156 mg, 0.300 mmol) were placed in a dry flask and dissolved in DMF (1 mL). 2-Amino-5-(4-pyridyl)-1,3,4-thiadiazole (4, 54 mg, 0.3 mmol) was added followed by NMM (33 μL, 0.30 mmol). The reaction was stirred overnight at room temperature. A light precipitate formed over the course of the reaction. This precipitate was collected via vacuum filtration and dried under vacuum to afford 54 mg (59 %) of 5 as a light yellow powder. IR (neat) 3058 cm-1, 2970 cm-1, 1772 cm-1, 1594 cm-1, 1505 cm-1; 1H NMR (DMSO-d6): δ = 13.72 (s, 1H), 12.10 (s, 1H), 8.77 (d, J = 8.0 Hz, 2H), 7.99 (d, J = 8.0 Hz, 2H), 7.73 (d, J = 8.0 Hz, 1H), 7.59 (t, J = 8.0 Hz, 1H), 7.40 (d, J = 8.0 Hz, 1H), 7.23 (t, J = 8.0 Hz, 1H), 6.91 (s, 1H); 13C (DMSO-d6): δ = 165.2, 160.4, 159.7, 150.8, 149.2, 147.5, 142.0, 136.9, 131.6, 128.5, 128.5, 125.2, 122.9, 121.7, 120.9; HRMS-ESI+ m/z [M + H]+ calc'd for C17H11N5O2S: 350.0712, found: 350.0714.
4-((5-(pyridin-4-yl)-1,3,4-thiadiazol-2-yl)carbamoyl)quinoline-2-carboxylic acid (6) and (Z)-2-(4-oxopent-2-enoyl)-N-(5-(pyridin-4-yl)-1,3,4-thiadiazol-2-yl)quinoline-4-carboxamide (8)
This procedure was adapted from published literature.50 To a mixture of 2-(5-methylfuran-2-yl)-N-(5-(pyridin-4-yl)-1,3,4-thiadiazol-2-yl)quinoline-4-carboxamide (1, 250.0 mg, 0.605 mmol) in CCl4/MeCN (6 mL, 1:1, v/v) was added NaIO4 (582.0 mg, 2.721 mmol) in H2O (6 mL) and RuO2·H2O (6.0 mg, 0.048 mmol).The biphasic mixture was stirred vigorously at rt exposed to ambient air. After 16 h, the resulting precipitate was removed by filtration and washed with MeOH (5 × 20 mL) to yield a brown powder. Purification of 25 mg crude product was performed on an Agilent 1200 series instrument equipped with a diode array detector and Zorbax SB-C18 column (4.6 × 150 mm, 3.5 μm, Agilent Technologies). The purification method (1 mL/min flow rate) involved isocratic 10% MeCN in ddH2O (both containing 0.1% TFA; 0 to 2 mins) followed by linear gradients of 10% to 25% MeCN in ddH2O (both containing 0.1% TFA; 2 to 12 mins), 25-35% MeCN in ddH2O (both containing 0.1% TFA; 12 to 27 min), 35-90% MeCN in ddH2O (both containing 0.1% TFA; 27 to 35 min), and isocratic 90% MeCN in ddH2O (both containing 0.1% TFA; 35-40 mins). Wavelengths monitored = 215 nm and 254 nm. Compound 6 (5.0 mg, 29%) was isolated as a pale yellow powder, and compound 8 (9.1 mg, 36%) was isolated as a brown powder. It is important to note that compound 8 is not stable for storage in organic solvents for significant periods of time. (6) 1H NMR (DMSO-d6): δ = 13.90 (bs, 1H), 8.77 (d, J = 5.0 Hz, 2H), 8.47 (s, 1H), 8.30 (d, J = 9.5 Hz, 1H), 8.28 (d, J = 8.5 Hz, 1H), 8.01 (d, J = 5.0 Hz, 2H), 7.97 (t, J = 7.5 Hz, 1H), 7.86 (t, J = 7.5 Hz, 1H); HRMS-ESI+ m/z [M + H]+ calc'd for C17H11N5O2S: 378.0655, found: 378.0662; (8) 1H (DMSO-d6): δ = 14.00 (bs, 1H), 8.80 (bd, 2H), 8.54 (s, 1H), 8.47 (d, J = 16.5 Hz, 1H), 8.41 (d, J = 8.5 Hz, 1H), 8.36 (d, J = 9.0 Hz, 1H), 8.04 (m, 3H), 7.93 (t, J = 8.0 Hz, 1H), 7.19 (d, J = 16.5 Hz, 1H), 2.56 (s, 3H); HRMS-ESI+ m/z [M + H]+ calc'd for C17H11N5O2S: 430.0968, found: 430.0957.
Synthesis of Mechanistic Probes (Compounds 12-32)
For synthetic procedures and compound characterizations pertaining to the small molecules in Table 1, see the SI, Section XVIII.
Expression and Purification of APOBEC3A and APOBEC3G
A3A and A3G were expressed and purified as previously described.18
DNA Deaminase Assay
The DNA deaminase assay was performed as previously described with the ssDNA oligomer 5′-6-FAM-AAA-TAT-TCC-CTA-ATA-GAT-AAT-GTG-A-TAMRA-3′.18 A detailed description of the fluorescence-based deamination assay can be found in the SI, Section I. None of the synthesized compounds inhibited uracil DNA glycosylase in the context of the in vitro assay.
Aging of 1 Under Ambient Conditions
To a scintillation vial containing 1 (62.0 mg, 0.15 mmol) was added DMSO (15 mL). An aliquot (1 mL) was dispensed into a separate scintillation vial and left uncapped, exposed to laboratory air. A second aliquot (1 mL) was transferred to a flame dried scintillation vial under N2. Aliquots (100 μL at time points 0, 3, 7, 14, and 21 days were diluted with MeCN (0.9 mL) and analyzed on an Agilent 1200 series instrument equipped with a diode array detector and Zorbax SB-C18 column (4.6 × 150 mm, 3.5 μm, Agilent Technologies). The analysis method (1 mL/min flow rate) involved isocratic 10% MeCN in ddH2O (both containing 0.1% TFA; 0 to 2 mins) followed by linear gradients of 10% to 85% MeCN in ddH2O (both containing 0.1% TFA; 2 to 24 mins) followed by 85% to 95% MeCN in ddH2O (both containing 0.1% TFA; 24-26 mins). Wavelengths monitored = 215 nm and 254 nm. The experiment was performed in triplicate. Similar data was obtained for each replicate.
Isolation of 5, 6, 7a-b, and 8 from DMSO Stock Solutions of Aged 1 (10 mM)
Purification was performed on an Agilent 1200 series instrument equipped with a diode array detector and Zorbax SB-C18 column (4.6 × 150 mm, 3.5 μm, Agilent Technologies). The purification method (1 mL/min flow rate) involved isocratic 10% MeCN in ddH2O (both containing 0.1% TFA; 0 to 2 mins) followed by linear gradients of 10% to 25% MeCN in ddH2O (both containing 0.1% TFA; 2 to 12 mins), 25-35% MeCN in ddH2O (both containing 0.1% TFA; 12 to 27 min), 35-90% MeCN in ddH2O (both containing 0.1% TFA; 27 to 35 min), and isocratic 90% MeCN in ddH2O (both containing 0.1% TFA; 35-40 mins). Wavelengths monitored = 215 nm and 254 nm.
Aging of 1 under Photochemical Conditions
Rose Bengal (25.0 mg, 0.0242 mmol, 10 mol %) and 1 (100.0 mg, 0.242 mmol) were suspended in DMSO (1 mL) and O2 was bubbled into the mixture throughout the course of the reaction. The mixture was irradiated with 300W of visible light. The temperature of the reaction vessel was modulated by a tepid water bath and light was focused on the reaction by surrounding the light bulb and vessel with aluminum foil. After 9 h, an aliquot (4.2 μL) of the reaction mixture was dissolved in MeCN (0.995 mL) and analyzed on an Agilent 1200 series instrument equipped with a diode array detector and Zorbax SB-C18 column (4.6 × 150 mm, 3.5 μm, Agilent Technologies). The analysis method (1 mL/min flow rate) involved isocratic 10% MeCN in ddH2O (both containing 0.1% TFA; 0 to 2 mins) followed by linear gradients of 10% to 50% MeCN in ddH2O (both containing 0.1% TFA; 2 to 10 mins) and 50-95% MeCN in ddH2O (both containing 0.1% TFA; 10 to 12 min), and isocratic 95% MeCN in ddH2O (both containing 0.1% TFA; 12-15 mins). Wavelengths monitored = 215 nm and 254 nm. Once established that 1 autosensitizes its own photooxidation, rose bengal was omitted from future mechanistic studies. The identical reaction setup was employed to further probe the mechanism of decomposition and was accomplished by exchanging individual components. Specifically, bubbled oxygen was replaced in one experiment by N2 and in another by 18O2. In the final experiment, 10% (of the total volume) H218O was added to the DMSO. For the experiments employing N2, 18O2, and H218O, three cycles of freeze-pump-thaw were employed to remove any dissolved oxygen in the DMSO. Each of these aging experiments (O2, N2, 18O2 and H218O) was performed in triplicate. Similar data was obtained for each replicate.
Aging of Mechanistic Probes and Structurally Analogous 2-furylquinolines
Three 10 mM DMSO stock solutions were prepared for each compound and the three samples were aged over 6 weeks in 2 mL microcentrifuge tubes. The stocks were exposed to laboratory air by piercing the cap of the microcentrifuge tube with a small hole. Aliquots were taken from each sample at time points 0, 2, 21, and 38 days and were diluted with MeCN to obtain 1 mM solutions, which were analyzed by reverse phase analytical HPLC. Analysis was performed on an Agilent 1200 series instrument equipped with a diode array detector and Zorbax SB-C18 column (4.6 × 150 mm, 3.5 μm, Agilent Technologies). The analysis method (1 mL/min flow rate) involved isocratic 10% MeCN in ddH2O (both containing 0.1% TFA; 0 to 2 mins) followed by linear gradients of 10% to 50% MeCN in ddH2O (both containing 0.1% TFA; 2 to 10 mins) and 50-95% MeCN in ddH2O (both containing 0.1% TFA; 10 to 12 min), and isocratic 95% MeCN in ddH2O (both containing 0.1% TFA; 12-15 mins). Wavelengths monitored = 215 nm and 254 nm. To quantify the amount of parent compound remaining, the area under the curve (AUC) of the parent compound was divided by the AUC of an internal standard. Rose bengal (2.5 μM) was used as the internal standard and was added during the MeCN dilution steps immediately before HPLC analysis. Aging experiments were performed in triplicate and values shown are the mean ± standard deviation (calculated in Microsoft Excel). Calibration curves to normalize for injection variances during HPLC analysis were generated for each compound (See SI: Sections XVII - XXXIX). For all 21 compounds, R2 > 0.95 (non-linear regression).
Protocol for LC-MS Analysis of Aged compounds
LC-MS analyses were performed on an Agilent 1100 series instrument equipped with an Agilent MSD SL Ion Trap mass spectrometer (positive-ion mode) and a Zorbax SB-C18 column (0.5 × 150 mm, 5 μm, Agilent Technologies). The analysis method (15 μL/min flow rate) involved isocratic 10% MeCN (containing 0.1% TFA) in ddH2O (containing 0.1% HCO2H; 0 to 2 mins) followed by a linear gradient of 10% to 90% MeCN (containing 0.1% TFA) in ddH2O (containing 0.1% HCO2H; 2 to 24 mins), and isocratic 90% MeCN (containing 0.1% TFA) in ddH2O (containing 0.1% HCO2H; 24-26 mins). The column was heated to 40 °C. Wavelengths monitored = 214 nm and 254 nm.
Supplementary Material
Acknowledgments
This work was funded by the National Institutes of Health (P01-GM091743), the V Foundation for Cancer Research (V Translational Award), the University of Minnesota (Innovation Grant and Academic Health Center Faculty Research Development Grant), and the Prospect Creek Foundation. M.E.O. thanks the American Chemical Society Division of Medicinal Chemistry (ACS MEDI) for a Predoctoral Fellowship and the NIH for a Ruth L. Kirschstein NRSA Predoctoral Fellowship (F31-CA183246). The authors acknowledge Dr. Gunda Georg for helpful discussions, and Dr. Michael Walters for critical reading of this manuscript and assistance with HTS library annotation, and the Analytical Biochemistry Core Facility of the Masonic Cancer Center (University of Minnesota) for mass spectrometry resources. The Analytical Biochemistry Core is supported in part by the NIH (P30-CA77598).
Abbreviations
- APOBEC3
apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3
- C-to-U
cytosine-to-uracil
- EDCI·HCl
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride
- EtOH
ethanol
- HOBt
hydroxybenzotriazole
- 6-FAM
6-carboxyfluorescein
- KOH
potassium hydroxide
- NIH MLPCN
National Institutes of Health Molecular Library Probe Production Centers Network
- NMM
N-methylmorpholine
- PAINS
pan-assay interference scaffolds
- PyBOP
(benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate
- RB
rose bengal
- ssDNA
single-stranded DNA
- SD
standard deviation
- TAMRA
tetramethylrhodamine
- UDG
uracil DNA glycosylase
Footnotes
Supporting Information: Experimental procedures for the synthesis of mechanistic probes 12 – 32, 1H and 13C NMR, HPLC, and LC-MS characterization data; and figures supporting the text. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Bibette J. Gaining confidence in high-throughput screening. Proc Natl Acad Sci USA. 2012;109:649–650. doi: 10.1073/pnas.1119350109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Inglese J, Shamu CE, Guy RK. Reporting data from high-throughput screening of small-molecule libraries. Nat Chem Biol. 2007;3:438–441. doi: 10.1038/nchembio0807-438. [DOI] [PubMed] [Google Scholar]
- 3.Baell JB. Observations on screening-based research and some concerning trends in the literature. Future Med Chem. 2010;2:1529–1546. doi: 10.4155/fmc.10.237. [DOI] [PubMed] [Google Scholar]
- 4.Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 5.Dahlin JL, Nissink JW, Strasser JM, Francis S, Higgins L, Zhou H, Zhang Z, Walters MA. PAINS in the assay: chemical mechanisms of assay interference and promiscuous enzymatic inhibition observed during a sulfhydryl-scavenging HTS. J Med Chem. 2015;58:2091–2113. doi: 10.1021/jm5019093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hussain J, Harper G, Blaxill Z, Areri I, Saremi-Yarahmadi F, Pickett S, S P. International Conference on Chemical Structures. Noordwijkerhout; The Netherlands: 2008. pp. 58–59. [Google Scholar]
- 7.Baell J, Walters MA. Chemistry: Chemical con artists foil drug discovery. Nature. 2014;513:481–483. doi: 10.1038/513481a. [DOI] [PubMed] [Google Scholar]
- 8.Feng BY, Simeonov A, Jadhav A, Babaoglu K, Inglese J, Shoichet BK, Austin CP. A high-throughput screen for aggregation-based inhibition in a large compound library. J Med Chem. 2007;50:2385–2390. doi: 10.1021/jm061317y. [DOI] [PubMed] [Google Scholar]
- 9.Zitha-Bovens E, Maas P, Wife D, Tijhuis J, Hu QN, Kleinoder T, Gasteiger J. COMDECOM: predicting the lifetime of screening compounds in DMSO solution. J Biomol Screen. 2009;14:557–565. doi: 10.1177/1087057109336953. [DOI] [PubMed] [Google Scholar]
- 10.Chen A, Zhao X, Mercer L, Su C, Zalameda L, Liu Y, Lembke A, Eastwood H, Dang S, Oung T, Xia X, Young SW, Xiao S, McCarter JD. Assessment of the integrity of compounds stored in assay-ready plates using a kinase sentinel assay. Comb Chem High T Scr. 2013;16:644–651. doi: 10.2174/13862073113169990145. [DOI] [PubMed] [Google Scholar]
- 11.Hermann JC, Chen Y, Wartchow C, Menke J, Gao L, Gleason SK, Haynes NE, Scott N, Petersen A, Gabriel S, Vu B, George KM, Narayanan A, Li SH, Qian H, Beatini N, Niu L, Gan QF. Metal impurities cause false positives in high-throughput screening campaigns. ACS Med Chem Lett. 2012;4:197–200. doi: 10.1021/ml3003296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnston PA. Redox cycling compounds generate H2O2 in HTS buffers containing strong reducing reagents—real hits or promiscuous artifacts? Curr Opin Cell Biol. 2011;15:174–182. doi: 10.1016/j.cbpa.2010.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Soares KM, Blackmon N, Shun TY, Shinde SN, Takyi HK, Wipf P, Lazo JS, Johnston PA. Profiling the NIH Small Molecule Repository for compounds that generate H2O2 by redox cycling in reducing environments. Assay Drug Dev Technol. 2010;8:152–174. doi: 10.1089/adt.2009.0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rishton GM. Reactive compounds and in vitro false positive in HTS. Drug Discov Today. 1997;2:382–384. [Google Scholar]
- 15.Kozikowski BA, Burt TM, Tirey DA, Williams LE, Kuzmak BR, Stanton DT, Morand KL, Nelson SL. The effect of freeze/thaw cycles on the stability of compounds in DMSO. J Biomol Screen. 2003;8:210–215. doi: 10.1177/1087057103252618. [DOI] [PubMed] [Google Scholar]
- 16.Kozikowski BA, Burt TM, Tirey DA, Williams LE, Kuzmak BR, Stanton DT, Morand KL, Nelson SL. The effect of room-temperature storage on the stability of compounds in DMSO. J Biomol Screen. 2003;8:205–209. doi: 10.1177/1087057103252617. [DOI] [PubMed] [Google Scholar]
- 17.Blaxill Z, Holland-Crimmin S, Lifely R. Stability through the ages: the GSK experience. J Biomol Screen. 2009;14:547–556. doi: 10.1177/1087057109335327. [DOI] [PubMed] [Google Scholar]
- 18.Li M, Shandilya SM, Carpenter MA, Rathore A, Brown WL, Perkins AL, Harki DA, Solberg J, Hook DJ, Pandey KK, Parniak MA, Johnson JR, Krogan NJ, Somasundaran M, Ali A, Schiffer CA, Harris RS. First-in-class small molecule inhibitors of the single-strand DNA cytosine deaminase APOBEC3G. ACS Chem Biol. 2012;7:506–517. doi: 10.1021/cb200440y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stenglein MD, Burns MB, Li M, Lengyel J, Harris RS. APOBEC3 proteins mediate the clearance of foreign DNA from human cells. Nat Struct Mol Biol. 2010;17:222–229. doi: 10.1038/nsmb.1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Albin JS, Harris RS. Interactions of host APOBEC3 restriction factors with HIV-1 in vivo: implications for therapeutics. Expert Rev Mol Med. 2010;12:e4. doi: 10.1017/S1462399409001343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Refsland EW, Stenglein MD, Shindo K, Albin JS, Brown WL, Harris RS. Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: implications for HIV-1 restriction. Nucleic Acids Res. 2010;38:4274–4284. doi: 10.1093/nar/gkq174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harris RS. Enhancing immunity to HIV through APOBEC. Nat Biotechnol. 2008;26:1089–1090. doi: 10.1038/nbt1008-1089. [DOI] [PubMed] [Google Scholar]
- 23.Burns MB, Lackey L, Carpenter MA, Rathore A, Land AM, Leonard B, Refsland EW, Kotandeniya D, Tretyakova N, Nikas JB, Yee D, Temiz NA, Donohue DE, McDougle RM, Brown WL, Law EK, Harris RS. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature. 2013;494:366–370. doi: 10.1038/nature11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sieuwerts AM, Willis S, Burns MB, Look MP, Meijer-Van Gelder ME, Schlicker A, Heideman MR, Jacobs H, Wessels L, Leyland-Jones B, Gray KP, Foekens JA, Harris RS, Martens JW. Elevated APOBEC3B correlates with poor outcomes for estrogen-receptor-positive breast cancers. Horm Canc. 2014;5:405–413. doi: 10.1007/s12672-014-0196-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burns MB, Temiz NA, Harris RS. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat Genet. 2013;45:977–983. doi: 10.1038/ng.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roberts SA, Lawrence MS, Klimczak LJ, Grimm SA, Fargo D, Stojanov P, Kiezun A, Kryukov GV, Carter SL, Saksena G, Harris S, Shah RR, Resnick MA, Getz G, Gordenin DA. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat Genet. 2013;45:970–976. doi: 10.1038/ng.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Giardina GAM, Sarau HM, Farina C, Medhurst AD, Grugni M, Raveglia LF, Schmidt DB, Rigolio R, Luttmann M, Vecchietti V, Hay DWP. Discovery of a novel class of selective non-peptide antagonists for the human neurokinin-3 receptor. 1. Identification of the 4-quinolinecarboxamide framework. J Med Chem. 1997;40:1794–1807. doi: 10.1021/jm960818o. [DOI] [PubMed] [Google Scholar]
- 28.Strigacova J, Hudecova D, Varecka L, Lasikova A, Vegh D. Some biological properties of new quinoline-4-carboxylic acid and quinoline-4-carboxamide derivatives. Folia Microbiol. 2000;45:305–309. doi: 10.1007/BF02817551. [DOI] [PubMed] [Google Scholar]
- 29.Cai X, Zhang J, Chen M, Wu Y, Wang X, Chen J, Shen X, Qu D, Jiang H. The effect of the potential PhoQ histidine kinase inhibitors on Shigella flexneri virulence. PLoS One. 2011;6:e23100. doi: 10.1371/journal.pone.0023100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shankerrao S, B YD, Mety SS. Synthesis, antioxidant, and antibacterial studies of phenolic esters and amides of 2-(1-benzofuran-2-yl. quinoline-4-carboxylic acid. Med Chem Res. 2013;22:1163–1171. [Google Scholar]
- 31.Spinks D, Shanks EJ, Cleghorn LA, McElroy S, Jones D, James D, Fairlamb AH, Frearson JA, Wyatt PG, Gilbert IH. Investigation of trypanothione reductase as a drug target in Trypanosoma brucei. ChemMedChem. 2009;4:2060–2069. doi: 10.1002/cmdc.200900262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matsuno K, Masuda Y, Uehara Y, Sato H, Muroya A, Takahashi O, Yokotagawa T, Furuya T, Okawara T, Otsuka M, Ogo N, Ashizawa T, Oshita C, Tai S, Ishii H, Akiyama Y, Asai A. Identification of a new series of STAT3 inhibitors by virtual screening. ACS Med Chem Lett. 2010;1:371–375. doi: 10.1021/ml1000273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lack NA, Axerio-Cilies P, Tavassoli P, Han FQ, Chan KH, Feau C, LeBlanc E, Guns ET, Guy RK, Rennie PS, Cherkasov A. Targeting the binding function 3 (BF3) site of the human androgen receptor through virtual screening. J Med Chem. 2011;54:8563–8573. doi: 10.1021/jm201098n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ghodsi R, Zarghi A, Daraei B, Hedayati M. Design, synthesis and biological evaluation of new 2,3-diarylquinoline derivatives as selective cyclooxygenase-2 inhibitors. Bioorg Med Chem. 2010;18:1029–1033. doi: 10.1016/j.bmc.2009.12.060. [DOI] [PubMed] [Google Scholar]
- 35.Yu P, Hu J, Zhou TY, Wang P, Xu YH. Synthesis, insecticidal evaluation of novel 1,3,4-thiadiazole chrysanthemamide derivatives formed by an EDCI/HOBt condensation. J Chem Res. 2011;35:703–706. [Google Scholar]
- 36.Olson ME, Li M, Harris RS, Harki DA. Small-molecule APOBEC3G DNA cytosine deaminase inhibitors based on a 4-amino-1,2,4-triazole-3-thiol scaffold. ChemMedChem. 2013;8:112–117. doi: 10.1002/cmdc.201200411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mirek J, Sygula A. Semiempirical MNDO and UV Absorption Studies on Tautomerism of 2-Quinolones. Z Naturforsch Pt A. 1982;37A:1276–1283. [Google Scholar]
- 38.Galstyan G, Knapp EW. Computations of 36 tautomer/isomer equilibria of different lactams. J Phys Chem A. 2012;116:6885–6893. doi: 10.1021/jp302569g. [DOI] [PubMed] [Google Scholar]
- 39.Gerega A, Lapinski L, Nowak MJ, Furmanchuk A, Leszczynski J. Systematic effect of benzo-annelation on oxo-hydroxy tautomerism of heterocyclic compounds. Experimental matrix-isolation and theoretical study. J Phys Chem A. 2007;111:4934–4943. doi: 10.1021/jp070408j. [DOI] [PubMed] [Google Scholar]
- 40.Kaila N, Janz K, DeBernardo S, Bedard PW, Camphausen RT, Tam S, Tsao DH, Keith JC, Jr, Nickerson-Nutter C, Shilling A, Young-Sciame R, Wang Q. Synthesis and biological evaluation of quinoline salicylic acids as P-selectin antagonists. J Med Chem. 2007;50:21–39. doi: 10.1021/jm0602256. [DOI] [PubMed] [Google Scholar]
- 41.Foote CS, Wexler S, Ando W, Higgins R. Chemistry of singlet oxygen. IV. Oxygenations with hypochlorite-hydrogen peroxide. J Am Chem Soc. 1968;90:975–981. [Google Scholar]
- 42.Schenck GO. Colloquium am 26. Juni 1944. Angew Chem. 1944;57:101–102. [Google Scholar]
- 43.Criegee R. Mechanism of ozonolysis. Angew Chem. 1975;14:746–752. [Google Scholar]
- 44.Wasserman HH, Liberles A. Formation of epoxides in the pyrrole and furan series by photooxidation. J Am Chem Soc. 1960;82:2086–2086. [Google Scholar]
- 45.Bailey PS. Ozonation in Organic Chemistry. Vol. 1 Academic Press; New York: 1978. [Google Scholar]
- 46.Matsuura T, Saito I. Photochemistry of Heterocyclic Compounds. Wiley; New York: 1976. [Google Scholar]
- 47.Adam W, Rodriguez A. On the question of carbonyl oxide intermediates in the oxygen transfer by furan endoperoxides and bicyclic ozonides: intramolecular trapping experiments. Tetrahedron Lett. 1981;22:3509–3512. [Google Scholar]
- 48.Graziano ML, Lesce MR, Scarpati R. Photosensitized oxidation of furans. Part 1. Synthesis and properties of furan endo-peroxides. J Chem Soc, Perkin Trans. 1980;1:1955–1959. [Google Scholar]
- 49.Dufraisse C, Ecary S. Photooxidation on the cyclopentane ring: photooxydi-phenylisobenzofuran. C R Acad Sci Paris. 1946;233:735–737. [Google Scholar]
- 50.Palacios F, Alonso C, Amezua P, Rubiales G. Synthesis of Aza Polycyclic Compounds Derived from Pyrrolidine, Indolizidine, and Indole via Intramolecular Diels–Alder Cycloadditions of Neutral 2-Azadienes. J Org Chem. 2002;67:1941–1946. doi: 10.1021/jo016325v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.